

ABSTRACT
Nasal type, extranodal nature killer (NK)/T cell lymphoma-associated hemophagocytic syndrome (NK/T-LAHS) is a rare and fatal disorder with extremely poor prognosis. To investigate its clinical characteristics and risk factors, we retrospectively analyzed 295 patients with nasal type, extranodal nature killer/T cell lymphoma, of which 21 were diagnosed with hemophagocytic syndrome, with a cumulative incidence of 7.1%. The most frequently clinical characteristics were fever, lymphadenopathy, hepatosplenomegaly, pancytopenia, hyperferritinemia, liver dysfunction, hypertriglyceridemia, hypofibrinogenemia and evaluated lactate dehydrogenase (LDH) level. After a median follow-up of 27 months, the 2-year survival for the 295 patients was 74.6%. Significant difference for 2-year survival was found between patients with and without hemophagocytic syndrome (4.8% vs. 80.0%, P<0.001). After developing hemophagocytic syndrome, all patients survived no more than 3 months, with a median survival of 35 days. Risk factors for NK/T-LAHS were bone marrow (BM) involvement (P = 0.019; relative risk, 13.649; 95% confidence interval (CI): 1.538–121.103), hepatosplenomegaly (P = 0.003; relative risk, 9.616; 95%CI: 2.154–42.918), and elevated LDH level (>314U/L) (P = 0.038; relative risk, 6.293; 95%CI: 1.108–35.735). We conducted a risk model for all 295 patients based on the 3 adverse factors as follows: low risk (233 cases, 79.0%), no factor; intermediate risk (43 cases, 14.6%), one factor; high risk (19 cases, 6.4%), 2 or 3 factors. The probabilities for developing LAHS were 0.9% for low-, 14.0% for intermediate-, and 68.4% for high-risk group. Significant differences in the 3 risk groups were observed (P<0.001).
KEYWORDS: Clinical characteristics, lymphoma-associated hemophagocytic syndrome, nature killer/T cell, prognosis, risk factor, survival, treatment
Introduction
Hemophagocytic syndrome (HPS), also named hemophagocytic lymphohistiocytosis (HLH), is a rare and fatal disorder caused by reactive hyperplasia of mononuclear/ macrohagocytic system resulting from a variety of pathogenic factors. This disease is characterized by fever, splenomegaly, cytopenia, coagulopathy, damaged liver function, and hemophagocytosis in bone marrow or other organs.1,2 HPS is divided into 2 types, namely primary and secondary HPS. Primary HPS is associated with genetic mutations, which are common reasons of HPS in pediatric patients, while infections, connective tissue diseases and hematological malignancies are commonly associated with secondary HPS in adults.3,4 Malignancies-associated HPS accounts for a significant percentage of secondary HPS cases (as much as 20%), and is usually discovered in non-Hodgkin lymphoma (NHL), particularly in nasal type, extranodal nature killer (NK)/T cell lymphoma(ENKTL).5
ENKTL is a unique subtype of NHLs according to the World Health Organization (WHO) classification and is a heterogeneous and life-threatening disease. The clinical outcome of this entity is quite poor. Although most of patients are in located stage, the prognosis is poorer than other types of early lymphoma. The outcome of ENKTL would be even worse when complicated with HPS. A study from Japan suggested poor outcome for patients with NK/T cell lymphoma-associated HPS (NK/T-LAHS), with a median survival time of 1–2 months.6
The optimal treatment of NK/T-LAHS is still unclear because of the rarity in occurrence, difficulty in diagnosis, rapid progression and the poor condition of patients at the time of diagnosis. Due to the rarity and heterogeneity of the disease, there are few investigations in NK/T-LAHS with a large sample size. It is indistinct that the reason why some patients suffered HPS while other peers with the similar clinical characteristics did not have. Although the characteristics have been largely studied, the discussion for risk factors for NK/T-LAHS is quite limited.7 Consequently, in this retrospective study, we tried to clarify the incidence, clinical and laboratory characteristics and to determine the risk factors for NK/T-LAHS.
