

Abstract
In an attempt to develop effective vaccines against central nervous system (CNS) tumors, we evaluated the ability of vaccines with standard dendritic cells (DC) versus type 1 polarizing DCs (DC1) to induce glioma-specific type 1 CTLs with CNS tumor-relevant homing properties and the mechanism of their action. C57BL/6 mouse-derived bone marrow cells were cultured with mouse granulocyte/macrophage colony-stimulating factor (GM-CSF) for 6 days, and CD11c+ cells were subsequently cultured with GM-CSF, rmIFN-γ, rmIFN-α, rmIL-4, and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose for 24 hours to generate DC1s. In analogy to their human counterparts, mouse DC1s exhibited surface marker profiles of mature DCs and produced high levels of IL-12 and CXCL10. Importantly for their application as cancer vaccines, such DC1s stably retained their type 1 phenotype even when exposed to type 2-promoting or regulatory T cell (Treg)-promoting environments. Consistently, mouse DC1s induced antigen-specific type 1 CTLs more efficiently than nonpolarized DCs in vitro. DC1s given s.c. migrated into draining lymph nodes, induced antigen-specific CTLs, and suppressed Treg accumulation. In addition, s.c. immunization with DC1s loaded with glioma-associated antigen (GAA)-derived CTL epitope peptides prolonged the survival of CNS GL261 glioma-bearing mice, which was associated with efficient CNS glioma homing of antigen-specific CTLs. Intratumoral injections of GAA peptide-loaded DC1s further enhanced the anti-CNS glioma effects of DC1-based s.c. immunization. Interestingly, the antitumor functions were abrogated with CXCL10−/− mouse-derived DC1s. Collectively, these findings show the anti-CNS glioma effects of DC1-based therapy and a novel role of CXCL10 in the immunologic and therapeutic activity of DC-based cancer vaccines.
Introduction
Malignant gliomas represent the most common primary central nervous system (CNS) tumors and exhibit a dismal prognosis despite recent advances made in surgical, radiological, and chemotherapeutic approaches (1). Development of novel, multi-modal therapeutic approaches is, thus, critical to improve the outcome of these deadly tumors (2). Among these approaches, immunotherapy can be promising for glioma treatment if we are able to induce potent immune responses directed against glioma-associated antigens (GAA; refs. 3–5). Indeed, several immunologic strategies have been evaluated in early-phase clinical trials; however, modest clinical outcomes of glioma immunotherapy trials call for further improvement of strategies (6).
The rejection of CNS gliomas by GAA-targeted vaccines is initiated by sufficient expansion of GAA-specific effector T cells. Vaccine-induced GAA-specific T cells are then required to home to CNS glioma sites in sufficient numbers, retain their functionality, and exert their antitumor activities. In this regard, we have previously shown that intratumoral (i.t.) injections of dendritic cells (DC) transduced with IFN-α cDNA enhance anti-CNS tumor immunity not only by promoting cross-presentation of tumor-associated antigens in draining lymph nodes (DLN; refs. 7–9) but also by enhancing CNS tumor homing of antigen-specific type 1 CTLs through the induction of CXCL10, a type 1 chemokine (10, 11). These observations point to the significance of promoting type 1 adaptive immunity for anti-CNS tumor immunotherapy.
In an attempt to develop effective vaccines against CNS tumors, we evaluated the ability of DC-based vaccines with standard DCs versus type 1 polarizing DCs (DC1) to induce glioma-specific type 1 CTLs with CNS tumor-relevant homing properties and the mechanism of their action in vivo.
Whereas human DC1s show an excellent ability to induce Th1 and CTL responses (12–14), the mechanism of their action remains unclear. In particular, although human DC1s have been recently shown to produce diminished levels of regulatory T cell (Treg)– attracting chemokines but enhanced levels of chemokines attracting Th1 cells, CTLs, and natural killer (NK) cells (15, 16), the role of chemokine production in the overall effectiveness of DC1-based vaccines has not been evaluated.
In the current study, mouse DC1s exhibit superior therapeutic activity against GL261 gliomas in vivo. Moreover, we show that the anti-CNS glioma effects by DC1-based therapy depend on CXCL10 production by DC1s. This novel mouse DC1 system allows us to address how DC1-based therapy can be applied for glioma patients and points to the significance of CXCL10 production as a critical release criterion for patients' autologous DC1s in vaccine trials.
Materials and Methods
Reagents
RPMI 1640, fetal bovine serum (FBS), l-glutamine, sodium pyruvate, β-mercaptoethanol, nonessential amino acids, antibiotics, and carboxyfluorescein diacetate succinimidyl ester (CFSE) were obtained from Invitrogen Life Technologies. Recombinant mouse granulocyte/macrophage colony-stimulating factor (rmGM-CSF), rmIFN-α, rmIL-10, rmIL-12, and rmCXCL10 were obtained from R&D Systems. Recombinant human IL-2 (rhIL-2), rmIFN-γ, and rmTGF-β1 were obtained from PeproTech. Lipopolysaccaride (LPS) was obtained from Sigma-Aldrich. Polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose (poly-ICLC) was kindly provided by Oncovir. The following peptides were synthesized by the automated solid-phase peptide synthesizer at University of Pittsburgh Peptide Synthesis Facility: H-2Kb-binding EphA2682-689 (VVSKYKPM), H-2Db-binding Garc177-85 (AALLNKLYA), H-2Db-binding human/mouse gp100 (h/mgp100)25-33 (KVPRNQDWL), and H-2Kb-binding ovalbumin (OVA)257-264 (SIINFEKL).
Cell culture
CD40L+ J558 mouse myeloma cell line was used in the current study (13). TAP2−/− RMA-S mouse thymoma cell line was kindly provided by Dr. Walter J. Storkus (University of Pittsburgh; ref. 17). GL261 mouse glioma cell line was kindly provided by Dr. Robert Prins (University of California-Los Angeles); GL261 cells are of C57BL/6 mice (H-2b)-background and express mgp100, EphA2, and Garc-1 as GAAs (18-20). GL261 culture supernatants were filtered and concentrated 10-fold by using an Amicon Ultra Filter (Millipore). GL261-conditioned medium was then prepared by mixing the concentrated supernatants and fresh medium at a ratio of 1:9. All cells were maintained in mouse complete medium (RPMI 1640 supplemented with 10% heat-inactivated FBS, 100 units/mL penicillin, 100 mg/mL streptomycin, and 10 μmol/L l-glutamine) in a humidified incubator in 5% CO2 at 37°C.
Animals
Wild-type C57BL/6 mice (H-2b) were obtained from Taconic Farms. Pmel-I, IL-12p35−/−, CXCL10−/−, and green fluorescent protein-transgenic (GFP-Tg) mice, which are all of C57BL/6-background, were obtained from The Jackson Laboratory. Pmel-I mice are transgenic for an hgp10025-33-specific TCR that cross-reacts to an mgp10025-33/H-2Kd complex (21).
Antibodies and gp100-specific tetramer
The following antibodies were obtained from BD Biosciences: anti-CCR5 (C34-3448), anti-CD11c (HL3), anti-CD40 (HM40-3), anti-CD8 (53-6.7), anti-CD80 (16-10A1), CD86 (GL1), H-2Kb (AF6-88.5), anti-I-Ab (AF6-120.1), anti-IFN-γ (XMG1.2, anti-IL- 4 (11B11), and isotype-matched controls. Anti-CCR7 (4B12) and anti-Gr-1 (RB6-8C5) antibodies were obtained from BioLegend. Anti-CXCR3 (220803) antibody was obtained from R&D Systems. Anti-CD11b (M1-70), anti-FoxP3 (FJK-16s), and anti-granzyme B (16G6) antibodies were obtained from eBioScience. H-2Db/hgp10025-33 tetramer was obtained from National Institute of Allergy and Infectious Disease Tetramer Facility at Emory University Vaccine Center.
