

Abstract
Polymers or hydrogels containing modified cyclodextrin (CD) are highly useful in drug delivery applications as CD is a cytocompatible amphiphilic molecule that can complex with a variety of hydrophobic drugs. Here, we designed modular photo-click thiol-ene hydrogels from derivatives of βCD and poly(ethylene glycol) (PEG), including βCD-allylether (βCD-AE), βCD-thiol (βCD-SH), PEG-thiol (PEGSH), and PEG-norbornene (PEGNB). Two types of CD-PEG hybrid hydrogels were prepared using radical-mediated thiol-ene photo-click reactions. Specifically, thiol-allylether hydrogels were formed by reacting multi-arm PEGSH and βCD-AE, and thiol-norbornene hydrogels were formed by cross-linking βCD-SH and multi-arm PEGNB. We characterized the properties of these two types of thiol-ene hydrogels, including gelation kinetics, gel fractions, hydrolytic stability, and cytocompatibility. Compared with thiol-allylether hydrogels, thiol-norbornene photo-click reaction formed hydrogels with faster gelation kinetics at equivalent macromer contents. Using curcumin, an anti-inflammatory and anti-cancer hydrophobic molecule, we demonstrated that CD-crosslinked PEG-based hydrogels, when compared with pure PEG-based hydrogels, afforded higher drug loading efficiency and prolonged delivery in vitro. Cytocompatibility of these CD-crosslinked hydrogels were evaluated by in situ encapsulation of radical sensitive pancreatic MIN6 β-cells. All formulations and crosslinking conditions tested were cytocompatible for cell encapsulation. Furthermore, hydrogels crosslinked by βCD-SH showed enhanced cell proliferation and insulin secretion as compared to gels crosslinked by either dithiothreitol (DTT) or βCD-AE, suggesting the profound impact of both macromer compositions and gelation chemistry on cell fate in chemically crosslinked hydrogels.
Graphical abstract
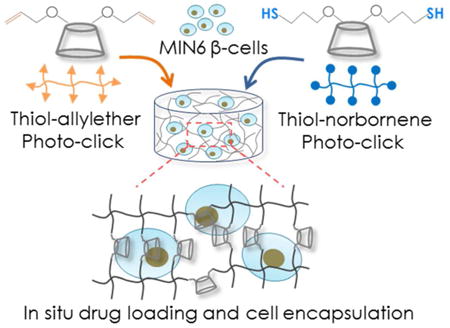
Introduction
As defined by the Food and Drug Administration (FDA), amphiphilic cyclodextrins (CDs) are a class of ‘Generally Recognized as Safe (GRAS)’ macromolecules 1-3. The hydrophobic cavity of CD can accommodate a broad range of poorly soluble drugs through host-guest complex formation, whereas the hydrophilic surface of CD facilitates the dissolution of the CD-drug complex in aqueous solutions 4. Three types of CD, α-, β-, and γ-CD (with 6, 7, and 8 repeating units of glycopyranose, respectively), are commonly used for increasing the solubility of hydrophobic drugs 5. In addition to serving as a drug-dissolving agent, CDs can be easily modified for forming multifunctional macromolecules, including linear polymers and network hydrogels 6-8. For example, hydroxyl groups of βCD could be functionalized with acrylate for crosslinking into hydrogel for binding and releasing anti-cancer drug, such as chlorambucil. The release rate was controlled by adjusting pH or degree of network crosslinking 9. CD could also be functionalized with azide for chemical crosslinking with PEG-di-alkyne via the copper-catalyzed azide-alkyne cycloaddition (CuAAC) 10. Unfortunately, copper is a cytotoxic metal that induces oxidative damage to cells 11. Hence, CuAAC is mostly not compatible with live cell experiments 12. Recently, Sanyal et al. reported the formation and characterization of Michael-type thiol-maleimide hydrogels using hepta-thiol-substituted βCD (βCD-SH7) and linear poly(ethylene glycol)-maleimide (PEG-maleimide) 13. However, maleimide group deprives topoisomerase II catalytic activity of cells that can lead to cell apoptosis 14 and the presence of unreacted maleimide might create adverse effects on cell viability 14, 15. In addition, thiol-maleimide gelation proceeds via a nucleophilic reaction that does not allow one to easily control polymerization kinetics. In this regard, Arslan et al. prepared CD-PEG hybrid hydrogels through photo-crosslinking of commercially available βCD-SH7 and PEG-allylether and the hydrogels were used to deliver hydrophobic puerarin 16. This hydrogel crosslinking, however, was carried out in an organic condition using dimethylformamide (DMF) as the solvent. As a result, this system was not compatible for in situ cell encapsulation.
In the past few years, we and other groups have explored the aqueous-based step-growth thiol-norbornene photo-click chemistry to prepare highly cytocompatible and tunable hydrogels for tissue engineering applications 17. The gelation of thiol-norbornene hydrogels can be initiated by type I photoinitiator (e.g., Irgacure-2959 or lithium arylphosphinate (LAP)) under long wavelength, low intensity ultraviolet (UV) light irradiation (365 nm, 5–10 mW/cm2). Thiol-norbornene gelation could also be initiated by visible light (400-700 nm) exposure with type II photoinitiator (e.g., eosin-Y or rose bengal) as the sole photosensitizer 18, 19. The advantages of step-growth thiol-norbornene photopolymerization include rapid, ambient, and aqueous reaction conditions, as well as spatial-temporal control over gelation kinetics 20-22. More importantly, thiol-norbornene hydrogels are highly cytocompatible even for radical-sensitive pancreatic β-cells 23, 24. In addition to PEG-norbornene, other macromers containing non-homopolymerizable ‘ene’ moieties have been explored for forming step-growth thiol-ene hydrogels. For example, Kloxin and colleagues recently reported an aqueous thiol-allylether reaction to form thiol-ene hydrogel for in situ cell encapsulation. The radical-mediated thiol-allylether hydrogel was formed between multi-arm PEG-thiol (PEGSH) and bis-allyloxycarbonyl functionalized peptides 25. This thiol-allylether gelation produced hydrolytically stable hydrogels that were cytocompatible for encapsulation of human mesenchymal stem cells (hMSCs).
Thiol-ene hydrogels have been exploited as platforms for encapsulating hydrophilic biomolecules, drugs, and cells. Since CDs are capable of increasing the solubility of poorly soluble molecules, a CD-crosslinked thiol-ene hydrogel can potentially be used to encapsulate and to deliver both hydrophilic and hydrophobic biomolecules. Furthermore, CD can be used to form supramolecular association with selected hydrophobic molecules or drugs, such as adamantane and curcumin. A CD-immobilized hydrogel formed by the aqueous thiol-ene photo-click reaction will not only permit in situ cell encapsulation, but may also allow researchers to create hydrogels with dynamically tunable properties in the presence of cells. As the first step toward achieving this goal, we report here the synthesis and characterization of βCD derivatives, the crosslinking of βCD-PEG hybrid hydrogels via radical-mediated thiol-ene photopolymerizations, the delivery of a model hydrophobic drug, and the cytocompatibility of these step-growth hydrogels. Specifically, a two-step synthesis procedure was developed for functionalizing βCD with the desired number of allylether, which could be used to form hydrolytically stable thiol-allylether photo-click hydrogel. Alternatively, allylether moieties could be further converted to thiols, which could be used to form thiol-norbornene hydrogels. Both thiol-ene gelation systems utilized a low concentration of LAP as the photoinitiator and the gelation was initiated via light exposure at 365 nm. We investigated the gelation kinetics and physical properties of these two types of thiol-ene hydrogels. Curcumin, a hydrophobic molecule with anti-inflammatory and anti-cancer potential, was used as a model drug to demonstrate the enhanced uptake and prolonged release of hydrophobic drug from CD-crosslinked thiol-ene hydrogels. Furthermore, the cytocompatibility of these step-growth CD-crosslinked thiol-ene hydrogels were evaluated via in situ encapsulation of radical-sensitive pancreatic MIN6 β-cells.
