

ABSTRACT
The prevalence of neuroendocrine tumors (NETs) has recently been increasing. Although various drugs such as Octreotide and its analogs show certain efficacy, NETs in many patients progress and metastasize. It is desirable to develop new interventions to improve the therapy. Here we show that human neuroendocrine tumor BON cells are resistant to several drugs commonly used for NET therapy, including Octreotide that activates somatostatin receptor-induced anti-proliferation, and Capecitabine and Temozolimide that damage DNA. In contrast, an inhibitor (IBET) to an epigenetic regulator, Brd4 that binds acetylated histones and upregulates transcription of multiple genes including protooncogene c-Myc, potently inhibited the NET cells. We found that IBET increased the protein levels of cyclin-dependent kinase (CDK) inhibitor p27kip/cip (or p27), but not its mRNA levels. Moreover, the p27 induction at protein level by IBET was at least partly through increasing the protein stability of p27. The increased protein stability of p27 likely resulted from IBET-mediated suppression of Skp2, an E3 ligase that can mediate p27 degradation by increasing its ubiquitinylation. These findings unravel a new mechanism whereby the IBET-induced repression of proliferation of neuroendocrine cells.
KEYWORDS: Brd4, IBET, p27, neuroendocrine tumor, Skp2
Introduction
Neuroendocrine tumors (NET) not only pose serious threats as neoplastic diseases but also exacerbate the underlying symptoms by secreting an excessive amount of hormones.1,2 The prevalence of NETs has recently substantially increased.3 Several organs, such as the pancreas, small intestine, and lungs, can develop carcinoid or neuroendocrine tumors, however, the genetic mechanisms regulating the development and progression of these tumors are not well understood.4 Several drugs, such as Octreotide and its analogs that mimic somatostatin (SST), bind somatostatin receptors (SSTR), G protein coupled receptors, to suppress protein kinase A (PKA) activity, leading to inhibition of hormone secretion and cell proliferation.5 While Octreotide and derivatives have been used to attenuate the symptoms of the diseases, the efficacy and specificity of such drugs require improvement.6 Recently, mTOR (mammalian Target Of Rapamycin) inhibitors such as Everolimus, has also been approved for treatment of neuroendocrine tumors.7 Other chemotherapy agents such as Capecitabine, a precursor of 5-Fluro-Uracil, and Temozolimide, a DNA-alkylating agent, have shown some promise in treating patients with metastatic NETs.8 However, many of these agents fail to suppress growth of NETs after long-term treatment, and many patients eventually succumb to the disease. As such, it is important to explore more avenues to inhibit NET cells to develop more effective therapy.
It has become increasingly clear that epigenetic regulation plays a very important role in tumorigenesis, maintenance of the cancer phenotype and cancer progression.9,10 One member of a family of acetylated histone binding proteins, Brd4, can specifically bind the modified histones and the chromatin, facilitating gene transcription. The crystal structure of Brd4 has been solved and it contains a cavity that can specifically bind to the acetylated histones.11-13 Based on this information, a specific inhibitor, JQ1, was synthesized to block Brd4 binding to the acetylated histones, potently suppressing expression of multiple of proproliferative or protooncogenes such as c-Myc, leading to tumor suppression.11-13 Later on, multiple inhibitors targetting Brd4 or some of its homologous proteins were developed, including IBET151, which potentially inhibits transcription of c-Myc and CDK4, resulting in potent suppression of leukemia cells.14
Brd4 inhibitor was also used to treat the cultured neuroendocrine tumor (NET) cells,15 such as BON cells, a human neuroendocrine tumor cell line derived from pancreatic NETs.16 Certain Brd4 inhibitor such as JQ1 can suppress NET cell proliferation, concomitant with reduced expression of c-Myc and cyclin D1, both pro-proliferative genes, and increased expression of certain anti-proliferative proteins.15 However, it was unclear that the Brd4 inhibitor also affects expression of tumor suppressors in NET cells.