Flow cytometry
The procedure used in the current study has been described previously (10). Briefly, single-cell suspensions were surface-stained with fluorescent dye-conjugated antibodies. For intracellular staining, cells were surface-stained, washed, fixed, permeabilized with Cytofix/Cytoperm buffer (BD Biosciences), and intracellularly stained. All stained cells were compared with those stained with isotype-matched control antibodies. Samples were examined by Coulter EPICS cytometer (Beckman Coulter); data were analyzed by the WinMDI software.8
Cytokine and chemokine release assay
The ELISA kits obtained are as follows: mIL-12p70 from eBioscience; mCXCL10, mCCL5, and mCCL22 from R&D Systems; and mIFN-γ from BD Biosciences. Culture supernatants were evaluated by specific ELISA kits.
In vitro DC preparation
The protocol used in the current study was established on the basis of our previous studies with additional procedures for DC1 generation (11, 22). Bone marrow cells (BMC) were cultured in complete medium supplemented with 10 ng/mL mGM-CSF. The medium and supplements were replaced every 3 d. On day 6, immature DCs (iDC) were purified by positive selection with CD11c magnetic beads (Miltenyi Biotec). Mature, standard DCs (sDC) were then generated by further 24-h cultivation of CD11c+ DCs with 250 ng/mL LPS and 10 ng/mL mGM-CSF. DC1s were generated from iDCs by further 24-h cultivation with 5 μg/mL poly-ICLC and 10 ng/mL each of rmIFN-γ, rmIFN-α, and rmIL-4.
In some experiments, to stimulate IL-12p70 production by DCs, CD40L+ J558 cells were added in culture at a J588/DC ratio of 1:5. In some experiments, DCs were loaded with 5 μg/mL hgp10025-33, Garc177-85, or a combination of Garc177-85 and EphA2682-689 peptides, incubated for 4 h at 37°C, and washed twice with PBS. In s.c. immunization experiments, on days 0 and 10, recipient mice received s.c. injections of 1 × 106 DCs loaded with GAA-derived peptides mentioned above with 100 μL PBS.
In vivo DC migration assay
DCs were generated from C57BL/6-background GFP-Tg mice; in some experiments, DC1s were generated from either wild-type or CXCL10−/− mice and labeled with 0.1 μmol/L CFSE. On day 0, recipient mice received s.c. injection of 3 × 106 DCs in 0.1 mL PBS without peptide loading. At 48 h, GFP+ or CFSE+ cells in DLNs were evaluated by flow cytometry.
In vitro CTL preparation
The procedure used in the current study has been described previously (23). Briefly, CD8+ T cells were enriched from Pmel-I mouse-derived spleen cells (SPC) by positive selection with CD8 magnetic beads (Miltenyi Biotec) and stimulated with DCs loaded with 5 μg/mL hgp10025-33 peptide in the presence of 100 units/mL rhIL-2. CD8+ T cells were cultured at 6 to 9 d for in vitro cytolytic assay and 8 to 12 d for i.v. injection.
In vitro cytolytic assay
The procedure used in the current study has been described previously (11). Briefly, target RMA-S cells were pulsed with 5 μg/mL of either mgp10025-33 or OVA257-264 peptide and labeled with 100 μCi (Amersham). The labeled cells (100 μL) were then incubated with effector cells (100 μL) in a U-bottomed 96-well microtiter plate (Corning). After a 4-h incubation at 37°C, 50 μL supernatants were harvested from each well and transferred to wells of a LumaPlate-96 (Packard, Inc.). The amount of 51Cr in each well was measured in a Microplate Scintillation Counter (Packard, Inc.). The percentage of antigen-specific cytolysis was calculated as follows: 100 × (experimental release − spontaneous release) / (maximum release − spontaneous release).
In vivo CTL proliferation assay
On day – 1, wild-type mice received i.v. injection of 5 × 106 Pmel-I mouse-derived, naive CD8+ T cells labeled with 1 μmol/L CFSE. On day 0, mice received s.c. immunization with DCs loaded with 5 μg/mL hgp10025-33 peptide. On day 6, proliferation of CFSE+ cells in DLNs was evaluated by flow cytometry.
In vivo cytolytic assay
The procedure used in the current study has been described previously (24). Briefly, on day 0, wild-type mice received s.c. immunization of DCs loaded with 5 μg/mL Garc177-85 peptide. On day 6, SPCs were obtained from naive C57BL/6 mice and divided into two groups: (a) a CFSEhi group, consisting of SPCs pulsed with 5 μg/mL Garc177-85 peptide, incubated for 1 h at 37°C, and labeled with 1 μmol/L CFSE, and (b) a CFSElo group, consisting of SPCs labeled with 0.1 μmol/L CFSE without peptide loading. Mice then received i.v. injection of 1 × 107 cells from each group. At 4 h, SPCs were evaluated by flow cytometry for the proportion of CFSElo and CFSEhi cells.
Intracranial injection of GL261 glioma cells, DCs, and rmCXCL10
The procedure used in the current study has been described previously (10,23, 24). Briefly, using a Hamilton syringe (Hamilton Company), 1 × 105 GL261 cells in 2 μL PBS, 1 to 3 × 106 DCs in 2 μL PBS, or 1 μg rmCXCL10 in 1 μL PBS were stereotactically injected through an entry site at the bregma, 3 mm to the right of sagittal suture, and 4 mm below the surface of the skull of anesthetized mice by using a stereotactic frame (Kopf). In some experiments, symptom-free survival was monitored as the primary end point; in other experiments, treated mice were sacrificed on indicated days to evaluate immunologic end points, such as brain infiltrating lymphocytes (BIL).
BIL isolation
The procedure used in the current study has been described previously (25). Briefly, mice were sacrificed by CO2 asphyxia and immediately perfused with PBS through the left cardiac ventricle. Brain tissues were mechanically minced, resuspended in 70% Percoll (Sigma-Aldrich), overlaid with 37% and 30% Percoll, and centrifuged for 20 min at 500 × g. Enriched BIL populations were recovered at the 70% to 37% Percoll interface.
Two-photon excitation microscopy
The Olympus FV1000MPE multi-photon laser scanning microscope (Olympus Corp.) was used to image GFP in sections of brain tissues and cervical lymph nodes (CLN). The Olympus Fluoview software controlled data acquisition. The mode-locked Chameleon Ultra Ti:Sapphire laser (Coherent, Inc.) produced illumination for two-photon excitation at 820 nm. The Olympus 60-fold objective with numerical aperture of 0.70 was used with a 2-fold digital zoom to optically section tissue at a 2-μm step size. Fluorescence emission was collected with an external photomultiplier tube using a 520/40 nm bandpass filter. Scan resolution was set to 1,024 × 1,024 pixels at 4,096 gray scales.