Materials and methods
Patients
A total of 295 patients diagnosed with ENKTL in West China Hospital of Sichuan University between August 2008 and May 2015 were accrued, and 21 patients were diagnosed with NK/T-LAHS. The diagnosis of ENKTL was ascertained pathologically and immunohistochemically according to the WHO classification.8 The definition of HPS was established on the basis of International Histiocyte Society HLH-2004 diagnostic criteria3: (1) fever, (2) splenomegaly, (3) hemocytopenia involving at least 2 lineages(hemoglobin<90 g per liter (/L); platelet count<100 × 109/L; neutrophils<1.0 × 109/L), (4) hypertriglyceridemia(≥ 3.0mmol/L) and/or hypofibrinogenemia(≤ 1.5 g/L), (5) hemophagocytosis in bone marrow (BM) or other organs, (6) hyperferritinemia (≥ 500ng/mL), (7) soluble interleukin-2 receptor (sIL-2R)≥ 2400 U/mL, (8) low or absent NK cell activity. The level of sIL-2R and NK cell activity were not detected because the 2 means were inapplicable in our hospital. Consequently, patients with ENKTL meeting 5 of the first 6 criteria were diagnosed with NK/T-LAHS.
All the patients underwent extensive inspections in terms of complete blood count, liver and kidney function, lactate dehydrogenase (LDH), coagulation function test etc. Lymphoma stage was assessed using the Ann Arbor Staging System. Computed tomography (CT) or magnetic resonance (MR) of nasopharynx, neck, chest, and whole abdomen or positron emission tomography/computed tomography (PET/CT) was used to evaluate patients' disease.
Treatment
One of the following therapeutic regimens was implemented: (1) HLH-2004 protocol3(n = 10); (2) combined chemotherapy (n = 8); (3) only supportive treatment (n = 3). The combined chemotherapy regimens included L-asparaginase-based and non–L-asparaginase–containing regimens. L-asparaginase-based regimens included etoposide, cisplatin, L-asparaginase and dexamethasone (VDLP, n = 2), L-asparaginase, vincristine and prednisone (LVP, n = 2) and gemcitabine, L-asparaginase, ifosfamide, etoposide and dexamethasone (GLIED, n = 1). The non–L-asparaginase–containing regimen was gemcitabine, ifosfamide, dexamethasone and etoposide (n = 1), single-agent etoposide (n = 2). Of the 10 patients receiving HLH-2004 protocol, 9 patients were implemented etoposide and dexamethasone, and only one had the combination of cyclosporine A. No patient received allogeneic haematopoietic stem cell transplantation (allo-HSCT) on account of poor treatment response.
Statistical analysis
The optimal cut-off value and its sensitivity and specificity for lactate dehydrogenase (LDH) were determined by Receiver Operating Curve (ROC) analysis. Overall survival (OS) was defined as the time from randomization to death from any cause. OS was evaluated according to the Kaplan-Meier method, and differences were compared using log-rank test. Logistic regression was used for evaluating significant risk factors for developing HPS. A P value less than 0.05 was considered statistically significant. SPSS 22.0 software was used for statistical analysis (IBM SPSS).
Results
Patient characteristics
The cumulative incidence of NK/T-LAHS was 7.1% (21/295). Clinical features and laboratory data of the 21 patients are displayed in Table 1. The median age was 35 y (range, 16–65 years). There was a male predominance. All patients had fever, hemocytopenia involving at least 2 lineages, ferritin>2000 ng/mL and liver dysfunction, 10 patients (47.6%) showed hemophagocytosis in bone marrow, 8 patients (38.1%) were found BM involvement, 14 patients (66.7%) exerted hepatosplenomegaly and 16 cases (76.2%) were classified into Ann Arbor stage III – IV.
Table 1.
Clinical characteristics of patients with NK/T-LAHS.