Materials and Methods
Materials
4-arm PEG (20 kDa), 8-arm PEG (20 kDa and 40 kDa) and 8-arm PEG-thiol (PEG8SH, 20 kDa) were purchased from JenKem Technology USA. βCD was purchased from TCI America. 5-norbornene-2-carboxylic acid, N,N′-dicyclohexylcarbodiimide (DCC), 4-(dimethylamino) pyridine (DMAP), sodium hydride, and allyl bromide were obtained from Sigma-Aldrich. Dialysis membranes with MWCO 500-1000 Da and 6000-8000 Da were obtained from Spectrum Laboratories and Fisher Scientific, respectively. Other chemicals were purchased from Fisher Scientific unless noted otherwise.
Synthesis of photoinitiator and PEG macromers
The synthesis of photoinitiator lithium arylphosphonate (LAP) was described as reported elsewhere 26. PEG-tetra-norbornene (PEG4NB, 20 kDa) and PEG-octa-norbornene (PEG8NB, 20 kDa and 40 kDa) were synthesized following an established protocol 20. In brief, 5-norbornene-2-carboxylic acid (5-fold excess of OH group) and coupling reagent DCC (2.5-fold excess of OH group) were added to anhydrous dichloromethane (DCM). The mixture was purged with nitrogen and stirred at room temperature for 1 hour. The resulting norbornene anhydride was filtered into an addition funnel and added slowly to a flask containing PEG (4-arm or 8-arm), DMAP (0.5-fold of OH group), and pyridine (0.5-fold of OH group) dissolved in anhydrous DCM. The flask was purged with nitrogen, kept on ice and allowed to react overnight in dark. The product was precipitated in cold ethyl ether and collected by filtration. PEGNB product was re-dissolved in double distilled water (ddH2O) and proceed with dialysis (MWCO 6000-8000 Da) for 2 days using slightly acidic ddH2O (pH 6.8) to prevent ester hydrolysis of PEGNB. The pure product was obtained from lyophilization (degree of norbornene substitution >85 %). PEGNB: 1H NMR (CDCl3, 500 MHz): δ – 4.25 (m, 2H), 5.98 (m, 1H), 6.23 (m, 1H). PEG-allylether (PEGAE) was synthesized following a published protocol. Briefly, linear PEG (3.4 kDa) was dissolved in anhydrous toluene and dried with evaporation by reduced pressure. Dried PEG was re-dissolved in anhydrous tetrahydrofuran (THF). Sodium hydride (1.5-fold excess of hydroxyl group) was added slowly to the PEG solution under continuous nitrogen purging at 40 °C. Upon complete liberation of hydrogen gas, allylbromide (6-fold excess of hydroxyl group) was added drop wise to the solution. After overnight reaction in dark, sodium bromide salt was filtered off to afford a PEG-allylether (PEGAE) solution. PEGAE was precipitated in cold ethyl ether, filtered and dried in vacuuo. The purity of PEGAE product was verified with H1 NMR in CDCl3 (96 %, Bruker 500). PEGAE: 1H NMR (CDCl3, 500 MHz): δ – 4.04 (m, 2H), 5.31 (m, 1H), 5.43 (m, 1H), 6.07 (m, 1H).
Synthesis of allylether or thiol functionalized βCD
βCD-AE was synthesized following a published protocol with slight modification 27. βCD was dried in vacuum oven overnight and dissolved in 40 mL of anhydrous dimethylformamide (DMF) in a flask. While purging the flask with nitrogen, desired amount of sodium hydride (i.e., 2.3, 4.5 or 11.3-fold of [NaH] and [allylbromide] to [βCD]) was added slowly and the mixture was stirred until no hydrogen gas bubbles were visible (Figure 1A, step i). Allylbromide (Figure S1A) was added drop wise to the βCD solution using an addition funnel and the reaction was allowed to continue overnight in dark (Figure 1A, step ii). Next, the volume of βCD-AE mixture was reduced using a rotary evaporator. βCD-AE was precipitated in cold acetone twice and dried in a vacuum oven. βCD-AE: 1H NMR ((CD3)2SO, 500 MHz): δ – 4.04 (m, 2H), 5.31 (m, 1H), 5.43 (m, 1H), 6.07 (m, 1H). The exact mass of βCD-AE was determined by using mass spectroscopy (Agilent Technologies 6520 Accurate-Mass Q-TOF LC/MS).
Figure 1.
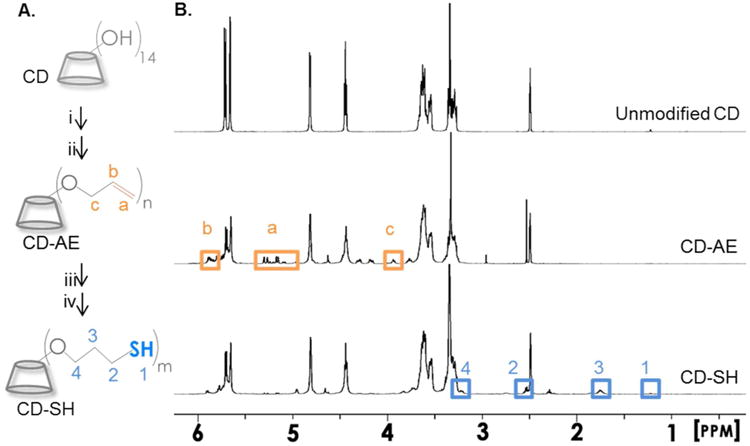
(A) Schematics of βCD modifications. (i) NaH, DMF, room temperature; (ii) allylbromide, DMF, room temperature; (iii) thioacetic acid, I-2959, UV (365 nm, 10 mW/cm2), 30 min; (iv) sodium hydroxide (aq.), followed by neutralization with hydrogen chloride. 1H NMR of (B) βCD, (C) βCD-AE, and (D) βCD-SH (dissolved in (CD3)2SO).
Next, thioacetic acid (Figure S1B, 2-fold excess to [βCD-AE]) was added slowly to 10 mL of DMF containing 1 g of βCD-AE and photoinitiator Irgacure I-2959 (0.5 wt%). Photo-conjugation was initiated by UV-light exposure (Omicure S1000, 365 nm and 10 mW/cm2) for 15 minutes and continued for another 30 minutes after supplementing with another portion of 0.5 wt% I-2959 (Figure 1A, step iii). βCD-thiolacetate (Figure S1C) was precipitated in cold acetone, filtered, and dried in vacuum oven. After re-dissolving βCD-thioacetate in ddH2O (10 mL), while purging with nitrogen, 5 mL of sodium hydroxide (2 N) was added to the solution for 5 minutes to hydrolyze the thioacetate group. The solution was neutralized by adding equal volume of hydrochloride acid (2 N) solution, followed by dialysis (MWCO 500-1000) for 2 days at room temperature (Figure 1A, step iv). The product βCD-thiol (βCD-SH) was obtained from lyophilization. βCD-SH: 1H NMR ((CD3)2SO, 500 MHz): δ – 1.3 (s, 1H), 1.76 (m, 2H), 2.53 (m, 2H), 3.35 (m, 2H). Ellman's assay was performed to determine the concentration of thiol in βCD-SH solution.
