

ABSTRACT
It's well known that microenvironment inflammatory signals could promote cancer development and progression. In colorectal cancer (CRC), chronic inflammation is a major driving mechanism for the development of CRC in patients having long-standing inflammatory bowel disease (IBD). Though it has been addressed that cancer cells ferment much of their glucose supply into lactate regardless of the presence of oxygen, it is unclear whether cell metabolism has been reprogramed during the process from IBD to CRC. Herein, with dextran sulfate sodium (DSS)-induced mouse colitis model, we found that inflammation upregulated key glycolytic enzymes expression via activation of STAT3/c-Myc signaling pathway. Interestingly, during the whole phase of chronic inflammation, the key metabolic enzymes demonstrated increased expression constantly, indicating the metabolic reprogramming was induced by long-term inflammatory signal. Moreover, either the inhibition of STAT3 signaling or c-Myc activity could block the glycolytic enzymes expression induced by interleukin 6 (IL-6). Thus, we presented the view that inflammation could induce the metabolic reprogramming and promote the progression from chronic colitis to colorectal cancer.
KEYWORDS: Chronic inflammation, c-Myc, IL-6, metabolic reprogramming, STAT3
Introduction
Recent years, more than 1 million new cases of colorectal cancer (CRC) are diagnosed worldwide every year, and there are accumulating evidences suggested that patients with inflammatory bowel disease (IBD), such as ulcerative colitis (UC) or Crohn's colitis are at high risk for developing CRC, indicating that chronic inflammation predisposing for cancer development. This increasing risk is more likely to result from chronic inflammation of the gastrointestinal mucosa than from any clear-cut genetic predisposition.1 Hence, colorectal cancer represents a paradigm for the connection between chronic inflammation and cancer.
Cancer development involves major alterations of cell metabolism. Multicellular organisms, even a single cell cannot live without metabolism, just not in the same manner. Reflecting this fundamental distinction in metabolic demands, cancer cells metabolize glucose in a manner that is distinct from that of cells in normal tissues, namely “Warburg effect,”2 or termed aerobic glycolysis.3 The existence of metabolic switch in cancer cells has been substantiated in the ensuing decades, such reprogramed energy metabolism as an emerging hallmark seems most appropriate.4 Most tumor cells are characterized by higher rates of glycolysis, lactate production, and macromolecules and lipids biosynthesis.5
Accordingly, this view of metabolic reprogramming in cancer cells seems more convincing and reasonable with growing number of study evidences. As mentioned earlier, chronic inflammation predisposed the carcinogenesis initiation, but the metabolic manner of inflammation and cancer transition process is not clear so far. Due to the close relationship between chronic colitis and neoplasia, we therefore supposed that the cellular metabolic profiles with long-term inflammatory microenvironment might distinct from the normal. In this study, we aimed to clarify whether the key metabolic enzymes changed in inflammatory tissue compared with the normal tissues. Considering this, we established the acute and chronic colitis mouse models with dextran sulfate sodium (DSS) treatment respectively. Meanwhile, we tested the enzyme activities, mRNA, and protein expression of the key metabolic enzymes respectively. Furthermore, we explored the main mechanism causing the metabolic alteration from chronic inflammation to tumorigenesis.
In our studies, we found that the key glycolytic enzymes were changed in DSS-induced chronic colitis model. Accordingly, IL-6 treatment also increased the key enzyme expressions in CRC cells. Besides, in acute inflammation model, expressions of the glycolytic enzymes increased much higher in the early steps than that in the late steps. Thus, the cellular metabolic stress can be triggered by the long-standing inflammatory signaling. Our data clearly provided the evidence that the Inflammation could accelerate the progression from chronic colitis to colorectal cancer through metabolic reprogramming.
Materials and methods
Animals and treatment
Male 10-week-old C57/BL6 mice were obtained from Fourth Military Medical University (FMMU). In parallel experiments, mice were respectively exposed to 2.5% and 4% DSS (dextran sulfate sodium) in the drinking water, leading to increased intestinal permeability and mucosal changes in the colon. In the acute DSS-induced colitis model, mice were respectively administered 2.5% and 4% DSS by adding it in drinking water for 7 d. But in the chronic model, DSS was applied in 3 cycles. Each cycle consisted of 7 d of DSS administration followed by a 10-day interval with normal drinking water. Mice were monitored daily for body weight.