| Characteristics | No. (n = 21, %) |
|---|---|
| Gender | |
| Male | 17/21(81.0) |
| Female | 4/21(19.0) |
| Age(years), median(range) | 35(16–65) |
| Ann Arbor stage | |
| I – II | 5/21(23.8) |
| III – IV | 16/21(76.2) |
| ECOG PS | |
| 0–1 | 9/21(42.9) |
| 2–4 | 12/21(57.1) |
| Fever | 21/21(100.0) |
| Hepatosplenomegaly | 14/21(66.7) |
| Lymphadenopathy | 13/21(61.9) |
| BM involvement | 8/21(38.1) |
| Lymphoma status | |
| At onset | 19/21(90.5) |
| At relapse | 2/21(9.5) |
| Triglyceride>3mmol/L | 12/21(57.1) |
| Fibrinogen<1.5 g/L | 16/21(76.2) |
| Ferritin>2000 ng/mL | 21/21(100) |
| Hemocytopenia involving at least 2 lineages | 21/21(100) |
| Hemophagocytosis in bone marrow | 10/21(47.6) |
| LDH>ULN | 18/21(85.7) |
| Liver dysfunction | 21/21(100) |
ECOG PS the Eastern Cooperative Oncology Group performance status, BM bone marrow, LDH lactate dehydrogenase, ULN upper limit of normal. The criteria for liver dysfunction is jaundice, alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) >ULN. Hepatits and drug related damage are not included.
Survival analysis
After a median follow-up of 27 months, an overall 2-year survival of the 295 patients was 74.6% (Fig. 1). In patients without NK/T-LAHS, the overall 2-year survival was 80.0%. The 2-year survival was 4.8% for patients with NK/T-LAHS. The 2-year OS for patients with NK/T-LAHS was obviously inferior when compared with those without NK/T-LAHS(Fig. 2, P<0.001). After the onset of HPS, all patients died within 3 months, with a median survival time of 35 days.
Figure 1.
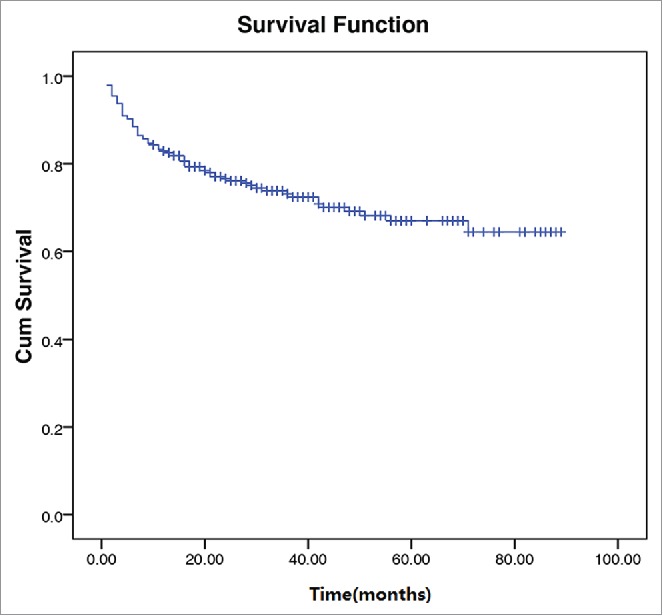
Survival curves of 295 patients with Extranodal natural killer/T-cell lymphoma.
Figure 2.
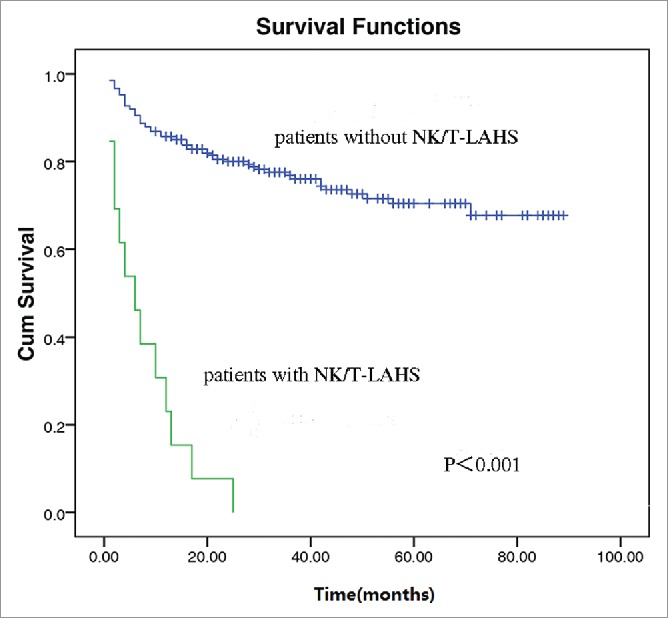
Overall survival curves for patients with and without Extranodal nature killer/T cell lymphoma-associated hemophagocytic syndrome.
