

Abstract
Macrophages accumulate with glioblastoma multiforme (GBM) progression, and can be targeted via inhibition of colony stimulating factor-1 receptor (CSF-1R) to regress high-grade tumors in animal models of this cancer. However, whether and how resistance emerges in response to sustained CSF-1R blockade is unknown. We show that while overall survival is significantly prolonged, tumors recur in >50% of mice. Gliomas re-establish sensitivity to CSF-1R inhibition upon transplantation, indicating that resistance is tumor microenvironment-driven. Phosphatidylinositol 3-kinase (PI3K) pathway activity was elevated in recurrent GBM, driven by macrophage-derived insulin-like growth factor (IGF-1) and tumor cell IGF-1 receptor (IGF-1R). Combining IGF-1R or PI3K blockade with CSF-1R inhibition in recurrent tumors significantly prolonged overall survival. Our findings thus reveal a potential therapeutic approach for treating resistance to CSF-1R inhibitors.
Therapies targeted against the tumor microenvironment (TME) represent a promising approach for treating cancer. This appeal arises in part from the decreased likelihood of acquired resistance through mutations in target TME cells, as is frequently observed with cancer cell-targeted therapies. As multiple TME-directed therapies are currently advancing through different clinical trials (1, 2), this necessitates an understanding of potential mechanisms of intrinsic or acquired resistance. We have focused on addressing this issue here by investigating whether resistance to a macrophage-targeted therapy emerges during the course of long-term trials in various preclinical models of high-grade glioma (glioblastoma multiforme; GBM).
GBM is the most common and aggressive adult primary brain tumor, and survival is only minimally prolonged by current standard of care treatment, including surgery, radiation and temozolomide chemotherapy (3). Accordingly, targeting the glioma TME is emerging as a promising alternative therapeutic strategy. In GBM, tumor-associated macrophages and microglia (TAMs) comprise up to 30% of the bulk tumor mass (4). In many cancers, including glioma, elevated TAM numbers are associated with high grade and poor patient prognosis (4–7). As such, targeting TAMs in GBM represents an attractive therapeutic approach.
Macrophages critically depend on colony stimulating factor-1 (CSF-1) for multiple functions; consequently, strategies to target TAMs often include CSF-1 receptor (CSF-1R) blockade (8–10). In clinical trials, several approaches to inhibit CSF-1R are currently being employed including antibodies and small molecules (7, 11, 12). However, the long-term effects of these agents on clinical outcome are still under evaluation, and thus gaining insight into potential mechanisms of drug resistance and/or inefficacy is now critical.
Here, we use a potent and highly selective small-molecule CSF-1R inhibitor, BLZ945. We have shown that BLZ945 blocks early gliomagenesis, while short-term treatment of advanced, high-grade glioma causes robust tumor debulking after just 7 days (8). Interestingly, CSF-1R inhibition has no direct effect on glioma cell viability, as these cells do not express CSF-1R in the models we have used. Instead, glioma TAMs remain abundant and become anti-tumorigenic in response to treatment, by downregulating markers of M2-like macrophage polarization/alternative activation and adopting a pronounced phagocytic phenotype (8). We now address herein the unanswered question of whether long-term CSF-1R inhibition in aggressive late-stage GBM has a sustainable anti-tumorigenic effect, or instead leads to acquired resistance.
A subset of GBMs develop resistance to CSF-1R inhibition in long-term preclinical trials
We first analyzed the kinetics of GBM response to continuous long-term BLZ945 treatment using a transgenic platelet-derived growth factor-driven glioma (PDG) model (RCAS-hPDGF-B/Nestin-Tv-a;Ink4a/Arf−/−) (8, 13) (Fig. 1A). Two weeks into the trial we observed maximal tumor regression with an average volume reduction of 62% (Fig. 1, B and C). At this time point, 8% of animals showed no evidence of residual tumor by MRI. By contrast, vehicle-treated tumors exhibited a 2522% increase in volume over the same period (Fig. 1C).
Figure 1. 56% of GBMs develop resistance to CSF-1R inhibition in long-term preclinical trials.
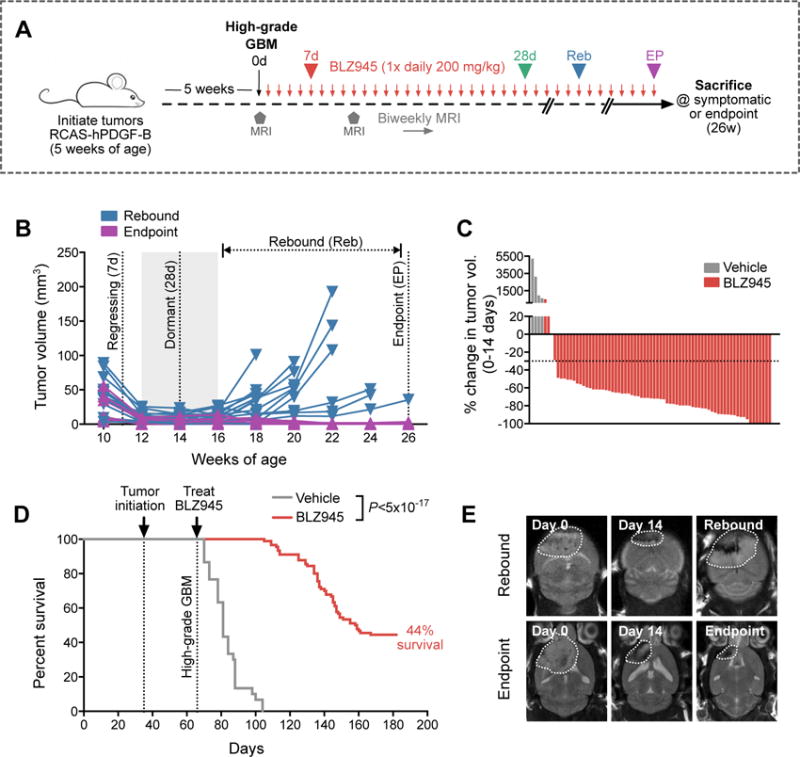
(A) Long-term trial design for testing BLZ945 efficacy in a PDGF-B-driven glioma (PDG) model. High-grade PDG tumors were treated with BLZ945 (200 mg/kg/d) or vehicle (20% captisol) and monitored by biweekly MRI for up to 26 weeks (defined endpoint; see methods) or until symptomatic. (B) Tumor volume curves from biweekly MRIs of long-term BLZ945 trials (n=90 animals treated, 23 representative curves are shown). Four key phases are indicated, including 7d (regressing tumor), 28d (dormant tumor), rebound (Reb; recurrent tumor, evaluated on a mouse-to-mouse basis by MRI), and endpoint (EP; stable regression at the 26-week endpoint). (C) Waterfall plots showing percent change in tumor volume between 0–14d in a representative subset of animals from Fig. 1D (BLZ945 n=71; vehicle n=4). (D) Kaplan-Meier of BLZ945-treated (n=90) versus vehicle-treated (n=30) mice bearing high-grade PDG tumors (Log-rank Mantel-Cox test, P<5×10−17). Median survival for vehicle-treated animals was 15d post-treatment initiation, while median survival for BLZ945-treated animals was 93d. (E) Representative MRI images over time of one mouse with a rebounded tumor (top row), and another mouse that had stable disease until EP (bottom row).
Following this regression phase, all BLZ945-treated tumors entered a dormancy phase, lasting for ≥4 weeks (Fig. 1B). 44% of treated animals remained symptom-free and survived to the trial endpoint of 26 weeks (Fig. 1D; P<5×10−17), with minimal or, in some cases, no evidence of residual tumor by MRI and histology (Fig. 1E). This is in stark contrast to vehicle-treated GBMs, which were purposely selected to be smaller in size upon treatment initiation (fig. S1A), yet median survival was only 15d post-treatment initiation (vs. 93d for BLZ945), and no animals survived beyond 6 weeks (Fig. 1D). Following the dormancy phase observed in all BLZ945-treated animals, however, 56% eventually developed resistance and tumors rebounded, despite effective, continued inhibition of CSF-1R phosphorylation in TAMs (Fig. 1, B and D, and fig. S1B).
We next focused on understanding how resistance to the CSF-1R inhibitor emerged, and chose several time points throughout the long-term trial for comparison, including Veh (vehicle; 20% Captisol until symptomatic), 7d (BLZ945-responsive, regressing), 28d (BLZ945-responsive, dormant), Reb (BLZ945-resistant, actively rebounding), and EP (26-week endpoint, stably regressed) (Fig. 1B). Histological analysis showed that after 7d of BLZ945, tumor grade was significantly reduced. At 28d and EP, histological grade remained low, with 33% and 50% of mice respectively showing no evidence of tumor by histology, and the remainder of animals exhibiting either residual disease or grade II tumors (fig. S1, C and D). By comparison, the majority of rebound tumors were grade III or IV, and similar in size to Veh tumors at sacrifice (fig. S1, C to E). Both Veh and Reb tumors also exhibited a high proliferation: apoptosis index (Ki67: CC3) indicating a state of rapid growth (fig. S1F).
Glioma cells resistant to CSF-1R inhibition in vivo exhibit elevated PI3K signaling
To determine the mechanism by which tumor cells acquire resistance, we first performed array comparative genomic hybridization (aCGH) analyses and found no copy number alterations in primary rebound glioma tumorsphere lines (passage 1; fig S2). To then assess which signaling pathways are altered specifically in recurrent tumors, we first FACS-purified glioma cells (PDGFRα+) from Veh, EP and Reb lesions, and performed RNA-sequencing. Glioma cells were isolated from EP lesions that were stably regressed, but still detectable by MRI. Gene ontology analysis demonstrated that Veh and Reb tumor cells showed an enrichment of cell cycle-related genes, compared to EP tumor cells (fig. S3A), corroborating the observed changes in Ki67 levels (fig. S1F), and supporting the notion that EP tumors were in a state of cell cycle dormancy. To interrogate which pathways were differentially regulated between the three groups, we used gene set variation analysis (14) for each pair-wise comparison. Nine gene sets in total were significantly enriched in Reb tumor cells compared to EP (fig. S3B), including a PI3K gene set (Fig. 2A), potentially explaining the robust differences in proliferation given the importance of PI3K signaling in cell cycle regulation. In accordance with this result, we found elevated phosphorylated (p)-AKT (a PI3K substrate) in Reb tissues compared to Veh and EP, using immunofluorescence staining and western blotting (Fig. 2B, and fig. S3, C and D).
Figure 2. Combined CSF-1R and PI3K inhibition improves survival in the PDG model.
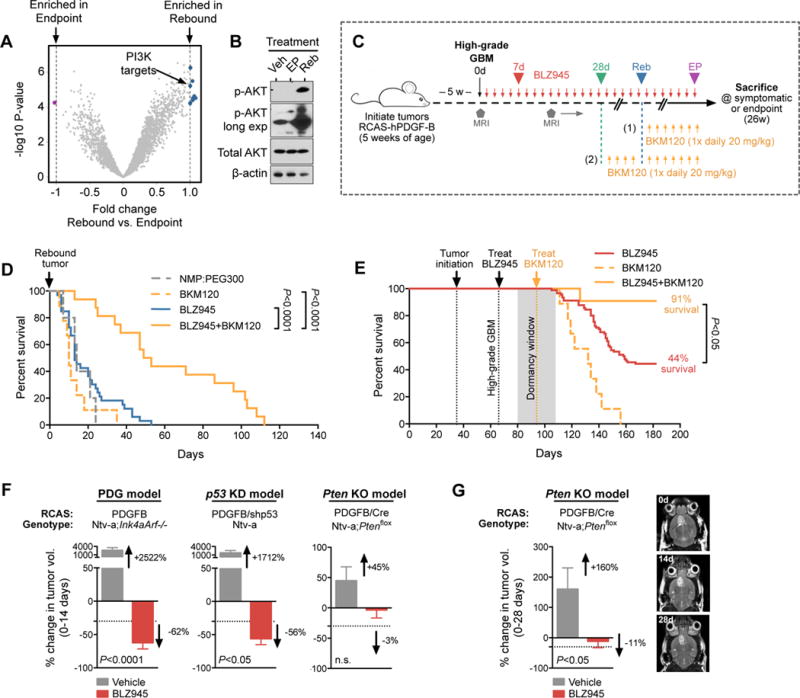
(A) Gene set variation analysis based on RNA-seq from FACS-purified EP and Reb tumor cells (PDGFRα+; see fig. S3B). Blue circles indicate gene sets significantly enriched in Reb tumor cells, while purple circles identify those enriched in EP tumor cells. The PI3K gene set is indicated with an arrow. Vertical lines indicate fold cutoff for significance (n=5–6 samples per group). (B) Immunoblot from snap-frozen Veh, EP and Reb tumors demonstrating elevated phospho (p)-AKT in Reb tumors compared to Veh and EP (n=3 experiments, one representative blot is shown). (C) Long-term trial design for evaluating BLZ945 and BKM120 combination therapy on PDG tumors. High-grade tumors were treated with BLZ945 until recurrent tumors developed (trial design 1) or until dormancy (28d, trial design 2), whereupon BKM120 was either added (with continuous BLZ945 treatment) or switched (discontinued BLZ945). (D) Survival of animals with recurrent tumors treated either with BLZ945 alone (n=33), BKM120 alone (n=9), or BLZ945 in combination with BKM120 (n=16; trial design 1 in Fig. 2C). Combination of BLZ945+BKM120 led to an increase in overall survival (Log-rank Mantel-Cox test, P<0.0001), and in median survival (51d) following recurrence compared to BLZ945 (13d) or BKM120 (10d) monotherapy. (E) Survival of animals with 28d dormant tumors treated either with BLZ945 alone (n=90; same cohort as presented in Fig. 1D), BKM120 alone (n=9), or BLZ945 in combination with BKM120 (n=11; trial design 2 in Fig. 2C). Combination of BLZ945+BKM120 led to increased overall survival compared to either monotherapy. Log-rank Mantel-Cox test was used to calculate significance. (F) Average percent change in tumor volume (0–14d) between vehicle- or BLZ945-treated tumors, in 3 different RCAS-PDGFB-HA Nestin-Tv-a GBM models (termed PDG, p53 knockdown (KD), and Pten knockout (KO) here; see methods). BLZ945 efficacy in the p53 KD model (BLZ945 n=8 mice; vehicle n=6 mice) was comparable to the PDG model (BLZ945 n=71 mice; vehicle n=4 mice) after 2 weeks (56% and 62% volume reduction, respectively); however, BLZ945 efficacy was less pronounced in the Pten KO model (n=5 mice per treatment group; 3% volume reduction). (G) Average percent change in tumor volume (0–28d) between vehicle- or BLZ945-treated Pten KO tumors (n=5 mice per group). BLZ945 caused 11% volume reduction after 4 weeks. Data were analyzed by Student’s t-test unless indicated otherwise.
