

Abstract
In neurons, intracellular membrane rafts are essential for specific actions of brain-derived neurotrophic factor (BDNF), which include the regulation of axon outgrowth, growth cone turning and synaptic transmission. Virtually all the actions of BDNF are mediated by binding to its receptor, TrkB. The association of TrkB with the tyrosine kinase, Fyn, is critical for its localization to intracellular membrane rafts. Here, we show that synapsins, a family of highly amphipathic neuronal phosphoproteins, regulate membrane raft lipid composition and consequently, the ability of BDNF to regulate axon/neurite development and potentiate synaptic transmission. In the brains of mice lacking all synapsins, the expression of both BDNF and TrkB were increased, suggesting that BDNF/TrkB-mediated signaling is impaired. Consistent with this finding, synapsin-depleted neurons exhibit altered raft lipid composition, deficient targeting of Fyn to rafts, attenuated TrkB activation, and abrogation of BDNF-stimulated axon outgrowth and synaptic potentiation. Conversely, overexpression of synapsins in neuroblastoma cells results in corresponding reciprocal changes in raft lipid composition, increased localization of Fyn to rafts, and promotion of BDNF-stimulated neurite formation. In the presence of synapsins, the ratio of cholesterol to estimated total phospholipids converged to 1, suggesting that synapsins act by regulating the ratio of lipids in intracellular membranes, thereby promoting lipid raft formation. These studies reveal a mechanistic link between BDNF and synapsins, impacting early development and synaptic transmission.
Keywords: lipid, mouse, signal transduction, neurodevelopment
Graphical Abstract
We investigated the role of synapsins in neuronal development. The results show that synapsins regulate intracellular membrane raft lipid composition by restricting the ratio of cholesterol to total phospholipids, thereby promoting the localization of Fyn/TrkB to rafts, and subsequent activation of TrkB by BDNF. Consistent with these results, synapsins are required for BDNF-stimulated axon outgrowth and synaptic potentiation, thus revealing mechanistic links between BDNF and synapsins.
Introduction
BDNF plays a central role in the regulation of neurodevelopment and synaptic plasticity in the central nervous system. Virtually all the actions of BDNF are mediated by binding to its cognate receptor, TrkB, a receptor tyrosine kinase that triggers multiple downstream signaling cascades such as phosphatidyl inositol-3 (PI3)-kinase, phospholipase C-γ1 (PLC-γ) and MAP kinases (Reichardt, 2006). How this receptor orchestrates the spatiotemporal activation of such diverse neural signaling pathways is poorly understood. Lipid membrane rafts, specialized microcellular domains enriched with cholesterol, sphingolipids, and gangliosides, have been proposed as important organizing platforms for signal transduction (Lingwood & Simons, 2010). Disruption of membrane rafts by cholesterol depletion or ganglioside competition interferes with the ability of BDNF to enable growth cone turning or stimulate axon outgrowth (Guirland et al., 2004), or potentiate synaptic transmission (Suzuki et al., 2004). While these observations imply a role for rafts in mediating the actions of BDNF, the specific mechanisms by which rafts function with respect to BDNF signaling have not been identified.
Synapsins are abundant neuronal phosphoproteins that are extraordinarily surface-active, highly amphipathic proteins (Ho et al., 1991) that bind with tight affinity to biological membranes (Benfenati et al., 1989) and stabilize phospholipid bilayers (Pera et al., 2004). All synapsins possess amphipathic lipid packing sensor motifs that permit insertion into curved membranes (Krabben et al., 2011). Significantly, synapsins have been implicated in the regulation of neurogenesis (Kao et al., 2008), neurite formation (Ferreira et al., 1994; Ferreira et al., 2000), axon elongation (Kao et al., 2002), synaptogenesis (Giachello et al., 2010) and synaptic maturation (Valtorta et al., 1995) – developmental processes also stimulated by BDNF. Accordingly, we hypothesized that synapsins and BDNF share a mechanistic link.
An important step in BDNF signaling is the localization of activated TrkB to intracellular membrane rafts, which requires the binding of TrkB to Fyn (Pereira & Chao, 2007). Given the strong affinity of synapsins to intracellular membranes, we began our investigations by examining the effect of synapsins on membrane rafts. We demonstrate that synapsins possess an intrinsic ability to regulate the lipid composition of membrane rafts, and consequently the downstream actions of BDNF.
Materials and methods
Mice
The generation of synapsin triple knockout mice was previously described (Gitler et al., 2004a) and the mice were maintained in the Animal Care Facility at Brown University or the Korea Institute of Science and Technology. Synapsin triple knockout mice have been deposited with the Mutant Mouse Resource and Research Center at the Jackson Laboratory (MMRRC # 41434). Mice that were used as controls (C57/BL6) were originally obtained from Jackson Laboratory (Bar Harbor, Maine) and maintained in the Animal Care Facilities. All experimental protocols involving mice were approved by the Institutional Animal Care and Use Committee at Brown University or at the Korea Institute of Science and Technology.
Cortical neuronal and glial cultures
Brains were dissected from newborn mice, their cortices isolated, and tissues dissociated with papain and DNase using previously published methods (Brewer, 1995). Dissociated cells were incubated in Hibernate medium (Life Technologies, Inc., Grand Island, New York) on uncoated tissue culture plates at 37°C for 1 hour to absorb glial cells. Neurons were harvested from the Hibernate supernatant after this panning step. For microscopy, neurons were grown on poly-D-lysine coated glass coverslips in tissue culture dishes. For the preparation of detergent-resistant membrane (DRM) raft fractions, neurons were plated on poly-D-lysine coated 85 mm tissue culture dishes. Cultured neurons were maintained in serum-free Neurobasal medium with B-27 supplement, 0.5 mM glutamine, 25 μM glutamate, and 25 U/ml penicillin/streptomycin (Life Technologies, Inc.) at 37°C in a 5% CO2-humidified incubator for up to 1 week prior to analyses or further experimentation. Glial cells that were absorbed on tissue culture plates during the panning enrichment step for the isolation of neurons were grown in DMEM supplemented with 10% FCS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Glial cultures were maintained at 37°C in a 5% CO2-humidified incubator for 1 week prior to further experimentation.
In various experiments, neurons or differentiated SH-SY5Y cells were treated with recombinant human BDNF (Promega Corporation, Madison, Wisconsin) diluted to a final concentration of 50 ng/mL with culture media as indicated in individual experiments. In western blots involving MAPK activation in cultured neurons, cells were incubated for 1 hour with 1 μM tetrodotoxin to reduce hyperexcitability prior to addition of BDNF.
Cell lines and transfection
SH-SY5Y neuroblastoma and HeLa cells (originally obtained from the American Type Culture Collection, Manassas, Virginia) were maintained at 37°C in a 5% CO2-humidified incubator. Both cell lines were fed with DMEM supplemented with 10% FCS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. To differentiate SH-SY5Y cells, the medium was replaced with DMEM supplemented with 2% FCS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 μM all-trans retinoic acid (Sigma-Aldrich, St. Louis, Missouri). SH-SY5Y cells were maintained in differentiation medium for three days prior to further analysis or experiments.
To express exogenous genes in SH-SY5Y cells, the cells were trypsinized, washed in DMEM, and resuspended in DMEM with DNA. 100 μL of cells (from one confluent 85 mm tissue culture dish) was resuspended with 5 μg DNA and transferred to an electroporation cuvette. Cells were electroporated using the program A-023 in the Amaxa Nucleofector (Lonza Inc, Allendale, New Jersey). After electroporation, the cells were plated and fed in differentiation buffer for at least 3 days prior to treatment with BDNF. About 50% of the cells express the exogenous gene with this procedure.
For expression of exogenous genes in HeLa cells, the cells were transfected with Fugene HD (Promega Corporation, Madison, Wisconsin) using DNA diluted in DMEM and specifications described by the manufacturer. More than 95% of the cells express the exogenous gene under these conditions.
DNA expression constructs
Cloning and characterization of synapsins Ia, IIa and IIIa cDNAs have been previously described (Südhof et al., 1989; Hosaka & Südhof, 1998; Kao et al., 1998). Relevant synapsin and eGFP sequences were amplified using the polymerase chain reaction (PCR) and cloned into pcDNA3.1 using conventional recombinant DNA technology (Sambrook et al., 1989). Each synapsin and eGFP construct was engineered with a carboxyl terminal FLAG tag (Malhotra, 2009). All assembled DNAs were confirmed by sequencing. All three synapsins were combined and expressed in equal amounts in cell culture. Synapsins or eGFP were detected in fixed cell culture using a monoclonal antibody directed towards the FLAG epitope.
Isolation of detergent-resistant membrane (DRM) raft fractions
Brain tissues were homogenized in lysis buffer (50 mM Tris-HCl, pH 8.0, 138 mM NaCl, 2.7 mM KCl, 5% glycerol, and 1% Triton X-100) supplemented with protease inhibitors (mammalian protease inhibitor cocktail; Sigma-Aldrich, St. Louis, Missouri) and phosphatase inhibitors (5 mM EDTA, 50 mM NaF, 5 mM Na3VO4) on ice. The homogenate was rotated at 4°C for 60 minutes. Optiprep (Sigma-Aldrich, St. Louis, Missouri) was added to 1000 μg of brain homogenate (protein) to a concentration of 40%, and Triton X-100 added to a concentration of 1% in a volume of 400 μl on ice. The homogenate formed the bottom of an optiprep step gradient (Fig. 1A), which was overlayed with 1.4 ml 30% optiprep and 0.2 ml of 0% optiprep (all in lysis buffer supplemented with protease and phosphatase inhibitors). The gradient was centrifuged in a TLS-55 rotor (Beckman Coulter, Inc., Brea, California) at 55K rpm for 2 hours at 4°C. Nine fractions were collected, with the top two fractions representing DRM raft fractions.
Figure 1.