Hydrogel fabrication and characterizations
Step-growth thiol-ene hydrogels were formed by light mediated photo-click reactions using the following macromers: (1) PEG8SH (20 kDa) and βCD-AE, (2) PEG8NB (20 kDa) and βCD-SH, or (3) PEG8NB (20 kDa) and DTT. To obtain the best crosslinking efficiency in each gelation formulation, the stoichiometric ratio of thiol to ene was maintained at one. The concentration of PEGNB was adjusted based on 1H NMR results to afford correct norbornene concentrations in the gelation mixtures. Photoinitiator LAP was added to the precursor solution (50 μL/gel) to afford a final concentration of 1 mM. The precursor solution was injected in between two glass slides separated by 1 mm thick spacers. Hydrogel slabs were prepared by exposing the solution to 365 nm light at 5 mW/cm2 for 5 minutes. To characterize gel fractions, hydrogels were immediately dried in vacuuo after gelation. Dry mass (WDry,1) was measured and the dried polymers were incubated in ddH2O at 37 °C on an orbital shaker for 24 hours to remove uncrosslinked (sol fraction) species. The swollen gels were dried again in vacuuo and weighed to obtain dried polymer weights (WDry,2). After swelling hydrogels in pH 7.4 PBS for two days, the swollen gel weights were measured (WSwollen). Gel fractions were obtained by the ratio of the two dry masses (i.e., WDry,1/WDry,2). Hydrogel swelling ratios (q) were defined as the ratio of swollen gel mass to the second dried polymer mass (i.e., WSwollen/WDry,2).
Crosslinking efficiency of thiol-ene hydrogel
The average molecular weights between crosslinks ( ) of thiol-ene hydrogels can be obtained from the structural information of the macromers (i.e., molecular weight and functionality) 28:
| (3) |
Here, MWA and MWB represent the molecular weight of PEGNB and βCD-SH, while fA and fB are the number of reactive functionality for PEGNB and βCD-SH, respectively. With a known , the ideal network crosslinking density or density of elastically active chains (vc) and polymer volume fraction (v2) can be calculated 29:
| (4) |
Here, is the specific volume of PEG (0.92 cm3/g at 37 °C), V1 is the molar volume of water (18 cm3/mole) and χ12 is the Flory-Huggins interaction parameter for a PEG-H2O system (0.45).
In situ photo-rheometry was performed at room temperature on a Bohlin CVO 100 digital rheometer equipped with a light cure cell. A macromer solution (100 μL) was placed on a quartz plate in the light cure cell and irradiated with light (Omnicure S1000, 365 nm, 10 mW/cm2) through a flexible light guide. Light was turned on 30 seconds after starting time-sweep measurement (10 % strain, 1 Hz frequency, and a gap size of 90 μm) using a 25 mm parallel plate geometry. Gel points (i.e., crossover time) were determined at the time when storage modulus (G′) surpassed loss modulus (G″).
Oscillatory rheometry in strain-sweep mode was used to obtain elastic modulus of the hydrogel. At equilibrium swelling (after 48 hours in pH 7.4 PBS), a biopsy punch was used to punch out circular gel discs (8 mm in diameter) from the gel slabs. Storage moduli of the hydrogels were measured using 8 mm parallel plate geometry with a gap size of 800 μm. The elastic moduli from at least three hydrogels for each gel formulation were recorded from the average of the linear viscoelastic region (LVR, linear portion of G′ plotting against % strain).
Uptake and release of curcumin
Thiol-ene hydrogel discs (25 μL; 4.8 mm dia. × 1.1 mm height) were formed as described above using the following formulations: (1) PEG8SH (20 kDa) and βCD-AE, or (2) PEG8SH (20 kDa) and PEGAE (3.4 kDa). After photo-crosslinking, hydrogels were incubated in 2 mL of curcumin suspended in ddH2O (2 mg/mL). After 8 hours, hydrogels were removed from the curcumin suspension, rinsed with ddH2O, and placed individually in 1.6 mL tube. Hydrogels were individually incubated in 500 μL of DMSO. A portion of DMSO (200 μL) was collected and fresh DMSO (200 μL) was added every 24 hours for one week. The amount of released curcumin in DMSO was characterized via measuring absorbance at 430 nm using a microplate reader (BioTek®, Synergy HT).
In another experiment, hydrogels were transferred to 4 mL scintillation vials containing 400 μL of curcumin solution, which was prepared by dissolving curcumin in 0.5 N sodium hydroxide, followed by dilution with pH 7.4 PBS to a final concentration of 2 mg/mL 30. After incubating the gels at room temperature for 36 hours, hydrogels were transferred to scintillation vials containing 400 μL of pH 7.4 PBS to rinse off the curcumin adsorbed on gel surface for 10 minutes. The solution was removed from the vials and 1 mL of pH 7.4 PBS was added to each vial for curcumin release study. Scintillation vials containing hydrogels were placed on an orbital shaker (60 rpm) at room temperature. At specific time points, 500 μL of solution was collected from each vial, and fresh 500 μL of pH 7.4 PBS was added back to the vial. After 48 hours, PBS was completely removed from the vials, and 1 mL of DMSO was added to each hydrogel to extract the remaining curcumin from the gels. At specific time points, 500 μL of DMSO was collected from each vial, and fresh DMSO was added back to the vial to maintain the total volume at 1 mL. The absorbance of all collected curcumin samples (from curcumin release and DMSO, 200 μL/sample) were measured using a microplate reader (BioTek®, Synergy HT) at 430 nm. Measured absorbance was blanked with either pH 7.4 PBS or DMSO, and correlated to standard curves with known concentrations of curcumin. The cumulative uptake of curcumin per gel was the sum of curcumin released in PBS and DMSO.
Cell encapsulation and viability assay
Mouse insulinoma cells (MIN6, final cell density in the gels at 2×106 cells/mL) were suspended in polymer solutions containing 1 mM LAP and macromers, including: (1) PEG8NB20kDa with DTT, (2) PEG8NB20kDa with βCD-SH or (3) PEG8SH20kDa with βCD-AE. Precursor solution was exposed to light (365 nm, 5 mW/cm2) for 5 minutes to obtain cell-laden hydrogels (25 μl/gel), which were maintained in high-glucose DMEM supplemented with 10 % fetal bovine serum (FBS), 50 μM β-mercaptoethanol, and 1× antibiotic-antimycotic. To characterize cell viability, cell-laden hydrogels were incubated in 500 μL Almarblue® reagent (10 % in cell culture medium) at 37 °C and 5 % of CO2. After 14 hours of incubation, 200 μl of media were transferred to a 96-well plate for fluorescence quantification (excitation: 560 nm and emission: 590 nm). To obtain qualitative cell viability, cells were stained with a live/dead staining kit (Calcein AM stained live cells green and Edithium homodimer stained dead cells red) and imaged with confocal microscopy. In each stained hydrogel, three images were taken at a step size of 10 μm for a total depth of 100 μm per image. The viability of MIN6 cells was quantified as the percent of live cells over the total number of live and dead cells.
Insulin secretion from MIN6 cells
Cell function was assayed by glucose stimulated insulin secretion (GSIS). Briefly, cell-laden hydrogels were rinsed with HBSS, followed by priming in Krebs-Ringer bicarbonate (KRB) buffer (23 mM sodium chloride, 1 mM potassium chloride, 4.8 mM sodium bicarbonate, 0.2 mM magnesium chloride hexahydrate, 0.2 mM calcium chloride dehydrate, 0.5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid and 0.1 vol% bovine serum albumin) containing 2.5 mM glucose for 1 hour at 37 °C and 5 % CO2. Then, the gels were incubated with 500 μL of low (2.5 mM) or high (25 mM) glucose KRB for 2 hours each. The high glucose buffer solution was collected and subjected to insulin ELISA (Mercodia).
Data analysis & statistics
All experiments were conducted independently for three times and results were reported as mean ± SD. Data was analyzed with student's t-test or two-way ANOVA analysis using GraphPad Prism 5.