Statistical analysis
The statistical significance of differences between groups was determined by one-way ANOVA with Holm's post-hoc test. Survival data were analyzed by log-rank test. We considered differences significant when P < 0.05. All data were analyzed by the R environment.9
Results
Requirements for cytokines and TLR ligands in the generation of mouse DC1s
We first determined the requirements for a combination of cytokines and TLR ligands to polarize mouse DC1s effectively. As DC maturation markers, we evaluated DC expression of the following molecules: MHC class II (I-Ab), costimulatory molecule CD80, and chemokine receptor CCR7, which primarily mediates DC migration toward DLNs (26). We also measured DC production of IL-12p70, which is a key cytokine for type 1 adaptive immunity (26). As shown in Supplementary Fig. S1, the expression of maturation markers and production of IL-12p70 depended greatly on TLR3 ligand poly-ICLC and IFN-γ; at lesser degrees on IFN-α and IL-4. Therefore, we used poly-ICLC, IFN-γ, IFN-α, and IL-4 for the generation mouse DC1s in the current study, as detailed in Materials and Methods.
Mouse DC1s stably retain their type 1 polarizing phenotype when exposed to type 2–promoting or Treg-promoting environments
We characterized mouse DC1s for their production of cytokines and chemokines, as well as their surface phenotype in vitro. As shown in Fig. 1A, compared with iDCs, both sDCs and DC1s exhibited higher levels of MHC class I (H-2Kb) and class II (I-Ab), costimulatory molecules CD80/86, and the chemokine receptor CCR7, which is consistent with mature DC phenotype. As shown in Fig. 1B, DC1s produced significantly higher levels of type 1 cytokine IL-12p70 and type 1 chemokines CXCL10 and CCL5 than sDCs or iDCs upon stimulation with CD40L. By contrast, DC1s suppressed the production of CCL22, a chemokine that primarily attracts Tregs (16, 27). As shown in Fig. 1C, when these DCs were subsequently exposed to type 2–promoting or Treg-promoting factors (IL-10, TGF-β, or GL261 glioma-conditioned media), only DC1s were able to maintain their production of IL-12p70 and CXCL10. Collectively, these data show that mouse DC1s stably retain their type 1 polarizing phenotype even when they are subsequently exposed to type 2–promoting or Treg-promoting environments.
Figure 1.
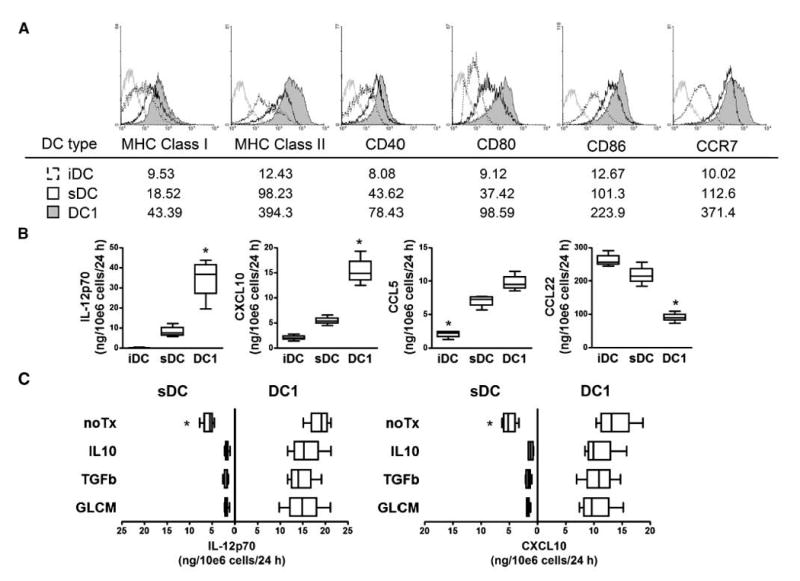
Mouse DC1s stably retain their type 1 polarizing phenotype when exposed to type 2–promoting or Treg-promoting environments. On day 0, C57BL/6 mouse–derived BMCs were cultured with 10 ng/mL rmGM-CSF. On day 6, iDCs were obtained by positive selection with CD11c magnetic beads; sDCs were then generated by further 24-h cultivation of CD11c+ DCs with 250 ng/mL LPS and 10 ng/mL rmGM-CSF. CD11c+ DC1s were generated from iDC by further 24-h cultivation with 5 μg/mL poly-ICLC and 10 ng/mL each of rmIFN-γ, rmIFN-α, and rmIL-4. A, on day 7, CD11c+ cells were evaluated by flow cytometry for DC maturation markers. Histograms represent the following DC types: DC1s (shaded), sDCs (open), iDCs (dashed line), and DCs stained with isotype control IgG (gray line). Numbers under each histogram represent mean fluorescent intensity (MFI) for each DC type. B, the amount of IL-12p70, CXCL10, CCL5, and CCL22 produced for the last 24 h was evaluated after stimulation of DCs by CD40L-transduced J558 cells. *, P < 0.05, compared with other groups with the same DC type. C, DC1s and sDCs were subsequently cultured in the presence of either 10 ng/mL rmIL-10, 10 ng/mL rmTGF-β1, or GL261-conditioned medium (GLCM) for another 24 h, followed by CD40L stimulation for another 24 h. The amount of IL-12p70 and CXCL10 produced for the last 24 h was evaluated. *, P < 0.05 compared with other groups in the same DC type.
Mouse DC1s efficiently induce antigen-specific type 1 CTLs in vitro
We then evaluated the ability of mouse DC1s to induce type 1 effector CTLs through their antigen presentation in vitro (Fig. 2). DC1s, control sDCs, or iDCs were loaded with hgp10025-33 peptide and cocultured with Pmel-I mouse-derived, gp10025-33-specific CD8+ T cells. DC1s induced superior levels of intracellular IFN-γ, granzyme B, and CXCR3 in responder CD8+ T cells when compared with sDCs or iDCs, whereas the expression levels of IL-4 and CCR5 were comparable in DC1-stimulated versus sDC-stimulated CD8+ T cells (Fig. 2A). Furthermore, DC1-stimulated T cells produced significantly higher levels of IFN-γ in culture supernatants (Fig. 2B) and exhibited a superior antigen-specific CTL activity against both antigen-pulsed RMA-S cells and syngeneic GL261 cells (Fig. 2C) when compared with sDC-stimulated or iDC-stimulated cells.
Figure 2.
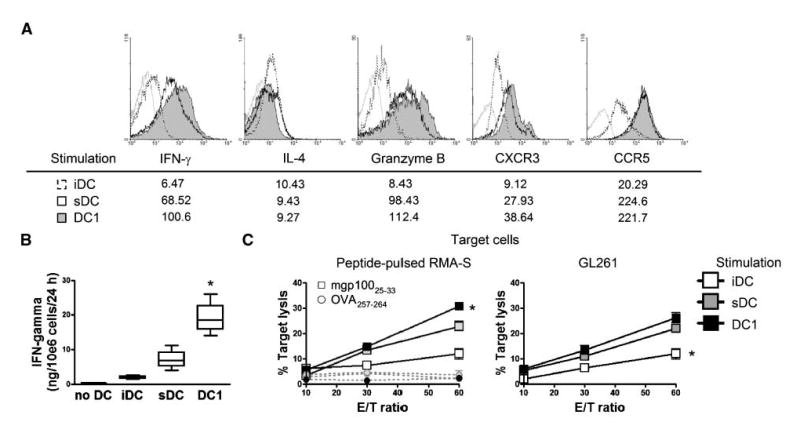
Mouse DC1s efficiently induce antigen-specific type 1 CTLs in vitro. Naive Pmel-I CD8+ T cells were stimulated with DC1s, sDCs, or iDCs loaded with 5 μg/mL hgp10025-33 peptide in the presence of 50 units/mL rhIL-2 for 6 d. A, CD8+ T cells were evaluated by flow cytometry for the intracellular expression of IFN-γ, IL-4, granzyme B, CXCR3, and CCR5. Histograms represent T cells stimulated with the following DC types: DC1s (shaded), sDCs (open), and iDCs (dashed line). Gray lines, T cells stained with isotype control IgG. Numbers indicated under histograms represent MFI for T cells stimulated with each DC type. B, IFN-γ secretion of T cells into the culture medium was evaluated by ELISA. *, P < 0.05 compared with other groups. C, cytolytic ability of effector T cells against target cells was evaluated by 51Cr releasing assay. Left, target RMA-S cells were pulsed with 5 μg/mL of either mgp10025-33 (square) or OVA257-264 (circle) peptide. Right, GL261 cells were used as target cells. *, P < 0.05 compared with other groups at the same E/T ratio.