Synapsins regulate the lipid composition of membrane rafts. The indicated tissues were homogenized in cold 1% triton X-100 lysis buffer, adjusted to 40% optiprep, and separated by optiprep density gradient ultracentrifugation. In all graphs depicted, significance was determined by Student’s two-sided t-test and error bars denote ± SEM. (A) Schematic diagram illustrating preparation of detergent-resistant membrane (DRM) raft fractions. After optiprep density gradient ultracentrifugation, 9 fractions were collected. Fractions 1 and 2 represent DRM raft fractions as indicated by cholera toxin staining for ganglioside GM1, which is enriched in lipid rafts. Graph shows typical fractionation of cholesterol and protein from brain tissue. (B) Concentrations of individual lipids and protein from DRM raft fractions of neonatal (P1) and postnatal day 7 (P7) and day 14 (P14) brains from wild-type (WT) and synapsin triple knockout (TKO) mice (n ≥ 4). (C–D) Concentrations of individual lipids (Choles=cholesterol; GM1=ganglioside GM1; PC=phosphatidylcholine; SM=sphingomyelin) and protein from DRM raft fractions of cultured neurons (C) and glia (D) of WT and synapsin TKO mice (n ≥ 4). (E) Western blot depicting relative quantities of synapsins Ia, IIa and IIIa from brain lysates derived from mice of the indicated age. 30 μg protein were loaded in each well. Note that synapsin IIIa is highly phosphorylated from ages P1–P7 (Porton et al., 2004), resulting in several higher molecular weight bands. (F) Concentrations of individual lipids and protein from DRM raft fractions of differentiated SH-SY5Y cells expressing all three synapsins (Syn) or pEGFP/pcDNA3.1 (Control) (n ≥ 5). (G) Western blot in which equal amounts of protein (30 μg) from differentiated SH-SY5Y cells expressing the indicated genes were probed with an antibody (G304) directed towards all three synapsin “a” isoforms (Pieribone et al., 1995). (H) Concentrations of individual lipids and protein from DRM raft fractions of HeLa cells transfected with a control plasmid (pEGFP) or all three synapsins (n ≥ 4). *P<0.05; **P<0.01.
Cell lines and neuronal cultures were harvested using a cell scraper and centrifuged in phosphate buffered saline (PBS). 500 μg of cell homogenate (protein) were used in the isolation of DRM raft fractions using the same method described above.
Quantitation of lipids and protein
10 μl of sample were assayed in each of the procedures below.
Cholesterol
Samples were incubated in a total volume of 80–110 μl containing 100 mM Tris-HCl, pH 7.4, 50 mM NaCl, 25 U/ml horse radish peroxidase, 1 U/ml cholesterol esterase, 1 U/ml cholesterol oxidase, and 50 μM Amplex Red (Molecular Probes, Eugene, Oregon). All enzymes were obtained from Sigma-Aldrich, St. Louis, Missouri. Dilutions of cholesterol (dissolved in ethanol) were used as standards, and were obtained from Cayman Chemical Company, Ann Arbor, Michigan. Negative control samples, which lack cholesterol oxidase and cholesterol esterase, were used in all assays. After the samples were incubated on a 96-well assay plate, they were read in a microplate reader equipped with a filter set for excitation and emission at 540±10 and 590±10 nm respectively. Cholesterol levels were determined using a standard curve generated by the standards. The assay is adapted from Amundsen and Zhou (Amundson & Zhou, 1999).
Phosphatidylcholine
Samples were incubated in a total volume of 80–110 μl containing 50 mM Tris-HCl, pH 7.0, 0.66 mM CaCl2, 1.2 U/ml choline oxidase (Sigma-Aldrich, St. Louis, Missouri), 25 U/ml horse radish peroxidase (Sigma-Aldrich), 72 U/ml phosphatidylcholine-specific phospholipase D (BML-SE302, from Enzo Life Sciences, Inc., Farmingdale, New York), and 50 μM Amplex Red (Molecular Probes). Dilutions of phosphatidylcholine (Sigma-Aldrich; dissolved in 2% Triton X-100 in ethanol) were used as standards. Negative control samples, which lack choline oxidase and phospholipase D, were used in all assays. After the samples were incubated on a 96-well assay plate, they were read in a microplate reader using excitation and emission at 540±10 and 590±10 nm respectively. The assay was adapted from a previously described method using Amplex Red (Hojjati & Jiang, 2006).
Sphingomyelin
Samples were incubated in a total volume of 80–110 μl containing 50 mM Tris-HCl, pH 8.0, 0.66 mM CaCl2, 1.2 U/ml choline oxidase (Sigma-Aldrich, St. Louis, Missouri), 25 U/ml horse radish peroxidase (Sigma-Aldrich), 0.6 U/ml sphingomyelinase (Sigma-Aldrich), 12 U/ml alkaline phosphatase (Sigma-Aldrich), and 50 μM Amplex Red (Molecular Probes). Dilutions of sphingomyelin (Biomol International - Enzo Life Sciences, Inc., Farmingdale, New York; dissolved in 2% TritonX-100 in ethanol) were used as standards. Negative control samples, which lack choline oxidase, sphingomyelinase, and alkaline phosphatase, were used in all assays. After the samples were incubated on a 96-well assay plate, they were read in a microplate reader using excitation and emission at 540±10 and 590±10 nm respectively. The assay was adapted from a previously described method using Amplex Red (Hojjati & Jiang, 2006).
Ganglioside GM1
Samples were applied to nitrocellulose filters using a Bio-dot apparatus (Bio-Rad, Hercules, California), according to instructions provided by the manufacturer. Dilutions of GM1 (Sigma-Aldrich) were used as standards on each nitrocellulose filter. The filters were blocked in Odyssey buffer (LI-COR Biosciences, Lincoln, Nebraska) overnight at 4°C, probed with 1 μg/ml biotinylated cholera toxin (Sigma-Aldrich) for 1 hour at room temperature, and revealed using IR800-conjugated streptavidin (Rockland Immunochemicals, Gilbertsville, Pennsylvania). Images were scanned using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, Nebraska).
Protein
Protein levels were quantitated with the BCA protein assay kit using procedures described by the manufacturer (Pierce Chemical Company – Thermo Scientific, Rockland, Illinois).
Western blot analyses
Proteins were resolved on 10% NuPage Bis-Tris mini gels and transferred to PVDF membranes (Millipore, Inc. Billerica, Massachusetts) using conditions described by the manufacturer (Life Technologies, Grand Island, New York). Native SDS-PAGE was carried out using conditions previously described (Nowakowski et al., 2014). Western blots were blocked in Odyssey buffer (LI-COR Biosciences, Lincoln, Nebraska) on a rotating platform overnight at 4°C, and probed sequentially with primary and secondary antibodies the next day.
The following primary antibodies were used: rabbit anti-BDNF (1:100; N-20; Santa Cruz Biotechnology, Santa Cruz, California), mouse anti-α-tubulin (1:500; Sigma, St. Louis, Missouri), rabbit anti-14-3-3ζ (1:500; Santa Cruz Biotechnology), rabbit anti-Fyn (1:1000; Cell Signaling Technology, Beverly, Massachusetts), mouse anti-TrkB (1:500; Becton-Dickinson, Franklin Lakes, New Jersey), rabbit anti-TrkB (1:200; #794; Santa Cruz Biotechnology), rabbit anti-phospho TrkB-Y706 (1:250; Cell Signaling Technology), rabbit anti-P75 (1:250; sc-8317; Santa Cruz Biotechnology), rabbit anti-pan-synapsin, G304 (1:1000; Pieribone et al., 1995), mouse anti-total Erk1/2 (1:1000; Cell Signaling Technology), rabbit anti-phospho-Erk1/2 (1:1000; Cell Signaling Technology), and mouse anti-GAP-43 (1:250; Chemicon-EMD Millipore, Billerica, Massachusetts). The primary antibodies were revealed by incubation with Alexa680-conjugated anti-rabbit (1:5000; Molecular Probes) or IR800-conjugated anti-mouse antibodies (1:5000; Rockland Immunochemicals). Images of western blots were captured using the Odyssey Infrared Imaging System (LI-COR Biosciences). Non-saturated protein bands were quantitated using ImageJ software (NIH freeware).
Confocal microscopy and quantitative colocalization
Neurons or SH-SY5Y cells were grown on poly-D-lysine treated glass coverslips and subjected to various treatments (e.g. differentiation, BDNF treatment). After treatment, cells were fixed in 4% paraformaldehyde in PBS, lysed with 0.3% Triton X-100 (5 minutes) and blocked in 10% donkey serum in Tris-buffered saline (pH 8.0) with 0.05% Tween 20 (TBST) overnight at 4°C. Fixed cells were then incubated overnight with antibodies (diluted in TBST) directed against various protein epitopes.
The following primary antibodies were used: mouse anti-Fyn (1:100; #15; Santa Cruz Biotechnology, Santa Cruz, California), rabbit anti-TrkB (1:100; #794; Santa Cruz Biotechnology), and mouse anti-FLAG (1:100; M2 monoclonal antibody, Stratagene, La Jolla, California). Coverslips were washed with TBST and the primary antibodies revealed by incubation with Alexa647-conjugated anti-mouse antibody (1:100) and Alexa555-conjugated anti-rabbit antibody (1:100; Invitrogen – Life Technologies). Images were captured using the Zeiss LSM 510 confocal microscope, using conditions to optimize the signal for each antibody used.
Confocal images were analyzed using ImageJ (NIH freeware) and the plug-in for colocalization analyses (Costes et al., 2004). The colocalization highlighter program is part of the same package and allows demarcation of colocalized regions under specified conditions.
Neurite length and formation
Phase-contrast images were captured using the Zeiss Axioplan epifluorescent microscope, and neurites were traced using Adobe Photoshop. Neurite lengths were determined using ImageJ.
To obtain a measure of neurite formation, nucleofected SH-SY5Y cells were plated on poly-D-lysine treated glass coverslips, differentiated with retinoic acid for 3 days, and treated with BDNF for 24 hours. The cells were then fixed with 4% paraformaldehyde in PBS, and cells expressing eGFP or Synapsins (Ia, IIa and IIIa in equimolar amounts) were identified by immunolabeling with mouse anti-FLAG (M2 monoclonal antibody, Stratagene, La Jolla, California). Neurites were traced using phase-contrast images captured with the Zeiss Axioplan epifluorescent microscope. Neurite lengths were traced from all cells (FLAG-positive and FLAG-negative cells). The proportion of FLAG-positive over FLAG-negative cells bearing neurites >50 microns in the same experiment (>100 cells analyzed in each condition per experiment) was used as an index of neurite formation.