Cell proliferation or cell cycling are tightly regulated by multiple factors, and a key class of proteins controlling cell cycle are cyclin-dependent kinases (CDKs).17 CDK4/6 are crucial for controlling G0 or G1 to S phase transition, yet CDK2 is crucial for S to G2 transition. Another class of cell cycle regulators is CDK inhibitors including p27kip/cip (p27 hereafter), which can bind and inhibit several CDKs including CDK4/6 as well as CDK2, thus potently inhibiting proliferation of multiple types of cells.17
Expression or function of p27 is compromised in neuroendocrine tumors.18-20 It is thus conceivable that the expression and function of p27 is tightly controlled by multiple factors. For instance, transcription of p27 is enhanced by anti-proliferative transcription factors such as FOXOs, which can directly bind and activate transcription of p27.21 Moreover, p27 is also regulated posttranscriptionally. P27 is ubiquitinylated by E3 ligase, Skp2, and the ubiquitinylated p27 is promptly delivered to proteosomes and is degraded in the proteosomes.22 p27 is reduced in expression or mutated in a subgroup of NETs, and Brd4 inhibitor JQ1 increased p27 expression in certain NET cells.15 But it is poorly understood how p27 is regulated in NETs by Brd4 inhibitors. In the current studies, we found that while neuroendocrine tumor BON cells are resistant to several commonly used drugs for NET therapy, they are quite sensitive to a Brd4 inhibitor IBET. Notably, IBET suppresses expression of Skp2, leading to increased stability of p27 and reduced cell proliferation. These findings unravel a new means for Brd4 inhibitors to repress neuroendocrine tumors.
Results
Neuroendocrine tumor (NET) BON cells are resistant to multiple drugs used to treat NETs, but are sensitive to Brd4 inhibitor IBET151 (IBET hereafter)
To determine the sensitivity of an established human neuroendocrine tumor cell line, BON cells, to the drugs commonly used to treat human NETs, first, we examined the impact of Everolimus,7 an inhibitor of mTOR kinase which is upregulated in certain human NETs,23 on BON cells. We found that Everolimus barely reduced cell proliferation in doses ranging from 0.01–100 nM (Fig. 1A), while longer treatment of 5 day slightly reduced cell number (Fig. 2B). We further examined the impact of another commonly used drug for treating NETs, octreotide, which is a synthetic cyclic peptide mimicking the human inhibitory hormone, somatostatin (SST).24 The results indicate that BON cells were resistant to Octreotide in doses ranging from 0.1–20 uM (Fig. 1B). We also found that Capecitabine and Temozolimide, a nucleotide analog and DNA alkylating agent, respectively, used to treat metastatic NETs,8 failed to suppress BON cells, in a wide range of doses from 0.005–5 uM of concentrations (Fig. 1C-D). It is likely that BON cells represent a type of metastatic NETs that have developed resistance to multiple drugs.
Figure 1.
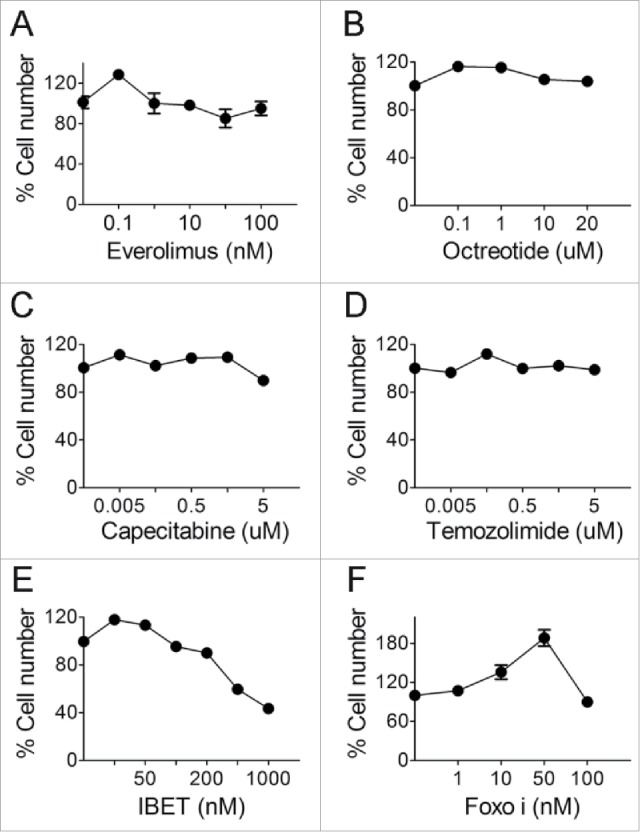
The effect of various drugs on BON cell growth. (A-D) BON cells were treated with various concentrations of everolimus (A), octreotide (B), capecitabine (C) and temozolimide (D). MTS assay was performed to assess cell proliferation after 3 d. (E) BON cells were treated with varying concentrations of IBET and MTS assay was performed to assess cell proliferation after 5 d of treatment. (F) BON cells were treated with various concentrations of Foxoi and cell counting was performed to assess cell proliferation after 5 d of the treatment.
Figure 2.