To investigate whether PI3K signaling is functionally important in driving recurrence, we performed a preclinical intervention trial. We treated PDG mice bearing high-grade gliomas with BLZ945 until they showed tumor rebound by MRI, at which point we intervened with BKM120 treatment (Fig. 2C, trial design 1, and fig. S3E) at an appropriate dose to avoid reported off-target effects (15). BKM120 was chosen because it is a brain-penetrant pan-Class 1 PI3K inhibitor that is currently being clinically evaluated in GBM patients with recurrent disease following standard therapy. Animals with rebound tumors treated with continued BLZ945 monotherapy led to a median survival of 13d post-recurrence, whereas rebound tumors treated with BLZ945+BKM120 extended median survival to 51d (Fig. 2D) and blocked tumor progression after 2 weeks of treatment (fig. S3F). By contrast, BKM120 monotherapy in rebound tumors (i.e. discontinued BLZ945) led to a median survival of 10d, which was indistinguishable from the vehicle control (Fig. 2D). Moreover, BKM120 was only modestly effective in treatment-naïve tumors (fig. S3, G and H). Collectively, these results indicate that continued CSF-1R inhibition is necessary to expose PI3K signaling dependency in rebound tumors and, consequently, a heightened sensitivity to pathway inhibition.
To determine if recurrence could be prevented by earlier PI3K inhibition, during the initial dormancy phase, we treated GBM-bearing PDG mice with BLZ945 alone for 28d, at which point we added BKM120 until the trial endpoint (Fig. 2C, trial design 2). With this early intervention, the percentage of animals that survived to endpoint increased substantially (91% BLZ945+BKM120) compared to single-agent treatments (44% BLZ945 alone and 0% BKM120 alone; Fig. 2E). Taken together, our results demonstrate that PI3K signaling is engaged during the acquisition of resistance to CSF-1R inhibition in the context of continued BLZ945 treatment.
Our finding that PI3K activation underlies resistance to CSF-1R inhibition was intriguing in light of the high frequency of mutations in the PI3K pathway in glioma patients (16). Therefore we also investigated whether genetic mutations in PI3K/PTEN would similarly confer a resistance-like phenotype in mouse models, which could potentially be informative in the clinical setting. To address this question, we compared BLZ945 efficacy in two additional RCAS-hPDGF-B/Nestin-Tv-a GBM models harboring distinct clinically-relevant oncogenic alterations besides Ink4a/Arf loss, including Pten deletion (Pten KO model) or p53 knockdown (p53 KD model; see methods). After 2 weeks of treatment, we found that BLZ945 efficacy in the p53 KD model (56% reduction of tumor volume) was comparable to that of the PDG model (62% reduction). However, CSF-1R inhibition was less potent in the Pten KO model over the same time period (3% reduction; Fig. 2F). Furthermore, although we did eventually observe a significant reduction in tumor volume in the Pten KO model after a prolonged treatment period of 4 weeks (11% reduction), this did not meet the response evaluation criteria in solid tumors (RECIST) standard for a partial response (Fig. 2G) (17). These results suggest that treatment efficacy of CSF-1R inhibitors may be blunted in patients with existing genetic alterations in the PTEN/PI3K pathway.
Resistance to CSF-1R inhibition is mediated by the microenvironment
We next investigated how PI3K was activated in rebound tumors, and first explored whether resistance to BLZ945 was tumor cell-intrinsic or -extrinsic. We previously established that BLZ945 does not directly affect glioma cell lines in culture (8), and demonstrate here that CSF-1R inhibition also has no direct effect on viability of a panel of primary cell lines derived from rebound tumors (fig. S4A). We designed an intracranial tumor transplantation model using early-passage Reb cells to address the following hypotheses: (i) resistance is tumor cell-intrinsic, therefore transplanted tumors will not respond to BLZ945, or (ii) resistance is mediated by the treatment-altered microenvironment, therefore transplanted tumors will re-establish sensitivity to CSF-1R inhibition in naïve animals. Interestingly, transplanted rebound tumors responded to BLZ945 treatment in the naïve setting (Fig. 3A, and fig. S4, B to F), indicating that resistance is likely mediated by the TME.
Figure 3. Resistance to CSF-1R inhibition is mediated by the microenvironment and rebound TAMs are alternatively activated.
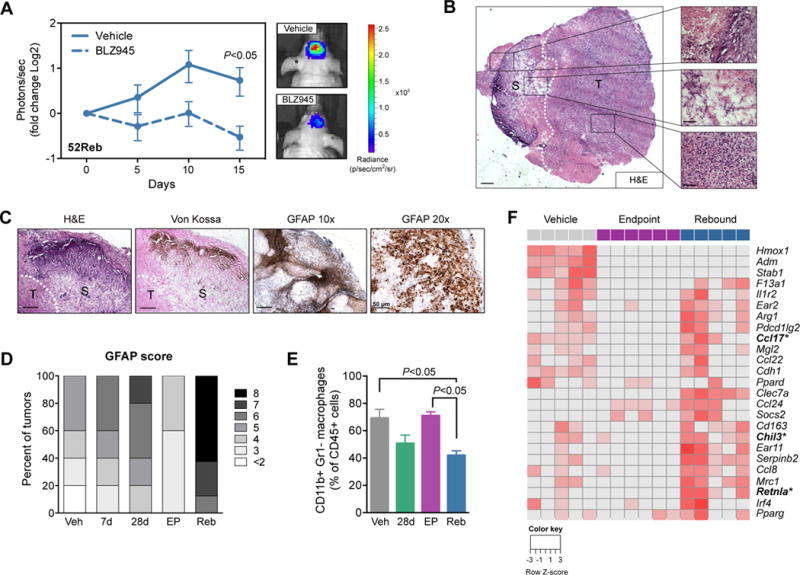
(A) Quantification of bioluminescent imaging (BLI) from intracranially transplanted 52Reb cells into naïve mice. Results show that 52Reb cells, isolated from a recurrent PDG tumor that developed resistance to BLZ945 treatment in vivo, reestablish sensitivity to BLZ945 in the naïve setting (Student’s t-test d15, P<0.05, n=10 mice). Representative BLI images at d15 are shown. (B) Left: H&E of a rebound tumor (T) adjacent to glial scarring (S). Scale bar = 500 μm. Right: Representative regions of calcification (top), reactive astrocytic barrier (middle), and recurrent tumor (bottom). Scale bars = 50 μm. (C) H&E, Von Kossa and GFAP staining on rebound tissue. Scale bars = 200 μm, except GFAP 20× = 50 μm as indicated. (D) Allred score for the astrocyte marker GFAP, showing a higher intensity and proportion of GFAP+ staining in Reb tissues (n=8 mice) compared to other treatment groups (n=5 mice per group). (E) Flow cytometry of TAMs (CD45+CD11b+Gr1−) in Veh, 28d, EP and Reb tumors (n=5–7). Data were analyzed by a one-way ANOVA and Tukey’s multiple comparisons test. (F) Heatmap showing RNA-seq expression changes of M2-like associated genes in Veh, EP and Reb TAMs (n=5–6 per group). Wound-associated genes are indicated in bold with asterisks.
We analyzed the TME in recurrent disease to determine how resistance to CSF-1R inhibition develops. We found that rebounding tumors always emerged adjacent to regions of glial scarring, characterized by reactive astrocytes, calcium deposition, and relatively low vascularity associated with elevated hypoxia (Fig. 3, B to D, and fig. S4, G to I). By contrast, scarring was infrequently observed in the 28d and EP tumors (fig. S4I). The scar tissue architecture was reminiscent of gliosis in response to neurodegeneration or physical injury (18). Given the parallels between a wound-associated microenvironment and tumorigenesis in epithelial tissues (19), we hypothesized that this brain injury response may likewise be contributing to a microenvironment that is potentially triggering recurrent disease.
Rebound TAMs adopt a wound-associated signature that is driven by enhanced interleukin-4 (IL-4) signaling
During gliosis, activated macrophages play a central role in providing growth factors and signaling molecules to nearby astrocytes and neurons, to form a reactive barrier that limits the extent of tissue damage in the brain (18, 20). Given that BLZ945 is a macrophage-targeted drug, we analyzed TAM numbers and phenotype in rebound tumors. We have previously shown that TAMs are not depleted in the glioma TME in a 1-week trial with BLZ945, but rather downregulate expression of M2-like genes and increase phagocytosis of tumor cells (8, 21). Consistently, we show here that TAMs are still present in 7d, 28d, EP or Reb tumors (Fig. 3E and fig. S5A). Interestingly, when we used flow cytometry to discriminate between CD45loCD11b+ cells (putative microglia) versus CD45hiCD11b+ cells (putative bone marrow-derived macrophages, BMDMs) (22–24) in Veh, EP and Reb tumors (fig. S5B; see methods for further discussion), we found that long-term BLZ945 treatment enriched for CD45loCD11b+ TAMs (fig. S5, C and D). This is potentially either a consequence of phenotypic mimicry between the macrophage populations, or the result of one macrophage population responding differently to CSF-1R inhibition than the other. Co-staining of CD68 or CD206 macrophage markers in combination with Ki67 demonstrated that a subset of remaining TAMs in rebound tumors were proliferating (fig. S5, E to G). Although these results can only be formally confirmed by lineage tracing experiments, they at least suggest that rebound TAMs (enriched for CD45loCD11b+ cells) may undergo a low level of replication, presumably as a means to compensate for the duress caused by prolonged CSF-1R blockade.
To assess potential differences in activation states, we FACS-purified TAMs from Veh, EP and Reb tumors, and performed RNA-seq. Principal component analysis confirmed distinct global gene expression profiles for Veh, EP and Reb TAMs (fig. S6A), and differential expression analysis revealed large numbers of differentially expressed genes between the three groups (Table S1). We first focused on a subset of M2-like genes previously identified as altered by CSF-1R inhibition (8, 25). We found that compared to Veh TAMs, alternative activation was suppressed in EP TAMs, while a subset of these genes were highly expressed in Reb TAMs (Fig. 3F). In accordance with previous findings (8), no inverse relationship was observed for M1-like markers such as tumor necrosis factor α (TNFα[Image] across the different treatment groups (25). Together these findings support our hypothesis that the Reb TME is pro-tumorigenic.