Univariate analysis was performed to evaluate the effect of treatment on survival for NK/T-LAHS. According to treatment classifications, no significant difference has been found between HLH-2004 protocol and combined chemotherapy (P = 0.231), or between HLH-2004 protocol and L-asparaginase–containing chemotherapy (P = 0.216).
The cutoff value of LDH
The most appropriate cut-off value for LDH selected by the ROC analysis was 314 U /L (area under the curve [AUC] value 0.821, 95% confidence interval (CI): 0.689–0.953; sensitivity 81.0%, specificity 88.0%). (Fig. 3).
Figure 3.
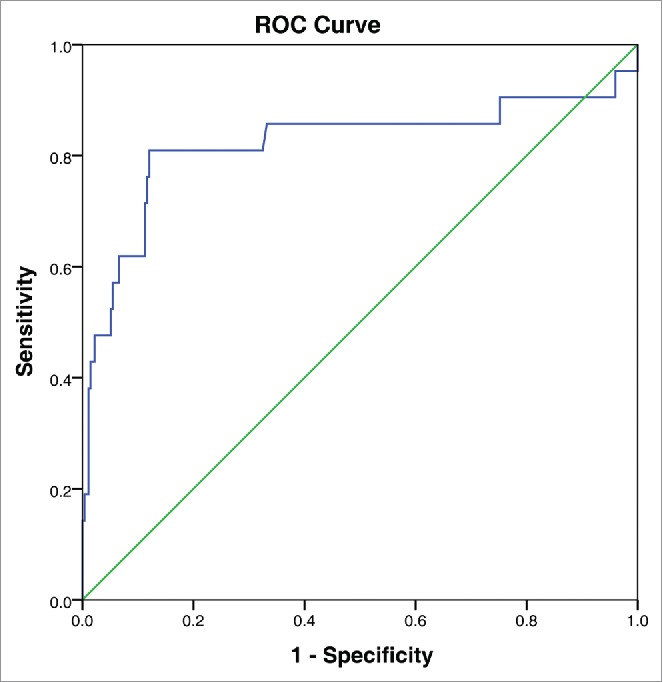
Receiver Operating Curve (ROC) for determining the optimal cut-off value of lactate dehydrogenase.
Risk factors
Risk factors for HPS were shown in Table 2. The result showed BM involvement (P = 0.019; relative risk, 13.649; 95% confidence interval (CI): 1.538–121.103), hepatosplenomegaly (P = 0.003; relative risk, 9.616; 95%CI: 2.154–42.918) and elevated LDH level (>314U/L) (P = 0.038; relative risk, 6.293; 95%CI: 1.108–35.735) were associated with NK/T-LAHS. Consequently, a new risk model for all 295 patients was performed by combining adverse factors as follows: low risk (233 cases, 79.0%), no factor; intermediate risk (43 cases, 14.6%), one factor; high risk (19 cases, 6.4%), 2 or 3 factors. The probabilities for developing LAHS were 0.9% for low-, 14.0% for intermediate-, and 68.4% for high-risk group (Table 3). Significant differences in the 3 risk groups were observed (P<0.001; Odds Ratio, 15.417; 95%CI: 6.890–34.498).
Table 2.
Risk factors for NK/T-LAHS.
| Parameters | p value |
|---|---|
| Gender | 0.814 |
| Age (> 60 years) | 0.863 |
| Ann Arbor (III∼IV) | 0.292 |
| ECOG PS (≥ 2) | 0.433 |
| B symptoms | 0.484 |
| Hepatosplenomegaly | 0.003 |
| Lymphadenopathy | 0.596 |
| BM involvement | 0.019 |
| Lymphoma status | 0.159 |
| LDH>314IU/L | 0.038 |
ECOG PS the Eastern Cooperative Oncology Group performance status, BM bone marrow, LDH lactate dehydrogenase.
Table 3.