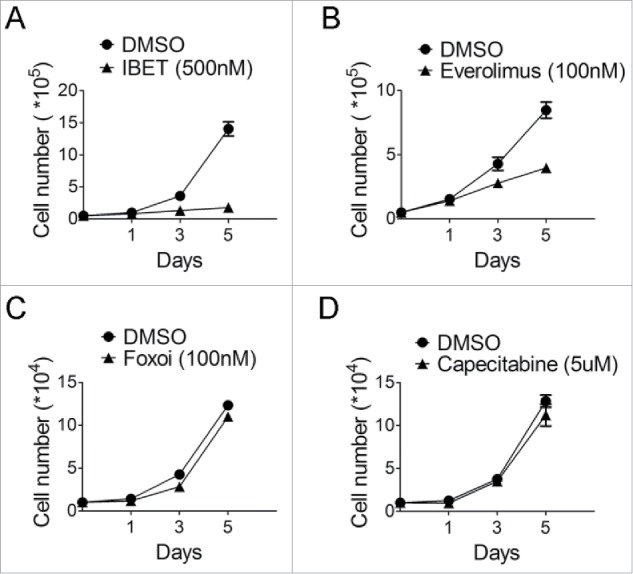
The time course of treatment of BON cells with various drugs. (A) Cell counting following treatment of the BON cells with IBET (500nM) for the indicated time. (B) Cell counting following treatment of the BON cells with everolimus (100nM) for the indicated time. (C) Cell counting following treatment of the BON cells with Foxoi(100nM) for the indicated time. (D) Cell counting following treatment of the BON cells with capecitabine (5μM) for the indicated time.
As such, we explored whether other new drug-like epigenetic inhibitors, such as IBET151 (IBET hereafter), which inhibits an acetylated histone-binding proteins such as Brd4, can affect growth of the neuroendocrine cells,14 and a previous report also indicates that this type of inhibitor can suppress certain NET cells.15 Our results indicate that indeed IBET substantially suppressed growth of BON cells (Fig. 1E). As a control, we also treated these cells with an inhibitor of FOXOs,25 and found that FOXOi did not suppress growth of BON cells (Fig. 1F).
To further examine the time-dependent effect of these various drugs or drug-like compounds on growth of the NET cells, we monitored the sensitivity of the cells to the various inhibitors in a timeframe up to 5 days, with daily replenishing of the drugs. The cells were treated with the various inhibitors, and counted to determine the impact of the drugs on cell growth. The results indicate that IBET markedly suppressed growth of the NET cells (Fig. 2A), and Everolimus only modestly suppressed growth of the cells after the second day of treatment (Fig. 2B), but the cells were resistant to Capecitabine treatment (Fig. 2C).
Together, our results indicate that BON cells, as a human NET cell line commonly used for cell biology studies, are resistant to multiple commonly used drugs, but that Brd4 inhibitor is effective in inhibiting growth of the cells. However, the underlying mechanism remains unclear.
IBET induces expression of p27 at the protein level, but does not affect the mRNA level
To determine how IBET suppressed growth of BON cells, we investigated whether IBET treatment affects various factors that regulate cell proliferation. As it has been reported that IBET decreases expression of cyclin D115 and c-Myc,12 we determined expression of these proteins in the control and IBET treated cells. As expected, IBET treatment reduced expression of cyclin D1 (Fig. S1), and c-Myc (Fig. 3, lane 2), in Western blotting analysis. We further examined whether IBET also affects expression of certain anti-proliferative factors such as p27 and FOXO1. Interestingly, IBET treatment also markedly increased the protein level of p27 (Fig. 3, lane 2), a cyclin dependent kinase inhibitor, and also FOXO1 (Fig. 3, lane 2).
Figure 3.
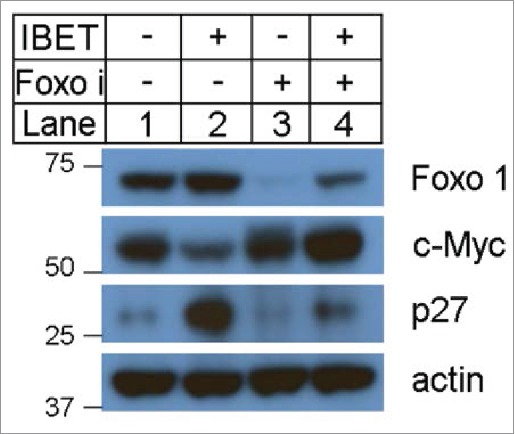
The effect of IBET treatment on the protein levels of p27, FOXO1 and c-Myc. The BON cells were treated with control DMSO or IBET (500 nM), for 5 days, followed by Western blotting analysis using the indicated antibodies.
We sought to determine whether IBET-induced expression results from increased mRNA level or transcription of p27 and FOXO1 or not. To this end, we examined the mRNA levels of the control and IBET treated cells using qRT-PCR analysis, and found that IBET treatment slightly increased the mRNA level of FOXO1 (Fig. 4A), but did not affect the mRNA level of p27 (Fig. 4B). Thus far, to our knowledge, the most biologic effect induced by Brd4 inhibitor was mediated by its impact on binding and suppressing Brd4 to the acetylated histones in the chromatin, resulting in suppression of gene transcription.12 Nevertheless, our findings regarding the impact of IBET on the protein level, but not the mRNA level, of p27 raises an intriguing possibility that IBET induces p27 epxression at a posttranstriptional level.