Given the similarities in alternative activation between Veh and Reb TAMs according to the M1/M2-like paradigm (26), yet clear differences in drug response between treatment-naïve and rebound tumors, we next employed a more fine-tuned approach to defining macrophage phenotype. We computationally interrogated a spectrum model of macrophage activation, defined by gene sets that are altered in response to different stimuli, including interferon γ (IFNγ), IL4, TNFα, transforming growth factor β1 (TGFβ1), IL1β, and two toll-like receptor (TLR) agonists specific for TLR2 (macrophage-activating lipopeptide 2; MALP2) and TLR9 (unmethylated CpG-containing oligonucleotide; CPG) (27, 28). We determined that IL4- and TGFβ1-targeted gene sets were significantly enriched in Reb TAMs compared to Veh TAMs (Fig. 4A), and also found a significant enrichment of these same gene sets in Reb TAMs versus EP TAMs (Fig. 4B). Given that IL4 is a known mediator of alternative activation associated with a wound-healing phenotype in macrophages (20, 29), and the role of TGFβ1 during wound-healing and tissue turnover in multiple contexts (30, 31), these results were consistent with our observation of glial scarring in association with rebound tumors. Indeed, a number of M2-like genes expressed by macrophages involved in wound repair and resolving inflammation (Retnla, Chil3, Ccl17) were enriched in Reb TAMs (Fig. 3F). By contrast, RNA-seq analyses of an independent set of Reb and 28d TAM samples revealed no significant differences in expression across this same gene set (fig. S6B), suggesting that a subset of 28d tumors may be rebound precursors, and that induction of M2-like gene expression precedes recurrence. When we investigated the ability of IL4 or TGFβ1 to regulate expression of these wound-associated genes in BMDMs in vitro, we found that IL4 was a potent inducer, whereas TGFβ1 was not (Fig. 4C, and fig. S6, C and D), indicating the specificity of IL4 for regulation of this particular gene set. Corroborating these findings, we confirmed increased expression of Il4 in rebound tumors by qRT-PCR (Fig. 4D), and elevated canonical downstream signaling by immunofluorescent staining for phosphorylated Stat6 (p-Stat6) in PDG tissue samples from the long-term trials (fig. S6, E and F). It should be noted that these results do not exclude the possibility that TGFβ may be playing a role in rebound tumors that is distinct from, or indirectly connected to, alternative activation of TAMs.
Figure 4. IL4 activates wound-associated genes in rebound TAMs and is produced by T cells in the rebound tumor microenvironment.
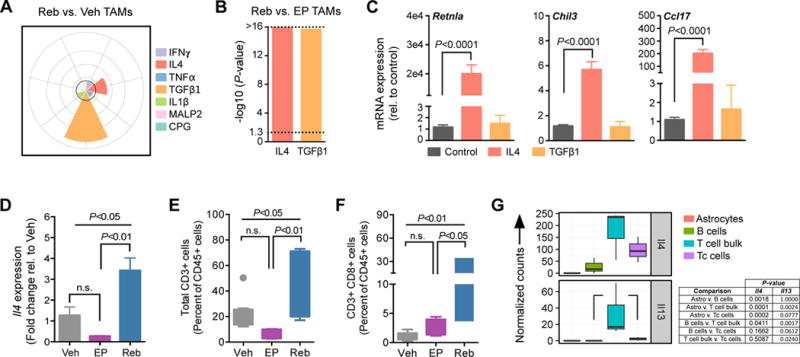
(A) Spectrum model of macrophage activation using gene set enrichment analysis (GSEA) of transcriptional programs regulated by the indicated cytokines (see methods). Reb TAMs exhibit activation of programs driven by TGFβ1 and IL4 compared to Veh TAMs (n=5 per group). The -log10 (P-value) is plotted for each gene set (0, 4, 8, 12, and >16 radially outward in grey concentric circles). The black line indicates a -log10 (P-value) cutoff of 3. (B) GSEA as in (A), confirming that Reb TAMs exhibit activation of programs driven by IL4 and TGFβ1 compared to EP TAMs (n=5–6 per group). Dotted line demarcates P<0.05. (C) qRT-PCR analysis demonstrates that IL4 induces expression of the wound-associated genes Retnla, Chil3 and Ccl17 in bone marrow-derived macrophages (BMDMs) in culture, while recombinant TGFβ1 treatment does not. Bone marrow isolate was obtained from 5 or more independent WT mice for replicate experiments. (D) qRT-PCR analysis of Il4 expression in snap frozen whole-tumor samples from Veh, EP or Reb treatment groups, demonstrating elevated expression in the Reb setting (n=4 tumors) compared to both Veh (n=4 tumors) and EP (n=4 tumors). (E) Flow cytometry of total CD3+ T cells (n=5–10) and (F) CD3+ CD8+ cytotoxic T cells (n=5–10) in Veh, EP and Reb tumors. (G) RNA-seq data from a panel of cell types (GFAP+ astrocytes, CD19+ B cells, CD3+ CD8+ cytotoxic T cells (Tc), and remaining CD3+ CD8- bulk T cells) isolated from rebound tumors by FACS (n=3,). Results show elevated Il4 expression in bulk T and Tc cells, and Il13 expression in bulk T cells. Significance values for C-F were calculated by one-way ANOVA and Tukey’s multiple comparisons test.
To determine the cellular source of IL4 in our model, we used multicolor flow cytometry to immunoprofile Veh, EP and Reb tumors using a panel of myeloid (CD11b, Gr1, Ly6G, Ly6C, CD11c, Tie2, MHCII) and lymphoid (CD19, CD3, CD4, CD8, FoxP3) cell markers. While we found few cell types that were uniquely enriched in rebound tumors (fig. S7, A to G), there was a significant increase in the proportion of CD3+ T cells in rebound tumors, driven by the CD8+ fraction (Fig. 4, E and F). FACS-purification of these cells from rebound tumors along with other putative contributors of IL4 (including astrocytes (Fig. 3D), B cells (32–34), and bulk T cells), revealed that Il4 expression was enriched in both bulk T and CD8+ T cell fractions (Fig. 4G and fig. S7H). By comparison, expression of Il13, a closely related cytokine that shares the IL4Rα subunit in its heterotypic receptor to initiate canonical Stat6 signaling, was enriched in the bulk T cell fraction (Fig. 4G). Expression of IL4 was assessed in a panel of human cell types, and detected in CD8+ T cells as expected, in addition to monocytes, eosinophils, astrocytes, and brain microvascular cells (fig. S7I). While additional analyses are needed to establish the precise cellular source of these cytokines in brain malignancies in patients, our data is consistent with IL4 being produced by multiple cell types.
The IGF-1/IGF-1R signaling axis is induced in rebound gliomas
We next investigated how IL4 and wound-associated gene expression might be connected to PI3K signaling in rebound tumors. Differential gene expression analysis revealed that TAM-derived Igf1 was one of the most significantly upregulated genes in Reb TAMs compared to EP TAMs, which we confirmed in comparisons with Veh TAMs or 28d TAMs (Fig. 5, A and B, fig. S8A, and Table S1). This was particularly interesting as Igf1 is an IL4 target gene in macrophages (fig. S8B) (35–37), it is a known mediator of tissue repair and neuroprotection (38–41), and importantly, one of its canonical downstream signaling pathways is PI3K/AKT (42). Congruent Igf1r upregulation was identified in glioma cells purified from rebound tumors (Fig. 5C, and Table S1), elevated p-IGF-1R was found in rebound tumors by immunostaining and western blotting (Fig. 5D, and fig. S8, C and D), and Igf1 upregulation was found in snap-frozen rebound tissue samples by qRT-PCR (fig. S8E). Additionally, levels of Igf1 expression were substantially higher in Reb TAMs than tumor cells, while Igf1r expression was enriched in tumor cells compared to TAMs (fig. S8F). Together these data demonstrate elevated IGF-1 signaling in recurrent disease.
Figure 5. The IGF-1/IGF-1R axis is induced in rebound gliomas.
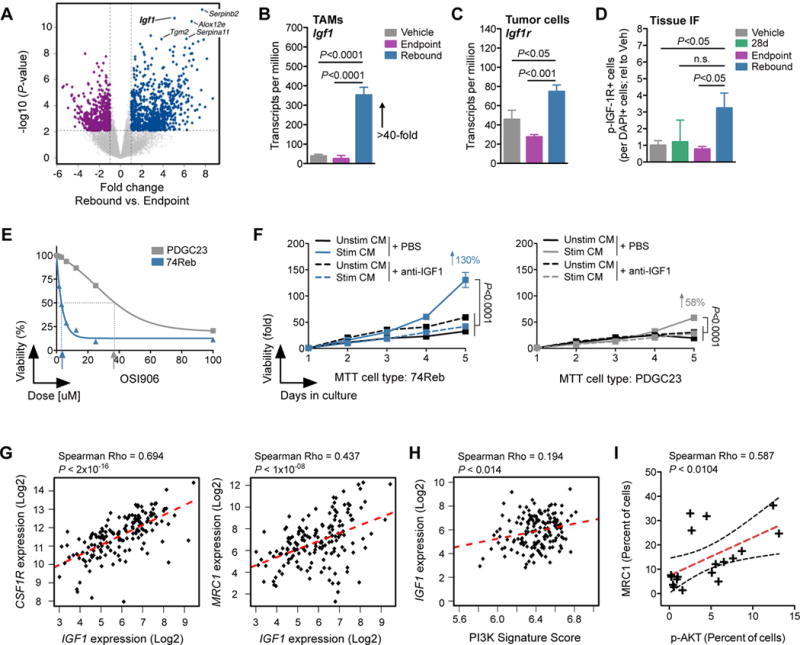
(A) Volcano plot showing the fold change (log2(fold)) between Reb (n=5) and EP (n=6) TAMs on the x-axis and the significance (−log10(P-value)) on the y-axis. Blue dots indicate genes upregulated in Reb TAMs, while purple dots indicate genes downregulated in Reb TAMs. Igf1 is labeled in the upper right quadrant. (B) RNA-seq barplot depicting Igf1 transcripts per million (TPM) in Veh, EP and Reb TAMs (n=5–6 per group; one-way ANOVA and Tukey’s multiple comparisons). (C) RNA-seq barplot depicting Igf1r TPM in Veh, EP and Reb tumor cells (n=5–6 per group; one-way ANOVA and Tukey’s multiple comparisons). (D) Quantification of immunofluorescent staining of phospho (p)-IGF-1R in Veh, 28d, EP and Reb tumor tissues. Results show pIGF-1R is elevated in rebound tumors compared to all other groups (n=5–8 per group; one-way ANOVA and Dunnett’s multiple comparisons to Reb). (E) MTT assay demonstrating higher sensitivity of an early-passage primary rebound PDG cell line (74Reb, blue) to IGF-1R inhibition with OSI906 compared to a primary treatment-naïve PDG cell line (PDGC23, grey) (n=3, one representative experiment shown). IC50 values are indicated with arrows. (F) MTT proliferation assays of 74Reb cells (left) compared to PDGC23 cells (right), treated with conditioned media (CM) from BMDMs that were stimulated with rebound glioma microenvironment culture CM (see fig. S8, I and J for details). Stimulated BMDM CM (Stim CM) induced growth of 74Reb cells more than PDGC23 cells (130% versus 58%, respectively), and this effect was blocked with an anti-IGF-1 neutralizing antibody (n=4 replicate experiments). A one-way ANOVA and Dunnett’s multiple comparisons to Stim CM + PBS was used to calculate differences at 5d (P<0.0001 for all). (G) Correlation between IGF1 and CSF1R or MRC1 expression from TCGA-GBM data. (H) Single sample GSEA for a hallmark PI3K signature was used to assign a pathway activity score (see methods) across patients from the TCGA-GBM dataset. PI3K signature scores were then correlated with IGF1 expression levels as shown. (I) Linear regression analysis of immunohistochemical staining for phospho (p)-AKT and MRC1 in serial sections from GBM patient tissue (n=18 patients). A significant correlation between MRC1 protein levels and AKT signaling was observed. For correlational analyses (G-I), a Spearman coefficient was used to assess significance, and a line of best fit is shown (red hatched). The 95% confidence band (black hatched) is also shown in (I).
To assay the effects of IGF-1 specific to the rebound setting, we used multiple approaches: first, through western blot analysis, we confirmed higher baseline levels of p-IGF-1R in early-passage rebound tumor cell lines compared to cell lines that we were able to propagate from dormant tumors in culture (fig. S8G; see methods). We determined that phosphorylation and downstream signaling could be reduced in rebound cell lines by using an inhibitor of IGF-1R (fig. S8G), and that early-passage rebound cells were more sensitive to IGF-1R blockade than naïve glioma cells in vitro using multiple pharmacological inhibitors (Fig. 5E and fig. S8H).
Next, to model the effects of macrophage-derived IGF-1 on rebound tumor cells, we designed an ex vivo culture system using primary glioma microenvironment cultures (GMECs). GMECs contain multiple cell types from the glioma TME when harvested at early-passage, including macrophages, T cells, astrocytes, among others (fig. S8I) (8). We hypothesized that rebound GMECs would be able to stimulate production of IGF-1 by macrophages, and subsequent growth of tumor cells. To test this, we collected conditioned media (CM) from rebound GMECs and applied it to wild-type (WT) BMDMs for 24h. After this treatment, we collected CM from the GMEC-stimulated BMDMs (Stim CM) and applied it to either rebound or naïve tumor cell lines in an MTT assay, +/− a neutralizing antibody against IGF-1 (see fig. S8J for experimental design). We found that Stim CM induced proliferation of rebound cell lines more effectively than naïve cell lines, and this effect was blocked by IGF-1 neutralization (Fig. 5F). These results suggest that the cells within a recurrent tumor are capable of stimulating production of IGF-1 by macrophages, which in turn gives a proliferative advantage to rebound tumor cells.