Electrophysiology
Cultures of hippocampal neurons were prepared as previously described (Gitler et al., 2004b) and were 12–15 days old for electrophysiological analyses. Whole-cell patch-clamp recordings were made from single neuron cell bodies in the cultures. Recording pipettes were filled with intracellular solution containing 130 mM K-gluconate, 2 mM NaCl, 4 mM MgCl2, 20 mM HEPES, 4 mM Na2ATP, 0.4 mM Na3GTP, 0.5 mM EGTA, and Na2-phosphocreatine (pH 7.25, 290 mOsm). The recording chamber was filled with external bath solution containing 150 mM NaCl, 3 mM KCl, 10 mM HEPES, 2 mM CaCl2, 2 mM MgCl2, and 20 mM D-glucose (pH 7.35, 310 mOsm). The tip of the puffing pipette was about 3–5 mm, and placed about 100 mm from the cell body of the neuron. The concentration of BDNF solution inside the puffing pipette was 100 ng/ml. Neurons were voltage clamped at −70 mV with a HEKA EPC-9D amplifier (HEKA, Lambrecht/Pfalz, Germany). All experiments were performed at room temperature. Electrophysiological data were sampled at 10 kHz and filtered at 10 kHz. To collect only mEPSCs (miniature excitatory postsynaptic currents), the external bath solution was added with 1 μM tetrodotoxin and 20 mM bicuculline. Spontaneous synaptic events were analyzed using the MiniAnalysis program (Synaptosoft, Decatur, Georgia).
Immuno-capture of synapsins
Brains were homogenized in mammalian protein extraction reagent (Pierce Chemical Company – Thermo Scientific, Rockland, Illinois) supplemented with mammalian protease inhibitors (Sigma-Aldrich), and pelleted in a microfuge at 14K rpm for 10 minutes at 4°C. Lysates were incubated sequentially on ice with biotinylated G304 (a pan-synapsin antibody recognizing the E-domain of all “a” isoforms), followed by incubation with Mag-Avidin beads (Pierce Chemical Company) on a rotator overnight at 4°C. The beads were captured using a magnet, and washed twice in Tris-buffered saline. Since synapsins are typically phosphorylated, the beads were incubated in alkaline phosphatase (Promega Corporation, using the manufacturer’s conditions) for 1 hour (rotated at 37°C), washed twice again in Tris-buffered saline. Synapsins were eluted by incubation in 100 mM EDTA, pH 8.0, 20% N-octyl-β-D-glucopyranoside at 95°C for 5 minutes. Immuno-captured synapsins were dialyzed in 10 mM Tris pH 7.4 at 4°C with 3 changes of buffer prior to use. In experiments involving MAP kinase phosphorylation, synapsins were incubated with MAP kinase (New England Biolabs, Ipswich, Massachusetts) for 4 hours at 37°C using the manufacturer’s conditions. For experiments that involve multimerization without protein kinases, the proteins were incubated in MAPK buffer and ATP in the absence of MAP kinase for 4 hours at 37°C.
Results
Lipid composition of membrane rafts is regulated by synapsins
We first asked whether synapsins play a role in the lipid composition of membrane rafts. Lipids were quantified from detergent-resistant membrane (DRM) fractions, a biochemical enrichment procedure for membrane rafts (Simons & Toomre, 2000) (Fig. 1A). These fractions were prepared from whole brain tissue or whole cells, thus all membranes including plasma and intracellular membranes were analyzed. Because synapsins are encoded by three genes (I, II, and III), we compared the lipid composition of membrane rafts prepared from wild-type (WT) mice to those from mice lacking all three synapsin genes (triple knockout or TKO). DRM raft fractions were analyzed from neonatal (P1), postnatal day 7 (P7), and postnatal day 14 (P14) brains (Fig. 1B). Concentrations of individual lipids that are enriched in rafts (cholesterol, phosphatidylcholine, sphingomyelin, and ganglioside GM1) were significantly different between DRM raft fractions derived from WT and TKO brains at specific ages of development (Fig. 1B). We also found significant differences in cholesterol and sphingomyelin between WT and TKO neuronal cultures, but not in glial cultures (Fig. 1C–D), suggesting that the DRM raft lipid changes observed in brain could be explained solely by changes in neurons rather than glial cells. Glia, which lack synapsins, also displayed a lipid composition that was very different from that of WT brain or WT neuronal cultures (Fig. 1D). During these phases of development, the expression of isoforms derived from each synapsin gene varies. In accord with previous studies (Lohmann et al., 1978; Porton et al., 2004; Perlini et al., 2015), synapsins I and II are expressed later in development (P7 to adulthood), while the peak of synapsin III expression occurs between P1–P7 (Fig. 1E). These findings indicate that synapsins could play a role in regulating neuronal membrane rafts, and that each gene may have specific effects on membrane composition.
To further examine the role of synapsins in lipid raft composition, we asked whether expression of exogenous synapsins alters the lipid composition of membrane rafts in non-neuronal cell lines. We used SH-SY5Y cells, a neuroblastoma cell line that expresses virtually undetectable levels of synapsins (Thiel et al., 1991; LoPresti et al., 1992), as well as HeLa cells, which are easily transfected. When differentiated by retinoic acid, SH-SY5Y cells resemble neurons, express TrkB, and respond to BDNF by forming neurites (Kaplan et al., 1993). DRM raft fractions from SH-SY5Y cells or from HeLa cells contain markedly different levels of lipids compared to neurons or brain tissue (Fig. 1F–H). Moreover, lipid content differed markedly between HeLa cells and SH-SY5Y cells as expected, as they are different cell lines. In both cases, exogenous expression of synapsins resulted in significant changes of lipids in DRM raft fractions. These data indicate that exogenous synapsins readily alters the composition of lipids in membrane rafts of non-neuronal cells.
Synapsins constrain the proportion of lipids in membrane rafts
Analyses of single lipid concentrations in DRM raft fractions did not reveal an underlying pattern to the effects of synapsins on membrane rafts (Fig. 1). To further investigate the mechanism underlying the lipid composition changes, we measured the ratio of cholesterol to phospholipids and sphingomyelin (Fig. 2). We observed that the most consistent ratio in WT tissue (i.e. P1, P7, P14 brain and neuronal cultures) was the proportion of cholesterol to phosphatidylcholine (acting as a proxy for total estimated phosphoglycerides) combined with sphingomyelin in raft fractions (Fig. 2A). This ratio was not conserved in synapsin TKO DRM raft fractions nor in WT or TKO non-raft fractions (Fig. 2B). These data suggest that synapsins play a role in modifying the lipid composition of rafts (but not in non-rafts) by maintaining the proportion of cholesterol to total phospholipids (including sphingomyelin).
Figure 2.

Synapsins restrict the proportion of cholesterol and phospholipids in membrane rafts. Graphs depicting the ratio of cholesterol to estimated total phospholipid in (A) DRM raft (# 1–2) or (B) non-raft (# 4–9) fractions (see Fig. 1A) for the indicated brain tissues, WT and synapsin TKO cultured neurons and glia, and differentiated SH-SY5Y cells and HeLa cells expressing control plasmids (No Synapsin) or a combination of all three synapsin (Synapsin) (n = 4–12). The total estimated phospholipid concentration was determined by the formula (4 x phosphatidylcholine) + sphingomyelin, since phosphatidylcholine represents approximately 25% of all glycerophospholipids in DRM raft fractions in a variety of cells and neural tissue (Pike et al., 2005). Significance was determined by Student’s two-sided t-test and error bars denote ± SEM. (n ≥ 4). *P<0.05; **P<0.01.
We also examined the scenario whereby synapsins are expressed in non-neuronal cells. When synapsins were expressed in differentiated SH-SY5Y cells, we found that the proportion of cholesterol to total phospholipids in DRM raft lipids became indistinguishable from that of WT neurons (Fig. 2A). We next examined the effect of expressing synapsins in HeLa cells, which have a radically different biology and lipid composition compared to neurons or neuroblastoma cells. Expression of synapsins in HeLa cells also converted the proportion of DRM raft lipids to one resembling neurons (Fig. 2A), highlighting the importance of synapsins in regulating the lipid content of neuronal total membrane rafts.
In the presence of synapsins, the molar ratio of cholesterol to total phospholipids in DRM raft fractions was close to 1 in brain, cultured neurons, SH-SY5Y cells and HeLa cells (Fig. 2A). Curiously, in vitro studies indicate that model detergent-resistant membranes are ideally formed from a lipid mixture comprising of 1:1:1 phosphatidylcholine/sphingomyelin/cholesterol molar quantities in the absence of proteins (Edidin, 2003). These data suggest that synapsins act by constraining the proportion of lipids in membranes to create suitable conditions for the natural formation of functional rafts.
Depletion of synapsins results in an alteration of the levels of BDNF/TrkB signaling proteins without affecting the canonical MAPK cascade
There is a large body of literature supporting a role for synapsins in the regulation of multiple neurodevelopmental processes, such as neurogenesis (Kao et al., 2008), neuronal polarity and migration (Perlini et al., 2015), axon outgrowth (Chin et al., 1995; Ferreira et al., 2000; Kao et al., 2002), synapse formation (Ferreira et al., 1995), and synaptic development (Lu et al., 1992; Han & Greengard, 1994; Chin et al., 1995; Valtorta et al., 1995), which parallel the actions of BDNF/TrkB. To determine if there is a potential link between synapsins and BDNF/TrkB signaling, we next examined levels of proteins relevant to BDNF/TrkB signaling (Fig. 3). We found that levels of both mature BDNF and full-length TrkB were significantly increased in brains from synapsin TKO mice compared to WT mice (Fig. 3A–B; at P1 for mature BDNF, 0.25 vs 0.16, t28=3.11, P=0.0043, and at P1 for full-length TrkB, 1.57 vs 1.18, t24=2.24, P=0.035). The results suggest that feedback regulation governing BDNF/TrkB signaling in synapsin TKO neurons could be impaired, resulting in elevated BDNF/TrkB.