Stratification and frequencies of onset for LAHS based on risk group as defined by the new risk index.
| Risk group | No. of factors (%) | No. of patients (%) | LAHS onset (%) |
|---|---|---|---|
| Low risk | 0 | 233(79.0%) | 2(0.9%) |
| Intermediate risk | 1 | 43(14.6%) | 6(14.0%) |
| High risk | 2-3 | 19 (6.4%) | 13(68.4%) |
Discussion
NK/T-LAHS is a rare and life-threatening entity with heterogeneous characteristics and quite poor outcome. Previous studies demonstrated that LAHS were mostly occurred in ENKTL. Tong et al.9 reported an incidence for T cell lymphoma-associated HPS was 24.7% and 12.5 % (2/16) for NK/T-LAHS. Jia et al.7 reported that the incidence of NK/T-LAHS was 11.4%. An analysis of 20 patients with NK/mature T-LAHS suggested that 70% of the cases were NK/T-LAHS.6 However, according to Yu' study,10 LAHS was chiefly found in peripheral T cell lymphoma, not otherwise specified. Data from the current study revealed the NK/T-LAHS rate was 7.1%, which was comparatively lower than the rates reported in the previous studies. The reason for the relatively lower occurrence rate can be partially explained by the statement that the inspections for sIL-2R level and NK cell activity were unavailable in our center, which might underestimate the incidence rate of NK/T-LAHS. However, owing to its rarity, no large sample size was performed. Therefore, no census on odds of NK/T-LAHS was established.
Clinical features of LAHS have been explored in abundance.6,9-15 Janka et al.12 reported typical manifestations of LAHS involve prolonged fever and hepatosplenomegaly, and lymphadenopathy, rash, icterus or neurologic symptoms are less found. Laboratory examinations including cytopenias, hyperferritinemia, liver dysfunction, hypofibrinogenemia, hypertriglyceridemia are primary abnormal indexes.13 Hemophagocytosis may be absent in the early stage of the disease, however, repetitive aspirations of bone marrow or other organs might find hemophagocytosis in disease progression.14 Our result showed that all of the 21 patients presented with fever, pancytopenia, hyperferritinemia, and liver dysfunction. Most of the cases exerted hepatosplenomegaly, hypertriglyceridemia, hypofibrinogenemia and elevated LDH level. 10/21 patients were found hemophagocytosis in BM, which may indicate that hemophagocytosis is not a sensitive index of diagnosis for NK/T-LAHS.
To the best of our knowledge, risk factors for developing LAHS were rarely explored in previous studies.7,15 Sano et al.15 included age, gender and lymphoma lineage (B lymphoma or NK/T lymphoma) into study to analyze risk factors of LAHS. The result showed that the incidence rate of LAHS was much higher in patients with NK/T-cell lymphoma than in those with B-cell lymphoma. An analysis of 23 patients with NK/ T-LAHS in China suggested that younger age, BM involvement, and reduced serum albumin generate a higher incidence of HPS.7 In present study, we attempted to develop a system to stratify patients with different risk index according to pretreatment clinical features. The results indicated that BM involvement, hepatosplenomegaly, and elevated LDH level result in a higher probability of developing HPS. A risk model was designed on basis of the 3 adverse factors. Therefore, we identified 3 categories of patients with significantly different prevalence risk of NK/T-LAHS.
The reason why patients with BM involvement, hepatosplenomegaly, and elevated LDH level run a higher risk of developing NK/T-LAHS is unclear. Tong et al.9 reported that patients with LAHS have a significantly higher percentage of BM involvement than those without LAHS, and this may indicate that the early BM involvement is a remarkable risk indicator in T-cell lymphoma with LAHS. Similar phenomena were noticed in ENKTL6,8 and large B cell lymphoma.16 In present study, the incidence of BM involvement was significantly higher in patients with NK/T-LAHS compared with those without NK/T-LAHS at baseline assessment (38.1% vs. 0.7%). BM invasion seems to be highly related to the development of LAHS though the precise mechanism is unclear. Hepatosplenomegaly are frequently found in HPS. Cytokines generated by activated T cells/ macrophages may destroy liver metabolism and produce the hepatic mitochondrial injury characteristic of the clinical features of HPS.17 Cytokine storm and overwhelming inflammation may associate with hepatosplenomegaly. In this study, 14 of 21 patients (66.7%) showed hepatosplenomegaly. LDH could be sensitively and comprehensively reflect the organ index of tissue damage. A high level of LDH was found in HPS in a series of studies.7,16-22 Through TH1 cell recruitment and activation at organs with inflammation, interleukin (IL)-16 exert influence in the pathogenesis of HPS. Serum IL-16 levels showed a significant positive correlation with serum LDH levels and other indicators.21 Furthermore, elevated LDH was demonstrated to be a poor prognostic factor for survival in LAHS, and results indicated that 1000U/L5,7 or 2000U/L22 was the threshold value for prognosis. However, the optimal value of LDH serving as a risk factor in LAHS was unclear, and Jia et al.7 put forward that LDH>240U/L was not a risk factor for developing NK/T-LAHS. Therefore, in this study, we conducted an ROC analysis, which suggested that 314U/L was the optimal cut-off value. Patients with LDH>314U/L might result in a higher probability of developing HPS.