To assess the relevance of the IGF-1/IGF-1R axis in human malignancy, we evaluated IGF1 expression in publicly available human GBM gene expression datasets. We found that IGF1 expression was significantly correlated with macrophage-specific genes (CSF1R, CD68 and AIF1) and with genes associated with an M2-like phenotype (CD163 and MRC1) in TCGA GBM samples (Fig. 5G and fig. S9A). No such correlations were found for genes enriched in astrocytes (GFAP, ALDH1L1; fig. S9A), another key cell type in the glial scar phenotype (18). We also confirmed that IGF1 expression was significantly correlated with a PI3K signature score (Fig. 5H) generated from single sample gene set enrichment analysis for hallmarks of PI3K signaling (43). Consistently, IHC quantitation on an independent set of human GBM tissue samples revealed a significant association between p-AKT and the M2-associated protein MRC1 (Fig. 5I, fig. S9B, and Table S2). Together these data support our hypothesis that high IGF-1 levels translate to elevated PI3K signaling in patients, and that this axis is associated with M2-like gene expression.
To determine if macrophages are the predominant source of IGF1 in humans, we used qRT-PCR to show that macrophages express high levels of IGF1 compared to different immune cell types, astrocytes, endothelial cells, and glioma cells (fig. S9C). Consistently, we found that IGF1 expression was enriched in TAMs compared to the tumor bulk in GBM (fig. S9D) (44), and in mesenchymal GBM compared to other molecular subtypes (fig. S9E) where high macrophage content is a hallmark histological feature. These data corroborate our findings in the PDG model by suggesting that macrophages are an important source of IGF1 in human malignancy.
Finally, we used publicly available datasets to assess survival correlations in patients. We first generated Kaplan Meier curves using a median cutoff for IGF1 expression levels, and found no significant difference in overall survival between IGF1high and IGF1low patients (fig. S9F). Given that baseline IGF-1 signaling is critical during normal homeostasis in the brain (41), and also the extremely rapid progression of GBM in patients, we also surveyed the top 10% of IGF1high patients (versus all remaining), and found a clear decrease in overall survival (fig. S9F). Interestingly, when we used this same stringent top-10% cutoff for a macrophage marker (AIF1; also known as Iba1 in mouse) that correlates significantly with IGF1 expression levels (fig. S9A), there was no separation of survival curves (fig. S9F), suggesting that differences in survival are not simply due to differences in macrophage abundance, but rather due to differences in degree of IGF1 expression.
NFAT and Stat6 cooperate to regulate Igf1 expression in rebound TAMs
We next utilized transcription factor (TF) activity analysis to identify putative transcriptional networks regulating Igf1 expression in rebound tumors. Seven TF families showed enriched activity in Reb TAMs compared to EP TAMs (Fig. 6A, fig. S10A, and Table S3). Three of these were found to regulate Igf1 (NFAT, MYF and HMGB families; Fig. 6A), of which the NFAT family showed enriched activity specifically in Reb TAMs compared to both EP and Veh TAMs (Fig. 6B). These results were particularly interesting given the cooperative relationship between NFAT and Stat6 (canonical IL4 pathway) in transcriptional regulation (45). Corroborating these results, we found that IL4, but not TGFβ1, induced Igf1 expression in BMDMs in vitro, which was reduced by an NFAT inhibitor (fig. S10B).
Figure 6. NFAT and Stat6 cooperate to regulate Igf1 expression in rebound TAMs.
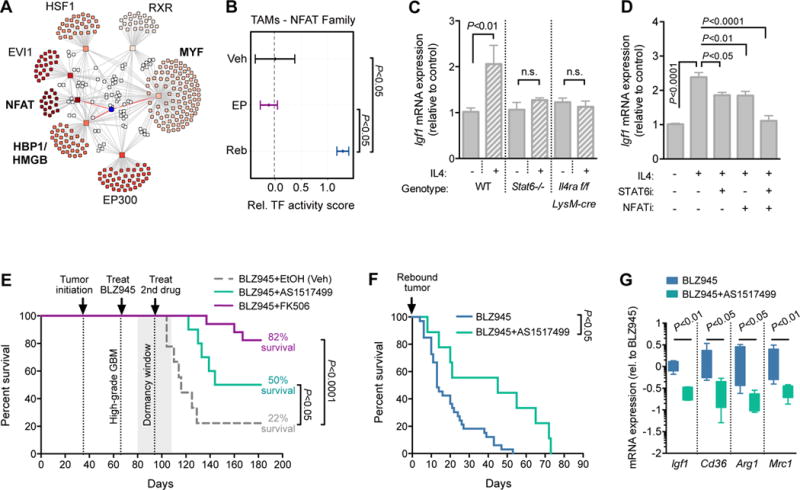
(A) Transcription factor (TF) network analysis from RNA-seq data showing enriched TF families (squares) connected with a line to target genes (circles). White circles indicate genes targeted by multiple TFs. Igf1 is shown as a blue circle connected by red lines to three regulating TF families. (B) Predicted NFAT TF activity in Veh, EP and Reb TAMs, showing a high score specifically in Reb TAMs (n=5–6 per group). (C) qRT-PCR analysis of Igf1 in BMDMs derived from WT, Stat6−/− or Il4ra flox; LysM-cre mice, treated +/− recombinant mouse IL4 (10 ng/ml; n=5 independent experiments). Student’s t-test was used for pairwise comparisons within each genotype. (D) qRT-PCR analysis of Igf1 in BMDMs derived from WT mice, treated +/− recombinant mouse IL4 (10 ng/ml), a Stat6 inhibitor (AS1517499, 50 nM) and/or an NFAT inhibitor (INCA-6, 40 μM; n=6 independent experiments). A one-way ANOVA and Dunnett’s multiple comparisons to the +IL4 condition was used to calculate significance. (E) Survival of PDG animals with high-grade tumors treated first with BLZ945 alone until dormancy (28d), and then enrolled on combination therapy with either AS1517499 (n=10), FK506 (NFAT inhibitor; n=17), or EtOH vehicle control (n=9). Combination therapy with either inhibitor led to a significant increase in overall survival (FK506= 82% survival P<0.0001; AS1517499= 50% survival P<0.05), compared to vehicle control (22% survival). Log-rank Mantel-Cox test was used to calculate significance. (F) Survival curve representing animals with recurrent tumors treated either with continuous BLZ945 alone (n=33) or BLZ945+AS1517499 (n=9). Combination therapy led to a significant increase in overall survival (Log-rank Mantel-Cox test, P<0.05), and in median survival following recurrence (45d) compared to BLZ945 monotherapy (13d). (G) qRT-PCR analysis of Igf1, CD36, Arg1 and Mrc1 levels in a subset of animals from (F). Results show a significant reduction of Igf1 expression in rebound tumors following Stat6 inhibition (n=5 per group, P<0.01), and a reduction of known IL4-Stat6 transcriptional targets (CD36 Pπ.05 Arg1 P<0.05; Mrc1 P<0.01), confirming drug efficacy in the brain (n=5 for all, Mann-Whitney test).
To further characterize the role of IL4-induced NFAT and/or Stat6 signaling in Igf1 regulation, we performed a series of in vitro and in vivo experiments. First, we confirmed that IL4 strongly induced expression of Igf1 and the three representative alternative activation/wound-associated genes (Retnla, Chil3 and Ccl17) in WT BMDMs, and that this capacity was reduced in BMDMs from either Stat6−/− or Il4ra flox; LysM-cre mice (Fig. 6C and fig. S10C). Furthermore, while pharmacological inhibition of either Stat6 or NFAT partially reversed the effects of IL4 on Igf1, Retnla, Chil3 and Ccl17 expression, dual inhibition of these pathways in WT BMDMs completely blocked the effects of IL4 on this gene set (Fig. 6D and fig. S10D). To validate the significance of these pathways in vivo, PDG mice with high-grade GBMs were treated continuously with BLZ945 alone until 28d, at which point FK506 (a NFAT-calcineurin inhibitor) or AS15171499 (a Stat6 inhibitor) was added until the trial endpoint. With addition of either of these inhibitors, the percentage of animals that survived to endpoint was significantly increased (22% BLZ945+Veh, 50% BLZ945+AS1517499, and 82% BLZ945+FK506; Fig. 6E). In accordance with these results, when we treated animals with AS1517499 in combination with continued BLZ945 treatment at a later time point, during the rebound phase, we were able to extend survival (Fig. 6F), and qRT-PCR analysis of these tumors confirmed reduced levels of Igf1 expression along with additional known targets of IL4-Stat6 signaling (CD36, Arg1 and Mrc1; Fig. 6G) (27). Collectively these data suggest that both NFAT and/or Stat6 signaling contribute to macrophage activation and IGF-1 regulation in rebound tumors, and that pharmacological blockade of either of these pathways is sufficient to reduce the incidence of disease recurrence. It is important to note, however, that these models are limited by their inability to distinguish cell type-specific pathway blockade. Furthermore, although FK506 is used in multiple clinical settings, targeted specificity of the Stat6 inhibitor in vivo is not fully characterized. In future studies, elucidating the effects of T cell-specific IL4 expression, or macrophage IL4Ra, on BLZ945 efficacy through the use of genetic models would provide additional insight into mechanistic communication between different TME cell types.
Combination of CSF-1R and IGF-1R inhibition improves outcome
To formally test our hypothesis that the IGF-1/IGF-1R axis may underlie resistance to CSF-1R inhibition, we targeted IGF-1R in vivo using both pharmacological and genetic approaches. First, we designed similar preclinical intervention trials as for BKM120, except with an inhibitor of IGF-1R (OSI906/Linsitinib). We treated PDG mice with BLZ945 until they showed signs of rebound by MRI, at which point we intervened with OSI906 (Fig. 7A, trial design 1). OSI906 was chosen because it is currently being clinically evaluated for multiple cancer types, its effect on BMDM viability in vitro was minimal compared to other IGF-1R inhibitors tested (fig. S11A), and we confirmed it is brain-penetrant in rebound tumors by showing reduced p-IGF-1R immunostaining (fig. S11B). In concordance with the BLZ945+BKM120 trial results, we found that rebound tumors treated with OSI906 and continuous BLZ945 significantly extended median survival to 63d (versus 13d post-recurrence for continuous BLZ945 monotherapy; Fig. 7B), and markedly reduced tumor progression and proliferation: apoptosis ratios after 2 weeks of treatment (Fig. 7, C and D, and fig. S11C). By contrast, OSI906 monotherapy in rebound tumors (i.e. discontinued BLZ945) led to a median survival of just 12d (Fig. 7B), and was only modestly effective in treatment-naïve tumors (fig. S11, D and E). Together these results mirror those from the BKM120 trials, and suggest that continued CSF-1R inhibition is necessary to drive IGF1R/PI3K signaling dependency, rendering recurrent tumors sensitive to pathway inhibition. Similar combination treatment efficacy and mechanistic commonalities (including immunofluorescence quantification of Ki67: CC3 ratios, CD206, and p-IGF-1R) were observed in orthotopic xenograft trials performed with patient-derived proneural tumorspheres, and with established human glioma cell lines (Fig. 7, E and F, and fig. S12, A to F).
Figure 7. Combination of CSF-1R inhibition and IGF-1R inhibition significantly improves outcome in preclinical models.