Mouse DC1s efficiently migrate into DLNs, induce antigen-specific CTLs, and suppress the accumulation of Tregs in vivo
We subsequently evaluated the ability of GAA peptide-loaded DC1s to induce antigen-specific CTLs in vivo (Fig. 3). As shown in Fig. 3A, at 48 h after s.c. immunization with DCs derived from GFP-Tg mice, GFP + DC1s were found in DLNs at higher numbers than sDCs or iDCs, which is consistent with the high-level expression of CCR7 on DC1s (Fig. 1A). As shown in Fig. 3B, CFSE-labeled, Pmel-I– derived CD8+ T cells exhibited a higher degree of proliferation after s.c. immunization with hgp10025-33-loaded DC1s than immunization with sDCs or iDCs. In vivo CTL assays with Garc177-85 peptide showed that DC1-based s.c. immunization induced higher levels of Garc177-85-specific CTL responses (Fig. 3C) than sDC-based or iDC-based immunization. DC1-based immunization also led to a reduction of FoxP3+CD4+ Tregs that accumulated in DLNs (Fig. 3D), suggesting that the DC1s that migrated into DLNs may modulate the overall immunologic milieu, perhaps as a result of their type 1 chemokine production profiles (Fig. 1). Collectively, these data show that mouse DC1s efficiently migrate into DLNs, induce antigen-specific CTLs, and may suppress the accumulation of Tregs in DLNs.
Figure 3.
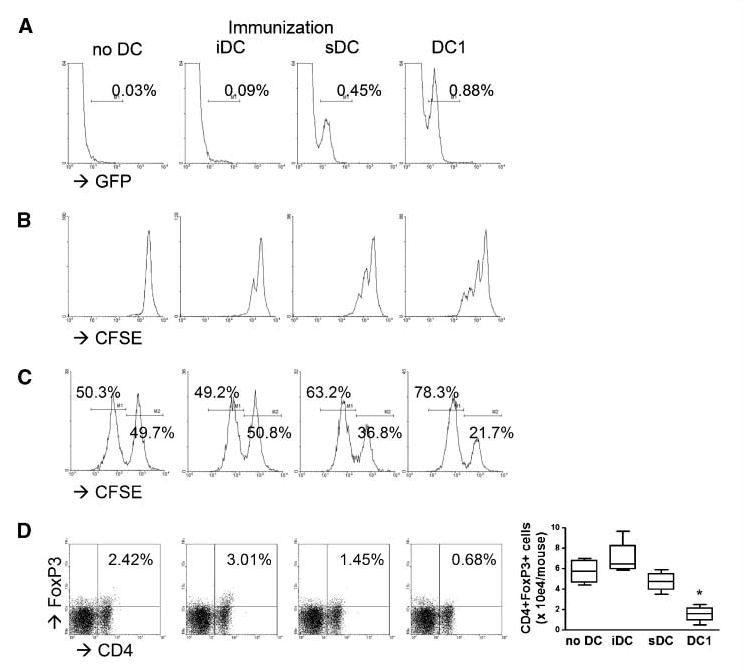
Mouse DC1s efficiently migrate into DLNs, induce antigen-specific CTLs, and suppress Treg accumulation in vivo. On day 0, wild-type mice received s.c. immunization with either DC1s, sDCs, or iDCs. A, mice received 3 × 106 GFP+ DCs. GFP+ cells in DLNs were evaluated by flow cytometry at 48 h. Numbers in each histogram represent the percentage of GFP+ cells in monocyte-gated populations. B, DCs were loaded with 5 μg/mL hgp10025-33 peptide. On day – 1, mice received i.v. injection of 5 × 106 naive Pmel-I CD8+ T cells labeled with CFSE. On day 6, proliferation of CFSE-positive subpopulations in iDLNs was analyzed. C, DCs were loaded with 1 × 106 Garc177-85 peptide. On day 6, cytolytic ability of effector T cells against target cells was evaluated by in vivo cytolytic assay, as described in Materials and Methods. D, on day 6, DLNs were evaluated by flow cytometry for FoxP3+CD4+ cells. Numbers in each dot plot represent the percentage of double-positive cells in lymphocyte-gated subpopulations in DLNs. Absolute numbers of double-positive cells are enumerated in the right box plots. *, P < 0.05 compared with other groups.
Peripheral immunization with DC1s prolongs survival of CNS glioma-bearing mice associated with the efficient CNS glioma homing of antigen-specific CTLs
We next evaluated whether DC1-based peripheral s.c. immunization would promote effective therapeutic immunity against CNS GL261 gliomas (Fig. 4). CNS GL261-bearing mice received s.c. immunization with 1 × 106 DC1s, sDCs, or iDCs loaded with GL261 glioma-derived CTL epitope peptides, Garc-177-85 (H-2Db), and EphA2682-689 (H-2Kb), on days 0 and 10 after the intracranial inoculation of GL261 glioma cells. As shown in Fig. 4A, immunization with GAA peptide-loaded DC1s prolonged the survival of CNS glioma-bearing mice significantly longer than that with sDCs, iDCs, or mock PBS treatment. When the immune cell infiltration in CNS glioma sites was examined using naive Pmel-I–derived CD8+ T cells adoptively transferred and their relevant hgp10025-33-loaded DCs (Fig. 4B–D), DC1-based s.c. immunization resulted in a significant increase of antigen-reactive CD8+ T cells but a decrease of FoxP3+CD4+ Tregs and Gr-1+CD11b+ myeloid suppressor cells in CNS glioma sites when compared with sDC-based or iDC-based immunization. Collectively, these data show that peripheral s.c. immunization with DC1s prolongs the survival of CNS glioma-bearing mice, which was associated with the efficient CNS glioma homing of antigen-specific CTLs.
Figure 4.