Results and Discussion
Preparation and characterization of βCD derivatives
A two-step process was employed to modify βCD with allylether moiety. First, NaH was added at different concentrations to activate hydroxyl groups on βCD (Figure 1A, step i). Following the secession of hydrogen gas, excess allyl bromide was added to the reaction mixture (Figure 1A, step ii) to afford βCD-AE. 1H NMR results (Figure 1B) showed that allylether group was successfully introduced to βCD. Unexpected peaks between 4.1 to 4.3 ppm were likely due to regioisomerism. Peaks of similar intensities (i.e., 3.8 or 4.6 ppm) were likely due to the shifting of protons on βCD as a result of allylether substitutions. The modification was confirmed by mass spectroscopy analysis. The difference of the average molecular mass of βCD-AE and βCD was divided by the molecular mass of allylether (i.e., 40 Da) to estimate the degree of allylether functionalization. As the concentration of NaH or allylbromide to βCD was increased (from 0 to 11.3-fold, Figure S1), so was the detected mass of the modified βCD and the estimated degree of substitution (0, 1.5, 2.4 and 4.5, respectively). To obtain thiol-substituted βCD, βCD-AE was first photoconjugated with thioacetic acid via radical-mediated thiol-allylether addition (Figure 1A, step iii). The intermediate product, βCD-thioacetate, was hydrolyzed with sodium hydroxide, neutralized with hydrochloric acid, and purified via dialysis to obtain the final product, βCD-SH (Figure 1A, step iv). 1H NMR results confirmed that allylether was replaced with thiol group (Figure 1B, bottom panel). The presence of thiol on modified βCD was further quantified by Ellman's assay (Figure S2). As the concentration of βCD-SH was increased from 0.02 to 1 mg/mL, the concentration of thiol detected increased linearly (R2 = 0.99) from 0.006 ± 0.001 to 0.362 ± 0.005 mM. As expected, no thiol was detected from unmodified βCD or βCD-AE.
Preparation of thiol-ene photo-click hydrogels
We next demonstrated the crosslinking of CD-PEG hybrid hydrogels using PEG8SH (M.W. 20 kDa) and βCD-AE through a radical-mediated thiol-allylether photo-click reaction (Figure 2A) and the gelation was evaluated via in situ photorheometry (Figure 2B, S3). Specifically, 5 wt% of PEG8SH was cross-linked with different concentrations of βCD-AE (9 to 24 mg/mL). The gel points of all formulations evaluated were similar regardless of βCD-AE concentration in the precursor solution (gel point ∼ 17 to 22 seconds, Figure S3). However, the formulation afforded the highest elastic modulus (∼2.5 kPa at 300 second) at 24 mg/mL of βCD-AE (Figure 2B). Further increasing βCD-AE concentration did not yield higher gel modulus (data not shown). Controlled experiment using unmodified βCD with PEG8SH showed no sign of gelation even after 300 seconds of light exposure in the presence of photoinitiator (Figure 2C), suggesting that the gelation was not due to disulfide bond formation. Since 24 mg/mL of βCD-AE yielded the highest shear modulus of thiol-allylether hydrogel, we examined the gelation of thiol-norbornene hydrogel (Figure 2D) using this concentration (24 mg/mL) of thiol-substituted βCD (i.e., βCD-SH) and 5 wt% of 8-arm PEG-norbornene (PEG8NB, M.W. 20 kDa). Even with slightly lower degree of functionalization (∼85 % on PEG8NB), the gelation of βCD-SH with PEG8NB still afforded a 5-fold faster gel point (Figure 2E, gel point ∼3 seconds). However, the final shear modulus of thiol-norbornene hydrogel formed with βCD-SH and PEG8NB only reached about 1.1 kPa (Figure 2E). Control experiment showed that gelation was not possible when unmodified βCD was mixed with PEG8NB (Figure 2F) and subjected to the same radical-initiation conditions, indicative of the lack of either PEG8NB homopolymerization or gelation due to supramolecular assembly (or threading) between multi-arm PEG and βCD.
Figure 2.
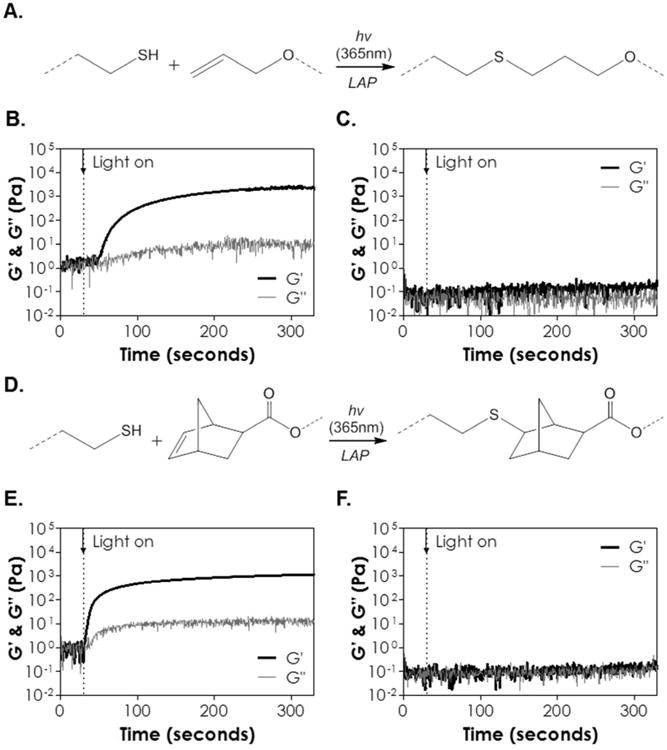
(A) Scheme of light and radical mediated thiol-allylether gelation. (B) In situ photorheometry of thiol-allylether photopolymerization (G′: storage modulus; G″: loss modulus) between PEG8SH and βCD-AE. (C) In situ photorheometry of solution containing PEG8SH and βCD. (D) Scheme of light and radical mediated thiol-norbornene gelation. (E) In situ photorheometry of thiol-norbornene photopolymerization PEG8NB and βCD-SH. (F) In situ photorheometry of solution containing PEG8NB and βCD (5 wt% PEG8SH or PEG8NB, 24 mg/mL of βCD-AE or βCD-SH, 1 mM LAP). Light (365 nm, 10 mW/cm2) was turned on at 30 seconds (dotted line). N = 3, error bars were omitted for clarity.
The fast thiol-ene gelation using macromers functionalized with norbornene, a strained ene, is well-documented in the literature. For example, we have shown that PEG-peptide thiol-norbornene gelation could be achieved rapidly (gel point ∼ 2-3 seconds) using low PEGNB and photoinitiator concentrations (e.g., 4 wt% PEG4NB20kDa with DTT or bis-cysteine peptides and 1 mM LAP) 31. On the other hand, Kloxin and colleague designed a radical-mediated (1.1 mM of LAP) thiol-allylether hydrogel system using 10 wt% of PEG4SH (M.W. 20 kDa) with alloc-functionalized peptides and obtained a gel point of about 15 seconds 25. While both allylether and norbornene react with thiol via a strictly step-growth mechanism (i.e., no homopolymerization between the ene groups) 32, the polymerization rate of thiol-norbornene is six times faster than that of thiol-allylether due to the unity ratio of thiyl radical propagation to chain transfer kinetic parameters (kpSC /kCT) 32. It is worth noting that, while thiol-norbornene gelation was one order of magnitude faster than thiol-allylether gelation, the final gel shear modulus of the former was only about half of the latter. This could be explained by the fact that PEG8NB, which was synthesized in house, was lower in the degree of functionalization (85%, per 1H NMR) as compared with commercially acquired PEG8SH (>95%, per manufacturer's certificate of analysis, respectively). Furthermore, since βCD-SH was modified from βCD-AE, it would make sense that gels crosslinked by βCD-SH would have a lower degree of crosslinking, and hence lower shear modulus, as compared with gels crosslinked by βCD-AE.