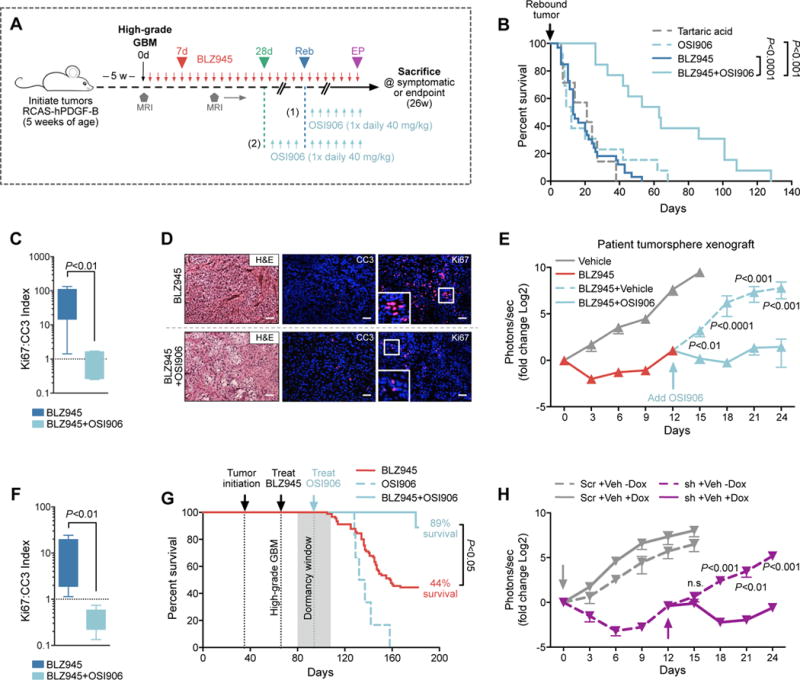
(A) Long-term trial design for testing BLZ945 and OSI906 combination therapy on PDG tumors. High-grade tumors were treated with BLZ945 until recurrent tumors developed (trial design 1) or until dormancy (28d, trial design 2), whereupon OSI906 was either added (with continuous BLZ945) or switched (discontinued BLZ945). (B) Survival of animals with recurrent tumors treated either with BLZ945 alone (n=33), OSI906 alone (n=13), or BLZ945 in combination with OSI906 (n=13; trial design 1 in Fig. 7A). Combination therapy of BLZ945+OSI906 led to an increase in overall survival (Log-rank Mantel-Cox test, P<0.001), and in median survival following recurrence (63d) compared to BLZ945 (13d), or OSI906 (12d) monotherapy. (C) Ki67:CC3 (cleaved caspase 3) proliferation:apoptosis index from immunofluorescent staining of recurrent tumors treated with BLZ945 alone versus BLZ945+OSI906 for 2 weeks (Mann-Whitney test, P<0.01, n=7–8 mice). (D) Representative H&E and immunofluorescent images corresponding to data in (C). Scale bars = 50 μm. (E) Bioluminescent imaging (BLI) from orthotopically xenografted patient-derived tumorspheres (TS573) that were subject to 24d of treatment with BLZ945 (red) versus vehicle control (grey). Results demonstrate that treatment with BLZ945+OSI906 blunts outgrowth of rebound tumors compared to BLZ945+vehicle. Mann-Whitney test was used to calculate P-values for each time point (n=5–20 mice). (F) Ki67:CC3 index from immunofluorescent staining of TS573 orthotopic xenograft tissues (Mann-Whitney test, P<0.01, n=5 mice per group). (G) Kaplan-Meier analysis of PDG animals treated either with BLZ945 alone (n=90; same cohort as presented in Fig. 1D), OSI906 alone (n=6) or with BLZ945 in combination with OSI906 during dormancy (n=9; trial design 2 in Fig. 7A). Combination therapy of BLZ945+OSI906 extended overall survival compared to either monotherapy. Log-rank Mantel-Cox test was used to calculate significance. (H) BLI of orthotopically xenografted U251 cells genetically engineered to express an IGF1R-targeted doxycycline (dox)-inducible shRNA (sh; n=15–16 mice; purple lines) or a scrambled control vector (Scr; n=4–5 mice; grey lines). Colored arrows indicate respective administration of dox. Graph shows two combined shRNAs; data for individual hairpins are shown in fig. S13E. Mann-Whitney test was used to calculate significance.
To determine if tumor outgrowth could be prevented by earlier IGF-1R inhibition, PDG mice with high-grade GBMs were treated continuously with BLZ945 alone until 28d, at which point OSI906 was added until the trial endpoint (Fig. 7A, trial design 2). With early combination treatment, we indeed extended overall survival and increased the percentage of animals that survived to endpoint (89% BLZ945+OSI906 versus 44% BLZ945 alone or 0% OSI906 alone; Fig. 7G). Together, our results suggest that targeting either IGF-1R or PI3K signaling in GBMs resistant to CSF-1R inhibition may interfere with disease progression and improve overall survival.
Given that pharmacological inhibition of IGF-1R using OSI906 cannot confirm whether tumor cell-specific blockade is sufficient to reduce recurrent disease, we utilized a genetic approach to target this receptor in glioma cells. U251 glioma cell lines were genetically engineered to express a doxycycline-inducible shRNA against IGF1R, and orthotopic xenograft experiments were performed (see fig. S13A for trial design). Two independent shRNAs targeting IGF1R were used, and a scramble-sequence shRNA was used as a control (fig. S13, B to D). We found in both cases, dox-induction of the shRNAs during the rebound phase of the trial (d12) mitigated tumor progression compared to no-dox control animals (Fig. 7H, and fig. S13E). These results support our hypothesis that tumor cell-specific IGF-1R contributes to BLZ945 resistance and disease recurrence.
Discussion
Here, we describe the development of acquired resistance to CSF-1R inhibition in mouse models of GBM. While initial therapeutic response to CSF-1R inhibition is robust, rapid, and completely penetrant, we found that approximately half of the animals eventually develop resistance, with rapidly progressing rebound tumors. In light of recent results from ongoing patient studies with CSF-1R inhibitors in glioma (46) and other cancers, our findings suggest the need to prepare for the emergence of therapeutic resistance to CSF-1R inhibitors in GBM in the clinical setting, and determine if other brain malignancies besides GBM will respond similarly to CSF-1R inhibition. While classical mechanisms of tumor cell-intrinsic resistance to cytotoxic and targeted agents have been well-defined, including aberrant drug metabolism and transport, drug target mutation, and activation of alternative survival pathways (47), it still remains unclear whether resistance to TME-directed therapies will follow similar principles. Given that TME-targeted agents are increasingly being evaluated in the clinic (1, 2), it will be critical to mechanistically define how resistance evolves in response to these therapies in order to provide long-term disease management for patients.
In light of this problem, we have now identified a mechanism of drug resistance that can circumvent therapeutic response to a TME-targeted therapy, and promote disease progression in the absence of tumor cell-intrinsic alterations. Specifically, we have uncovered a heterotypic paracrine signaling interaction that is initiated by the TME, and drives resistance to CSF-1R inhibition. In rebound tumors, we found that IGF-1 is upregulated in TAMs in response to IL4 (possibly supplied by T cells or alternative cell types), in part via NFAT activation. IGF-1 secretion into the extracellular environment results in activation of IGF-1R and PI3K signaling in glioma cells, supporting tumor growth and malignancy (see model in Fig. 8). Multiple nodes in this signaling loop can be targeted therapeutically by agents that are currently used clinically, including OSI906, BKM120, or FK506, resulting in a substantial improvement in survival in the preclinical setting when combined with CSF-1R inhibition. Indeed, given that PI3K signaling is aberrantly activated in a substantial proportion of GBM patients (16), and recent clinical trial results showing limited efficacy in recurrent (albeit very advanced) GBM (46), it is possible that this pathway could similarly contribute to intrinsic resistance to CSF-1R inhibition, and consequently those patients may benefit from combinatorial inhibition of these pathways from the outset.
Figure 8. Working model for mechanism of resistance to CSF-1R inhibition in glioma.
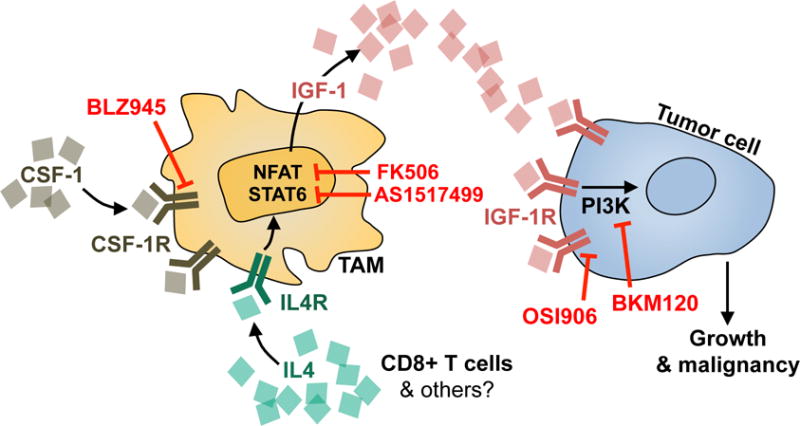
IGF-1 is significantly upregulated in TAMs in response to long-term CSF-1R inhibition in GBM. IGF-1 secretion into the extracellular environment results in activation of IGF-1R on tumor cells, and downstream PI3K signaling to support tumor regrowth during continuous BLZ945 treatment. Upstream of IGF-1 in TAMs, NFAT and/or Stat6 transcriptional activity regulate its expression. This is thought to be initiated in response to IL4/IL4Ra pathway activation, feeding in from other cell types in the TME, including T cells (and possibly others). Multiple nodes in this signaling loop can be targeted therapeutically, including OSI906, BKM120, AS1517499, or FK506, resulting in a substantial improvement in survival in preclinical trials when combined with CSF-1R inhibition.
Importantly, our findings underscore the importance of bidirectional feedback between cancer cells and their microenvironment, and support the notion that while stromal cells are less susceptible to genetic mutation than cancer cells, a tumor can nonetheless persist by exploiting its extracellular environment to acquire a resistant phenotype. Thus we propose that an integrated analysis of cancer cells with their microenvironment will be critical to understand both their parallel evolution during tumor progression, and their capacity for adaptation in the context of therapeutic intervention and the development of resistance.
MATERIALS AND METHODS
Study design
The overall objective of our study was to understand how resistance to CSF-1R inhibitors develops in high-grade glioma using various in vivo and in vitro models. Within the animal studies, mice were randomly assigned to different therapy groups, which included treatment with BLZ945 (a CSF-1R inhibitor) in combination with inhibitors of putative resistance pathways (e.g. IGF-1R or PI3K), versus single-agent or vehicle controls. Survival and disease progression were monitored using a combination of MRI, histology, flow cytometry, and gene expression analyses throughout all trials. To power our studies, sample size was predefined as at least n=3 independent experiments, replicates or samples for in vitro and in vivo experiments, and up to a maximum of n=90 mice for survival analyses. Replicate values are indicated for each experiment in the figure legend. All analyses were calculated in a blinded manner through numerical coding of samples. For all long-term survival trials, 26 weeks was selected as a predefined endpoint because mice in the Ink4a/Arf−/− background develop spontaneous lymphomas and sarcomas beginning at ~30 weeks of age. For in vivo trials, mice were considered outliers if they developed (i) spontaneous lymphomas (given this disposition in the Ink4a/Arf−/− background), (ii) early symptoms of tumor burden >1 week prior to trial enrollment, or (iii) development of hydrocephalus prior to trial enrollment.
Biologicals and pharmaceuticals
BLZ945 (8) was obtained from Novartis for both in vitro and in vivo use. 6,700 nM of BLZ945 was used for all in vitro experiments in tumor cells, versus an equal percent DMSO as a vehicle control. This concentration represents 100× the IC50 for BLZ945 in macrophages (8). For in vivo experiments, BLZ945 was obtained pre-formulated at 12.5 mg/ml. BLZ945 was administered p.o. 1× daily at 200 mg/kg, and 20% Captisol was used as a vehicle control. BKM120 (Buparlisib; 1 μM unless indicated otherwise), AEW541, ADW742, BMS754807 and OSI906 (Linsitinib; 10 μM unless indicated otherwise) were purchased from Selleckchem for in vitro use, and were used up to 100 μM for dose response assays versus an equal percent DMSO as a vehicle control. BKM120 and OSI906 were purchased from ChemieTek for in vivo use. BKM120 was formulated by dissolving 52 mg into 500 ml NMP, boiling, and then adding 9.5 ml of PEG300. BKM120 was administered p.o. 1× daily at 20 mg/kg, and NMP:PEG300 (1:19) was used as a vehicle control. Animals were dosed at 20 mg/kg to avoid off-target effects, as BKM120 binds tubulin at concentrations above 50 mg/kg in subcutaneous tumor models, but not below 40 mg/kg (15). OSI906 was formulated daily at 4 mg/ml in 25 mM tartaric acid with shaking and sonication for ~15 min. OSI906 was administered p.o. 1× daily at 40 mg/kg, and 25 mM tartaric acid was used as a vehicle control. We chose to treat at 40 mg/kg as the maximum tolerated dose for OSI906, 75 mg/kg (48), was found to be toxic within 4 days in our studies. For all combination trials, BLZ945 was administered in the morning, and BKM120 or OSI906 was administered in the evening. The NFAT inhibitor, INCA-6, was purchased from Tocris, and was used at a concentration of 40 μM for in vitro use (49). For in vivo inhibition of NFAT signaling, FK506 was used to inhibit the activating interaction between calcineurin and NFAT, at a dose of 10 mg/kg (administered i.p. every 3 days) (50). The Stat6 inhibitor, AS1517499, was purchased from Axon Medchem, and was used at a concentration of 50 nM for in vitro use, and dosed at 10 mg/kg for in vivo use (administered i.p. 2× weekly) (51). The vehicle control for FK506 and AS1517499 in vivo was 10% EtOH and 1% Tween-80 in PBS. For in vitro PCR assays, recombinant mouse IL4 (R&D Systems) was used at a concentration of 10 ng/ml, recombinant mouse TGFβ1 (R&D Systems) was used at a concentration of 50 ng/ml, and the TGFβ1 type 1 receptor inhibitor, SB431542 (Tocris), was used at a concentration of 10 μM. For ex vivo glioma microenvironment culture (GMEC) assays, a neutralization antibody against IGF-1 (R&D Systems) was used at a concentration of 0.5 μg/ml. For culture of macrophages in vitro, recombinant mouse CSF-1 was used at a concentration of 10 ng/ml. For in vitro assays using bone marrow-derived macrophages (BMDMs), CSF-1 supplementation was excluded from all experimental conditions.