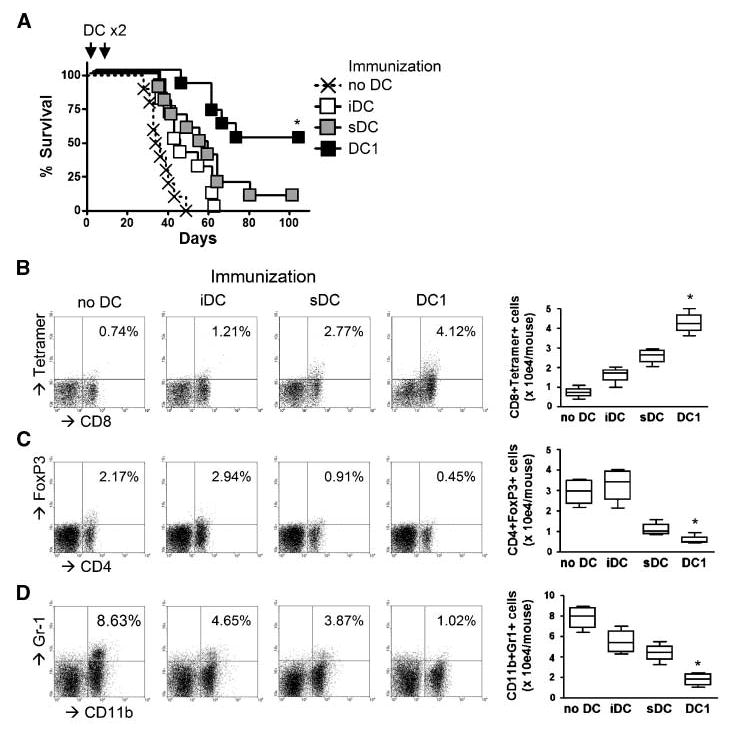
Peripheral immunization with DC1s prolongs survival of CNS glioma-bearing mice, associated with the efficient CNS glioma homing of antigen-specific CTLs. On day 0, each syngeneic mouse received a stereotactic injection with 1 × 105 GL261 glioma cells into the right basal ganglia. On days 0 and 10, mice received s.c. immunization with 1 ×106 GAA-loaded DC1s, sDCs, or iDCs. A, DCs were loaded with a combination of Garc177-85 and EphA2682-689 peptides. Symptom-free survival was monitored. *, P < 0.05 compared with other groups. B-D, DCs were loaded with hgp10025-33 peptide. On day –1, mice received i.v. infusion of 5 × 106 naive Pmel-I CD8+ T cells. On day 16, BILs were evaluated by flow cytometry for the following subpopulations: gp100-reactive CD8+ cells (B), FoxP3+CD4+ Tregs (C), and Gr-1+CD11b+ myeloid suppressor cells (D). Numbers in each dot plot represent the percentage of double-positive cells in lymphocyte-gated subpopulations in BILs. Absolute numbers of double-positive cells are enumerated in the right box plots. *, P < 0.05 compared with other groups.
Intratumoral injection of DC1s enhances anti-CNS glioma effects of s.c. immunization with DC1s
We subsequently addressed whether i.t. injections of DC1s would promote or enhance the therapeutic efficacy against CNS GL261 gliomas. To this end, we first examined the persistence of DC1s after i.t. injections. As shown in Fig. 5A, i.t. injected GFP+ DC1s seemed to persist better than sDCs in both the CNS glioma sites and CLNs, which are regional DLNs of the brain. We then addressed whether i.t. injections of DC1s would prolong the survival of CNS glioma-bearing mice. As shown in Fig. 5B, both s.c. and i.t. DC1 injections prolonged the survival of CNS glioma-bearing mice better than mock PBS injections. Remarkably, the combination of both s.c. and i.t. DC1 injections resulted in the highest long-term survival rate with 5 of 10 mice surviving at day 100 after tumor inoculation. When we examined the infiltration of antigen-reactive CD8+ T cells in CNS glioma sites (Fig. 5C), s.c. injections of DC1s recruited significantly higher numbers of antigen-reactive CD8+ T cells when compared with i.t. injections alone. Addition of i.t. DC1 injections in the s.c. DC1 regimen led to a further improvement of T cell homing to the CNS glioma sites. Collectively, these data show that i.t. injections of DC1s enhance anti-CNS glioma effects of DC1-based s.c. immunization.
Figure 5.
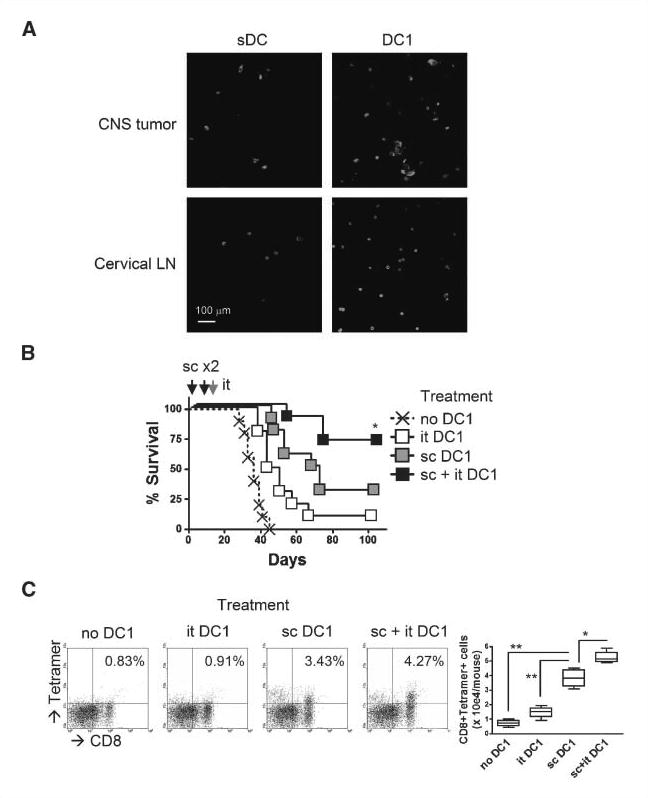
Intratumoral injection of DC1s enhances anti-CNS glioma effects of s.c. immunization with DC1s. A mice bearing day 21 CNS GL261 glioma received i.t. injection of 3 × 106 GFP+ DC1s or GFP+ sDCs. On day 27, brain sections and ipsilateral CLNs were evaluated by two-photon microscopy for GFP+ cells. Original magnification, 60×. B and C, mice received either one of the following combinations: (a) s.c. PBS and i.t. PBS, (b) s.c. PBS and i.t. GAA-loaded DCs, (c) s.c. GAA-loaded DCs and i.t. PBS, or (d) s.c. GAA-loaded DCs and i.t. GAA-loaded DCs. For s.c. immunization on days 0 and 10, 1 × 106 GAA-loaded DC1s were injected with 100 μL PBS; for i.t. injections on day 16, 1 × 106 GAA-loaded DC1s were injected with 2 μL PBS. B, DCs were loaded with a combination of Garc177-85 and EphA2682-689 peptides. Symptom-free survival was monitored. *, P < 0.05 compared with other groups. C, DCs were loaded with hgp10025-33 peptide. On day –1, mice received i.v. injection of 5 × 106 naive Pmel-I CD8+ T cells. BILs were evaluated by flow cytometry on day 21 for gp100-reactive CD8+ cells. Numbers in each dot plot represent the percentage of double-positive cells in lymphocyte-gated subpopulations in BILs. Absolute numbers of double-positive cells are enumerated in the right box plots. *, P < 0.05 compared with sDC group; **, P < 0.01 compared with no DC1 and i.t. DC1 groups.
DC1-derived CXCL10 plays a significant role in both induction of antigen-specific CTLs and their CNS glioma homing
Based on our previous observations (10, 11), we hypothesized that CXCL10 produced by i.t. injected DC1 would play a major role in CNS glioma homing of antigen-specific CTLs. We generated CXCL10−/− mouse-derived DC1s that exhibited similar surface expression levels of MHC class II (I-Ab), CD80, and CCR7 compared with wild-type DC1s (Fig. 6A, top). CXCL10−/− DC1s were able to migrate to DLNs after s.c. injections as efficiently as wild-type DC1s (Fig. 6A, bottom).