CD has been shown to assist the assembly of physical ‘pseudo-polyrotaxane’ hydrogel through interactions with PEG or block co-polymers of polypropylene oxide (PPO) and polyethylene oxide (PEO) (e.g., Pluronic). Previously, Cooper-White and colleagues reported supramolecular gelation of αCD with Pluronic co-polymers and concluded that the ‘threading’ induced gelation would only occur in the presence of ‘poly-CD’ aggregates, which was only apparent at a sufficiently high concentration of CD (at least 50-70 mg/mL) 33. The gelation in the work presented here was purely based on thiol-ene photochemistry since the CD concentrations used in this study were less than 40 mg/mL and the fact that no gelation was observed by simply mixing unmodified βCD with PEGNB (Figure 2C). Another relevant work reported by Elisseeff and colleagues showed a tunable CD/PEG hybrid hydrogel crosslinked by chain-growth photopolymerization. It is worth noting that the functionalized βCD motifs were ‘threaded’ onto PEG-diacrylate after overnight incubation34. We believe that the threading of CD to PEG chains was minimal, if any, since we added βCD into the mixture right before photopolymerization in all experiments. Another potential complication is the formation of LAP/βCD complex. We performed isocratic HPLC using acetonitrile/phosphate buffer (35/65) containing βCD (0 to 10 mM) as the mobile phase. While the elution time of LAP shifted slightly (Figure S4A), the peaks were not significantly different from each other regardless of βCD concentration (Figure S4B). Furthermore, since the gelation was accomplished within 5 minutes, it is unlikely that the association between LAP and βCD, if any, would impede light-induced initiator breakdown and gelation. If association between LAP and βCD does exist, it would also not be a concern because upon light irradiation the photoinitiators would be photolysed into smaller fragments and readily diffuse out from the CD cavity.
Effects of macromer functionality and molecular weight on gelation kinetics
Since thiol-norbornene reaction exhibits faster gelation kinetics than thiol-allylether reaction, we evaluated the effects of PEGNB macromer functionality and molecular weight on the gelation of CD-based thiol-norbornene hydrogels (Figure 3). PEG4NB (M.W. 20 kDa, Figure 3A) and PEG8NB (M.W. 40 kDa, Figure 3B) were selected as these two macromers provided the same total concentration of norbornene at the same weight percentage (i.e., 10 mM at 5 wt%). Note that the molar ratio of norbornene to thiol was maintained at one, whereas the concentration of thiol from βCD-SH was determined by Ellman's assay (Figure S2). Since the average molecular weight per arm in these two macromers was the same (i.e., 5 kDa), the average molecular weight between crosslinks ( ) of the two was also the same (Eq. 3). This implied that the two sets of hydrogels would have the same ideal crosslinking efficiency (Eq. 4). Experimentally, however, we found that thiol-norbornene hydrogels cross-linked by PEG4NB (20 kDa) had a slightly slower gel point (∼6 seconds) and a lower shear modulus (∼1 kPa) when compared with gels crosslinked by PEG8NB (40 kDa) (∼4 seconds and ∼3 kPa). These results was consistent with the trend found in previously reported step-growth Michael-type hydrogels 35. Theoretically, step-growth hydrogels with the same (Eq. 3) should have identical mesh size (Eq. 4) regardless of the macromer concentration. Experimentally, however, macromer with lower functionality formed hydrogel with higher swelling ratio compared with macromer with higher functionality 28, 31. The phenomenon was attributed to network defects and inefficient cross-linking at lower macromer functionality. We also compared gelation kinetics of CD-based thiol-norbornene hydrogels using macromer PEG8NB with different molecular weight (Figure 3B & 3C). The gel point of hydrogel cross-linked by 20 kDa PEG8NB was 2.2–fold faster, and the elastic modulus was 1.5–fold higher, than the gels cross-linked by 40 kDa PEG8NB. These results were also expected because of the higher molarity of norbornene groups on 20 kDa PEG8NB at equivalent polymer weight content.
Figure 3.
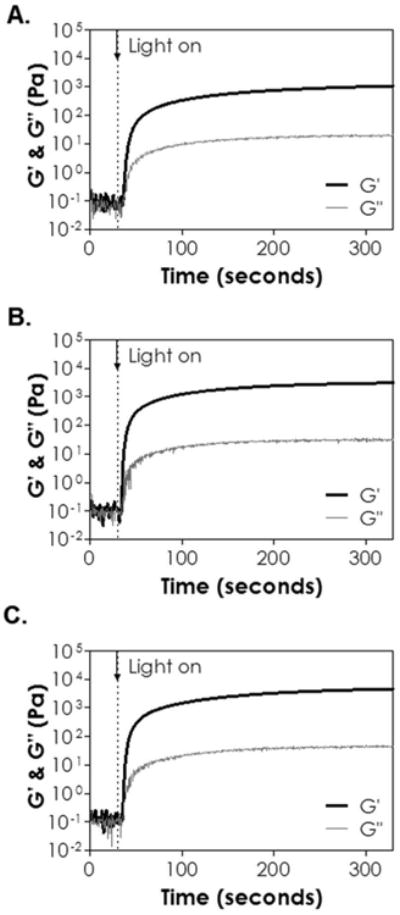
In situ photorheometry of thiol-ene hydrogels crosslinked by βCD-SH and (A) PEG4NB (20 kDa), (B) PEG8NB (40 kDa) or (C) PEG8NB (20 kDa). (5 wt% PEGNB, [thiol] = [norbornene], 1 mM LAP). Light (365 nm, 10 mW/cm2) was turned on at 30 seconds (dotted line). N = 3, error bars were omitted for clarity.
Effects of cross-linker on hydrogel properties
After examining the effect of CD-PEG macromer formulations on gelation, we evaluated the influence of crosslinker on the physical properties of selective groups of thiol-norbornene hydrogels. Three sets of macromers were used: (1) DTT with PEG4NB (labelled as DTT in Figure 4), (2) βCD-SH with PEG4NB (labelled as CD-SH), and (3) PEG4SH with βCD-AE (labelled as CD-AE). As shown in Figure 4A, the gel fraction was not significantly different between the three groups (86 ± 3 %, 79 ± 8 %, and 74 ± 6 % for DTT, CD-SH, and CD-AE gel, respectively). However, the DTT gel exhibited the highest equilibrium shear modulus when compared with gels crosslinked with CD-SH or CD-AE (Figure 4B, ∼7.1, 3.4, and 2.4 kPa for DTT, CD-SH and CD-AE gel, respectively). The differences in equilibrium shear moduli reflected the variation in network crosslinking density, which was affected by the types of thiol (i.e., DTT, CD-SH, or PEG4SH) and ene (i.e., norbornene or allylether) used in the formulations 31. The lowest crosslinked CD-AE gels also had highest swelling ratio among the three groups (Figure 4D). Interestingly, even though the modulus of DTT gel was higher than that of CD-SH gel (Figure 4B & 4C), the swelling ratio of DTT gel was also 2-fold higher than that of CD-SH gel (Figure 4D). This result was contradictory to the inverse scaling of modulus and swelling ratio in chemically crosslinked hydrogels. A significantly lower swelling ratio for CD-SH gel might be a result of higher CD-SH concentration and mass (48 mg/mL) required for this formulation. As a comparison, the concentration of DTT used was only 1.5 mg/mL. Inclusion of higher content of amphiphilic CD in the gel formulations might profoundly impact the ability of these gels to imbibe water.
Figure 4.

Effect of crosslinker on (A) gel fraction, (B) elastic modulus at equilibrium gel swelling, (C) shear modulus as a function of time, and (D) swelling ratio as a function of time. Asterisks indicate statistical significance (*p < 0.05, 5 wt% 20 kDa PEG8SH or PEG8NB, 1 mM LAP, 365 nm light, intensity at 5 mW/cm2, mean ± SD, N = 3).