Histology and immunohistochemistry
Following measure of the colon length, the distal colon was taken and fixed in 4% paraformaldehyde in phosphate-buffered saline (PFA) for H&E staining and scored by the histological damage. And reminder of colon tissues had been embedded in paraffin or saved for RNA and protein extraction.
Quantitative real-time PCR
Total RNA was extracted from colon tissues using Trizol (Invitrogen, Carlsbad, CA, USA). Reverse transcription of total RNA for cDNA synthesis was synthesized using the qPCR-RT kit (TaKaRa, Dalian, China). PCR amplification conditions were as follows: 1 cycle of 95°C for 3 min, followed by 40 cycles of 95°C for 10 s and 60°C for 22 s. Primers were as follows:
hMycF: 5′-GGAGGAACAAGAAGATGAGGAAG-3′;
hMycR: 5′-AGGACCAGTGGGCTGTGAGG-3′;
hHK2F: 5′-GATTGTCCGTAACATTCTCATCGA-3′;
hHK2R: 5′-TGTCTTGAGCCGCTCTGAGAT-3′;
hPKM2F: 5′-GACTGCCTTCATTCAGACCCA-3′;
hPKM2R:5′- GGGTGGTGAATCAATGTCCAG-3′;
hLDH-AF: 5′-CCCCAGAATAAGATTACAGTTATTG-3′;
hLDH-AR: 5′-GAGCAAGTTCATCTGCCAAGT-3′;
hPFKF: 5′-GGAGAGCGTTTCGATGATGC-3′;
hPFKR: 5′-TCGGAGTCGTCCTTCTCGTT-3′;
hActinF: 5′-CCTGGGCATGGAGTCCTGTG-3′;
hActinR: 5′-TCTTCATTGTGCTGGGTGCC-3′;
mActinU: 5′-ATGCCATCCTGCGTCTGGACCTGGC-3′;
mActinD: 5′-AGCATTTGCGGTGCACGATGGAGGG-3′;
mMycU: 5′-TCTCCATCCTATGTTGCGGTC-3′;
mMycD: 5′-TCCAAGTAACTCGGTCATCATCT-3′;
mHK2U: 5′-TGATCGCCTGCTTATTCACGG-3′;
mHK2D: 5′-AACCGCCTAGAAATCTCCAGA-3′;
mPKM2U: 5′-TCGCATGCAGCACCTGATT-3′;
mPKM2D: 5′-CCTCGAATAGCTGCAAGTTGGTA-3′;
mLDH-AU: 5′-GCTCCCCAGAACAAGATTACAG-3′;
mLDH-AD: 5′-TCGCCCTTGAGTTTGTCTTC-3′;
Expression analysis of the reported genes was performed by real-time PCR by PCR detection kit (TaKaRa, China) and BioRad real-time PCR Detection Systems.
Western blot
Proteins were separated on a 10% SDS-PAGE gels, transferred to NC membrane, and incubated overnight at 4°C with antibodies directed against the interesting proteins respectively. After washing blots to remove excessive primary antibody binding, the membrane was incubated for 1 hour with secondary antibody at room temperature. The results were visualized with BioRad.
Cell culture and experiments in vitro
The human colorectal cancer cell lines HT-29, SW480, SW620 and human intestinal epithelial cell lines (HIEC), were respectively maintained in McCoy's 5A, L-15, and DMEM, all supplemented with 10% fetal bovine serum at 37°C in humidified tissue culture incubator of 5% CO2.
Proliferation assay
To confirm whether IL-6-induced inflammatory environment has a direct stimulatory impact on CRC and normal cells proliferation and/or viability, cells were allowed to attach over 24 hours in 96-well plates. Then the cells were cultured with serum-free DMEM and incubated respectively for 1, 3, 5 d with 30 ng/ml IL-6. Next, MTT assay was used to detect the cell viability and proliferation. Meanwhile, cells were seeded in culture dishes with appropriate dilutions to form colonies after 7–9 day incubations, and finally analyzed the colony formation difference.