Figure 3.

Depletion of synapsins results in up-regulation of TrkB and BDNF. (A) Representative western blots of whole brain homogenates from WT or synapsin TKO mice, at the designated age [neonatal (P1), postnatal day 7 (P7), and postnatal day 14 (P14)], probed with antibodies to the indicated protein. (B) Relative quantitation of the indicated protein from western blots of whole brain homogenates from the indicated mice and age (n ≥ 3). Quantities for each set of WT and TKO lystates were normalized to α-tubulin for proteins > 40K and 14-3-3ζ for proteins <40K. (C) Representative western blots of lysates from neuronal cultures from the indicated mice, treated ±BDNF for 1 hour, and probed with the indicated antibody. (D) Relative quantitation of phospho-Erk1/2 over total Erk1/2 (P/T ratio) from western blots (n ≥ 4). Significance was determined by Student’s two-sided t-test and error bars denote ± SEM. *P<0.05; **P<0.01.
In addition to the full-length form of TrkB, there are three other spliced variants: TrkB.T1 (Klein et al., 1990), TrkB.T2 (Middlemas et al., 1991), and TrkB.shc (Stoilov et al., 2002), all of which lack an intracellular tyrosine kinase domain but are still capable of binding to BDNF (Baxter et al., 1997). Little is known about TrkB.T2 or TrkB.shc, but TrkB.T1 is abundantly expressed in neurons, and increases during development to become the most abundant TrkB isoform expressed in the adult brain (Fryer et al., 1996). We observed that levels of TrkB.T1 were significantly raised in brain extracts derived from synapsin TKO mice (at P14, 1.49 vs 0.61, t6=3.15, P=0.02). As TrkB.T1 is thought to act as a dominant negative inhibitor of TrkB function (Eide et al., 1996), this finding suggests that reduction of TrkB.T1 production is one mechanism by which synapsins could exert their effects.
Not all signaling molecules associated with BDNF/TrkB signaling were altered by depletion of synapsins. The mature 14 kD form of BDNF is derived from the proteolytic processing of a 32 kD precursor, pro-BDNF (Mowla et al., 2001). The 32 kD pro-BDNF has been shown to undergo N-terminal cleavage to produce a 28 kD truncated BDNF, which is not further cleaved (Seidah et al., 1999). In general, deletion of synapsins did not result in a consistent, significant difference in levels of either pro-BDNF (Fig. 3A–B, at P1, 0.55 vs 0.63, t26=0.1.17, P=0.25) or truncated BDNF (Fig. 3A–B, at P1, 0.23 vs 0.23, t21=0.33, P=0.74). In addition to binding to TrkB, BDNF associates with a low affinity pan-neurotrophin receptor, p75NTR, which regulates neuronal survival (Huang & Reichardt, 2003). Again, levels of p75NTR in brain were not significantly different between synapsin TKO versus wild-type mice (Fig. 3A–B, at P1, 0.87 vs 0.79, t14=0.73, P=0.48).
The up-regulation of specific proteins involved in BDNF signaling, namely TrkB, TrkB.T1, and mature BDNF, in the absence of synapsins, raises the question as to how BDNF/TrkB signaling could be disrupted. The canonical MAPK cascade is one of the best studied pathways known to be activated by BDNF, and thought to represent a major mechanism by which TrkB exerts its actions (Segal & Greenberg, 1996). However, in cultured neurons, there was no apparent significant difference in the activation of MAPK by BDNF between synapsin TKO and wild-type mice (Fig. 3C–D, 4.61 vs 2.64, t6=1.92, P=0.10). Similarly, the lack of changes in levels of pro-BDNF [which induces apoptosis by binding to p75NTR (Teng et al., 2005)] or p75NTR would appear to preclude the involvement of synapsins in apoptotic pathways. These negative findings prompted us to investigate other routes by which synapsins target BDNF/TrkB signaling.
Synapsins promote localization of Fyn to membrane rafts, thereby promoting TrkB activation
Given that synapsins regulate lipid composition in rafts, we next asked whether synapsins also regulate protein targeting to rafts. Previous studies have shown that palmitoylation of Fyn enables partitioning of this protein to DRM raft fractions (Wolven et al., 1997). Fyn also facilitates the translocation of activated TrkB to lipid rafts (Suzuki et al., 2004; Pereira & Chao, 2007). Levels of Fyn increases with age (Fig. 3A–B), but the majority of Fyn does not appear in WT rafts fractions until postnatal day 14 (P14) (Fig. 4A). By contrast, levels of Fyn were not maintained in synapsin TKO P14 raft fractions (Fig. 4A–B). These findings imply that deficient membrane raft targeting of Fyn occurs in synapsin TKO mouse brains. To confirm that localization of Fyn to membrane rafts is regulated by synapsins, we overexpressed synapsins in differentiated SH-SY5Y cells (Fig. 4C–D). Synapsins increased levels of Fyn in DRM raft fractions in these cells, while expression of eGFP or negative controls had no effect (Fig. 4C–D). These findings indicate that synapsins promote the localization of Fyn to rafts; a likely mechanism is the maintenance of lipid proportions in membranes by synapsins (Fig. 2A), thereby enabling targeting of palmitoylated proteins, such as Fyn, to DRM raft fractions (Moffett et al., 2000).
Figure 4.

Synapsins promote the localization of Fyn to DRM raft fractions. (A) Representative western blot depicting equal volumes of DRM raft (#1–2) and non-raft (#4–9) fractions (see Fig. 1A) derived from postnatal P14 brains of WT or synapsin TKO mice, probed with antibodies to Fyn. Line indicates the band that was quantitated in (B). (B) Percentage of Fyn in DRM brain raft fractions of postnatal P14 mice, quantitated by western blot (n ≥ 4). (C) Representative western blot of DRM raft and non-raft fractions from synapsin-expressing (Syn) and eGFP-expressing differentiated SH-SY5Y cells, probed with antibodies directed towards Fyn. Line indicates the band that was quantitated in (D). (D) Percentage of Fyn in DRM raft fractions from western blots of electroporated (Syn, eGFP; n ≥ 9) differentiated SH-SY5Y cells and of non-electroporated (no DNA, n = 3) differentiated SH-SY5Y cells. Significance was determined by Student’s two-sided t-test and error bars denote ± SEM. *P<0.05.
Autophosphorylation of TrkB is activated by BDNF and is required for interaction of TrkB with Fyn (Iwasaki et al., 1998) and consequent translocation of TrkB to membrane rafts (Suzuki et al., 2004; Pereira & Chao, 2007). Since the role of specific TrkB phosphorylation sites in raft-mediated signaling is poorly understood, we examined tyrosine phosphorylation at Y706, because this site plays a critical role in downstream PLC-γ activity, which has been shown to be particularly sensitive to membrane raft disruption (Suzuki et al., 2004; Pereira & Chao, 2007). In WT DRM raft fractions, levels of TrkB phosphorylation at Y706 are high (Fig. 5). Synapsins regulate TrkB phosphorylation: the total quantity of phospho-TrkB as well as the ratio of Phospho/Total protein at Y706 in DRM raft fractions was significantly decreased in postnatal P14 brains in the absence of synapsins (Fig. 5B). A major effect of synapsins is to promote localization of Fyn to membrane rafts (Fig. 4) and Fyn directly phosphorylates TrkB in vitro (Rajagopal & Chao, 2006). Phosphorylation of TrkB at Y706 is required for the docking of adaptor proteins, which in turn governs downstream TrkB/PLC-γ signaling (Reichardt, 2006). Thus, these findings raise the possibility that synapsins regulate downstream TrkB signaling at the level of the membrane raft by promoting TrkB-Fyn interactions, which in turn affects TrkB phosphorylation.
Figure 5. Depletion of synapsins results in decreased TrkB activation on rafts.

(A) Representative western blots of DRM raft and non-raft fractions from brains of postnatal P14 mice, probed with phospho-Y706 antibodies to TrkB. The numbering of Y706 refers to the human TrkB sequence and is equivalent to Y785 in TrkA (Reichardt, 2006). (B) Relative quantitation of phospho-Y706 in DRM raft or non-raft fractions of WT or synapsin TKO postnatal P14 mice, quantitated by western blot (n ≥ 3). Significance was determined using Student’s two-sided t-test. Error bars denote ± SEM. *P<0.05; **P<0.01.
Synapsins promote the association of TrkB and Fyn
To test whether synapsins affect the interaction between TrkB and Fyn, we quantified the colocalization of TrkB and Fyn in cortical neurons (Fig. 6A–C). In WT neurons, BDNF significantly stimulated TrkB-Fyn colocalization (Fig. 6A–C). In contrast, BDNF had no effect on TrkB-Fyn colocalization in synapsin TKO neurons. The effects of synapsin depletion are similar to those observed when WT neurons were treated with the lipid raft disruptors, methyl-β-cyclodextrin or GM1 (Fig. 6C). These agents have different mechanisms of action: methyl-β-cyclodextrin depletes cholesterol from cell membranes, thereby eliminating membrane rafts (Simons & Toomre, 2000), while GM1 displaces glycophosphatidylinositol-anchored proteins (Simons et al., 1999) but leaves rafts intact (Simons & Toomre, 2000; Pereira & Chao, 2007).
Figure 6.

Depletion of synapsins in neurons abrogates BDNF-dependent TrkB-Fyn colocalization. (A) Confocal images depicting localization of TrkB and Fyn in cortical neurons derived from WT or synapsin TKO brains under control conditions (no BDNF) or BDNF treatment for 1 hour. The signals in each image were optimized to enable colocalization analyses. (B) To facilitate visualization of TrkB-Fyn colocalized regions in (A), the regions were highlighted against the outline of each cell using ImageJ highlighter software (identical conditions were used for all panels in A). (C) Magnified image of the region shown in the 25 x 25 micron box of panel (B). (D) Quantitation of colocalization for TrkB and Fyn in control (no BDNF) WT and synapsin TKO neurons, and in WT neurons treated with the raft disruptors methyl-β-cyclodextrin (MCD) or GM1 (n =14–33; Student’s two-sided t-test). Error bars denote ± SEM. ***P<0.001.