The step-growth thiol-norbornene hydrogels were known to degrade hydrolytically if ester bonds were presence in the PEGNB macromers 31. We evaluated the hydrolytic stability of the three groups of thiol-ene hydrogels (i.e., DTT, CD-SH, and CD-AE gel). The hydrolytic degradation of these thiol-ene hydrogels in neutral buffer solution (pH 7.4) was not significant as the modulus and swelling ratio remained relatively stable over 10 days (Figure 4C & 4D). We reason that this was because these gels were either highly crosslinked (G′ ∼7.1 kPa and 3.4 kPa for DTT and CD-SH gel, respectively) or contained no hydrolytically labile bonds (e.g., CD-AE thiol-allylether gel). We also conducted an accelerated hydrolysis study (Figure S5) by incubating these hydrogels in basic buffer (pH 9). As expected, a decline in G′ over time was observed for DTT and CD-SH gels (both crosslinked with PEG4NB), indicative of base-catalysed hydrolysis of ester bonds in PEG4NB macromer (Figure S5). On the other hand, CD-AE gel remained intact over time (i.e., constant G′) since thiol-allylether hydrogels did not contain hydrolytic degradable moieties.
Uptake and release of hydrophobic curcumin from thiol-allylether hydrogels
One potential application of the CD-PEG hydrogel system is to deliver hydrophobic drugs. Many anti-inflammatory drugs, such as curcumin 36, are hydrophobic and hence have lower bioavailability in vivo. Curcumin has also been a subject of intensive research interest in diabetes because of its blood glucose lowering effect 37. Since thiol-norbornene hydrogels would be hydrolyzed in basic buffer 31, we chose thiol-allylether hydrogels crosslinked by CD-AE and PEG4SH as a model gel system to demonstrate the enhanced uptake and sustained release effect of curcumin from CD-PEG hybrid hydrogels. Pure PEG hydrogels crosslinked by PEGAE and PEG4SH under the same gelation conditions were used as control. The two sets of gels (labeled as PEGAE and CD-AE in Figure 5) were incubated in ddH2O suspended with hydrophobic curcumin for 8 hours. As shown in the top panel of Figure 5A, hydrogels crosslinked with CD-AE appeared more yellowish when compared to hydrogels crosslinked with PEGAE, indicative of higher amount of curcumin (yellow in color) uptake due to the presence of chemically crosslinked CD in the gel. Subsequently, these gels were immersed in DMSO to completely liberate the curcumin. As shown in the bottom panel of Figure 5A, both sets of gels appear transparent and identical, indicating that all curcumin were released from the gels. The liberated curcumin was quantified via UV/vis absorbance measurement (Figure 5B). Compared to pure PEG hydrogels crosslinked with PEGAE and PEG4SH, hydrogels crosslinked with CD-AE were capable of loading twice as much curcumin (∼9 and ∼20 μg/gel for PEGAE and CD-AE gel, respectively).
Figure 5.
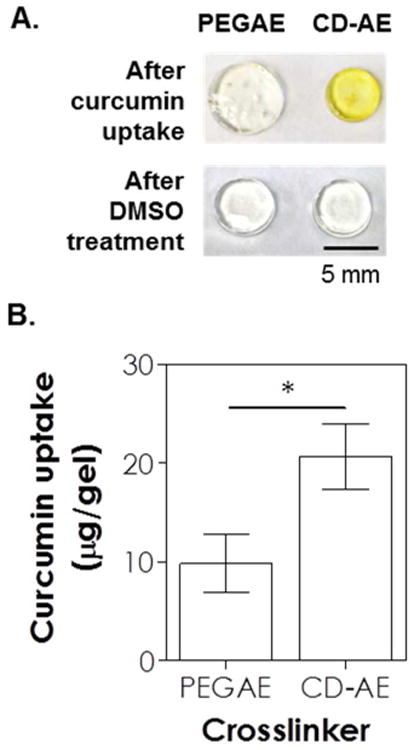
(A) Photographs of thiol-allylether hydrogels incubated in curcumin suspended ddH2O for 8 hours (top) and after DMSO-treatment (bottom). (B) Curcumin uptake in hydrogels formed by 5 wt% PEG8SH with 3.4 kDa of PEGAE or 24 mg/mL βCD-AE (1 mM LAP, 365 nm light at 10 mW/cm2).
Because the amount of curcumin loading in the control PEGAE gel was much lower than that in the CD-AE gel, the release of curcumin from PEGAE hydrogel was barely detectable and it would be difficult to compare the release of curcumin from these two sets of gels. Therefore, we used an alkaline solution to dissolve and complex curcumin with βCD (curcumin was dissolved in 0.5 N NaOH first, followed by diluting with pH 7.4 PBS). This method was used previously to form CD-curcumin complex without affecting the association of the two molecules 30. Figure 6A shows that prior to drug loading the two sets of gels were equally transparent (panel 1). After drug loading (panel 2), the two sets of thiol-allylether hydrogels became equally opaque, suggestive of similar curcumin intake. The similar amount of curcumin absorbed in the gels were quantified by DMSO treatment as described earlier (Figure 6B, 6.6 ± 1.1 and 6.9 ± 0.4 μg/gel for PEGAE and CD-AE gel, respectively). The gels were then incubated in buffer solution for curcumin release. Significantly more curcumin was released from hydrogels crosslinked by PEGAE than by CD-AE (Figure 6C, 85 % and 42 % of curcumin was released from PEGAE and CD-AE hydrogels after 10 hours, respectively). Over the course of first 100 hours, a 1.8-fold higher amount of curcumin was released from PEGAE-crosslinked gels than from CD-AE crosslinked gels (Figure 6C, 87 % and 49 %, respectively\). After the release study, CD-AE gel appeared slightly darker than PEGAE gel (Figure 6A, panel 3), suggesting that a higher amount of curcumin was still retained in this gel. However, after DMSO treatment for an additional 100 hours, the remaining curcumin were completely released from both sets of hydrogels (Figure 6C) and the appearances of the gels were similar in color (Figure 6A, panel 4).
Figure 6.
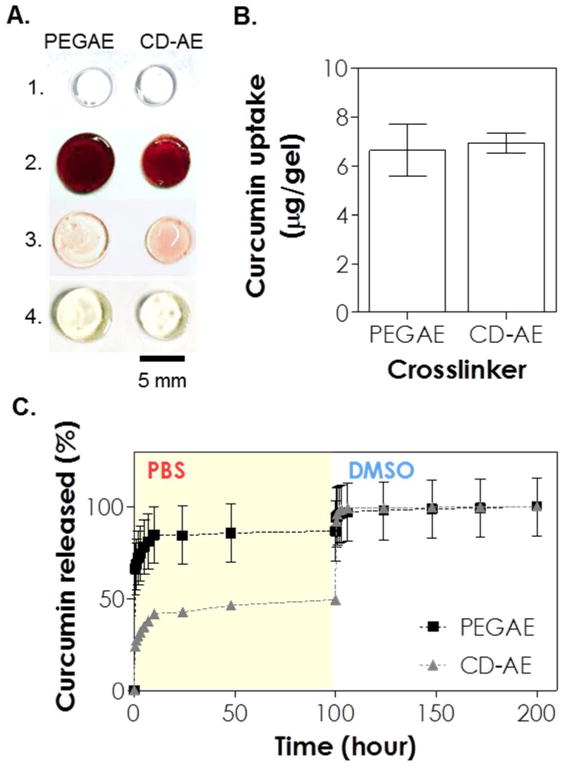
(A) Photographs of thiol-allylether hydrogels: (1) immediately after photopolymerization, (2) after incubating in curcumin solution for 36 hours, (3) after 100 hours of release in pH 7.4 PBS, and (4) after an additional 100 hours of release in DMSO (scale: 5 mm). (B) Effect of crosslinker on curcumin uptake in the gel. (C) Curcumin release from thiol-allylether hydrogel formed by 5 wt% PEG8SH with 3.4 kDa of PEGAE or 24 mg/mL βCD-AE (1 mM LAP, 365 nm light at 10 mW/cm2).