Animals
Crl:NU(NCr)-Foxn1nu immunodeficient mice were purchased from Charles River Laboratories for orthotopic transplantation studies. NOD/CB17-Prkdcscid immunodeficient mice were purchased from The Jackson Laboratory for orthotopic implantation of human cells. Three different transgenic mouse models expressing the avian tv-a receptor under the control of the nestin (N) promoter in either mixed strain or BL6 backgrounds were used (Ntv-a;Ink4a/Arf−/−, Ntv-a, and Ntv-a;Ptenflox), all previously described (13, 52–57) and bred within the MSKCC animal facility. Stat6−/−, Il4raflox;LysM-cre, and wild-type (WT) C57BL/6 (BL6) mice were also bred within the MSKCC animal facility, and were used for bone marrow isolations. All animal studies were approved by the Institutional Animal Care and Use Committee of Memorial Sloan-Kettering Cancer Center.
PDG mouse model
The initiation of PDGF-driven gliomas (PDG) with RCAS-hPDGF-B-HA in adult mice has been previously described (8, 13, 58). Briefly, Ntv-a;Ink4a/Arf−/− mice were fully anesthetized with ketamine/xylazine prior to surgery. Pain management included a 50 μl subcutaneous injection of bupivacaine (0.25%) at the surgical site prior to surgery, and an intraperitoneal injection of buprenorphine immediately following surgery. Mice were intracranially injected with DF-1:RCAS-hPDGF-B-HA cells (200,000 cells/1 μl) between 5–6 weeks of age using a fixed stereotactic apparatus (Stoelting). Injections were made into the right frontal cortex, approximately 1.5 mm lateral and 1 mm caudal from bregma, and at a depth of 2 mm into the subventricular zone (SVZ). In this model, injection into the SVZ induces tumors with low latency (4–5 weeks), 100% penetrance, and histological features characteristic of patient GBM including microvascular proliferation and pseudopalisading necrosis (13). The incision was sealed using Vetbond tissue adhesive (3M). Tumors were imaged by MRI after 5 weeks, and drug intervention was initiated for tumors ≥2 mm3. A total of 90 animals were treated in long-term experiments with BLZ945 alone, which represented 5 independent cohorts. These data are compiled and presented in Fig. 1D, 2E, and 7G (red lines).
p53 KD mouse model
Injections were performed as described for the PDG mouse model above, except Ntv-a mice were used (i.e. WT Ink4a/Arf). Mice were intracranially injected with a 1:1 ratio of DF-1:RCAS-hPDGF-B-HA cells and DF-1:RCAS-shp53 cells (total of 300,000 cells/2 μl) between 5–6 weeks of age. Injection into the SVZ in the p53 knockdown (KD) model induces high-grade tumors with low latency (6–7 weeks), 100% penetrance, and histological features of human GBM (13, 54, 55). Tumors were detected by MRI after 6–7 weeks, at which point drug intervention with BLZ945 was initiated (see Fig. 2F).
Pten KO mouse model
Injections were performed as described for the PDG mouse model above, except Ntv-a;Ptenflox mice were used (i.e. WT Ink4a/Arf). Mice were intracranially injected with a 1:1 ratio of DF-1:RCAS-hPDGF-B-HA cells and DF-1:RCAS-Cre cells (total of 300,000 cells/2 μl) between 5–6 weeks of age. Injection into the SVZ in the Pten knockout (KO) model induces tumors with moderate latency (8–12 weeks) and penetrance (~20–30%). Tumors that form harbor key characteristics of human GBM including highly infiltrative histology (13, 54, 56, 57). Tumors were detected by MRI after 8–12 weeks, at which point drug intervention with BLZ945 was initiated (see Fig. 2, F and G).
Derivation of mouse primary glioma cell lines from PDG tumors
To derive rebound or dormant cell lines from BLZ945-treated PDG tumors, MRI was used to confirm whether a particular tumor was in rebound or dormancy phase. Macrodissected rebound or dormant lesions from the BLZ945-treated PDG mouse model were manually dissociated and filtered through a 40 μm mesh filter. The cell suspension was washed 2× with PBS, and cultured in Mouse Neural Stem Cell (mNSC) Basal Media (Stem Cell Technologies) containing mNSC proliferation supplement, 1 mg/ml Heparin (Stem Cell Technologies), 10 ng/ml recombinant epidermal growth factor (rEGF; Invitrogen), and 20 ng/ml recombinant basic-fibroblast growth factor (rbFGF; Sigma). To generate cell lines in monolayer, tumorsphere cultures were expanded and dissociated, and transferred to culture with DMEM + 10% FBS (59). In total, 5 rebound cell lines were derived (89AReb, 89BReb, 74Reb, 48Reb, 52Reb), and 1 cell line was derived from the 28d dormant timepoint. The 28d dormant cells (fig. S8G) took several weeks before starting to proliferate in culture, and upon transplantation into naïve animals, the cells did not give rise to growing tumors (BLI signal remained stable and was monitored up to 22d; data not shown). Derivation of the PDGC23 primary glioma cell line from an untreated/naïve mixed-background PDG mouse was described previously (8).
Human cell lines
Human umbilical vein endothelial cells (HUVEC) were purchased from ATCC. Human brain microvascular endothelial cells (HBMEC) and human astrocytes were purchased from Sciencell. Astrocytes were cultured on poly-L-lysine-coated plates, and both HUVECs and HBMECs were cultured on gelatin-coated plates with endothelial cell media (ECM, Sciencell) + 10% FBS + an endothelial cell growth factor supplement. The U251 (commercially available) and TS573 (patient-derived) cell lines were selected based on our previously published work, which showed efficacy in response to BLZ945 in orthotopic xenograft trials (8). The patient-derived TS573 glioma tumorsphere line was derived from a consenting patient under Institutional Review Board (IRB)-approved protocols for the banking of excess tumor tissue during routine surgical resection (MSKCC IRB# 99-125A(2) and 06–107), as previously described (8, 60). Tumorspheres were maintained in Human Neural Stem Cell (hNSC) Basal Media (Stem Cell Technologies) containing hNSC proliferation supplement, 1 mg/ml Heparin (Stem Cell Technologies), 10 ng/ml rEGF (Invitrogen), and 20 ng/ml rbFGF (Sigma). Tumorspheres were passaged by dissociation with Accutase cell detachment solution (Millipore). Characterization and molecular subtyping by Sequenom, Nanostring and aCGH were performed as previously described (8). Briefly, aCGH on primary spheroids showed a high level amplification of PDGFRA and CDK6 loci, and a regional chromosome 5 loss. Nanostring analysis confirmed PDGFRA overexpression, and sequenom analyses were negative for IDH1/2 mutations.
Isolation of bone marrow-derived macrophages (BMDMs)
To generate macrophages from bone marrow, femurs and tibiae from Stat6−/−, Il4raflox;LysM-cre, or WT BL6 mice were flushed and cells harvested under sterile conditions. The isolate was filtered through a 40 μm mesh filter and cultured in 30 ml Teflon bags (PermaLife PL-30) for 5–7 days in DMEM + 10% FBS + 10 ng/ml recombinant mouse CSF-1 (R&D Systems). Media were changed every other day.
TGL infections
Cell lines were labeled with a triple-imaging vector (TK-GFP-Luc; TGL) (61) for use in orthotopic in vivo experiments. The TGL vector was developed to enable non-invasive in vivo imaging of tumor growth over time. A standard protocol for retroviral infection was used. Briefly, GP2-293T cells were transfected with the TGL construct and pCL-Ampho at a 1:1 ratio, using Fugene (Promega) and OptiMEM (Gibco). 12 h later, media was replaced with complete antibiotic-free DMEM, and collected for 3 consecutive days for infection of target cells.
Orthotopic transplantation experiments
TGL-labeled cells were resuspended in antibiotic-free serum-free DMEM for all orthotopic injections. Mice were fully anesthetized with ketamine/xylazine prior to surgery. Pain management included a 50 μl subcutaneous injection of bupivacaine (0.25%) at the surgical site prior to surgery, and an intraperitoneal injection of buprenorphine immediately following surgery. Mice were intracranially injected with glioma cells between 5–6 weeks of age using a fixed stereotactic apparatus (Stoelting). The number of cells injected was as follows: mouse glioma lines 2.5–5×104 cells/2 μl, human U251 cells 2.5×105 cells/2 μl, and patient-derived TS573 cells 5×104 cells/2 μl. Injections were made to the right frontal cortex, approximately 1.5 mm lateral and 1 mm caudal from bregma, and at a depth of 2 mm. Hydrogen peroxide was used to clean the hole made by the surgical drill, and bone wax was used to close the hole. The incision was sealed using Vetbond tissue adhesive (3M). One week following injections, mice were randomly assigned to Vehicle or BLZ945 treatment groups, and dosed 1× daily by oral gavage. Bioluminescence imaging (BLI; Xenogen IVIS-200 Optical In Vivo Imaging System) was performed every 3–5 days over the course of the experiment to monitor tumor progression and response to therapy. Once BLZ945-treated tumors reached 3× the volume of their lowest BLI measurement, tumors were considered ‘resistant’, and mice were randomly assigned to combination treatment with BLZ945 + OSI906, or BLZ945 + vehicle. For doxycycline (dox)-inducible IGF1R shRNA experiments with U251 cells, mice were additionally assigned to either +/− dox groups (doxycycline hyclate diet formulated at 2,500 mg/kg, Envigo).
Animal sacrifice and tissue harvest
Mice were euthanized at the defined 26 week endpoint, or when symptomatic (poor grooming, lethargy, weight loss, hunching, macrocephaly/hydrocephalus, seizures). 26 weeks was selected because mice in the Ink4a/Arf−/− background develop spontaneous lymphomas and sarcomas beginning at ~30 weeks of age (62). Euthanasia was performed by either carbon dioxide asphyxiation or anesthesia (avertin; 2,2,2-tribromoethanol; Sigma) followed by cervical dislocation. For snap freezing of whole tumor samples, mice were euthanized 1 h following the last treatment dose, and tissues were collected, frozen immediately in liquid nitrogen, and stored at -80°C for subsequent applications (e.g. RNA isolation, protein extraction). For isolation of whole tissues for histology, mice were fully anesthetized with avertin, transcardially perfused with 10 ml of PBS, followed by 10 ml of paraformaldehyde (PFA; 4% in PBS). Tissues were incubated in PFA overnight at 4 °C, rinsed in PBS, and then transferred to sucrose (30%) for 2–3 days at 4 °C. For hypoxia analysis, mice were injected intraperitoneally with 60 mg/kg of pimonidazole (hypoxyprobe-1; HPI) ~20 min prior to sacrifice and tissue collection. All tissues were embedded and frozen in Optimal Cutting Temperature (OCT) compound (Tissue-Tek®), and 10 μm cryostat tissue sections were used for all subsequent staining and analyses.
Immunofluorescence (IF)
For IF staining, 10 μm frozen sections were thawed and dried at room temperature, and rinsed in PBS. Tissue sections were blocked in 0.5% PNB (1 h at room temperature or overnight at 4 °C), followed by incubation in primary antibody (2 h at room temperature or overnight at 4 °C). All primary antibodies and dilution factors are listed in Table S4. Sections were then washed in PBS and incubated with the appropriate fluorophore-conjugated secondary antibody (Molecular Probes) at a dilution of 1:500 in 0.5% PNB (1 h at room temperature). DAPI was used as a counterstain prior to mounting with fluorescent mounting media (Dako).
Immunohistochemistry (IHC) and Von Kossa staining
For manual IHC, tissue sections were first subjected to citrate buffer based antigen retrieval by submerging in antigen unmasking solution (0.94% v/v in distilled water; Vector Laboratories) and microwaving for 10 min, followed by cooling to room temperature for ≥30 min. Endogenous peroxidases were blocked for 10 min with Dual Endogenous Enzyme Block (Dako). Slides were incubated with serum-free protein block (Dako) for 1 h at room temperature, and then incubated with primary antibody in a humidity chamber overnight at 4 °C. The following day, slides were washed and incubated with HRP-conjugated secondary antibodies (Jackson Immunoresearch) for 1 h at room temperature, and positive staining was detected using diaminobenzidine (DAB) substrate-chromogen. Haematoxylin was used as a counterstain and slides were mounted with VECTASHIELD® mounting media (Vector). As an alternative to manual staining, a Ventana autostainer was used for staining of mouse GFAP, human phospho (p)-AKT, and human MRC1, which included automated deparaffinization, citrate buffer-based antigen retrieval, non-specific protein and endogenous peroxidase block, antibody incubation, and DAB detection. All primary antibodies and dilution ratios for both manual staining and autostaining are listed in Table S4. For visualization of calcium deposition in tissue sections, a Von Kossa staining kit was used as per manufacturer’s instructions (Abcam). Briefly, tissues are treated with a silver nitrate solution, which is deposited by replacing calcium reduced by UV light. Nuclear fast red was used as a counterstain (Vector).
Histology and grading
For analysis of tissue histology and grading of tumor malignancy, haematoxylin and eosin (H&E) staining was performed using a Tissue-Tek® automated slide stainer, and slides were mounted with VECTASHIELD® mounting media (Vector). Tissues were blindly graded by a neuropathologist (Dr. Jason Huse, MSKCC), according to standard WHO criteria (63). For CNS tumors, this grading system is based on a malignancy scale, where tumors that are minimally proliferative and infiltrative are considered grade I, while the most histologically aggressive, infiltrative, and incurable tumors (glioblastomas) are grade IV (63). GFAP quantification was performed by the Allred scoring method, which is the sum of a proportion score (0–5) and an intensity score (0–3) for a given marker (64).