Figure 4.
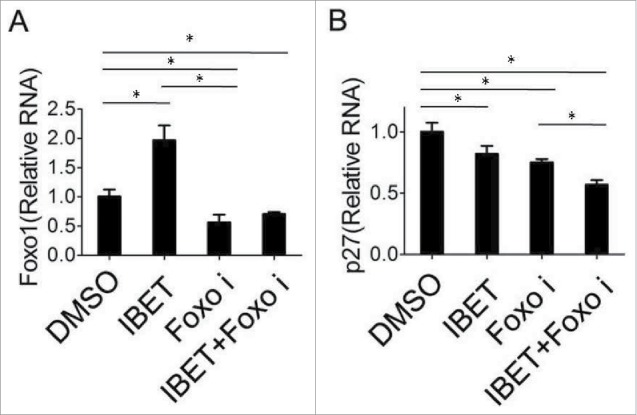
The effect of IBET treatment on the mRNA level of p27cip/kip. BON cells were treated with either control DMSO or IBET (500 nM) for 5 days, followed by isolation of RNA from the cells, and qRT-PCR analysis for the FOXO1 and p27 (B). The means of the different groups in the mRNA level were compared using one-way ANOVA test. Differences were considered statistically significant when P < 0.05(*P < 0.05).
Treatment of NET cells with IBET substantially increases the stability of p27
To determine whether IBET treatment influences the protein stability, we first treated the cells with DMSO or IBET for 3 days, followed by treatment with cycloheximide, an inhibitor of protein translation in cells,26 for various periods of time before Western blot analysis. The results indicate that in control cells, p27 protein degraded quickly 2 hr after the treatment with cycloheximide (Fig. 5A, lane 1 vs 2). In contrast, treatment of the cells with IBET markedly delayed the reduction of p27 protein, even maintaining approximately 60% of the protein after 6 hr of IBET treatment (Fig. 5A, lanes 5–8 and 5B). In control cells, after 6 hr treatment, only less than 20% of p27 was detectable. These results indicate that IBET treatment substantially reduced degradation of p27, an anti-proliferative protein that may contribute to IBET-induced suppression of BON cell growth.
Figure 5.
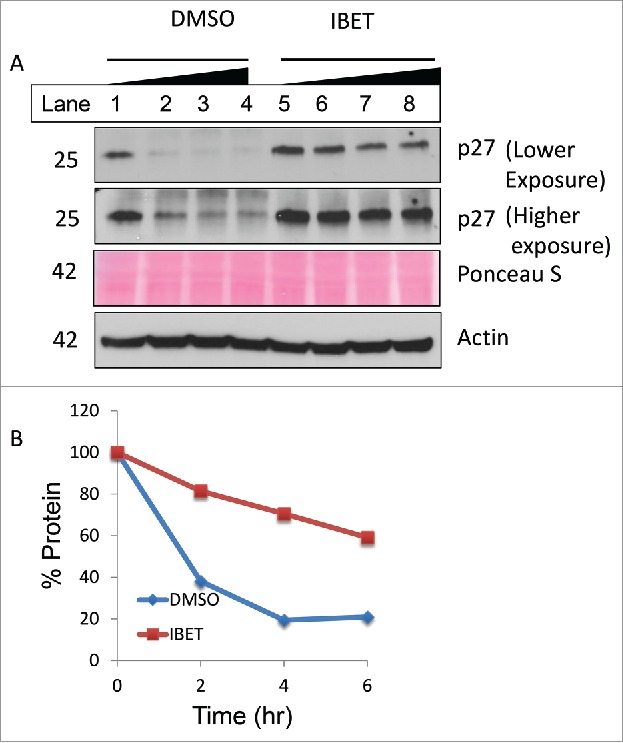
IBET treatment increases p27 protein stability. (A) BON cells were treated with DMSO or IBET for 3 days, followed by addition of 20 μg/ml cycloheximide in the respective medium, and the cells were collected at 0, 2, 4 and 6 hr after the cycloheximide treatment of Western blotting with the indicated antibodies. (B) p27 protein band in (A) was scanned, and was quantitated using Image J and plotted as % protein relative to the signal value at 0 min.