The increase of curcumin solubility via guest-host interactions with CD is well documented 38-40. In our study, CD-AE was employed as both a hydrogel crosslinker and a carrier for hydrophobic drug (i.e., curcumin). Compared to thiol-allylether hydrogels crosslinked by PEGAE, gels crosslinked by CD-AE accommodated significantly more curcumin due to the formation of inclusion complex. The presence of CD/curcumin inclusion complex also resulted in delayed/sustained release of curcumin from the CD-AE hydrogel. Since all curcumin absorbed in the gel could be liberated by DMSO, we believe that the retention of curcumin was purely due to inclusion complex formation but not Michael-type reaction occurred between curcumin and nucleophilic thiols in the gels 41,42.
Cytocompatibility of CD-crosslinked thiol-ene hydrogels
Next, we evaluated the cytocompatibility of CD-crosslinked thiol-ene hydrogels using in situ encapsulation of pancreatic MIN6 β-cells at a relatively low cell density (2×106 cells/mL) 24. Thiol-norbornene hydrogels crosslinked by DTT and PEG8NB (M.W. 20 kDa) were used as a control group as similar gelation formulation has been shown to be cytocompatible for radical-sensitive MIN6 β-cells 23. One day after cell encapsulation, live/dead staining was performed and the results show that thiol-norbornene hydrogels crosslinked by CD-SH were highly cytocompatible for MIN6 cells (Figure 7A, ∼83 % live cells), while hydrogels crosslinked by DTT or by CD-AE contained slightly more dead cells (Figure 7A, ∼72 % and 73 % live cells for gels crosslinked by DTT and CD-AE, respectively). Regardless of the cross-linker used, MIN6 β-cells formed spherical aggregates in all hydrogels 10 days after cell encapsulation (Figure 7A, right panel). The average diameters of the spheroids were about 18 μm, 26 μm, and 32 μm for hydrogels crosslinked by DTT, CD-SH and CD-AE, respectively. We also quantified cell metabolic activity using CellTiter-Glo® (for measuring intracellular ATP content) and AlamarBlue® regent (for measuring cell metabolic activity). In DTT or CD-AE crosslinked hydrogels, the intracellular ATP content increased from ∼13 to 200 pmol of ATP per sample (Figure S7) from day 1 to day 10. The difference between DTT and CD-AE crosslinked hydrogels was not significantly on either day 1 or day 10. On the other hand, the amount of ATP increased from ∼70 to 390 pmol per sample in hydrogels crosslinked by CD-SH (Figure S7). The increased in intracellular ATP was most likely due to the cell proliferation and the formation of multi-cell aggregates. Cell proliferation in CD-PEG hydrogels was verified by Alamarblue® reagent assay (Figure 7B) and the trends were consistent with live/dead staining and ATP assay.
Figure 7.
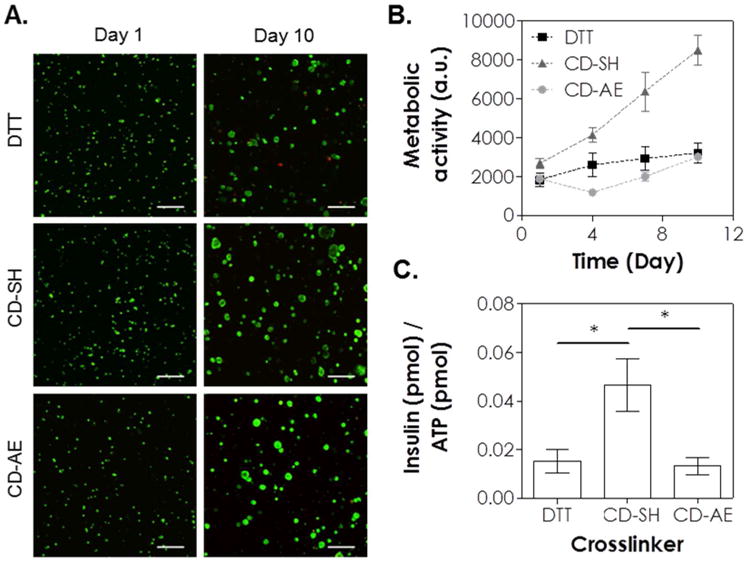
Effect of crosslinker on the cytocompatibility of thiol-ene hydrogels on MIN6 β-cells. (A) Representative confocal z-stack images of MIN6 cells stained with live/dead staining kit on day 1 and day 10. MIN6 cells were encapsulated (2×106 cells/mL) in thiol-ene hydrogels crosslinked by DTT, CD-SH or CD-AE. (B) Cells viability as assessed by Alamarblue® reagent. (C) Normalized insulin secretion. All gel formulations contained 5 wt% 20 kDa PEG8SH or PEG8NB, 1 mM LAP, and 365 nm light at 5 mW/cm2 (Scales: 100 μm).
Studies have shown that softer matrix and hydrolytic gel degradation promoted the phenotype, function, and proliferation of MIN6 βcells in 3D 23, 43. The thiol-norbornene hydrogels in the current study were slightly stiffer (Figure 4B) and relatively stable at pH 7.4 over the course of 10 days (Figure 4C & 4D). Consistent with our published work using PEG-based thiol-norbornene hydrogels 31, the hydrolysis rate of CD-PEG hybrid hydrogels could be accelerated under basic condition (e.g., pH 9, Figure S5). On the other hand, thiol-allylether hydrogels remained hydrolytically stable (Figure S5). Compared to CD-SH crosslinked hydrogels, the slightly higher initial cell death (Figure 7A to 7C) in DTT-crosslinked hydrogels might be a result of higher gel stiffness. When we replaced PEG8NB with PEG4NB without changing macromer molecular weight (20 kDa) or content (5 wt%), the modulus of the hydrogels decreased by half (Figure S7, 3.1 kPa and 1.2 kPa for DTT and CD-SH cross-linked gels, respectively). Additional cell encapsulation studies using these softer gels showed improved viability of encapsulated MIN6 cells (Figure S9, top panel, >98 % viable cells). In addition to gel stiffness, the presence of active radicals could induce cellular damage during photo-encapsulation 44. While thiol-allylether hydrogels crosslinked by CD-AE were softer than thiol-norbornene hydrogel crosslinked by DTT (Figure 4B, ∼2.4 kPa and 7.1 kPa for CD-AE and DTT gel, respectively), the viability of MIN6 cells was not much higher (Figure 7). We reason that the radical-sensitive MIN6 cells were exposed to active radicals for a longer time during the thiol-allylether gelation because of a much lower chain transfer kinetic constant, and hence the benefits of a softer gel on cell viability was offset by longer exposure of the cells to radical species.
Finally, we evaluated function of encapsulated cells with static glucose stimulated insulin secretion (GSIS, Figure 7C). Since the viability of MIN6 cells varied among different conditions, the amount of insulin was normalized to the amount of intracellular ATP, which scales with cell number at day 10. Compared with thiol-ene hydrogels crosslinked by DTT or CD-AE, the secretion of insulin by MIN6 cells encapsulated in thiol-ene hydrogel crosslinked by CD-SH was about 3-fold higher than thiol-ene hydrogel crosslinked by DTT and CD-AE (Figure 7C, about 0.04 CD-SH, and 0.01 for DTT and βCD-AE). This functional assay result also concurs with the notion that softer gel matrix promotes phenotype of insulin secreting cells.
Conclusion
In summary, we have synthesized step-growth thiol-ene photopolymerized hydrogel using derivatives of βCD and PEG. We found that thiol-norbornene hydrogels crosslinked by CD-SH and PEGNB exhibited faster gelation kinetics and higher crosslinking efficiency when comparing with thiol-allylether hydrogels crosslinked by PEGSH and CD-AE. Furthermore, the gelation kinetics of thiol-norbornene depended highly on the functionality of macromer used. In addition, CD/PEG hydrogels afforded higher loading, as well as prolonged and sustained delivery, of hydrophobic curcumin. The results of live/dead staining, cell viability assay, and GSIS demonstrated that cell viability was maintained in these hydrogels, but in some cases affected by hydrogel properties (e.g., stiffness). Future work will focus on utilizing this CD-based radical-mediated thiol-ene photopolymerized hydrogel as a delivery vehicle of hydrophobic biomolecules.