NK/T-LAHS is a frequently fatal disorder with a high rate of mortality. In our study, patients with NK/T-LAHS had an inferior 2-year survival when compared with those without NK/T-LAHS (4.8% vs. 80.0%, P<0.001, Fig. 2). After the onset of HPS, all cases died no more than 3 months, with a median survival time of 35 days, which is consistent with previous report.6
Optimal treatment of NK/T-LAHS remains to be investigated owing to the rarity in occurrence and rapid progression of disease. It is generally accepted that HLH-2004 protocol3 is suitable for all patients with HPS, including LAHS. Song et al.23 suggested plasma exchange was a useful remedy for patients with HPS. Allo-HSCT has also been proved to be successful in some cases.12,24 Chemotherapy in combination with high dose of corticosteroids and immunosuppressive therapy are regarded as an effective treatment of LAHS.25,26 However, no conformity has been established. Han et al. indicated that pegaspargase was effective to improve survival in NK/T-LAHS.27 However, our results demonstrated that the application of L-asparaginase in our patients did not lead to a significantly longer survival, in agreement with a prior report.7 Data from our study did not evaluate the role of allo-HSCT or plasma exchange because none of the patients obtained these treatments.
There are some potential limitations in this retrospective study. Firstly, the single-institutional, retrospective nature of this study is a limitation. Secondly, the incidence rate of NK/T-LAHS may be underestimated for 2 indicator used for the diagnosis of HPS cannot be detected.
In conclusion, patients with NK/T-LAHS showed higher complication rates and mortality rates than those without NK/T-LAHS. Risk factors for this entity have been investigated and a risk model was conducted in present study, which suggested a promising result. However, further studies with larger sample are indispensable to confirm the result.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the medical, nursing, and clinical laboratory staff in West China Hospital of Sichuan University for their commendable contributions to this study.
Author contributions
Li-qun Zou designed the research. Na Li collected and analyzed data, performed research and wrote the paper. Li Zhang contributed vital new reagents and analyzed data. Jie Liu performed statistical analysis. Jing Zhang collected data. Hua-wei Weng and Hong-yu Zhuo analyzed and interpreted data.
References
- 1.Allory Y, Challine D, Haioun C, Copie-Bergman C, Delfau-Larue MH, Boucher E, Charlotte F, Fabre M, Michel M, Gaulard P. Bone marrow involvement in lymphomas with hemophagocytic syndrome at presentation: A clinicopathologic study of 11 patients in a western institution. Am J Surg Pathol 2001; 25:865-74; PMID:11420457; http://dx.doi.org/ 10.1097/00000478-200107000-00004 [DOI] [PubMed] [Google Scholar]
- 2.Kumakura S. Hemophagocytic syndrome. Intern Med 2005; 44:278-80; PMID:15897634; http://dx.doi.org/ 10.2169/internalmedicine.44.278 [DOI] [PubMed] [Google Scholar]
- 3.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, et al.. HLH- 2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48:124-31; PMID:16937360; http://dx.doi.org/ 10.1002/pbc.21039 [DOI] [PubMed] [Google Scholar]
- 4.Szyper-Kravitz M. The hemophagocytic syndrome/macrophage activation syndrome: A final common pathway of a cytokine storm. Isr Med Assoc J 2009; 11:633-4; PMID:20077953 [PubMed] [Google Scholar]