Figure 6.
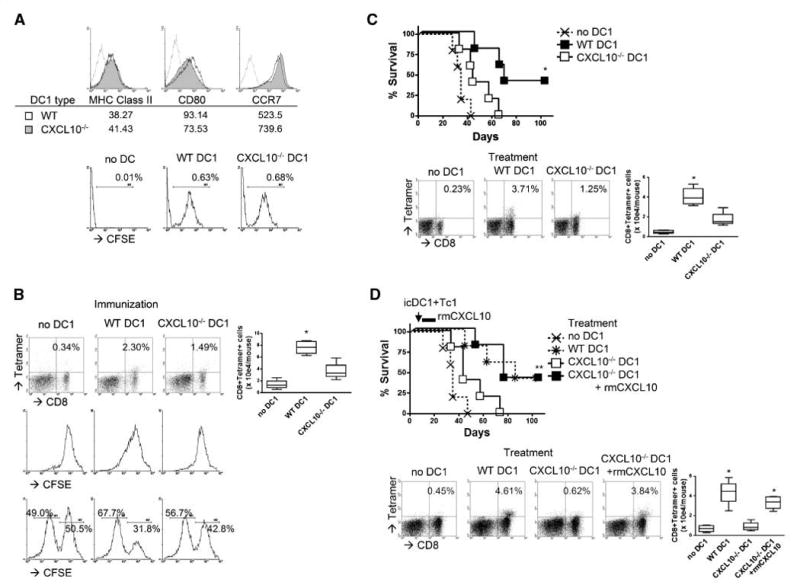
DC1-derived CXCL10 plays a significant role in both induction of antigen-specific CTLs and their CNS glioma homing. DC1s were generated from either wild-type or CXCL10−/− mice. A, top, CD11c+ cells were evaluated by flow cytometry for DC maturation markers. Histograms represent the following DC types: CXCL10−/− DC1s (shaded), wild-type DC1s (open), and DCs stained with isotype control IgG (gray line). Numbers indicated under each histogram represent MFI for each DC type. Bottom, on day 0, wild-type mice received s.c. immunization with 3×106 CFSE-labeled DC1s. CFSE+ cells in DLNs were evaluated by flow cytometry at 48 h. Numbers in each dot plot represent the percentage of double-positive cells in monocyte-gated subpopulations in DLNs. B, on day 0, wild-type mice received s.c. immunization with GAA-loaded DC1s. Top and center, DCs were loaded with 5 μg/mL hgp10025-33 peptide. On day –1, mice received i.v. injection of naive Pmel-I CD8+ T cells. Top, on day 6, DLNs were evaluated by flow cytometry for gp100-reactive CD8+ cells. Numbers in each dot plot represent the percentage of double-positive cells in lymphocyte-gated subpopulations. Center, T cells injected were labeled with CFSE. On day 6, proliferation of CFSE+ cells were evaluated by flow cytometry. Bottom, DCs were loaded with 1 × 106 Garc177-85 peptide. On day 6, cytolytic ability of effector T cells against target cells was evaluated by in vivo cytolytic assay, as described in Materials and Methods. C, on days 0 and 10, CNS glioma-bearing mice received s.c. immunization with GAA-loaded DC1s. Top, DCs were loaded with a combination of Garc177-85 and EphA2682-689 peptides. Symptom-free survival was monitored. *, P < 0.05 compared with other groups. Bottom, DCs were loaded with hgp10025-33 peptide. On day –1, mice received i.v. injection of naive Pmel-I CD8+ T cells. On day 16, BILs were evaluated by flow cytometry for gp100-reactive CD8+ cells. D, on day 12, CNS glioma-bearing mice received i.t. injection of hgp10025-33-loaded DC1s and i.v. injection of Pmel-I–derived, ex vivo–expanded type 1 CTLs. For a group of CXCL10−/− DC1s, 1 μg rmCXCL10 in 1 μL PBS was i.t. injected on days 12, 15, and 18. Top, symptom-free survival was monitored. **, P < 0.05 both wild-type (WT) DC1 and CXCL10−/− DC1 with rmCXCL10groups compared with either no DC control or CXCL10−/− DC1 groups. Bottom, BILs were evaluated by flow cytometry on day 21 for gp100-reactive CD8+ cells. B–D, numbers in each dot plot represent the percentage of double-positive cells in lymphocyte-gated subpopulations in BILs. Absolute numbers of double-positive cells are enumerated in the right box plots. *, P < 0.05 compared with other groups (B and C) and compared with no DC1 and CXCL10−/− DC1 groups (D).
To our surprise, as shown in Fig. 6B, s.c. immunization with GAA peptide-loaded CXCL10−/− DC1s induced decreased levels of antigen-reactive CD8+ T cells, as shown by enumeration in DLNs (top), proliferation (center), and antigen-specific cytolytic ability (bottom) of in vivo induced CTLs compared with wild-type DC1s, indicating that CXCL10 plays a role in the induction of antigen-specific CTLs.
We subsequently examined the significance of CXCL10 produced by DC1s in the s.c. immunization model. As shown in Fig. 6C, lack of DC1-derived CXCL10 partially abrogated the therapeutic effects of DC1-based s.c. immunization (top; P = 0.0936 compared with the no DC control group), which was associated with the decreased CNS glioma homing of antigen-reactive CD8+ T cells (bottom).
We then examined the significance of CXCL10 produced by DC1s in the i.t. injection model. As shown in Fig. 6D, DC1-derived CXCL10 was at least partially responsible in the therapeutic effects and CNS glioma homing of antigen-reactive CD8+ T cells, as shown by injections of CXCL10−/− DC1s. In a group of mice receiving CXCL10−/− DC1s, coinjections of rmCXCL10 recovered the therapeutic efficacy and CNS glioma homing of antigen-specific CD8+ cells.
Collectively, these data show a major role of DC1-derived CXCL10 both in the systemic induction of antigen-specific CTLs and their CNS glioma homing.
Discussion
In the current study, we focused on the development of DC1-based therapeutic strategies for gliomas. The results shown here support our hypothesis that immunization with GAA peptide-loaded DC1s promotes desirable type 1 adaptive immune responses against CNS gliomas, with the effects being significantly dependent on the DC production of CXCL10. In particular, a combination approach of s.c. immunization with and i.t. injection of DC1s showed remarkable anti-CNS glioma effects (Fig. 5), indicating the possibility to use DC1s to jointly target the afferent and efferent phases of immune responses.
Several protocols to generate DC1s from mouse BMCs have been proposed previously (28, 29). Compared with other mouse DC1s, our DC1s generated in the current study exhibited the following unique and novel biological properties: (a) high-level production of not only IL-12 but also type 1 chemokines, such as CXCL10 (Fig. 1B) and (b) commitment to produce IL-12 and CXCL10 even when they are exposed to IL-10, TGF-β, or glioma-conditioned medium (Fig. 1C). These data indicate that mouse DC1s share the hallmark characteristics of human αDC1 in terms of their ability to produce type 1 cytokines and chemokines at high levels (12, 15).