Apoptosis assay
To investigate IL-6 plays a key role in cell survival, cells were treated with 5-Fu as positive control, cells untreated as negative control. Flow cytometry analysis was used to analyze the apoptosis.
Statistics
Results are reported as means ± SEM. Differences in mean value between each group were analyzed by 2-tailed Student's t-test analysis, or multiple comparisons were performed by one-way analysis of variance followed by S-N-K method. p ≤ 0.05 was considered statistically significant.
Results
Glycolytic enzymes were upregulated in DSS-induced acute colitis model
To uncover the possible alterations of the key metabolic enzymes under inflammation microenvironment, we established both acute and chronic colitis mouse models with DSS treatment. In the acute DSS-induced colitis model, mice were administered 2.5% or 4% DSS respectively by adding it in drinking water. Body weight loss, colon length and histological analysis were observed after 7 d from first administration (Fig. 1A). Following DSS treatment, both the body weight and colon length of colitis mice were dramatically decreased, though there is only slight difference between 2.5% and 4% groups. In addition, we evaluated the inflammation status of colonic mucosa by HE staining and the colon histological score (Fig. 1B and C). A large number of lobular necroinflammatory cells were observed in the DSS group. Considering the dramatic loss of body weight, we propose that metabolic hallmarks might be affected following this acute inflammation. Expectedly, both mRNA and protein levels of hexokinase-2 (HK2) and PKM2 were increased in colon epithelial cells following 7-day DSS treatment, accompanying with the induction of pho-STAT3 and c-Myc (Fig. 1 E and F). But phosphate fructose kinase (PFK), pyruvate (PKM2), and lactate dehydrogenase (LDH-A) have not obviously elevated (data not shown). Furthermore, Image analysis of the immunohistochemical staining revealed almost the same results relative to Western and qPCR data (Fig. 1F and G).
Figure 1.
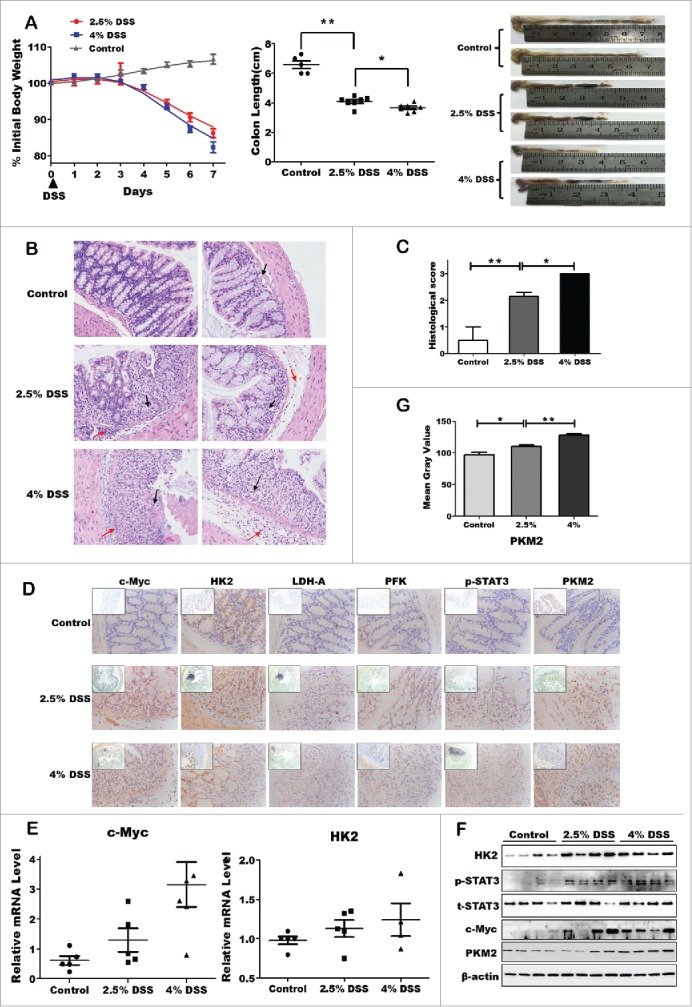
Upregulation of glycolytic enzymes expression in DSS-induced acute colitis model. (A) Body weight change percentage was calculated in control WT (gray) and DSS-treated group, namely 2.5% DSS-induced mice (red) and 4% DSS (blue). Colonic length ratio, and photographs of large intestine, oriented from cecum to rectum, was presented. Data expressed as mean ± SEM. Statistical analysis was performed using 2-way ANOVA with Bonferroni posttest to calculate p values. Significant resistance to