Supplementary Material
Acknowledgments
This project was funded in part by the National Institutes of Health (R21CA188911), IUPUI Office of the Vice Chancellor for Research (OVCR) through a FORCES grant, Purdue Research Foundation through a Purdue Summer Faculty Research Grant, Indiana Diabetes Research Center through a Pilot & Feasibility grant, and the Department of Biomedical Engineering at IUPUI through a faculty start-up grant. The authors thank Dr. Karl Dria for his technical assistant with mass spectroscopy.
Footnotes
Supporting Information Available: This information is available free of charge via the internet at http://pubs.acs.org/.
References
- 1.Vyas A, Saraf S, Saraf S. J Inclusion Phenom Macrocyclic Chem. 2008;62:23–42. [Google Scholar]
- 2.Tiwari G, Tiwari R, Rai AK. J Pharm BioAllied Sci. 2010;2:72–9. doi: 10.4103/0975-7406.67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.GRAS Notices. Aug 7; http://www.accessdata.fda.gov/scripts/fdcc/?set=GRASNotices&sort=GRN_No&order=DESC&startrow=1&type=basic&search=cyclodextrin.
- 4.Savjani KT, Gajjar AK, Savjani JK. ISRN Pharm. 2012;2012:195727. doi: 10.5402/2012/195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szejtli J. Chem Rev. 1998;98:1743–1754. doi: 10.1021/cr970022c. [DOI] [PubMed] [Google Scholar]
- 6.Douhal A. Cyclodextrin Materials Photochemistry, Photophysics and Photobiology. Elsevier; Italy: 2006. [Google Scholar]
- 7.Li J, Loh XJ. Adv Drug Delivery Rev. 2008;60:1000–1017. doi: 10.1016/j.addr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ZX, Liu KL, Li J. Angew Chem Inter Ed. 2013;52:6180–6184. doi: 10.1002/anie.201301956. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y, Fan XD. J Appl Polym Sci. 2009;113:3068–3077. [Google Scholar]
- 10.Tan S, Blencowe A, Ladewig K, Qiao GG. Soft Matter. 2013;9:5239–5250. [Google Scholar]
- 11.Gaetke LM, Chow CK. Toxicology. 2003;189:147–163. doi: 10.1016/s0300-483x(03)00159-8. [DOI] [PubMed] [Google Scholar]
- 12.Soriano del Amo D, Wang W, Jiang H, Besanceney C, Yan AC, Levy M, Liu Y, Marlow FL, Wu P. J Am Chem Soc. 2010;132:16893–16899. doi: 10.1021/ja106553e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arslan M, Gevrek TN, Sanyal A, Sanyal R. RSC Adv. 2014;4:57834–57841. [Google Scholar]
- 14.Jensen LH, Renodon-Corniere A, Wessel I, Langer SW, Sokilde B, Carstensen EV, Sehested M, Jensen PB. Mol Pharmacol. 2002;61:1235–43. doi: 10.1124/mol.61.5.1235. [DOI] [PubMed] [Google Scholar]
- 15.Machado KE, de Oliveira KN, Andreossi HMS, Bubniak LdS, de Moraes ACR, Gaspar PC, Andrade EdS, Nunes RJ, Santos-Silva MC. Chem Res Toxicol. 2013;26:1904–1916. doi: 10.1021/tx400284r. [DOI] [PubMed] [Google Scholar]
- 16.Arslan M, Gevrek TN, Sanyal R, Sanyal A. Eur Polym J. 2015;62:426–434. [Google Scholar]
- 17.Lin CC, Ki CS, Shih H. J Appl Polym Sci. 2015;132:41563. doi: 10.1002/app.41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih H, Fraser AK, Lin CC. ACS Appl Mater Interfaces. 2013;5:1673–80. doi: 10.1021/am302690t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shih H, Lin CC. Macromol Rapid Commun. 2013;34:269–73. doi: 10.1002/marc.201200605. [DOI] [PubMed] [Google Scholar]
- 20.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bryant SJ, Nuttelman CR, Anseth KS. J Biomater Sci Polym Ed. 2000;11:439–57. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 22.Williams CG, Malik AN, Kim TK, Manson PN, Elisseeff JH. Biomaterials. 2005;26:1211–8. doi: 10.1016/j.biomaterials.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 23.Lin CC, Raza A, Shih H. Biomaterials. 2011;32:9685–9695. doi: 10.1016/j.biomaterials.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin CC, Anseth KS. Proc Natl Acad Sci. 2011;108:6380–6385. doi: 10.1073/pnas.1014026108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawicki LA, Kloxin AM. Biomater Sci. 2014;2:1612–1626. doi: 10.1039/c4bm00187g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Biomaterials. 2009;30:6702–7. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fulton DA, Stoddart JF. Org Lett. 2000;2:1113–1116. doi: 10.1021/ol005668x. [DOI] [PubMed] [Google Scholar]
- 28.Metters A, Hubbell J. Biomacromolecules. 2005;6:290–301. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 29.Flory P. Principles in Polymer Chemistry. Cornell University Press; Ithaca, NY: 1953. [Google Scholar]
- 30.Yadav VR, Prasad S, Kannappan R, Ravindran J, Chaturvedi MM, Vaahtera L, Parkkinen J, Aggarwal BB. Biochem Pharmacol. 2010;80:1021–1032. doi: 10.1016/j.bcp.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Shih H, Lin CC. Biomacromolecules. 2012;13:2003–12. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cramer NB, Reddy SK, O'Brien AK, Bowman CN. Macromolecules. 2003;36:7964–7969. [Google Scholar]
- 33.Pradal C, Jack KS, Grøndahl L, Cooper-White JJ. Biomacromolecules. 2013;14:3780–3792. doi: 10.1021/bm401168h. [DOI] [PubMed] [Google Scholar]
- 34.Singh A, Zhan J, Ye Z, Elisseeff JH. Adv Funct Mater. 2013;23:575–582. doi: 10.1002/adfm.201201902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metters A, Hubbell J. Biomacromolecules. 2004;6:290–301. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 36.Amoli MM, Mousavizadeh R, Sorouri R, Rahmani M, Larijani B. Transplant Proc. 2006;38:3035–3038. doi: 10.1016/j.transproceed.2006.08.172. [DOI] [PubMed] [Google Scholar]
- 37.Srinivasan M. Indian Journal of Medical Sciences. 1972;26:269–270. [PubMed] [Google Scholar]
- 38.Tønnesen HH, Másson M, Loftsson T. Inter J Pharm. 2002;244:127–135. doi: 10.1016/s0378-5173(02)00323-x. [DOI] [PubMed] [Google Scholar]
- 39.El-Kemary M, Douhal A. Chapter 4 - Photochemistry and Photophysics of Cyclodextrin Caged Drugs: Relevance to Their Stability and Efficiency. In: Douhal A, editor. Cyclodextrin Materials Photochemistry, Photophysics and Photobiology. Vol. 1. Elsevier; Amsterdam: 2006. pp. 79–105. [Google Scholar]
- 40.Yallapu MM, Jaggi M, Chauhan SC. Macromol Biosci. 2010;10:1141–51. doi: 10.1002/mabi.201000084. [DOI] [PubMed] [Google Scholar]
- 41.Jung Y, Xu W, Kim H, Ha N, Neckers L. Biochim Biophys Acta, Mol Cell Res. 2007;1773:383–390. doi: 10.1016/j.bbamcr.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 42.Rachmawati H, Edityaningrum CA, Mauludin R. AAPS PharmSciTech. 2013;14:1303–12. doi: 10.1208/s12249-013-0023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nyitray CE, Chavez MG, Desai TA. Tissue Eng Part A. 2014;20:1888–95. doi: 10.1089/ten.tea.2013.0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin CC, Anseth KS. Biomacromolecules. 2009;10:2460–2467. doi: 10.1021/bm900420f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.