IBET reduces expression of Skp2, an E3 ubiquitin ligase that enhances degradation of p27
It has been reported that Skp2, an E3 ubiquitin ligase can catalyze protein ubiqutination and promote subsequent proteosome-mediated degradation.22 As such, we investigated the potential impact of IBET treatment on the protein level of Skp2. To this end, we treated BON cells with either control vehicle or IBET, and then determined the amount of Skp2 protein using Western blot. The results indicate that indeed treatment with IBET substantially reduced Skp2 protein, in contrast to increased expression of p27 and FOXO1 in protein levels (Fig. 6A, lane 2 vs 1), while the control actin levels remained the same. Moreover, treatment of the BON cells with FOXOi, which blocked IBET-induced increase of p27 protein level (Fig. 3, lane 4), also upregulated the protein level of Skp2 (Fig. 6B). Together, these results indicate that IBET can repress expression of Skp2, leading to increased stability and protein level of p27, thus suppressing NET cell proliferation (Fig. 6C), a novel mechanism whereby IBET inhibits cell proliferation.
Figure 6.
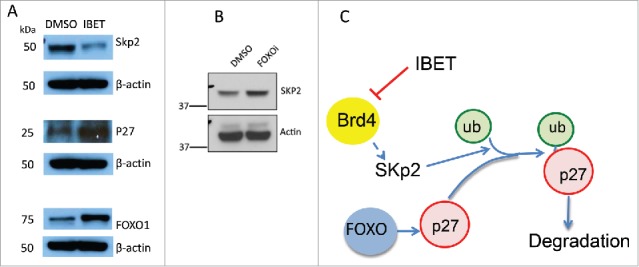
The effect of IBET and FOXOi treatment on the protein level of Skp2. (A) BON cells were treated with either DMSO or IBET (0.5uM) for 3 days, followed by Western blotting analysis to detect the protein levels with the indicated antibodies. (B) BON cells were treated with DMSO or FOXOi (100nM) for 3 days, followed by Western blotting with the indicated antibodies. (C) A working model to explain IBET-induced upregulation of p27 protein level via suppressing expression of Skp2.
Discussion
One major challenge in cancer therapy is the development of de novo or acquired resistance.27 Certain cancer cells develop drug resistance via traceable mutations in the target genes, such as the BCR-ABL kinase mutations in chronic myeloid leukemia (CML) in response to BCR-ABL inhibitor Gleevec.28 Other secondary mutations in EGFR, whose mutation is a driver in a subset of non-small cell lung cancer (NSCLC), such as T790M that reduces the mutant EGFR binding to its kinase inhibitor in patients, occur in cancer cells, causing drug resistance.27 On the other hand, the de novo resistance, sometime similar to reduced sensitivity, to drugs of targeted therapy, such as PI3K/AKT inhibitors, takes place by activation of the feedback loop of the kinase pathways such as activation of pro-proliferative receptor tyrosine kinases (RTK).29 In other cases, the certain adaptive and compensatory pathways are activated epigenetically, in a time dependent manner, resulting reduced drug and failure of therapy. One example is that treatment of human HER2+ breast cancer cells with Lapatinib, a small molecular HER2/HER1 kinase inhibitor, leads to a biphasic response of the cancer cells to the treatment.30 In the first phase, Lapatinib suppresses HER1/2, eventually leads to inhibition of AKT, reducing the phosphorylation of FOXOs, thus activating FOXOs and cell apoptosis. However, several days later, in the second phase, the remaining cells became quiescent, but markedly upregulating FOXOs, resulting in enhanced trascription of c-Myc via paradoxical FOXO/c-Myc axis and eventual loss of the sensitivity of the HER2+ cancer cells.
In neuroendocrine tumors, it is even more challenging to uncover the pathways that can cause effective resistance to the drugs as only a few pathways are found to be mutated in the cancer, and no drug resistance-causing mutations in the driver genes have yet been found.23 As such, we evaluated the impact of the commonly used drugs such as Everolimus, Octreotide, Capacitabine, and Temozolimide on the growth of BON cells. It is not surprising that many of these drugs were not effective to suppress the human NET cells.
IBET can effectively suppress transcription of several key pro-proliferative oncogenes or proto-oncogenes such as c-Myc, CDK4, and cyclin D1, in several types of leukemias or solid tumors.12,14,15 However, little is known as to how the Brd4 inhibitors affect anti-proliferative genes. Our findings indicate that Brd4 inhibitor not only suppresses cell proliferation via inhibiting pro-proliferative genes, but also coordinately increases anti-proliferative tumor suppressing proteins such as p27. These findings provide new insights into understanding how the Brd4 inhibitor suppresses NET cells via upregulating anti-proliferative proteins.