colitis, as established by percent of weight loss (p < 0.01) and colonic weight/length ratio (p < 0.001) were observed in DSS-treated mice, including 2.5% and 4%, as compared with WT mice. *p < 0.05, **p < 0.01, ***p < 0.001. (B) Representative H&E-stained colon sections (magnification *200). (C) Histology score. Values were expressed as means ± SD (n = 10). *p < 0.05, **p < 0.01, ***p < 0.001 vs. control group. (D) Inmmunohistochemical staining showing expression of the key enzymes in colon mucosa of DSS-induced colitis and control groups. (E and F) The mRNA and protein expression of the key enzymes of aerobic glycolysis. (G) Quantitative analysis of PKM2 protein level.
Chronic inflammation conferred to a constant induction of the metabolic hallmarks associated with cancer
Besides the acute inflammation model, we further analyzed the metabolic hallmarks alteration in chronic colitis model. For induction of chronic colitis, mice received 3 cycles of treatment with DSS. Each cycle consisted of 2.5% DSS in drinking water for 5 days, followed by a 10-day interval with normal drinking water, as described.6 Histological evaluation of intestinal mucosa samples did not reveal any histopathological changes in the control group, but there are obvious abnormalities like distortion or inflammatory infiltration in the DSS group (Fig. 2A and B). Immunohistochemical staining showed that c-Myc, HK2, PFK, PKM2 and pho-STAT3 expression increased significantly under chronic inflammation condition (Fig. 2C). Interestingly, the key metabolic enzymes like LDH-A and PKM2, which playing critical roles in the later stage of aerobic glycolysis pathway, increased dramatically in chronic DSS-induced colitis mice, but failed be observed in acute colitis model (Fig. 2D and E). These results suggest that body itself has a capability to resist and adapt the inflammatory microenvironment in short time of acute inflammation, but this might be attenuated while inflammation prolonged and exacerbated. Therefore, normal cells with chronic inflammation might finally turn to metabolic reprogramming and early-stage tumor development.
Figure 2.
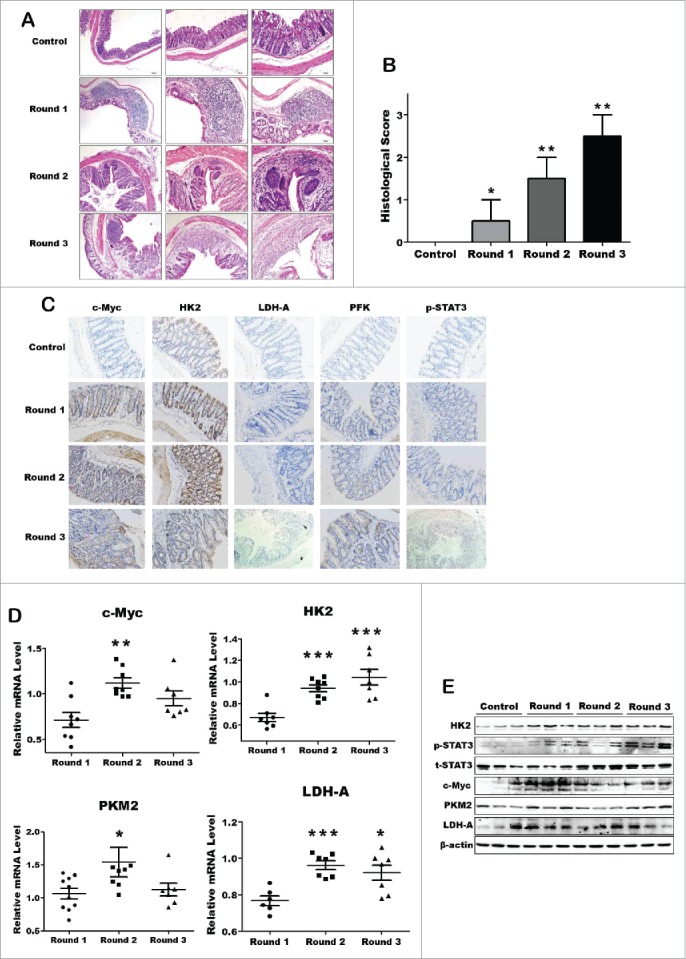
The key metabolic enzymes expression significantly increased in DSS-induced chronic colitis model. (A) Representative H&E-stained colon sections (magnification *200). (B) Histology score. Values were expressed as means ± SD (n = 10). *p < 0.05, **p < 0.01, ***p < 0.001 vs. control group. (C) Inmmunohistochemical staining showing expression of the key enzymes in colon mucosa of each DSS-induced colitis groups and control group. (D and E) The mRNA and protein expression of the key enzymes of aerobic glycolysis.