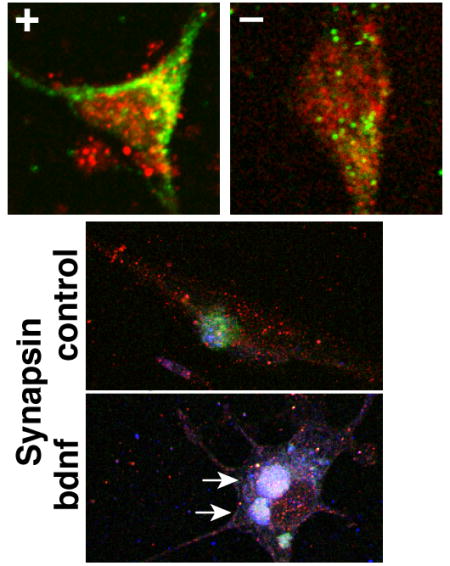
Conversely, overexpressing synapsins in differentiated SH-SY5Y cells produced reciprocal effects on TrkB-Fyn colocalization (Fig. 7). Expression of synapsins, but not eGFP, was significantly correlated with the degree of TrkB-Fyn colocalization (Fig. 7C). Moreover, BDNF induced a significant increase in TrkB-Fyn colocalization in synapsin-expressing cells, but not in eGFP-expressing cells (Fig. 7C–D). Exogenous synapsins also appeared to induce the formation of large clusters in SH-SY5Y cells that were immunoreactive to TrkB, Fyn and synapsins combined (Fig. 7A–B, Synapsin-bdnf). Together with the results in cortical neurons, these data establish that synapsins promote TrkB-Fyn colocalization. These data also suggest that synapsins could regulate downstream actions of BDNF by promoting TrkB-Fyn interactions in membrane rafts.
Figure 7. Synapsins promote BDNF-dependent TrkB-Fyn colocalization in SH-SY5Y cells.

(A) Confocal images depicting localization of TrkB and Fyn in differentiated SH-SY5Y cells expressing eGFP or synapsins (Synapsin), under control conditions (no BDNF) or treated with BDNF for 1 day. The signals in each image were optimized to enable colocalization analyses. Arrows point to clusters immunoreactive to TrkB, Fyn and synapsins in BDNF-treated synapsin-expressing cells. (B) Colocalized regions for TrkB and Fyn were highlighted against the outline of each cell in panel (A) using identical conditions for each panel with ImageJ highlighter software. (C) Magnified image of the region shown in the 30 x 30 micron box of panel (B). (D) Graphs depict correlations between TrkB-Fyn colocalization and expression of either eGFP or Synapsin in differentiated SH-SY5Y cells under control conditions (no BDNF) or treated with BDNF for the indicated days. The correlations between TrkB-Fyn colocalization and eGFP expression were not significant [Pearson correlation; Control–r25=0.06, P=0.8; BDNF 1 day–r39=0.13, P=0.4; BDNF 2 day–r44=0.22, P=0.15]. By contrast, the correlations between TrkB-Fyn colocalization and Synapsin expression were all highly significant [Control–r23=0.66, P=0.0003; BDNF 1 day–r46=0.69, P<0.0001; BDNF 2 day–r61=0.69, P<0.0001]. (D) Quantitation of TrkB-Fyn colocalization in differentiated SH-SY5Y cells expressing eGFP or Synapsin, untreated (control) or treated with BDNF for the indicated days in culture (Student’s two-sided t-test; n > 24 for each condition). Error bars denote ± SEM. **P<0.01.
Synapsins are required for BDNF-mediated axon elongation and neurite formation
We next tested whether synapsins have functional consequences with respect to BDNF signaling. Early in neural development, lipid rafts (Guirland et al., 2004) and synapsins (Ferreira et al., 1994; Ferreira et al., 2000) are concentrated in growth cones. Because both BDNF and synapsins regulate axon outgrowth in neurons (Valtorta et al., 2011), we asked whether BDNF-stimulated axon outgrowth occurs via lipid rafts. As expected, there was a significant increase in axon elongation when cortical neurons were exposed to BDNF for 24 hours (Fig. 8A). This effect was abolished by treatment with the raft-disrupting agent, GM1 (Fig. 8A). BDNF also failed to stimulate axon outgrowth in neurons derived from synapsin TKO mice (Fig. 8A), similar to the effect of GM1 treatment. When synapsins were overexpressed in differentiated SH-SY5Y cells, which express TrkB and increase neurite formation in response to BDNF (Kaplan et al., 1993), neurite formation was greatly enhanced (Fig. 8B). In contrast, neurite formation in control cells expressing eGFP was similar to that of non-electroporated cells (Fig. 8B). The effect of synapsin overexpression was markedly reduced by treatment with the raft disruptor, GM1, again implicating lipid rafts in this effect (Fig. 8B). Taken together, these data support a role for synapsins in the regulation of axon/neurite outgrowth by modulating BDNF signaling via lipid rafts.
Figure 8.

Synapsins mediate BDNF-induced axon elongation and neurite formation. (A) Representive phase contrast images of 3-day old WT or synapsin TKO cortical neurons under control conditions (no BDNF) or treated with BDNF for 24 hr. (B) Graph depicting axon length under control conditions (no BDNF) or in 24 hr BDNF-treated 3-day old WT or synapsin TKO cortical neuronal cultures, or in 3-day old WT neurons treated concomitantly with 100 μM GM1, a raft disruptor (n≥68 for each condition). Methyl-β-cyclodextrin was not used because it is lethal when exposed to neurons for 24 hours. (C) Representive fluorescence and phase contrast images of differentiated SH-SY5Y cells expressing exogenous eGFP or all three synapsins (Syn) under control conditions (no BDNF) or treated with BDNF for 24 hours. Cells were fixed and fluorescently stained with FLAG monoclonal antibody, which is specific for the exogenous protein as both eGFP and synapsins were tagged with this epitope. Arrows in the phase contrast image indicate cells that are fluorescent. Bar = 100 microns and applies to all panels in (A) and (C). (D) Graph depicting neurite formation in differentiated SH-SY5Y cells expressing exogenous eGFP or all three synapsins (Syn) under control conditions (no BDNF) or treated with BDNF for 24 hours. Neurite formation that is stimulated by synapsins and BDNF was abolished by concomitant incubation of the cells with 100 μM GM1. Methyl-β-cyclodextrin was not used because it is lethal when exposed to SH-SY5Y cells for 24 hours. Significance was determined using Student’s two-sided t-test. Error bars denote ± SEM. *P<0.05; ***P<0.001.
Synapsins are required for BDNF-mediated enhancement of synaptic transmission
BDNF is known to enhance transmission at many excitatory synapses (Lohof et al., 1993; Li et al., 1998; Valenti et al., 2012) and plays an important role in higher-order neuronal processes such as synaptic plasticity (Minichiello et al., 2002; Valenti et al., 2012). This regulation of synaptic transmission involves both synapsins (Jovanovic et al., 2000; Valenti et al., 2012) and lipid rafts (Suzuki et al., 2004). Therefore, we examined the role of synapsins in BDNF-induced synaptic enhancement in cultured hippocampal neurons from WT or synapsin TKO mice. After BDNF treatment of WT neurons, there was a robust increase in spontaneous neurotransmitter release (measured as the frequency of miniature excitatory postsynaptic currents, mEPSCs), consistent with previous reports (Li et al., 1998; Suzuki et al., 2004) (Fig. 9A–B). Pretreatment of WT neurons with the lipid raft disruptors, methyl-β-cyclodextrin or GM1, abolished the ability of BDNF to stimulate spontaneous release (Fig. 9A–C). Similarly, treatment of synapsin TKO neurons with BDNF did not increase mEPSC frequency (Figs. 9B–C). This is consistent with previous results showing that genetic deletion of synapsins I (Valenti et al., 2012) or both synapsins I and II (Jovanovic et al., 2000) reduces the ability of BDNF to enhance depolarization-evoked glutamate release. It is noteworthy that the PLC-γ pathway, which is dependent on phosphorylation at Y706, is critically important for raft-mediated synaptic potentiation (Suzuki et al., 2004) and BDNF/TrkB-mediated long-term potentiation (Minichiello et al., 2002). The PLC-γ pathway is also particularly sensitive to chemical disruption of membrane rafts (Pereira & Chao, 2007) and phosphorylation at Y706 is disrupted by synapsin depletion (Fig. 5). Thus, abrogation of BDNF-dependent synaptic potentiation by both synapsin removal and lipid raft disruption is consistent with a role for synapsins in regulating BDNF-mediated potentiation via lipid rafts.
Figure 9.

Synapsins are required for BDNF-induced synaptic potentiation in hippocampal neurons. (A) mEPSC frequency was increased by BDNF in WT hippocampal neurons, while concomitant treatment with MCD or GM1 inhibited this increase. (B) Plot depicting BDNF-induced increase in mEPSC frequency in WT hippocampal neurons, which was abolished when the neurons were treated with the lipid raft disruptors methyl-β-cyclodextrin (MCD) or GM1, and not present in TKO hippocampal neurons. (C) Change in frequency of mEPSCs after the addition of BDNF is depicted for WT hippocampal neurons, WT hippocampal neurons treated concomitantly with the indicated raft disruptors, and TKO hippocampal neurons. Significance was determined using Student’s two-sided t-test. Error bars denote ± SEM. **P<0.01.
The actions of synapsins are potentiated by expression of all three synapsin genes
Previous studies concerning the developmental actions of synapsins have generally focused on the properties of a single synapsin gene. Our observations suggest that synapsins accelerate the development of SH-SY5Y cells, thereby promoting BDNF/TrkB signaling. To achieve a better understanding of how synapsins act, we expressed individual synapsins in SH-SY5Y cells, and asked if this altered the development of these cells.