p27 is regulated by multiple mechanisms including gene transcription inducd by FOXOs21 and protein degradation mediated by Skp2.22 However, thus far, it is not clear whether IBET affects p27 protein degradation. Our current studies found that IBET increases p27 expression and protein stability at least partly via reducing expression of E3 ligase Skp2, which has been shown to mediate p27 degradation. While FOXOs can increase transcription of p27 in certain cells,21 our results show IBET can also increase FOXO1 protein as well as mRNA levels, however, it is not yet clear whether IBET-mediated regulation of FOXOs plays any role in regulating p27 stability. On the other hand, we found that IBET increased the p27 protein level but did not affect the mRNA level of p27. IBET treatment markedly increased the stability of p27 in the NET cells. IBET increases the p27 stability and protein level, at least partly via repressing Skp2, which can normally degrade p27 protein by increasing p27 ubiquitinylation, a signal triggering proteasome-mediated protein degradation. Thus, IBET-mediated increase in p27 protein level may contribute to the suppression of proliferation of BON cells.
Consistent with these findings, Brd4 inhibitor JQ1 also upregulates p27 protein in hepatocellular carcinoma cells.31 Moreover, Brd4 inhibitor also reduced Skp2 but increased expression of p27 protein levels in melanoma cell lines,32 even though the precise p27 stability was not examined. As it has been reported that Brd4 inhibitor IBET like compound also reduces the mRNA level of Skp2 in melanoma cells,1 and FOXOs can directly bind to the promoter of the p27 gene to unpregulate its expression, it is likely that IBET can inhibit the transcription-activating aceylated histone binding proteins such as Brd4 at the Skp2 promoter to reduce its expression, resutling in reduction of Skp2 but upregulation of p27 protein level. Even at the basal level without marked repression of AKT and upregulation of FOXO activities, it is possible that FOXOi can still reduce the basal transcription of p27, so that IBET-induced suppression of Skp2 could no longer lead to marked increase in the p27 protein level (Fig. 3, lane 4). Nevertheless, the detailed mechnisms regarding the role of FOXO1 in the process is complex as regulation of the ubiquitin-proteosome system can be mTOR-FOXO1 dependent or independent,2 and further work is required to elucidte futher detailed mechanisms. Together, our findings, coupled with the results from other studies, indicate that IBET mediates suppression of BON cells at least partly by suppressing Skp2 expression and thus leading to increased p27 protein level, contributing to reduced cell proliferation (Fig. 6C).
Materials and methods
Cell culture
The human pancreatic neuroendocrine cell lines BON cells were obtained from
Dr. Courtney Townsend from The University of Texas Medical Branch at Gavelston. BON cells were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 Units/mL penicillin, and 100 mg/mL streptomycin at 37°C in a humidified 5% CO2.
Chemical inhibitors
IBET151 (abbreviated as IBET)14 was synthesized by Chemizon and purity was>98%. FOXO inhibitor, AS1842856, was purchased from EMD Biosciences. Everolimus was obtained from LC Laboratories (#E-4040). Octreotide was obtained from Sigma (#O-1014). Capecitabine (#SML-0653) and Temozolimide (#T2577) were also purchased from from Sigma. IBET, Foxoi and Everolimus were prepared as 10 mM stock solutions in DMSO, and Capecitabine and Temozolimide were prepared as 50 mM stock solutions in DMSO. Octreotide was prepared as 10 mM stock solution in water. All the stock solutions were stored at −20°C.
Cell proliferation assays
For the MTS assay, BON cells were plated in 96-well plates at a density of 5 × 103 cells/well. After overnight incubation, the cells were treated with either DMSO (control), or various concentrations of Everolimus, Octreotide, Temozolimide or Capecitabinefor 72 hr or with IBET for up to 120 hr. The MTS [3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assay kit (Promega) was used to assess cell proliferation and was performed according to the manufacturer's instructions. Absorbance of each well was recorded at 490 nm using an ELISA plate reader, and after subtracting a background values, these results were normalized to the value from the control wells. The DMSO treated group was normalized as 100% of proliferation in Fig. 1. Each experiment was performed in triplicate, with mean values ± SD reported for each treatment group. For cell counting experiments BON cells were plated in 6-well plates at a density of 5 × 104 and 104 cells/well, respectively. After overnight incubation, the cells were then treated with DMSO (control), IBET, Foxoi or a combination of IBET and Foxoi for 5 d. The attached cells were trypsinized, stained with tryptan blue, and the live cells were counted using a hemocytometer. Each experiment was performed in duplicate, withmean values ± SD reported for each treatment group.