IL-6 increased glycolytic enzyme expressions and lactate production
Recent studies suggest that pro-inflammatory cytokine IL-6 is an important mediator within the cancer microenvironment to promote cancer cells proliferation. Thus, we propose that IL-6 might be able to alter the cell metabolic program and behaviors. Herein, we used human intestinal epithelial cell lines (HIEC) and colorectal cancer cells to determine the biologic role of IL-6. As shown in Fig. 3A, IL-6 treatment increased cell viability in each cell lines, especially in HIEC cells. Accordingly, flow cytometry analysis and colony formation assay further confirmed that IL-6 had a direct growth stimulatory impact on colon cells, including HIEC or CRC cells (Fig. 3B). Moreover, IL-6 inhibited the late apoptosis of HIEC cells induced by 5-Fu (Fig. 3C). Notably, IL-6 treatment significantly increased both mRNA and protein levels of the key glycolytic enzymes in HIEC/HT-29 cells (Fig. 3D and E). Further, the enzyme activity of LDH-A and PKM2 showed comparatively high levels under the stimulation of IL-6 (Fig. 3F and G). Expectedly, IL-6 treatment caused a relatively higher enzyme activity in SW620 than HIEC cells, indicating that malignant and wide-spread metastasis cancer cells seem to be more competent to finish glycolysis process compared with the normal cells. In addition, IL-6 also increased the lactate production ability of each cells (Fig. 3H). Therefore, IL-6 induced inflammatory environment has a direct stimulatory effect on aerobic glycolysis in both human normal and colon cancer cells.
Figure 3.

Pro-inflammatory cytokine IL-6 promoted the expression and activity of glycolytic enzymes in CRC and normal cells. (A) Both CRC and normal cells proliferation and / or viability using MTT assay. Error bars represent cell numbers ± SD for triplicate experiments. (B) Colony- formation assay with HIEC cells treated with IL-6 and 5-Fu as indicated. (C) Cell apoptosis was detected of HIEC cells treated with IL-6 and 5-Fu using FITC-Annexin V / PI staining kit and analyzed using flow cytometry. Representative flow cytometry results are shown. (D and E) mRNA and protein expression levels of key glycolytic enzymes in HIEC cell tread with IL-6. (F and G) Enzyme activity analysis of LDH-A and PKM2 in HIEC, SW480 and SW620 cells respectively. (H) Detection of lactate production with IL-6 treatment. Data was shown as mean ± SE of the above independent experiments.