To facilitate this analyses, we examined a well-characterized marker of neuronal differentiation, GAP-43, in which expression is restricted to growth cones (Goslin et al., 1988), is correlated with axon outgrowth (Kalil & Skene, 1986), and increases in SH-SY5Y cells treated with retinoic acid (Encinas et al., 2000). In accord with previous studies (Encinas et al., 2000; Edsjo et al., 2003; Cuende et al., 2008), we found that GAP-43 protein expression was induced in SH-SY5Y cells treated with all-trans retinoic acid, but only after differentiation in culture over a prolonged period of time [>5 days] (Fig. 10A–B). We then examined the effect of expressing the major isoform of each synapsin gene and all three synapsin genes combined. We found that GAP-43 levels were significantly different when synapsins were expressed in SH-SY5Y cells [Fig. 10C–D, one-way ANOVA, F4, 24=9.12, P<0.001]. Post-hoc analyses using Tukey’s test revealed that GAP-43 expression in the presence of all three synapsins was significantly greater than nontransfected or single synapsin-transfected cells [No treatment vs All; Q24=7.14, P=0.0010; Syn1 vs All, Q24==6.0662, P=0.0021, Syn2 vs All, Q24==5.39, P=0.0069; Syn3 vs All, Q24=6.85, P=0.0010]. The results suggest that the effect of expressing all three synapsin genes is much greater than the effect of expressing a single synapsin gene. Moreover, GAP43 protein induction using retinoic acid required prolonged differentiation (6–12 days) while GAP43 induction was apparent after just 3 days with synapsin expression, highlighting the ability of synapsins to accelerate differentiation in these cells.
Figure 10.

All three synapsins potentiate the induction of GAP-43 and the formation of higher molecular weight complexes. (A) Representative western blot depicting GAP-43 and total Erk (T-Erk) protein in SH-SY5Y cells treated with retinoic acid for the indicated days. (B) Quantitation of relative GAP-43 protein levels in retinoic acid-treated SH-SY5Y cells. GAP-43 protein levels were normalized to that of total Erk (T-Erk). A significant, approximately 2-fold induction in GAP-43 protein levels was observed after 9–12 days of treatment (one-way ANOVA, F1, 11=5.14, P=0.018; post-hoc Tukey’s test reveals that for 0 days vs 12 days, Q11=4.81, *P=0.026) (C) Representative western blots of lysates derived from SH-SY5Y cells expressing no synapsin (0), the indicated single synapsin gene (I, II, or III), or combination of all three synapsins (I+II+II), probed with GAP-43 antibody. (D) Graph depicting quantitation of GAP-43 in SH-SY5Y cells expressing no synapsin (0), the indicated single synapsin gene (I, II, or III), or combination of all three synapsins (I+II+II). GAP-43 protein levels were normalized to that of total Erk (T-Erk). GAP-43 levels were significantly different when synapsins were expressed in SH-SY5Y cells (one-way ANOVA, F4, 24=9.12, P<0.001). Post-hoc analyses using Tukey’s test revealed that GAP-43 expression in the presence of all three synapsins (I+II+III) was significantly greater than nontransfected or single synapsin-transfected cells (0 vs I+II+III; Q24=7.14, **P=0.0010; I vs I+II+III, Q24==6.0662, **P=0.0021, II vs I+II+III, Q24==5.39, **P=0.0069; III vs I+II+III, Q24=6.85, **P=0.0010]. (E) Western blot of immuno-captured synapsins derived from adult WT, synapsin II KO (S2K) and P7 synapsin II KO (P7-S2K), resolved by standard SDS-PAGE and probed with a pan-synapsin antibody, G304. Since adult brains express much less synapsin III than I or II, the final preparation from WT adult brain consists mainly of synapsins I and II. Likewise, preparations from adult synapsin II KO brain contain almost exclusively, synapsin I. Preparations from P7 synapsin II KO brain contain both synapsins I and III (synapsin III is abundantly expressed at that time). (F) Western blot of immuno-captured proteins resolved by native SDS-PAGE (Nowakowski et al., 2014) and probed with a pan-synapsin antibody, G304, which recognizes the “a” isoform from each synapsin gene. The various combinations were derived from immuno-captured synapsins described in (D). Note that “b” synapsin isoforms are not detected by this antibody, and are capable of binding to “a” synapsin isoforms, which could account for multiple bands detcted by this method of protein resolution. Peak intensities were identified by ImageJ and indicated by red dots to the right of each lane. (G) Relative quantities of complexed and monomeric synapsin proteins were quantitated from the native SDS-PAGE western blot depicted in (F) using plot profiles in ImageJ. A higher ratio of complexed/monomeric synapsin is observed when all three synapsins are combined.
To determine if there is a potential underlying mechanism to explain why expression from all three synapsin genes appears to be more potent than single synapsin genes, we examined the ability of synapsins to form higher molecular weight complexes using native SDS-PAGE (Nowakowski et al., 2014). Previous studies have shown that synapsins form homodimers and heterodimers (Hosaka & Sudhof, 1999), as well as higher-order multimers (Brautigam et al., 2004). Synapsins were isolated from mouse brain using a biotinylated pan-specific synapsin antibody. Our experiments revealed that immuno-capture using this antibody was effective only for synapsin Ia or synapsin Ia dimerized to another synapsin isoform. Nonetheless, we found that the combination of all three synapsins appeared to be more capable of forming high molecular weight multimers than synapsin Ia alone or a combination of synapsin Ia with another synapsin (Fig. 10E–G). Thus, this finding is consistent with the notion that all three synapsins have a more potent effect than a single synapsin on development, and may be a potential clue to the mechanism of synapsin action.
Discussion
Synapsins act on membrane lipid rafts
Here, we show that synapsins maintain the functional integrity of membrane lipid rafts, which is required for BDNF-mediated synaptic and neurodevelopmental processes. Depletion of synapsins in neurons results in aberrant raft lipid composition, deficient targeting of Fyn to rafts, decreased association of Fyn with TrkB, and abrogation of BDNF-stimulated axon outgrowth and synaptic potentiation. Conversely, expression of synapsins in neuroblastoma cells results in corresponding reciprocal changes in raft lipid composition, increased localization of Fyn to rafts, increased association of Fyn with TrkB, and promotion of BDNF-stimulated neurite formation. Thus, synapsins regulate raft lipid composition, and consequently, BDNF-mediated axon/neurite outgrowth and synaptic potentiation.
Our experiments examined total membranes of cells, and previous studies have shown that the surface of intracellular membranes exceeds that of the plasma membrane by more than an order of magnitude (Luby-Phelps, 2000). Since synapsins are not associated with the plasma membrane (Huttner et al., 1983), but are abundantly associated with synaptic vesicles (Huttner et al., 1983) and possibly other intracellular membranes (Ferreira et al., 2000), it is likely that synapsins influence a class of intracellular membrane rafts, thereby affecting BDNF signaling.
This report, as well as others (Guirland et al., 2004; Suzuki et al., 2004; Pereira & Chao, 2007), demonstrates that intracellular membrane rafts are crucial for BDNF-mediated axon outgrowth and synaptic potentiation. Consistent with these actions, rafts are specifically concentrated in growth cones and synapses (Guirland et al., 2004). Despite the importance of raft-dependent signaling, the amount of TrkB residing on rafts represents a small proportion of total TrkB (Fig. 5A) (Suzuki et al., 2004; Pereira & Chao, 2007), and disruption of rafts has little effect on the activation of downstream cascades (Pereira & Chao, 2007). These observations suggest that BDNF/TrkB signaling is organized into non-raft and raft-mediated events. For instance, non-raft BDNF signaling promotes cell survival while raft-dependent BDNF signaling does not (Suzuki et al., 2004; Pereira & Chao, 2007). In contrast, raft-dependent BDNF signaling is required for BDNF-stimulated axon/neurite outgrowth (Fig. 8), growth cone turning (Guirland et al., 2004), and synaptic potentiation (Fig. 9) (Suzuki et al., 2004). Lipid rafts are sites of rapid protein movement (Kenworthy et al., 2004) and highly concentrated kinase activity (Fig. 5) (Suzuki et al., 2004; Pereira & Chao, 2007). This is consistent with the idea of rafts serving as membrane microdomains that organize signal transduction by recruiting signaling proteins to enable spatiotemporal regulation of downstream pathways. Within this framework, we propose that synapsins regulate the level of raft versus non-raft BDNF signaling.
Synapsins affect specific BDNF downstream pathways
Our studies reveal that synapsins likely act on only a subset of BDNF downstream pathways. For example, the canonical MAPK cascade appears to be intact in neurons depleted of synapsins, and this pathway is thought to be important for many of the actions of BDNF (Segal & Greenberg, 1996). Indeed, the responsiveness of the MAPK pathway has been shown to be independent of membrane rafts and disruption of rafts has little effect on the activation of other downstream cascades (Pereira & Chao, 2007).
Our findings differ from a recent study examining the effects of synapsin depletion on BDNF signaling (Marte et al., 2016). Marte et al (2016) report a significant decrease in MAPK activation by BDNF in synapsin TKO neurons, although MAPK activation was still very robust in these cells. The difference in results could be attributed to our use of pretreating all neuronal cultures with tetrodotoxin to inhibit the hyperexcitability of neurons, particularly those depleted of synapsins. Despite these differences, both our study and that of Marte et al (2016) observe that BDNF-mediated axonal outgrowth is abrogated in synapsin TKO neurons, leading these authors, as well as us, to conclude that synapsins are inextricably linked to BDNF signaling pathways.
Another finding that pertains to TrkB signaling is the robust elevation of TrkB.T1 in brains of synapsin TKO mice. Other groups have recently reported raised levels of TrkB.T1 in neurons derived from synapsin III KO (Piccini et al., 2015), and synapsin I KO and TKO (Marte et al., 2016) mice. While this finding appears highly reproducible, the significance of this finding as it pertains to BDNF/TrkB signaling remain to be determined.
We did not observe differences in the expression of p75NTR or pro-BDNF [which induces apoptosis by binding to p75NTR (Teng et al., 2005)] between WT and synapsin TKO brains. These results suggest that synapsins do not partake in apoptotic pathways. However, recently, p75NTR was reported to be increased in cultured synapsin-depleted neurons (Marte et al., 2016), and the survival of synapsin III KO neurons was also reported to be reduced in cultured neurons (Piccini et al., 2015). The differences reported by these groups could be attributed to the stress that neurons undergo when they are dissociated from their natural environment and placed in culture. Indeed, we observed that Fyn undergoes degradation in synapsin TKO cultured neurons (Fig. 4A), which is consistent with cleavage by caspase 3, an effector of apoptosis (Ricci et al., 1999). Thus, synapsin-depleted neurons may indeed be prone to apoptosis, but this is only apparent once neurons are placed in cell culture.