Western blotting
Bon cells were plated in 10 cm plates at a density of 1 × 106 cells/plate respectively and then treated with DMSO (control), IBET or Foxoi, or the combination of IBET and Foxoi. After 5 days, attached cells were collected and then lysed with RIPA buffer containing protease inhibitors. Protein concentrations were determined using a BCA assay kit (Thermo Scientific). Cell lysates were subjected to polyacrylamide gel electrophoresis on Novex gels (Life Technologies), and protein was transferred to PVDF membranes (Life Technologies). Blocking was performed in PBST containing 5% non-fat dry milk.33 Foxo1 antibody (#2880) was purchased from Cell Signaling Technology. C-Myc (#32072) antibody was purchased from Abcam. P27 (#528) was purchased from Santa Cruz Biotechnology. The antibody for actin (#A5441) waspurchased from Sigma. Anti-rabbit and anti-mouse secondary antibodies were purchased from Bio-Rad. The proteins were visualized by detection with Amersham ECL Western blotting detection reagents (GE Healthcare).
RT-PCR
RNA was extracted from cultured cells with Trizol, and subsequently isolated through the use of an RNeasy Mini Kit (Qiagen). RNA (1.0μg) was transcribed into cDNA, and real-time PCR (RT-PCR) was performed using a Quantitative SYBR Green PCR Kit (Qiagen) and a 7500 Fast Real Time PCR System (Applied Biosystems).
Foxo1 primers included: forward 5′- CACCATGATGCAGCAGACGC-3′ and reverse 5′-CAACTCCTTCAAGCCTCCAG-3′.
C-Myc primers included: forward 5′-CTGGTGCTCCATGAGGAGA-3′ and reverse 5′-CCTGCCTCTTTTCCACAGAA-3′.
P27primersincluded:forward 5′-GCCCTCCCCAGTCTCTCTTA-3′ and reverse 5′-ACACAGCCCGAAGTGAAAAGA-3′.
Actin primers included: forward 5′-GGTCATCACCATTGGCAATGA-3′ and reverse 5′-GCACTGTGTTGGCGTACA-3′.
Foxo1, c-Myc and P27 transcript levels were normalized to actin transcript levels, with mean values +/− SD reported for each group.
Protein stability assay
BON cells were treated with either DMSO or IBET for 3 days, followed by addition of 20 μg/ml cycloheximide. The resulting cells were collected at 0, 2, 4 and 6 hrs after the treatment of Western blotting with the indicated antibodies. The density of the protein band was quantified and analyzed with Image J program and plotted as % protein relative to the value at 0 min.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank the laboratory members for stimulating discussions and critical reading of the manuscript.
Funding
This work was supported in part by grants from the NIH (1-R01-CA-178856 and R01 DK097555), a CFCF grant, and an AACR-NETRF grant, a Pennsylvania Breast Cancer Refunds for Research (PABCC), and China Scholarship Council for awarding stipend to Lei Wang to pursue his study in University of Pennsylvania.
References
- 1.Kulke M. Advances in the treatment of neuroendocrine tumors. Curr Treat Options Oncol 2005; 6:397-409; PMID:16107243; http://dx.doi.org/ 10.1007/s11864-005-0043-9 [DOI] [PubMed] [Google Scholar]
- 2.Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology 2008; 135:1469-92; PMID:18703061; http://dx.doi.org/ 10.1053/j.gastro.2008.05.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, et al.. One hundred years after “carcinoid:” epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008; 26:3063-72; PMID:18565894; http://dx.doi.org/ 10.1200/JCO.2007.15.4377 [DOI] [PubMed] [Google Scholar]
- 4.Leotlela PD, Jauch A, Holtgreve-Grez H, Thakker RV. Genetics of neuroendocrine and carcinoid tumours. Endocr Relat Cancer 2003; 10:437-50; PMID:14713256; http://dx.doi.org/ 10.1677/erc.0.0100437 [DOI] [PubMed] [Google Scholar]
- 5.Eigler T, Ben-Shlomo A. Somatostatin system: molecular mechanisms regulating anterior pituitary hormones. J Mol Endocrinol 2014; 53:R1-19; PMID:24780840; http://dx.doi.org/ 10.1530/JME-14-0034 [DOI] [PubMed] [Google Scholar]
- 6.Strosberg JR, Kvols LK. A review of the current clinical trials for gastroenteropancreatic neuroendocrine tumours. Expert Opin Investig Drugs 2007; 16:219-24; PMID:17243941; http://dx.doi.org/ 10.1517/13543784.16.2.219 [DOI] [PubMed] [Google Scholar]
- 7.Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, et al.. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011; 364:514-23; PMID:21306238; http://dx.doi.org/ 10.1056/NEJMoa1009290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fine RL, Gulati AP, Krantz BA, Moss RA, Schreibman S, Tsushima DA, Mowatt KB, Dinnen RD, Mao Y, Stevens PD, et al.. Capecitabine and temozolomide (CAPTEM) for metastatic, well-differentiated neuroendocrine cancers: The Pancreas Center at Columbia University experience. Cancer Chemother Pharmacol 2013; 71:663-70; PMID:23370660; http://dx.doi.org/ 10.1007/s00280-012-2055-z [DOI] [PubMed] [Google Scholar]