IL-6 induces cell metabolic reprogramming via STAT3/c-Myc axis
All detection and results mentioned above were just functional information related to aerobic glycolysis, but underlying mechanism of inflammatory microenvironment reprogrammed the metabolic process of normal tissue cells toward fast-proliferating cells or cancer cells have remained elusive. Recent studies demonstrating the significance of IL-6/STAT3 signaling in promoting colitis-associated cancer, namely IL-6 signal may activate STAT3 and has direct stimulatory impact on CRC cell proliferation.7,8 Accordingly, we treated cells with S3I-201, a small-molecule inhibitor of STAT3 activation.9 As expected, the STAT3 phosphorylation was suppressed after cells treated with S3I-201 accompanying with the reduction of c-Myc and HK2 expression (Fig. 4A). Additionally, S3I-201 could further block IL-6 induced c-Myc and glycolytic enzyme expression (Fig. 4B). Owing to the mRNA and protein levels of c-Myc were both increased once stimulated by inflammatory signal, we then inhibited c-Myc expression via treating cells with c-Myc inhibitor JQ1. Interestingly, JQ1 could also interrupt the upregulation of glycolytic enzyme expression induced by IL-6 without altering phos-stat3 level (Fig. 4C and D). In addition, S3I-201 could block c-Myc and glycolytic enzyme expressions within a short time after cells pre-treated IL-6 in 12 hours (Fig 4E). Our data demonstrated that STAT3/c-Myc axis played a critical role in inflammation induced metabolic reprogramming.
Figure 4.

Protein expression analysis of the key glycolytic enzymes after inhibition of stat3/c-Myc signaling. (A) The expression of phos-STAT3, c-Myc, LDHA and HK2 were inhibited by STAT3 inhibitor S3I-201. (B) HT-29 cells first treated with IL-6 in advance to activate c-Myc and phos-STAT3 expression. Then glycolytic enzymes protein expression was detected after S3I-201 treatment. (C and D) c-Myc expression was inhibited by Myc inhibitor JQ1 in HIEC, then analyzed the key enzymes expression with JQ1 only or combined with IL-6 respectively. (E) S3I-201 blocked c-Myc and glycolytic enzymes expression within a short time after cells pre-treated IL-6 in 12 hours. (F) A scheme of the mechanism involved in the inflammation-induced metabolic reprogramming.
Discussion
Chronic inflammation is a well-known risk factor for colorectal cancer, and the mechanism by which inflammation contributes to tumorigenesis is rapidly coming into focus.1,10,11 Accordingly, within the tumor tissue, the localized inflammatory microenvironment can promote accumulation of additional mutations and epigenetic alterations.11 It seems that cancer cells become addicted to inflammatory signaling, in which inflammatory cytokines and chemokines perturb the differentiation and promote the growth and survival of cancer cells.12,13
The role for inflammation in tumorigenesis is now generally accepted, and it has become evident that inflammatory microenvironment is an essential component of all tumors, even including some of which a direct causal relationship with inflammation is not yet determined.14 Hence, cells stimulated by inflammatory signal in chronic inflammation might alter their metabolic manner to adapt inflammatory microenvironment, and cancer cells are no exception. IL-6 belongs to a large family of cytokines and binds with the IL-6R receptor to activate the down-stream effector STAT3.15 Early in 1998, the linkage between a member of the STAT3 family and the c-Myc gene activation had been first proposed, showing that upon stimulation of IL-6, STAT3 mostly mediates the rapid activation of c-Myc via binding to a region overlapping with the E2F binding site of c-Myc promoter.16 The IL-6/STAT3 signaling regulates the survival and proliferation of intestinal epithelial cells and plays an important role in the pathogenesis of inflammatory bowel disease and colorectal cancer. When exploring the mechanism of metabolism alteration stimulated with inflammation stress, we used inhibitors to block STAT3 and c-Myc signaling respectively. The results indicate that the key metabolic enzymes have decreased to varying degrees with STAT3/c-Myc signaling blocking. Thereby, we first found that chronic inflammation could alter the metabolic manner through STAT3/c-Myc axis to upregulate their downstream metabolic enzymes expression (Fig. 4F).