We consistently observe that levels of both mature BDNF and full-length TrkB were significantly increased in brains from synapsin TKO mice compared to WT mice. Previous studies in neurons showed that increasing BDNF down-regulates levels of TrkB (Frank et al., 1996), while knocking out BDNF results in increased TrkB levels (Martin et al., 2008). Similarly, up-regulation of TrkB down-regulates BDNF expression (Johansson et al., 2009) while inhibition of TrkB function by dominant-negative truncated TrkB isoforms (Eide et al., 1996) potentially leads to increased BDNF expression (Rudge et al., 1998; Venero & Hefti, 1998; Pillai & Mahadik, 2008). These findings suggest that the up-regulation of both mature BDNF and full-length TrkB in the absence of synapsins during early development is a manifestation of disrupted BDNF/TrkB signaling.
Potential mechanism of synapsin action on membrane rafts
A major effect of synapsins is to promote localization of Fyn to membrane rafts (Fig. 4) and Fyn directly phosphorylates TrkB in vitro (Rajagopal & Chao, 2006). Phosphorylation of TrkB at Y706 is required for the docking of adaptor proteins, which in turn governs downstream TrkB/PLC-γ signaling (Reichardt, 2006). Thus, these findings raise the possibility that synapsins regulate downstream TrkB signaling at the level of the membrane raft by promoting TrkB-Fyn interactions, which in turn affects TrkB phosphorylation.
While the mechanisms by which synapsins exert their effects on membrane rafts remains unknown, their ability to restrict the proportion of lipids in this fraction could be a clue to their action. Another relevant property of synapsins is their ability to form homodimers and heterodimers (Hosaka & Sudhof, 1999), and higher-level multimers (Brautigam et al., 2004). This may allow the coalescence of rafts into larger membrane subdomains that is important for their function (Lingwood & Simons, 2010). In support of the connection to lipid rafts, we also observed that expression of all three synapsins results in a robust induction of GAP-43 (Fig. 10C–D), which is associated with lipid rafts (Arni et al., 1998) and localizes to growth cones (Goslin et al., 1988). Finally, synapsins are abundant substrates for MAP kinase, the primary enzyme activated by the Ras/MAP kinase cascade downstream of BDNF/TrkB (Valtorta et al., 2011). Thus, synapsins have the capacity to serve as both downstream mediators and upstream regulators of lipid raft mediated BDNF/TrkB signaling. In this manner, synapsins may act as mediators of a feedback mechanism by which substrates of the MAPK cascade can influence activation of TrkB. Thus, our studies reveal a potentially novel mechanism for mediating BDNF action in early development and synaptic transmission.
Acknowledgments
Research was supported by internal funds from Butler Hospital and partly conducted at the Genomics Core Facility at Brown University [partial support from NIGMS P30GM103410, NCRR (P30RR031153, P20RR018728 and S10RR02763), and NSF (EPSCoR 0554548)], NIH NS047209, and by the World Class Institute (WCI) Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology of Korea (NRF grant number WCI 2009-003).
Abbreviations
- BDNF
brain-derived neurotrophic factor
- DRM
detergent-resistant membrane
- TKO
triple knockout
- WT
wild-type
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Footnotes
Where the work originated: Brown University, Korea Institute of Science and Technology
Conflict of Interest Statement
All authors indicate no potential conflicts of interest.
Author Contributions
HTK, BP designed the study. HTK, KR, SRJ, AL, BP carried out the experiments and analyses. HTK, GA, BP drafted the paper. All authors reviewed the paper and provided feedback.
Data Accessibility Statement
All data are summarized in the results section.
References
- Amundson DM, Zhou M. Fluorometric method for the enzymatic determination of cholesterol. J Biochem Biophys Methods. 1999;38:43–52. doi: 10.1016/s0165-022x(98)00036-0. [DOI] [PubMed] [Google Scholar]
- Arni S, Keilbaugh SA, Ostermeyer AG, Brown DA. Association of GAP-43 with detergent-resistant membranes requires two palmitoylated cysteine residues. J Biol Chem. 1998;273:28478–28485. doi: 10.1074/jbc.273.43.28478. [DOI] [PubMed] [Google Scholar]
- Baxter GT, Radeke MJ, Kuo RC, Makrides V, Hinkle B, Hoang R, Medina-Selby A, Coit D, Valenzuela P, Feinstein SC. Signal transduction mediated by the truncated trkB receptor isoforms, trkB. T1 and trkB.T2. J Neurosci. 1997;17:2683–2690. doi: 10.1523/JNEUROSCI.17-08-02683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Greengard P, Brunner J, Bahler M. Electrostatic and hydrophobic interactions of synapsin I and synapsin I fragments with phospholipid bilayers. J Cell Biol. 1989;108:1851–1862. doi: 10.1083/jcb.108.5.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA, Chelliah Y, Deisenhofer J. Tetramerization and ATP binding by a protein comprising the A, B, and C domains of rat synapsin I. J Biol Chem. 2004;279:11948–11956. doi: 10.1074/jbc.M312015200. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- Chin LS, Li L, Ferreira A, Kosik KS, Greengard P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci USA. 1995;92:9230–9234. doi: 10.1073/pnas.92.20.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuende J, Moreno S, Bolaños JP, Almeida A. Retinoic acid downregulates Rae1 leading to APC(Cdh1) activation and neuroblastoma SH-SY5Y differentiation. Oncogene. 2008;27:3339–3344. doi: 10.1038/sj.onc.1210987. [DOI] [PubMed] [Google Scholar]
- Edidin M. The state of lipid rafts: from model membranes to cells. Annu Rev Biophys Biomol Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- Edsjo A, Lavenius E, Nilsson H, Hoehner JC, Simonsson P, Culp LA, Martinsson T, Larsson C, Pahlman S. Expression of trkB in human neuroblastoma in relation to MYCN expression and retinoic acid treatment. Lab Invest. 2003;83:813–823. doi: 10.1097/01.lab.0000074895.48776.d8. [DOI] [PubMed] [Google Scholar]
- Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Cena V, Gallego C, Comella JX. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem. 2000;75:991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Han HQ, Greengard P, Kosik KS. Suppression of synapsin II inhibits the formation and maintenance of synapses in hippocampal culture. Proc Natl Acad Sci USA. 1995;92:9225–9229. doi: 10.1073/pnas.92.20.9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Kao HT, Feng J, Rapoport M, Greengard P. Synapsin III: developmental expression, subcellular localization, and role in axon formation. J Neurosci. 2000;20:3736–3744. doi: 10.1523/JNEUROSCI.20-10-03736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Kosik KS, Greengard P, Han HQ. Aberrant neurites and synaptic vesicle protein deficiency in synapsin II-depleted neurons. Science. 1994;264:977–979. doi: 10.1126/science.8178158. [DOI] [PubMed] [Google Scholar]
- Frank L, Ventimiglia R, Anderson K, Lindsay RM, Rudge JS. BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. Eur J Neurosci. 1996;8:1220–1230. doi: 10.1111/j.1460-9568.1996.tb01290.x. [DOI] [PubMed] [Google Scholar]
- Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Giachello CN, Fiumara F, Giacomini C, Corradi A, Milanese C, Ghirardi M, Benfenati F, Montarolo PG. MAPK/Erk-dependent phosphorylation of synapsin mediates formation of functional synapses and short-term homosynaptic plasticity. J Cell Sci. 2010;123:881–893. doi: 10.1242/jcs.056846. [DOI] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004a;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, Augustine GJ. Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci. 2004b;24:3711–3720. doi: 10.1523/JNEUROSCI.5225-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Schreyer DJ, Skene JH, Banker G. Development of neuronal polarity: GAP-43 distinguishes axonal from dendritic growth cones. Nature. 1988;336:672–674. doi: 10.1038/336672a0. [DOI] [PubMed] [Google Scholar]
- Guirland C, Suzuki S, Kojima M, Lu B, Zheng JQ. Lipid rafts mediate chemotropic guidance of nerve growth cones. Neuron. 2004;42:51–62. doi: 10.1016/s0896-6273(04)00157-6. [DOI] [PubMed] [Google Scholar]
- Han HQ, Greengard P. Remodeling of cytoskeletal architecture of nonneuronal cells induced by synapsin. Proc Natl Acad Sci USA. 1994;91:8557–8561. doi: 10.1073/pnas.91.18.8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MF, Bahler M, Czernik AJ, Schiebler W, Kezdy FJ, Kaiser ET, Greengard P. Synapsin I is a highly surface-active molecule. J Biol Chem. 1991;266:5600–5607. [PubMed] [Google Scholar]
- Hojjati MR, Jiang XC. Rapid, specific, and sensitive measurements of plasma sphingomyelin and phosphatidylcholine. J Lipid Res. 2006;47:673–676. doi: 10.1194/jlr.D500040-JLR200. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Sudhof TC. Homo- and heterodimerization of synapsins. J Biol Chem. 1999;274:16747–16753. doi: 10.1074/jbc.274.24.16747. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC. Synapsin III, a novel synapsin with an unusual regulation by Ca2+ J Biol Chem. 1998;273:13371–13374. doi: 10.1074/jbc.273.22.13371. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki Y, Gay B, Wada K, Koizumi S. Association of the Src family tyrosine kinase Fyn with TrkB. J Neurochem. 1998;71:106–111. doi: 10.1046/j.1471-4159.1998.71010106.x. [DOI] [PubMed] [Google Scholar]