- 5.Li F, Li P, Zhang R, Yang G, Ji D, Huang X, Xu Q, Wei Y, Rao J, Huang R, et al.. Identification of clinical features of lymphoma-associated hemophagocytic syndrome (LAHS): An analysis of 69 patients with hemophagocytic syndrome from a single-center in central region of China. Med Oncol 2014; 31:902; PMID:24610542; http://dx.doi.org/ 10.1007/s12032-014-0902-y [DOI] [PubMed] [Google Scholar]
- 6.Takahashi N, Miura I, Chubachi A, Miura AB, Nakamura S. A clinicopathological study of 20 patients with T/natural killer (NK)-cell lymphoma-associated hemophagocytic syndrome with special reference to nasal and nasal-type NK/T-cell lymphoma. Int J Hematol 2001; 74:303-8; PMID:11721967; http://dx.doi.org/ 10.1007/BF02982065 [DOI] [PubMed] [Google Scholar]
- 7.Jia J, Song YQ, Lin NG, Liu W, Ping L, Zheng W, Wang X, Xie Y, Tu M, Zhang C, et al.. Clinical features and survival of extranodal natural killer/T cell lymphoma with and without hemophagocytic syndrome. Ann Hematol 2016; 95(12):2023-31; PMID:27595760; http://dx.doi.org/ 10.1007/s00277-016-2805-9 [DOI] [PubMed] [Google Scholar]
- 8.Sabattini E, Bacci F, Sagramoso C, Pileri SA. WHO classification of tumors of haematopoietic and lymphoid tissues in 2008: An overview. Pathologica 2010; 102:83-7; PMID:21171509. [PubMed] [Google Scholar]
- 9.Tong H, Ren Y, Liu H, Xiao F, Mai W, Meng H, Qian W, Huang J, Mao L, Tong Y, et al.. Clinical characteristics of T-cell with hemophagocytic syndrome: Comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma 2008; 49:81-7; PMID:18203016; http://dx.doi.org/ 10.1080/10428190701713630 [DOI] [PubMed] [Google Scholar]
- 10.Yu JT, Wang CY, Yang Y, Wang RC, Chang KH, Hwang WL, Teng CL. Lymphoma-associated hemophagocytic lymphohistiocytosis: Experience in adults from a single institution. Ann Hematol 2013; 92:1529-36; PMID:23700280; http://dx.doi.org/ 10.1007/s00277-013-1784-3 [DOI] [PubMed] [Google Scholar]
- 11.Han AR, Lee HR, Park BB, Hwang IG, Park S, Lee SC, Kim K, Lim HY, Ko YH, Kim SH, et al.. Lymphoma-associated hemophagocytic syndrome: Clinical features and treatment outcome. Ann Hematol 2007; 86:493-8; PMID:17347847; http://dx.doi.org/ 10.1007/s00277-007-0278-6 [DOI] [PubMed] [Google Scholar]
- 12.Janka GE. Hemophagocytic syndromes. Blood Rev 2007; 21:245-53; PMID:17590250; http://dx.doi.org/ 10.1016/j.blre.2007.05.001 [DOI] [PubMed] [Google Scholar]
- 13.Chandrakasan S, Filipovich AH. Hemophagocytic lymphohistiocytosis: Advances in pathophysiology, diagnosis, and treatment. J Pediatr 2013; 163(5):1253-9; PMID:23953723; http://dx.doi.org/ 10.1016/j.jpeds.2013.06.053 [DOI] [PubMed] [Google Scholar]
- 14.Aricò M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, Martinetti M, Rusca MP. Hemophagocytic lymphohistiocytosis. Report of 122 children from the international registry. FHL study group of the histiocyte society. Leukemia 1996; 10(2):197-203; PMID:8637226. [PubMed] [Google Scholar]
- 15.Sano H, Kobayashi R, Tanaka J, Hashino S, Ota S, Torimoto Y, Kakinoki Y, Yamamoto S, Kurosawa M, Hatakeyama N, et al.. Risk factor analysis of non-Hodgkin lymphoma-associatedhaemophagocytic syndromes: A multicentre study. Br J Haematol 2014; 165:786-92; PMID:24606577; http://dx.doi.org/ 10.1111/bjh.12823 [DOI] [PubMed] [Google Scholar]