However, very few studies showed the evidence of metabolic reprogramming during the process from chronic inflammation to cancer with in vivo experiments. To address this issue, we first established 2 DSS-induced models, namely acute and chronic colitis mice model, aiming exactly to explore series of metabolic enzymes alteration and functional indexes. Our studies demonstrated that either in DSS-induced acute inflammation model or chronic one, the metabolic program were changed at different levels, which might be relate to colitis associated CRC. In addition, we also confirmed the functional indexes in vitro following the treatment with IL-6. Interestingly, there are some distinctions between DSS-induced acute colitis model and chronic inflammation. During the early stage of acute colitis, only HK2 was shown increased. However, in chronic colitis model, more key glycolytic enzymes were shown highly expressed constantly, indicating the metabolic reprogramming was induced by long-term inflammatory signaling.
From the above, our study showed that cancer associated metabolic reprogramming can be triggered during the process of chronic inflammation, termed as colitis-associated cancer in clinical diagnosis. These metabolism alterations are associated with the character of “Warburg effect” or aerobic glycolysis, such as enhanced glycolytic capability and lactate production. Perhaps, this metabolic characteristic differing from normal cells, might be novel early diagnosis biomarkers of chronic colitis associated colorectal cancer, or even as novel potential therapeutic targets for chronic inflammation.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National High-tech R&D Program of China for Young Scientist (863 Program, 2014AA020517), and National Natural Science Research Program of China (31401161, 81372390, 31571437, 81230043, 81421003, 81672751).
References
- 1.Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology 2011; 140:1807-16; PMID:21530747; http://dx.doi.org/ 10.1053/j.gastro.2011.01.057 [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science 1956; 123:309-14; PMID:13298683; http://dx.doi.org/ 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029-33; PMID:19460998; http://dx.doi.org/ 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 5.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell 2008; 13:472-82; PMID:18538731; http://dx.doi.org/ 10.1016/j.ccr.2008.05.005 [DOI] [PubMed] [Google Scholar]
- 6.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990; 98:694-702; PMID:1688816; http://dx.doi.org/ 10.1016/0016-5085(90)90290-H [DOI] [PubMed] [Google Scholar]
- 7.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, et al.. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009; 15:103-13; PMID:19185845; http://dx.doi.org/ 10.1016/j.ccr.2009.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, et al.. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009; 15:91-102; PMID:19185844; http://dx.doi.org/ 10.1016/j.ccr.2009.01.002 [DOI] [PubMed] [Google Scholar]
- 9.Sen N, Che X, Rajamani J, Zerboni L, Sung P, Ptacek J, Arvin AM. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc Natl Acad Sci U S A 2012; 109:600-5; PMID:22190485; http://dx.doi.org/ 10.1073/pnas.1114232109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol 2009; 9:405-10; PMID:19589728; http://dx.doi.org/ 10.1016/j.coph.2009.06.006 [DOI] [PubMed] [Google Scholar]
- 11.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology 2010; 138:2101-14 e5; PMID:20420949; http://dx.doi.org/ 10.1053/j.gastro.2010.01.058 [DOI] [PubMed] [Google Scholar]
- 12.Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: inflammatory bowel disease and colitis-associated colon cancer. Front Immunol 2012; 3:107; PMID:22586430; http://dx.doi.org/ 10.3389/fimmu.2012.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klampfer L. Cytokines, inflammation and colon cancer. Curr Cancer Drug Targets 2011; 11:451-64; PMID:21247378; http://dx.doi.org/ 10.2174/156800911795538066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140:883-99; PMID:20303878; http://dx.doi.org/ 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell 2009; 15:79-80; PMID:19185839; http://dx.doi.org/ 10.1016/j.ccr.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiuchi N, Nakajima K, Ichiba M, Fukada T, Narimatsu M, Mizuno K, Hibi M, Hirano T. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J Exp Med 1999; 189:63-73; PMID:9874564; http://dx.doi.org/ 10.1084/jem.189.1.63 [DOI] [PMC free article] [PubMed] [Google Scholar]