Of great interest is the observation that CXCL10 is required in the CNS glioma homing, as well as the induction of effector CTLs (Fig. 6A and B), because its receptor CXCR3 is expressed on activated T cells at high levels (30). In this regard, Dufour and colleagues have shown the role of CXCL10 in both the induction and homing of effector T cells using CXCL10−/− mice (31). They hypothesized two possible mechanisms by which CXCL10 influences the induction of effector T cells. One possibility is that CXCL10 may influence DC migration to DLNs, thereby affecting CTL induction. However, our observation does not support this possibility because both wild-type and CXCL10−/− DC1s migrate to DLNs at a comparable efficiency (Fig. 6A). The other possibility is that CXCL10 may costimulate T-cell activation in an autocrine loop because activated effector T cells also release CXCL10 (32). In our current models with CXCL10−/− DC1s, T cells in the recipient wild-type mice should be able to produce CXCL10. Nevertheless, s.c. immunization with CXCL10−/− DC1s failed to induce antigen-specific CTLs, pointing the significance of DC1-derived CXCL10 for induction of adaptive immune responses. Gustasfsson and colleagues have reported recently that αDC1s attract NK cells in vitro in a manner dependent on CXCR3, which is a receptor for CXCL10, leading to IFN-γ production from NK cells (15). Therefore, it may be due to the lack of NK-cell recruitment to the DLNs and their IFN-γ production that s.c. immunization with CXCL10−/− DC1s failed in CTL induction (Fig. 6B). We have recently reported that effector CTLs, but not memory T cells, kill antigen-loaded DCs in DLNs in an antigen-specific manner, thereby diminishing the effect of DC-based immunization (33). However, this is not likely to be the case in the current study because the CXCL10−/− DCs might not be able to attract effector CXCR3+ CTLs efficiently. Precise mechanisms by which DC1-derived CXCL10 promotes the induction of effector CTLs DC1s need to be elucidated.
In addition to CXCL10, other DC1-derived factors may play a pivotal role in inducing type 1 immune response. We and others have described DC-derived IL-12 as a critical factor in the induction of type 1 immunity (26, 34). To corroborate this point in our current mouse DC1 model, we generated DC1s from IL-12p35−/− mice (Supplementary Fig. S2). Absence of functional IL-12p70 production almost completely abrogated abilities of DC1s to induce type 1 CD8+ T-cell response in vitro and therapeutic efficacy, as well as T-cell trafficking to the CNS tumors in vivo. With regard to other DC1-derived chemokines, it was recently reported that human αDC1s overproduce CXCL9-11 and CCL5, which altogether contribute to the induction of type 1 immune response (15). It is, therefore, possible that these chemokines contribute to the observed beneficial effects in our glioma-bearing mice as well. Further studies are clearly warranted to elucidate the roles of these chemokines. Human αDC1s produce reduced levels of CCL22, thereby diminishing attraction of Tregs (16). These may explain our observation in the current study that immunizations with mouse DC1s resulted in reduced accumulation of Treg cells in DLNs (Fig. 3D).
DCs need to be licensed by CD40-CD40L engagement to induce a burst of IL-12, as shown by our previous study (13). However, in the current study, we did not use CD40L stimulation of DCs to evaluate their ability to induce CTL responses in vitro. This is because, in our preliminary studies, in vitro stimulation of DC1s with CD40L+ J558 cells resulted in the death of both DC1s and T cells even when relatively few CD40L+ J558 cells were used. We reason that supraphysiologic levels of CD40-CD40L engagement in mouse DC/T-cell interaction in vitro may induce excessive production of IL-12 followed by IFN-γ, leading to the overproduction of nitric oxide, to which mouse immune cells are highly susceptible (35-37). However, our in vivo DC1 administrations consistently resulted in superior levels of CTL induction compared with other DC types, suggesting that the excessive CD40-CD40L interaction may not be a concern in vivo.
Our data in Fig. 5 suggest that i.t.-given DC1s may induce antigen-specific CTL responses in draining CLNs. Based on our data with s.c. injected CXCL10−/− DC1s, we presume that i.t. injected DC1s also depend on endogenous CXCL10 for the induction of antigen-specific CTLs. Accordingly, i.t. injection of CXCL10−/− DC1s did not induce significant therapeutic responses (Fig. 6C). On the other hand, complementary administration of rmCXCL10 along with i.t. CXCL10−/− DC1 delivery recovered the therapeutic effects of i.v. type 1 CTL transfer (Fig. 6D). We think this was because the adoptive transfer of in vitro activated type 1 CTLs bypassed the necessity of endogenous CTL induction, which might have been diminished in mice receiving i.t. CXCL10−/− DC1s. Therefore, the observed recovery of CTL infiltration to the glioma site is likely due to the local chemoattractive effects of rmCXCL10.
In our experiments with i.t. DC1 injections, we loaded DC1s ex vivo with synthetic GAA-derived peptides. Based on our previous study (8) and the current data (Fig. 5A), i.t. injected DC1s seem to migrate into CLNs, regional DLNs of the brain, wherein they present antigen to reactive T cells. One may raise a question whether DC1s needed to be loaded with exogenous peptides to elicit therapeutic responses in spite of abundant availability of tumor antigens in situ. Immature DCs are highly phagocytic. During their maturation process, however, DCs lose the ability to engulf antigens while they gain the ability to migrate into DLNs and present antigens to naive T cells efficiently (26). Our DC1s exhibited mature DC phenotype (Fig. 1A) and, thus, might not be highly phagocytic. Therefore, in the current study, we elected to load DC1s ex vivo with synthetic GAA-derived peptides. However, it remains to be determined whether DC1s are able to phagocytose and present tumor-derived antigens efficiently.
In summary, the current study shows that mouse DC1s can induce superior therapeutic activity against CNS GL261 gliomas in vivo and that CXCL10 production is significant to the in vivo effectiveness of cancer vaccines.
Supplementary Material
Acknowledgments
Grant support: Grants 1R01NS055140-01, 1P01CA100327, and 1P01NS40923 (H. Okada); 1R01CA095128 (P. Kalinski).