- 9.Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, Luo JL, Patel DJ, Allis CD. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 2009; 459(7248):847-51; PMID:19430464; http://dx.doi.org/19075677 10.1038/nature08036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, Hua X. Menin, histone h3 methyltransferases, and regulation of cell proliferation: current knowledge and perspective. Curr Mol Med 2008; 8:805-15; PMID:19075677; http://dx.doi.org/ 10.2174/156652408786733702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al.. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011; 146:904-17; PMID:21889194; http://dx.doi.org/ 10.1016/j.cell.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al.. Selective inhibition of BET bromodomains. Nature 2010; 468:1067-73; PMID:20871596; http://dx.doi.org/ 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al.. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011; 478:524-8; PMID:21814200; http://dx.doi.org/ 10.1038/nature10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al.. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011; 478:529-33; PMID:21964340; http://dx.doi.org/ 10.1038/nature10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong C, Laddha SV, Tang L, Vosburgh E, Levine AJ, Normant E, Sandy P, Harris CR, Chan CS, Xu EY. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis 2014; 5:e1450; PMID:25299775; http://dx.doi.org/ 10.1038/cddis.2014.396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evers BM, Ishizuka J, Townsend CM Jr, Thompson JC. The human carcinoid cell line, BON. A model system for the study of carcinoid tumors. Ann N Y Acad Sci 1994; 733:393-406; PMID:7978888; http://dx.doi.org/ 10.1111/j.1749-6632.1994.tb17289.x [DOI] [PubMed] [Google Scholar]
- 17.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res 2000; 60:3689-95; PMID:10919634 [PubMed] [Google Scholar]
- 18.Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev 1998; 12:2899-911; PMID:9744866; http://dx.doi.org/ 10.1101/gad.12.18.2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A 2005; 102:14659-64; PMID:16195383; http://dx.doi.org/ 10.1073/pnas.0503484102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, et al.. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A 2005; 102:749-54; PMID:15640349; http://dx.doi.org/ 10.1073/pnas.0408836102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000; 404:782-7; PMID:10783894; http://dx.doi.org/ 10.1038/35008115 [DOI] [PubMed] [Google Scholar]
- 22.Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004; 6:661-72; PMID:15130491; http://dx.doi.org/ 10.1016/S1534-5807(04)00131-5 [DOI] [PubMed] [Google Scholar]
- 23.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, et al.. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011; 331:1199-203; PMID:21252315; http://dx.doi.org/ 10.1126/science.1200609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, Mayer C, Aminossadati B, Pape UF, Bläker M, et al.. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 2009; 27:4656-63; PMID:19704057; http://dx.doi.org/ 10.1200/JCO.2009.22.8510 [DOI] [PubMed] [Google Scholar]
- 25.Nagashima T, Shigematsu N, Maruki R, Urano Y, Tanaka H, Shimaya A, Shimokawa T, Shibasaki M. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Mol Pharmacol 2010; 78:961-70; PMID:20736318; http://dx.doi.org/ 10.1124/mol.110.065714 [DOI] [PubMed] [Google Scholar]
- 26.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 2010; 6:209-17; PMID:20118940; http://dx.doi.org/ 10.1038/nchembio.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014; 25:282-303; PMID:24651011; http://dx.doi.org/ 10.1016/j.ccr.2014.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 2003; 112:831-43; PMID:12654249; http://dx.doi.org/ 10.1016/S0092-8674(03)00190-9 [DOI] [PubMed] [Google Scholar]
- 29.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011; 19:58-71; PMID:21215704; http://dx.doi.org/ 10.1016/j.ccr.2010.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matkar S, Sharma P, Gao S, Gurung B, Katona BW, Liao J, Muhammad AB, Kong XC, Wang L, Jin G, et al.. An epigenetic pathway regulates sensitivity of breast cancer cells to HER2 inhibition via FOXO/c-Myc axis. Cancer cell 2015; 28:472-85; PMID:26461093; http://dx.doi.org/ 10.1016/j.ccell.2015.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li GQ, Guo WZ, Zhang Y, Seng JJ, Zhang HP, Ma XX, Zhang G, Li J, Yan B, Tang HW, et al.. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 2016; 7:2462-74; PMID:26575167; http://dx.doi.org/ 10.18632/oncotarget.6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, González-Gomez P, Morante M, Jubierre L, Zhang W, et al.. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res 2013; 73:6264-76; PMID:23950209; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0122-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He X, Wang L, Yan J, Yuan C, Witze ES, Hua X. Menin localization in cell membrane compartment. Cancer Biol Ther 2016; 17:114-22; PMID:26560942; http://dx.doi.org/ 10.1080/15384047.2015.1108497 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.