- 16.Yeh YM, Chang KC, Chen YP, Kao LY, Tsai HP, Ho CL, Wang JR, Jones D, Chen TY. Large B cell lymphoma presenting initially in bone marrow, liver and spleen: An aggressive entity associated frequently with haemophagocytic syndrome. Histopathology 2010; 57:785-95; PMID:21166693; http://dx.doi.org/ 10.1111/j.1365-2559.2010.03709.x [DOI] [PubMed] [Google Scholar]
- 17.Imashuku S, Teramura T, Morimoto A, Hibi S. Recent developments in the management of haemophogocytic lymphohistiocytosis. Expert Opin Pharmacother 2001; 2(9):1437-48; PMID:11585022; http://dx.doi.org/ 10.1517/14656566.2.9.1437 [DOI] [PubMed] [Google Scholar]
- 18.Lam-Tse WK, van den Berg L, Dawson L, van Ravenswaay Claasen HH, Hart W, Maartense E. Clinical reasoning and decision-making in practice. A patient with fever and pancytopenia. Ned Tijdschr Geneeskd 2008; 15; 152(11):606-14 [PubMed] [Google Scholar]
- 19.Suzuki Y, Takahashi N, Yada Y, Koike Y, Matano M, Nishimura H, Kono Y. Hemophagocytic lymphohistiocytosis in a newborn infant born to a mother with Sjögren syndrome antibodies. J Perinatol 2013; 33(7):569-71; PMID:23803677; http://dx.doi.org/ 10.1038/jp.2012.147 [DOI] [PubMed] [Google Scholar]
- 20.Okuda T, Yumoto Y. Reactive hemophagocytic syndrome responded to combination chemotherapy with steroid pulse therapy. Rinsho Ketsueki 1995; 36(11):1316-20; PMID:8691575. [PubMed] [Google Scholar]
- 21.Takada H, Ohga S, Mizuno Y, Nomura A, Hara T. Increased IL-16 levels in hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 2004; 26(9):567-73; PMID:15342983; http://dx.doi.org/ 10.1097/01.mph.0000134465.86671.2e [DOI] [PubMed] [Google Scholar]
- 22.Wang YR, Qiu YN, Bai Y, Wang XF. A retrospective analysis of 56 children with hemophagocytic lymphohistiocytosis. J Blood Med 2016; 11(7):227-31; PMID:27785117; http://dx.doi.org/ 10.2147/JBM.S92438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song KS, Sung HJ. Effect of plasma exchange on the circulating IL-6 levels in a patient with fatal hemophagocytic syndrome associated with bile ductopenia. Ther Apher Dial 2006; 10:87-9; PMID:16556142; http://dx.doi.org/ 10.1111/j.1744-9987.2006.00347.x [DOI] [PubMed] [Google Scholar]
- 24.Ohga S, Kudo K, Ishii E, Honjo S, Morimoto A, Osugi Y, Sawada A, Inoue M, Tabuchi K, Suzuki N, et al.. Hematopoietic stem cell transplantation for familial hemophagocytic lymphohistiocytosis and Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis in Japan. Pediatr Blood Cancer 2010; 54:299-306; PMID:19827139; http://dx.doi.org/ 10.1002/pbc.22310 [DOI] [PubMed] [Google Scholar]
- 25.Xie W, Hu K, Xu F, Zhou D, He J, Shi J, Luo Y, Zhu J, Zhang J, Lin M, et al.. Clinical analysis and prognostic significance of lymphoma-associated hemophagocytosis in peripheral T cell lymphoma. Ann Hematol 2013; 92:481-6; PMID:23238896; http://dx.doi.org/ 10.1007/s00277-012-1644-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han L, Zhang M, Li L, Zhang L, Wu J, Li X, Wang X, Young KH, Fu X, Ma W, et al.. Natural killer/T-cell lymphoma-associated hemophagocytic syndrome: A case report. Oncol Lett 2014; 8:886-90; PMID:25013513; http://dx.doi.org/ 10.3892/ol.2014.2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han L, Li L, Wu J, Li X, Zhang L, Wang X, Fu X, Ma W, Sun Z, Zhang X, et al.. Clinical features and treatment of natural killer/T cell lymphoma associated with hemophagocytic syndrome: Comparison with other T cell lymphoma associated with hemophagocytic syndrome. Leuk Lymphoma 2014; 55:2048-55; PMID:24359240; http://dx.doi.org/ 10.3109/10428194.2013.876629 [DOI] [PubMed] [Google Scholar]