Footnotes
Note:Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozo-lomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Selznick LA, Shamji MF, Fecci P, Gromeier M, Friedman AH, Sampson J. Molecular strategies for the treatment of malignant glioma-genes, viruses, and vaccines. Neurosurg Rev. 2008;31:141–55. doi: 10.1007/s10143-008-0121-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eguchi J, Hatano M, Nishimura F, et al. Identification of interleukin-13 receptor α2 peptide analogues capable of inducing improved antiglioma CTL responses. Cancer Res. 2006;66:5883–91. doi: 10.1158/0008-5472.CAN-06-0363. [DOI] [PubMed] [Google Scholar]
- 4.Okano F, Storkus WJ, Chambers WH, Pollack IF, Okada H. Identification of a novel HLA-A*0201-restricted, cytotoxic T lymphocyte epitope in a human glioma-associated antigen, interleukin 13 receptor α 2 chain. Clin Cancer Res. 2002;8:2851–5. [PubMed] [Google Scholar]
- 5.Hatano M, Eguchi J, Tatsumi T, et al. EphA2 as a glioma-associated antigen: a novel target for glioma vaccines. Neoplasia. 2005;7:717–22. doi: 10.1593/neo.05277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpentier AF, Meng Y. Recent advances in immunotherapy for human glioma. Curr Opin Oncol. 2006;18:631–6. doi: 10.1097/01.cco.0000245321.34658.f4. [DOI] [PubMed] [Google Scholar]
- 7.Tsugawa T, Kuwashima N, Sato H, et al. Sequential delivery of interferon-α gene and DCs to intracranial gliomas promotes an effective antitumor response. Gene Ther. 2004;11:1551–8. doi: 10.1038/sj.gt.3302300. [DOI] [PubMed] [Google Scholar]
- 8.Okada H, Tsugawa T, Sato H, et al. Delivery of interferon-α transfected dendritic cells into central nervous system tumors enhances the antitumor efficacy of peripheral peptide-based vaccines. Cancer Res. 2004;64:5830–8. doi: 10.1158/0008-5472.CAN-04-0130. [DOI] [PubMed] [Google Scholar]
- 9.Kuwashima N, Nishimura F, Eguchi J, et al. Delivery of dendritic cells engineered to secrete IFN-α into central nervous system tumors enhances the efficacy of peripheral tumor cell vaccines: dependence on apopto-tic pathways. J Immunol. 2005;175:2730–40. doi: 10.4049/jimmunol.175.4.2730. [DOI] [PubMed] [Google Scholar]
- 10.Nishimura F, Dusak JE, Eguchi J, et al. Adoptive transfer of type 1 CTL mediates effective anti-central nervous system tumor response: critical roles of IFN-inducible protein-10. Cancer Res. 2006;66:4478–87. doi: 10.1158/0008-5472.CAN-05-3825. [DOI] [PubMed] [Google Scholar]
- 11.Fujita M, Zhu X, Sasaki K, et al. Inhibition of STAT3 promotes the efficacy of adoptive transfer therapy using type-1 CTLs by modulation of the immunological microenvironment in a murine intracranial glioma. J Immunol. 2008;180:2089–98. doi: 10.4049/jimmunol.180.4.2089. [DOI] [PubMed] [Google Scholar]
- 12.Vieira PL, de Jong EC, Wierenga EA, Kapsenberg ML, Kalinski P. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J Immunol. 2000;164:4507–12. doi: 10.4049/jimmunol.164.9.4507. [DOI] [PubMed] [Google Scholar]
- 13.Mailliard RB, Wankowicz-Kalinska A, Cai Q, et al. α-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004;64:5934–7. doi: 10.1158/0008-5472.CAN-04-1261. [DOI] [PubMed] [Google Scholar]
- 14.Wesa A, Kalinski P, Kirkwood JM, Tatsumi T, Storkus WJ. Polarized type-1 dendritic cells (DC1) producing high levels of IL-12 family members rescue patient TH1-type antimelanoma CD4+ T cell responses in vitro. J Immunother. 2007;30:75–82. doi: 10.1097/01.cji.0000211316.15278.6e. [DOI] [PubMed] [Google Scholar]
- 15.Gustafsson K, Ingelsten M, Bergqvist L, Nystrom J, Andersson B, Karlsson-Parra A. Recruitment and activation of natural killer cells in vitro by a human dendritic cell vaccine. Cancer Res. 2008;68:5965–71. doi: 10.1158/0008-5472.CAN-07-6494. [DOI] [PubMed] [Google Scholar]
- 16.Muthuswamy R, Urban J, Lee JJ, Reinhart TA, Bartlett D, Kalinski P. Ability of mature dendritic cells to interact with regulatory T cells is imprinted during maturation. Cancer Res. 2008;68:5972–8. doi: 10.1158/0008-5472.CAN-07-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Townsend A, Ohlen C, Bastin J, Ljunggren HG, Foster L, Karre K. Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature. 1989;340:443–8. doi: 10.1038/340443a0. [DOI] [PubMed] [Google Scholar]
- 18.Prins RM, Odesa SK, Liau LM. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res. 2003;63:8487–91. [PubMed] [Google Scholar]
- 19.Hatano M, Kuwashima N, Tatsumi T, et al. Vaccination with EphA2-derived T cell-epitopes promotes immunity against both EphA2-expressing and EphA2-negative tumors. J Transl Med. 2004;2:40. doi: 10.1186/1479-5876-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iizuka Y, Kojima H, Kobata T, Kawase T, Kawakami Y, Toda M. Identification of a glioma antigen, GARC-1, using cytotoxic T lymphocytes induced by HSV cancer vaccine. Int J Cancer. 2006;118:942–9. doi: 10.1002/ijc.21432. [DOI] [PubMed] [Google Scholar]
- 21.Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self ”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–86. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada H, Tahara H, Shurin MR, et al. Bone marrow-derived dendritic cells pulsed with a tumor-specific peptide elicit effective anti-tumor immunity against intracranial neoplasms. Int J Cancer. 1998;78:196–201. doi: 10.1002/(sici)1097-0215(19981005)78:2<196::aid-ijc13>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 23.Sasaki K, Zhu X, Vasquez C, et al. Preferential expression of very late antigen-4 on type 1 CTL cells plays a critical role in trafficking into central nervous system tumors. Cancer Res. 2007;67:6451–8. doi: 10.1158/0008-5472.CAN-06-3280. [DOI] [PubMed] [Google Scholar]
- 24.Zhu X, Nishimura F, Sasaki K, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007;5:10. doi: 10.1186/1479-5876-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker PR, Calzascia T, Schnuriger V, et al. The brain parenchyma is permissive for full antitumor CTL effector function, even in the absence of CD4 T cells. J Immunol. 2000;165:3128–35. doi: 10.4049/jimmunol.165.6.3128. [DOI] [PubMed] [Google Scholar]
- 26.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 27.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 28.Hokey DA, Larregina AT, Erdos G, Watkins SC, Falo LD., Jr Tumor cell loaded type-1 polarized dendritic cells induce Th1-mediated tumor immunity. Cancer Res. 2005;65:10059–67. doi: 10.1158/0008-5472.CAN-05-1692. [DOI] [PubMed] [Google Scholar]
- 29.Feili-Hariri M, Falkner DH, Morel PA. Polarization of naive T cells into Th1 or Th2 by distinct cytokine-driven murine dendritic cell populations: implications for immunotherapy. J Leukoc Biol. 2005;78:656–64. doi: 10.1189/jlb.1104631. [DOI] [PubMed] [Google Scholar]
- 30.Loetscher M, Gerber B, Loetscher P, et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–9. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 32.Gattass CR, King LB, Luster AD, Ashwell JD. Constitutive expression of interferon γ-inducible protein 10 in lymphoid organs and inducible expression in T cells and thymocytes. J Exp Med. 1994;179:1373–8. doi: 10.1084/jem.179.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura Y, Watchmaker P, Urban J, et al. Helper function of memory CD8+ T cells: heterologous CD8+ T cells support the induction of therapeutic cancer immunity. Cancer Res. 2007;67:10012–8. doi: 10.1158/0008-5472.CAN-07-1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eguchi J, Kuwashima N, Hatano M, et al. IL-4-transfected tumor cell vaccines activate tumor-infiltrating dendritic cells and promote type-1 immunity. J Immunol. 2005;174:7194–201. doi: 10.4049/jimmunol.174.11.7194. [DOI] [PubMed] [Google Scholar]
- 35.Yang T, Witham TF, Villa L, et al. Glioma-associated hyaluronan induces apoptosis in dendritic cells via inducible nitric oxide synthase: implications for the use of dendritic cells for therapy of gliomas. Cancer Res. 2002;62:2583–91. [PubMed] [Google Scholar]
- 36.Nishioka Y, Wen H, Mitani K, et al. Differential effects of IL-12 on the generation of alloreactive CTL mediated by murine and human dendritic cells: a critical role for nitric oxide. J Leukoc Biol. 2003;73:621–9. doi: 10.1189/jlb.0402205. [DOI] [PubMed] [Google Scholar]
- 37.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–54. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.