Image analysis
Stained tissue sections were visualized under a Carl Zeiss Axioimager Z1 microscope equipped with an ApoTome.2, or a Carl Zeiss Axioimager M1 epifluorescence and brightfield microscope. Staining analyses were performed using a TissueGnostics slide scanning platform, and TissueQuest analysis software. This automated analysis platform was used to quantify number of positive counts, area of positive staining, and/or microvascular density within stained tissue sections in an unbiased manner (8).
Patient GBM tissue samples
Patient GBM tissue samples used for staining and analysis (Fig. 5I and fig. S9B) were obtained from the Brain Tumor Center, MSKCC. 18 patient tissue samples were used in total. IHC staining for MRC1 and p-AKT was performed as described above (see “Immunohistochemistry and Von Kossa staining” section), and quantitation was performed using the Axioimager Z1 scanning microscope (see “Image analysis” section). Correlational analysis across patients was performed using GraphPad Prism 6.0 (see “Data presentation and statistical analysis” section). Patient information for all samples can be found in Table S2.
Protein isolation and immunoblotting
To evaluate phospho-protein status of IGF-1R and AKT in culture, cells were seeded at 70–80% confluence, serum-starved for ~12 h, and then treated with OSI906 for 30 min. To evaluate phosphorylation of IGF-1R or AKT in snap-frozen tumor samples, tissues were dissociated with a glass homogenizer on ice in the presence of lysis buffer. All protein lysates were prepared in RIPA lysis buffer supplemented with Halt™ protease inhibitor (1:100; Thermo Scientific) and Halt™ phosphatase inhibitor (1:100; Thermo Scientific), and protein was quantified using a BCA protein assay kit (Pierce). Equal amounts of protein (20 μg/lane for cell lines, and 100 μg/lane for tissue samples) were loaded onto SDS-PAGE precast gels (Invitrogen) and transferred to PVDF membranes for immunoblotting. Membranes were blocked in 5% milk, incubated with primary antibodies (Table S4) for 1 h at room temperature or overnight at 4°C, washed with 0.1% TBS-T, and incubated with HRP-conjugated secondary antibodies (Jackson Immunoresearch) for 1 h at room temperature. SuperSignal West Femto or Pico chemiluminescent substrate and CL-XPosure Film (Pierce) were used for signal detection.
RNA isolation, reverse transcription, and quantitative real-time PCR
RNA was isolated with Trizol, DNase treated, and 1 μg of RNA was used for cDNA synthesis using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Mouse taqman probes (Applied Biosystems) were used for quantifying expression of Igf1 (Mm00439560_m1), Chil3 (Mm00657889_mH), Ccl17 (Mm01244826_g1), Retnla (Mm00445109_m1), Il4 (Mm00445260_m1), Arg1 (Mm00475988_m1), Cd36 (Mm00432403_m1), Mrc1 (Mm01329362_m1), Ubc (Mm02525934_g1; housekeeping), and Hprt (Mm01545339_m1; housekeeping). Human taqman probes (Applied Biosystems) were used for quantifying expression of IL4 (Hs00174122_m1), IL13 (Hs00174379_m1), IGF1 (Hs01547656_m1) and HPRT1 (Hs02800695_m1; housekeeping).
MTT assays
Cell growth rate was determined using an MTT cell proliferation kit (Roche). Briefly, cells were plated in ≥triplicate in 96-well plates. 1×103 cells/well were plated for mouse glioma cell lines, and 5×103 cells/well were plated for BMDMs. For BLZ945 time course experiments, cells were grown in the presence of 6,700 nM of BLZ945 versus an equal percent DMSO, media was changed every 48 h, and viability measurements were taken every 24 h. For dose response experiments, cells were grown in the presence of an IGF-1R inhibitor (AEW541, ADW742, BMS754807, or OSI906) versus an equal percent DMSO at the doses indicated (see “Biologicals and pharmaceuticals” section above), and viability measurements were taken after 24 h. Reduction of the MTT substrate was detected by colorimetric analysis using a plate reader as per the manufacturer’s protocol. 10 μl of MTT labeling reagent was added to each well and then incubated for 4 h at 37 °C, followed by the addition of 100 μl MTT solubilization reagent overnight. The mixture was gently resuspended and absorbance was measured at 595 nm and 750 nm on a spectraMax 340pc plate reader (Molecular Devices).
Ex vivo GMEC assays
Glioma microenvironment cultures (GMECs) are early-passage heterotypic cell cultures harvested directly from PDG mouse primary tumors. Flow cytometry characterization of these cultures revealed that passage 1 GMECs contain a high abundance of tumor cells, astrocytes, and macrophages, as well as smaller proportions of myeloid progenitors, T cells and B cells (fig. S8I). In our ex vivo assays, we derived conditioned media (CM) from passage 1 rebound GMECs, and used this CM to treat naïve BMDMs in vitro (see fig. S8J for experimental design). We collected CM from these GMEC-stimulated BMDMs (Stim CM), and applied it to either rebound glioma cell lines (highly sensitive to IGF-1R inhibition) or naïve glioma cell lines (less-sensitive to IGF-1R inhibition), +/− a neutralizing antibody for IGF-1. An MTT assay was used to assess changes in growth in response to each treatment condition, as described above.
Flow cytometry and fluorescence-activated cell sorting (FACS) on primary mouse tissues
Mice were fully anesthetized with avertin and transcardially perfused with 20 ml of PBS. The brain was then isolated and the tumor was macrodissected from the surrounding normal tissue. Tissues were mechanically dissociated and filtered into a single cell suspension. For flow cytometry, cells were counted and incubated with Fc block for 1 h (BD Biosciences; 1:100/106 cells), followed by a 30 min incubation with LIVE/DEAD® fixable dead cell kit (Invitrogen), and then a 1 h incubation with conjugated antibodies for extracellular markers. For FACS, cells were counted and incubated with Fc block for 1 h, followed by a 1 h incubation with conjugated antibodies, and then stained with DAPI for dead cell exclusion. All antibodies and dilution ratios used for these experiments are listed in Table S4. OneComp eBeads (eBioscience), ArC™ Amine Reactive Compensation Beads (Invitrogen), and/or cell suspensions from spleen were used for compensation controls. A BD LSRFortessa™ was used for flow cytometry, and a BD FACSAria III™ was used for cell sorting.
Distinguishing between putative BMDMs and microglia by flow cytometry
Flow cytometry was used to evaluate the proportions of peripherally derived BMDMs versus resident microglia in PDG tumors across different treatment groups, according to published methods (22–24). Briefly, after gating on live cells, we used cell surface expression of CD45 and CD11b to distinguish between the two populations, where CD45lo CD11b+ defined putative microglia, while CD45hi CD11b+ defined putative BMDMs. In using this method, we acknowledge its limitations, and recognize that these two populations cannot be definitively distinguished without lineage tracing experiments that specifically label yolk sac-derived microglia or peripherally recruited BMDMs.
FACS purification of human peripheral immune cell types
Human buffy coats from three consenting healthy donors were obtained from the New York Blood Center. For isolation of neutrophils and eosinophils, buffy coats were directly RBC lysed (BD PharmLyse) for 15 minutes at room temperature. All other cell types were isolated from the top layer of a Ficoll gradient separation (HistoPaque, Sigma). Cells were pelleted for 10 minutes at 300×G and washed twice with FACS buffer (PBS+2% fetal bovine serum) and Fc blocked (Biolegend TruStain FcX). Cells were incubated with the appropriate antibodies for 15 minutes (Table S4). Cells were FACS purified on an Aria III (BD). For human macrophage differentiation, PBMCs were isolated from buffy coats following a Ficoll gradient. Monocytes were further purified from the interphase of a 70%/30% Percoll gradient. Monocytes were then washed twice with PBS, and cultured in Teflon bags (Origin) for 7 days in DMEM+2% human serum + recombinant human CSF-1 (10 ng/ml; R&D Systems). CSF-1 and media were replaced every 48 h.
Array comparative genomic hybridization (aCGH)
All RNA-sequencing and quality control was performed at the Integrated Genomics Operation, MSKCC. DNA was isolated from passage 1 PDG neurospheres from rebound tumors or corresponding liver tissue using TRIzol as per manufacturer instructions (Invitrogen). 3 ug of DNA was used with an Agilent standard cy5/cy3 labeling protocol. Briefly, Agilent Mouse CGH 180k arrays were hybridized at 65 °C and 20 rpm for 40 h. Slides were then scanned using the Agilent scanner according to the manufacturer’s instructions. The raw data were extracted with Feature Extraction using Agilent default analysis settings. Subsequent analyses were performed in R v3.1.0 using the “DNAcopy” package (65).
RNA-sequencing
Three RNA-seq experiments were performed in total: (i) FACS-purified tumor cells (CD45-PDGFRα+) and TAMs (CD45+CD11b+Gr1−) from Veh, EP and Reb tumors, (ii) FACS-purified astrocytes (CD45-GLAST+), B cells (CD45+CD19+), Tc cells (CD45+CD3+CD8+) and bulk T cells (CD45+CD3+CD8−) from Reb tumors, (iii) FACS-purified TAMs (CD45+CD11b+Gr1−) from 28d and Reb tumors. RNA-sequencing and quality control was performed at the Integrated Genomics Operation, MSKCC, or GENEWIZ, NJ. In all cases, RNA was isolated using TRIzol as per manufacturer instructions (Invitrogen), and RNA integrity was assessed by an Agilent Bioanalyzer 2100. RNA-sequencing libraries were prepared using the SMART-Seq library preparation kit and 2×50 or 2×100 base pair sequencing was performed on an Illumina HISeq 2000. Sequencing quality was assessed with FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were mapped to the mouse genome (mm10) using STAR 2.3.0e (66) with the default parameters, a minimum intron length of 70 base pairs and a maximum of 100,000 base pairs. BAM files were generated and sorted, duplicate reads were then removed using SAMTOOLS (67). Read counts were tabulated with HT-Seq using “union” mode and the iGenomes GFF file as a reference (Illumina) (68).
Gene expression analyses
Raw count data from HT-Seq was imported into R (v3.1.0) and normalized using limma voom (69). Principal component analysis was completed using the princomp function on mean centered data. A log2 fold change cutoff of 1 and a false discovery rate of 10% were applied for all differential gene expression analyses (Table S1). Significantly upregulated genes from these lists were used in gene ontology analyses using DAVID (70). Gene set variation analysis (GSVA) was performed on RNA-seq data from FACS-purified PDGFRα+ tumor cells, using the GSVA package (14) with gene sets from the C2 group from MSigDB (71). A log2 fold change cutoff of 1 and a false discovery rate cutoff of 10% were used to determine differentially enriched gene sets (fig. S3B). The spectrum model of macrophage activation was assessed with gene set enrichment analysis (GSEA), using the gsea function from the phenoTest package in R (http://rpackages.ianhowson.com/bioc/phenoTest/). A minimum P-value of <1×10−16 was used to represent significance values that were reported as 0.0 and outside of the determined distribution. Gene sets were adapted from a previous study where murine macrophages were stimulated with IFN-gamma, IL-4, TNF-alpha, TGF-beta, IL-1beta, MALP2 or CPG (27). A literature-derived IL-4 responsive gene set was generated through the use of QIAGEN’s Ingenuity iReport (www.qiagen.com/ingenuity).
Transcription factor activity analysis
Transcription factor (TF) activity analysis was performed as an adaptation of previously published methods: ISMARA (72) and RegulatorInference (73). Briefly, transcription start sites and Motevo predictions (74) of binding sites were downloaded from the Swiss Regulon (http://swissregulon.unibas.ch/fcgi/sr/downloads#). These were used to determine the number of predicted binding sites for 185 transcription factor families across all mouse promoters. Promoters were designated as 2 kilobases upstream and downstream of transcription start sites. This tabulated matrix was then used in a ridge regression to model log2 gene expression values generated by voom. Ridge regression was performed with the glmnet function in R (75). The regularization parameter, lambda, was identified for each sample through 10-fold cross validation. The coefficients for each TF family were z-scored and used as relative TF activity scores in subsequent analyses. Differentially enriched TFs were identified by using the z-scored values in limma with a log2 fold change cutoff of 1 and a false discovery rate of 10% (Table S3).
External data set analysis
RNA-seq expression data from the TCGA glioblastoma patient data set was downloaded using TCGA-assembler (76). For survival analyses, these data were filtered for patients with updated clinical information from the Broad Firehose. Correlations between IGF1 and macrophage markers (CD163, MRC1, CSF1R, CD68, AIF1) or astrocyte markers (GFAP and ALDH1L1) were assessed using a Spearman correlation coefficient. Normalized gene expression data for human bulk tumor versus tumor-associated macrophage fraction was downloaded from the GEO under accession number GSE16119 (44). Subtype calls for patients were obtained from GlioVis (http://gliovis.bioinfo.cnio.es). PI3K signature scores were tabulated with a single sample gene set enrichment, as used for macrophage activity analysis. The gene set was from the Hallmark collection from MsigDB, systematic name M5923 (43).