- Johansson J, Formaggio E, Fumagalli G, Chiamulera C. Choline up-regulates BDNF and down-regulates TrkB neurotrophin receptor in rat cortical cell culture. Neuroreport. 2009;20:828–832. doi: 10.1097/WNR.0b013e32832b7324. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Kalil K, Skene JH. Elevated synthesis of an axonally transported protein correlates with axon outgrowth in normal and injured pyramidal tracts. J Neurosci. 1986;6:2563–2570. doi: 10.1523/JNEUROSCI.06-09-02563.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Kao HT, Li P, Chao HM, Janoschka S, Pham K, Feng J, McEwen BS, Greengard P, Pieribone VA, Porton B. Early involvement of synapsin III in neural progenitor cell development in the adult hippocampus. J Comp Neurol. 2008;507:1860–1870. doi: 10.1002/cne.21643. [DOI] [PubMed] [Google Scholar]
- Kao HT, Porton B, Czernik AJ, Feng J, Yiu G, Haring M, Benfenati F, Greengard P. A third member of the synapsin gene family. Proc Natl Acad Sci USA. 1998;95:4667–4672. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HT, Song HJ, Porton B, Ming GL, Hoh J, Abraham M, Czernik AJ, Pieribone VA, Poo MM, Greengard P. A protein kinase A-dependent molecular switch in synapsins regulates neurite outgrowth. Nat Neurosci. 2002;5:431–437. doi: 10.1038/nn840. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ. Induction of TrkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Eukaryotic Signal Transduction Group. Neuron. 1993;11:321–331. doi: 10.1016/0896-6273(93)90187-v. [DOI] [PubMed] [Google Scholar]
- Kenworthy AK, Nichols BJ, Remmert CL, Hendrix GM, Kumar M, Zimmerberg J, Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- Krabben L, Fassio A, Bhatia VK, Pechstein A, Onofri F, Fadda M, Messa M, Rao Y, Shupliakov O, Stamou D, Benfenati F, Haucke V. Synapsin I senses membrane curvature by an amphipathic lipid packing sensor motif. J Neurosci. 2011;31:18149–18154. doi: 10.1523/JNEUROSCI.4345-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Lohmann SM, Ueda T, Greengard P. Ontogeny of synaptic phosphoproteins in brain. Proc Natl Acad Sci USA. 1978;75:4037–4041. doi: 10.1073/pnas.75.8.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- LoPresti P, Poluha W, Poluha DK, Drinkwater E, Ross AH. Neuronal differentiation triggered by blocking cell proliferation. Cell Growth Differ. 1992;3:627–635. [PubMed] [Google Scholar]
- Lu B, Greengard P, Poo MM. Exogenous synapsin I promotes functional maturation of developing neuromuscular synapses. Neuron. 1992;8:521–529. doi: 10.1016/0896-6273(92)90280-q. [DOI] [PubMed] [Google Scholar]
- Luby-Phelps K. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int Rev Cytol. 2000;192:189–221. doi: 10.1016/s0074-7696(08)60527-6. [DOI] [PubMed] [Google Scholar]
- Malhotra A. Tagging for protein expression. Methods Enzymol. 2009;463:239–258. doi: 10.1016/S0076-6879(09)63016-0. [DOI] [PubMed] [Google Scholar]
- Marte A, Messa M, Benfenati F, Onofri F. Synapsins Are Downstream Players of the BDNF-Mediated Axonal Growth. Mol Neurobiol. 2016 doi: 10.1007/s12035-015-9659-3. (epub) [DOI] [PubMed] [Google Scholar]
- Martin MG, Perga S, Trovó L, Rasola A, Holm P, Rantamaki T, Harkany T, Castrén E, Chiara F, Dotti CG. Cholesterol loss enhances TrkB signaling in hippocampal neurons aging in vitro. Mol Biol Cell. 2008;19:2101–2112. doi: 10.1091/mbc.E07-09-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlemas DS, Lindberg RA, Hunter T. trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Moffett S, Brown DA, Linder ME. Lipid-dependent targeting of G proteins into rafts. J Biol Chem. 2000;275:2191–2198. doi: 10.1074/jbc.275.3.2191. [DOI] [PubMed] [Google Scholar]
- Mowla SJ, Farhadi HF, Pareek S, Atwal JK, Morris SJ, Seidah NG, Murphy RA. Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J Biol Chem. 2001;276:12660–12666. doi: 10.1074/jbc.M008104200. [DOI] [PubMed] [Google Scholar]
- Nowakowski AB, Wobig WJ, Petering DH. Native SDS-PAGE: high resolution electrophoretic separation of proteins with retention of native properties including bound metal ions. Metallomics. 2014;6:1068–1078. doi: 10.1039/c4mt00033a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pera I, Stark R, Kappl M, Butt HJ, Benfenati F. Using the atomic force microscope to study the interaction between two solid supported lipid bilayers and the influence of synapsin I. Biophys J. 2004;87:2446–2455. doi: 10.1529/biophysj.104.044214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira DB, Chao MV. The tyrosine kinase Fyn determines the localization of TrkB receptors in lipid rafts. J Neurosci. 2007;27:4859–4869. doi: 10.1523/JNEUROSCI.4587-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlini LE, Szczurkowska J, Ballif BA, Piccini A, Sacchetti S, Giovedi S, Benfenati F, Cancedda L. Synapsin III acts downstream of semaphorin 3A/CDK5 signaling to regulate radial migration and orientation of pyramidal neurons in vivo. Cell Rep. 2015;11:234–248. doi: 10.1016/j.celrep.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccini A, Perlini LE, Cancedda L, Benfenati F, Giovedi S. Phosphorylation by PKA and Cdk5 Mediates the Early Effects of Synapsin III in Neuronal Morphological Maturation. J Neurosci. 2015;35:13148–13159. doi: 10.1523/JNEUROSCI.1379-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Pike LJ, Han X, Gross RW. Epidermal growth factor receptors are localized to lipid rafts that contain a balance of inner and outer leaflet lipids: a shotgun lipidomics study. J Biol Chem. 2005;280:26796–26804. doi: 10.1074/jbc.M503805200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai A, Mahadik SP. Increased truncated TrkB receptor expression and decreased BDNF/TrkB signaling in the frontal cortex of reeler mouse model of schizophrenia. Schizophr Res. 2008;100:325–333. doi: 10.1016/j.schres.2007.11.030. [DOI] [PubMed] [Google Scholar]
- Porton B, Ferreira A, DeLisi LE, Kao HT. A rare polymorphism affects a mitogen-activated protein kinase site in synapsin III: possible relationship to schizophrenia. Biol Psychiatr. 2004;55:118–125. doi: 10.1016/j.biopsych.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Rajagopal R, Chao MV. A role for Fyn in Trk receptor transactivation by G-protein-coupled receptor signaling. Mol Cell Neurosci. 2006;33:36–46. doi: 10.1016/j.mcn.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci JE, Maulon L, Luciano F, Guerin S, Livolsi A, Mari B, Breittmayer JP, Peyron JF, Auberger P. Cleavage and relocation of the tyrosine kinase P59FYN during Fas-mediated apoptosis in T lymphocytes. Oncogene. 1999;18:3963–3969. doi: 10.1038/sj.onc.1202782. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Mather PE, Pasnikowski EM, Cai N, Corcoran T, Acheson A, Anderson K, Lindsay RM, Wiegand SJ. Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not neuroprotective. Exp Neurol. 1998;149:398–410. doi: 10.1006/exnr.1997.6737. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Seidah NG, Mowla SJ, Hamelin J, Mamarbachi AM, Benjannet S, Toure BB, Basak A, Munzer JS, Marcinkiewicz J, Zhong M, Barale JC, Lazure C, Murphy RA, Chretien M, Marcinkiewicz M. Mammalian subtilisin/kexin isozyme SKI-1: A widely expressed proprotein convertase with a unique cleavage specificity and cellular localization. Proc Natl Acad Sci USA. 1999;96:1321–1326. doi: 10.1073/pnas.96.4.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Simons M, Friedrichson T, Schulz JB, Pitto M, Masserini M, Kurzchalia TV. Exogenous administration of gangliosides displaces GPI-anchored proteins from lipid microdomains in living cells. Mol Biol Cell. 1999;10:3187–3196. doi: 10.1091/mbc.10.10.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov P, Castren E, Stamm S. Analysis of the human TrkB gene genomic organization reveals novel TrkB isoforms, unusual gene length, and splicing mechanism. Biochem Biophys Res Commun. 2002;290:1054–1065. doi: 10.1006/bbrc.2001.6301. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, Greengard P. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Numakawa T, Shimazu K, Koshimizu H, Hara T, Hatanaka H, Mei L, Lu B, Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167:1205–1215. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpankowski L, Encalada SE, Goldstein LS. Subpixel colocalization reveals amyloid precursor protein-dependent kinesin-1 and dynein association with axonal vesicles. Proc Natl Acad Sci USA. 2012;109:8582–8587. doi: 10.1073/pnas.1120510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel G, Greengard P, Südhof TC. Characterization of tissue-specific transcription by the human synapsin I gene promoter. Proc Natl Acad Sci USA. 1991;88:3431–3435. doi: 10.1073/pnas.88.8.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente P, Casagrande S, Nieus T, Verstegen AM, Valtorta F, Benfenati F, Baldelli P. Site-specific synapsin I phosphorylation participates in the expression of post-tetanic potentiation and its enhancement by BDNF. J Neurosci. 2012;32:5868–5879. doi: 10.1523/JNEUROSCI.5275-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtorta F, Iezzi N, Benfenati F, Lu B, Poo MM, Greengard P. Accelerated structural maturation induced by synapsin I at developing neuromuscular synapses of Xenopus laevis. Eur J Neurosci. 1995;7:261–270. doi: 10.1111/j.1460-9568.1995.tb01062.x. [DOI] [PubMed] [Google Scholar]
- Valtorta F, Pozzi D, Benfenati F, Fornasiero EF. The synapsins: multitask modulators of neuronal development. Semin Cell Dev Biol. 2011;22:378–386. doi: 10.1016/j.semcdb.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Venero JL, Hefti F. Regionally specific induction of BDNF and truncated trkB. T1 receptors in the hippocampal formation after intraseptal injection of kainic acid. Brain Res. 1998;790:270–277. doi: 10.1016/s0006-8993(98)00071-7. [DOI] [PubMed] [Google Scholar]
- Wolven A, Okamura H, Rosenblatt Y, Resh MD. Palmitoylation of p59fyn is reversible and sufficient for plasma membrane association. Mol Biol Cell. 1997;8:1159–1173. doi: 10.1091/mbc.8.6.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]