Data availability
All sequencing data has been deposited to the GEO under the accession number GSE69104. All code used in this project can be found at https://bitbucket.org/bowmanr/joycelab-brain-tme.
Data presentation and statistical analysis
GraphPad Prism 6.0 or R Studio was used for all data analysis. Parametric data are presented as mean ± standard error (s.e.m.) and were analyzed by an unpaired two-tailed Student’s t-test. For multiple comparisons, a one-way ANOVA with Tukey’s or Dennett’s correction was used as noted in the figure legend. Non-parametric data were analyzed by a Mann-Whitney test on ranks. For survival curves, P-values were obtained using the Log Rank (Mantel-Cox) test. Fisher’s exact test was used for histological tumor grading. A pairwise Spearman correlation test was used for correlational analyses. P<0.05 was considered as statistically significant in all cases. Principal component analyses, correlation plots, Volcano plots, heatmaps, and network plots were plotted in R Studio using base graphics (http://www.R-project.org/), rgl (http://CRAN.R-project.org/package=rgl), gplots (http://CRAN.R-project.org/package=gplots), ggplot2 (77) and qgraph packages (78).
Supplementary Material
Acknowledgments
We thank X. Chen and N. Shah for excellent technical support. We are grateful to C. Brennan for generously providing the TS573 patient tumorsphere line. We thank R. Benezra for critically reading the manuscript, and O. Olson, P. Castel and S. Schwartz for experimental advice. We thank the Animal Imaging Core, the Antitumor Assessment Core, Flow Cytometry Core, and Integrated Genomics Operation at MSKCC for assistance. The data presented in this manuscript are tabulated in the main paper and in the supplementary materials. All sequencing data has been deposited to the GEO under the accession number GSE69104, and aCGH data under the accession number GSE80399. BLZ945 is available from Novartis under a material transfer agreement with Dr. James Sutton, Novartis. DFQ, RLB and JAJ are inventors on a patent application filed by MSKCC related to the use of combination therapies with CSF-1R inhibitors for gliomas. This research was supported by the US National Cancer Institute (R01CA148967, J.A.J.), the Ludwig Institute for Cancer Research (J.A.J.), fellowships from Canadian Institutes of Health Research (D.F.Q.), NCI (F31CA167863, R.L.B.), American Brain Tumor Association (L.A.), and Obra Social Ibercaja (A.J.S.), and the MSK Cancer Center Support Grant from the NCI (P30 CA008748).
Footnotes
References
- 1.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, R. European Organisation for, T. Treatment of Cancer Brain, G. Radiotherapy, G. National Cancer Institute of Canada Clinical Trials Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008;216:15–24. doi: 10.1002/path.2370. [DOI] [PubMed] [Google Scholar]
- 5.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 6.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neoro Oncol. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruffell B, Coussens LM. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell. 2015;27:462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, Segall JE. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18:519–527. doi: 10.2119/molmed.2011.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel S, Player MR. Colony-stimulating factor-1 receptor inhibitors for the treatment of cancer and inflammatory disease. Curr Top Med Chem. 2009;9:599–610. doi: 10.2174/156802609789007327. [DOI] [PubMed] [Google Scholar]
- 11.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord JP, Levitsky H, Blay JY, Ruttinger D. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, Chmielowski B, Staddon AP, Cohn AL, Shapiro GI, Keedy VL, Singh AS, Puzanov I, Kwak EL, Wagner AJ, Von Hoff DD, Weiss GJ, Ramanathan RK, Zhang J, Habets G, Zhang Y, Burton EA, Visor G, Sanftner L, Severson P, Nguyen H, Kim MJ, Marimuthu A, Tsang G, Shellooe R, Gee C, West BL, Hirth P, Nolop K, van de Rijn M, Hsu HH, Peterfy C, Lin PS, Tong-Starksen S, Bollag G. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N Engl J Med. 2015;373:428–437. doi: 10.1056/NEJMoa1411366. [DOI] [PubMed] [Google Scholar]
- 13.Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol. 2009;2:89–95. doi: 10.1593/tlo.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brachmann SM, Kleylein-Sohn J, Gaulis S, Kauffmann A, Blommers MJ, Kazic-Legueux M, Laborde L, Hattenberger M, Stauffer F, Vaxelaire J, Romanet V, Henry C, Murakami M, Guthy DA, Sterker D, Bergling S, Wilson C, Brummendorf T, Fritsch C, Garcia-Echeverria C, Sellers WR, Hofmann F, Maira SM. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol Cancer Ther. 2012;11:1747–1757. doi: 10.1158/1535-7163.MCT-11-1021. [DOI] [PubMed] [Google Scholar]
- 16.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, T. R. Network The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Glabbeke MVan, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 18.Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012;12:170–180. doi: 10.1038/nrc3217. [DOI] [PubMed] [Google Scholar]
- 20.Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. 2015;17:34–40. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- 21.Bowman RL, Joyce JA. Therapeutic targeting of tumor-associated macrophages and microglia in glioblastoma. Immunotherapy. 2014;6:663–666. doi: 10.2217/imt.14.48. [DOI] [PubMed] [Google Scholar]
- 22.Locatelli G, Wortge S, Buch T, Ingold B, Frommer F, Sobottka B, Kruger M, Karram K, Buhlmann C, Bechmann I, Heppner FL, Waisman A, Becher B. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat Neurosci. 2012;15:543–550. doi: 10.1038/nn.3062. [DOI] [PubMed] [Google Scholar]
- 23.Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PloS One. 2011;6:e23902. doi: 10.1371/journal.pone.0023902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, McEvoy J, Roussel MF, Dyer MA, Qualls JE, Murray PJ. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Reports. 2015;12:1902–1914. doi: 10.1016/j.celrep.2015.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 27.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 28.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 30.Faler BJ, Macsata RA, Plummer D, Mishra L, Sidawy AN. Transforming growth factor-beta and wound healing. Perspect Vasc Surg Endovasc Ther. 2006;18:55–62. doi: 10.1177/153100350601800123. [DOI] [PubMed] [Google Scholar]
- 31.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 32.Lund FE, Garvy BA, Randall TD, Harris DP. Regulatory roles for cytokine-producing B cells in infection and autoimmune disease. Curr Dir Autoimmun. 2005;8:25–54. doi: 10.1159/000082086. [DOI] [PubMed] [Google Scholar]
- 33.Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM, Johnson LL, Swain SL, Lund FE. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol. 2000;1:475–482. doi: 10.1038/82717. [DOI] [PubMed] [Google Scholar]
- 34.Johansson-Lindbom B, Borrebaeck CA. Germinal center B cells constitute a predominant physiological source of IL-4: implication for Th2 development in vivo. J Immunol. 2002;168:3165–3172. doi: 10.4049/jimmunol.168.7.3165. [DOI] [PubMed] [Google Scholar]
- 35.Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci. 2005;29:381–393. doi: 10.1016/j.mcn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Forbes SJ, Rosenthal N. Preparing the ground for tissue regeneration: from mechanism to therapy. Nat Med. 2014;20:857–869. doi: 10.1038/nm.3653. [DOI] [PubMed] [Google Scholar]
- 37.Wynes MW, Riches DW. Induction of macrophage insulin-like growth factor-I expression by the Th2 cytokines IL-4 and IL-13. J Immunol. 2003;171:3550–3559. doi: 10.4049/jimmunol.171.7.3550. [DOI] [PubMed] [Google Scholar]
- 38.Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 39.Zheng WH, Kar S, Dore S, Quirion R. Insulin-like growth factor-1 (IGF-1): a neuroprotective trophic factor acting via the Akt kinase pathway. J Neural Transm Suppl. 2000:261–272. doi: 10.1007/978-3-7091-6301-6_17. [DOI] [PubMed] [Google Scholar]
- 40.Dupraz S, Grassi D, Karnas D, Nieto Guil AF, Hicks D, Quiroga S. The insulin-like growth factor 1 receptor is essential for axonal regeneration in adult central nervous system neurons. PloS One. 2013;8:e54462. doi: 10.1371/journal.pone.0054462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandez AM, Torres-Aleman I. The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci. 2012;13:225–239. doi: 10.1038/nrn3209. [DOI] [PubMed] [Google Scholar]
- 42.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 43.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murat A, Migliavacca E, Hussain SF, Heimberger AB, Desbaillets I, Hamou MF, Ruegg C, Stupp R, Delorenzi M, Hegi ME. Modulation of angiogenic and inflammatory response in glioblastoma by hypoxia. PloS One. 2009;4:e5947. doi: 10.1371/journal.pone.0005947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hermann-Kleiter N, Baier G. NFAT pulls the strings during CD4+ T helper cell effector functions. Blood. 2010;115:2989–2997. doi: 10.1182/blood-2009-10-233585. [DOI] [PubMed] [Google Scholar]
- 46.Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, Huse J, Phillips J, West BL, Nolop KB, Hsu HH, Ligon KL, Molinaro AM, Prados M. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2015 doi: 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 48.Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, O’Connor M, Pirritt C, Sun Y, Yao Y, Arnold LD, Gibson NW, Ji QS. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem. 2009;1:1153–1171. doi: 10.4155/fmc.09.89. [DOI] [PubMed] [Google Scholar]
- 49.Roehrl MH, Kang S, Aramburu J, Wagner G, Rao A, Hogan PG. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc Natl Acad Sci U S A. 2004;101:7554–7559. doi: 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loser K, Balkow S, Higuchi T, Apelt J, Kuhn A, Luger TA, Beissert S. FK506 controls CD40L-induced systemic autoimmunity in mice. J Invest Dermatol. 2006;126:1307–1315. doi: 10.1038/sj.jid.5700185. [DOI] [PubMed] [Google Scholar]
- 51.Chiba Y, Todoroki M, Nishida Y, Tanabe M, Misawa M. A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am J Respir Cell Mol Biol. 2009;41:516–524. doi: 10.1165/rcmb.2008-0163OC. [DOI] [PubMed] [Google Scholar]
- 52.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tchougounova E, Kastemar M, Brasater D, Holland EC, Westermark B, Uhrbom L. Loss of Arf causes tumor progression of PDGFB-induced oligodendroglioma. Oncogene. 2007;26:6289–6296. doi: 10.1038/sj.onc.1210455. [DOI] [PubMed] [Google Scholar]
- 54.Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell. 2010;18:619–629. doi: 10.1016/j.ccr.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7:356–368. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH, Sohn-Lee C, le Sage C, Agami R, Tuschl T, Holland EC. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327–1337. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004;64:4783–4789. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- 59.Jensen JB, Parmar M. Strengths and limitations of the neurosphere culture system. Mol Neurobiol. 2006;34:153–161. doi: 10.1385/MN:34:3:153. [DOI] [PubMed] [Google Scholar]
- 60.Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, Leversha MA, Mikkelsen T, Brennan CW. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A. 2012;109:3041–3046. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ponomarev V, Doubrovin M, Serganova I, Vider J, Shavrin A, Beresten T, Ivanova A, Ageyeva L, Tourkova V, Balatoni J, Bornmann W, Blasberg R, Tjuvajev JGelovani. A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur J Nucl Med Mol Imaging. 2004;31:740–751. doi: 10.1007/s00259-003-1441-5. [DOI] [PubMed] [Google Scholar]
- 62.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 63.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155–168. [PubMed] [Google Scholar]
- 65.Seshan VE, Olshen A. DNAcopy: DNA copy number data analysis. R package version 1.44.0 [Google Scholar]
- 66.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, S. Genome Project Data Processing The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 71.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Balwierz PJ, Pachkov M, Arnold P, Gruber AJ, Zavolan M, van Nimwegen E. ISMARA: automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res. 2014;24:869–884. doi: 10.1101/gr.169508.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Setty M, Helmy K, Khan AA, Silber J, Arvey A, Neezen F, Agius P, Huse JT, Holland EC, Leslie CS. Inferring transcriptional and microRNA-mediated regulatory programs in glioblastoma. Mol Syst Biol. 2012;8:605. doi: 10.1038/msb.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pachkov M, Erb I, Molina N, van Nimwegen E. SwissRegulon: a database of genome-wide annotations of regulatory sites. Nucleic Acids Res. 2007;35:D127–131. doi: 10.1093/nar/gkl857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu Y, Qiu P, Ji Y. TCGA-assembler: open-source software for retrieving and processing TCGA data. Nat Methods. 2014;11:599–600. doi: 10.1038/nmeth.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wickham H. ggplot2: Elegant graphics for data analysis. Springer; New York: 2009. [Google Scholar]
- 78.Epskamp S, Cramer AOJ, Waldorp LJ, Schmittmann VD, Borsboom D. qgraph: Network visualizations of relationships in psychometric data. J Stat Softw. 2012;48:1–18. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data has been deposited to the GEO under the accession number GSE69104. All code used in this project can be found at https://bitbucket.org/bowmanr/joycelab-brain-tme.