

Abstract
Establishing and maintaining the appropriate number of GABA synapses is key for balancing excitation and inhibition in the nervous system, though we have only a limited understanding of the mechanisms controlling GABA circuit connectivity. Here, we show that disrupting cholinergic innervation of GABAergic neurons in the C. elegans motor circuit alters GABAergic neuron synaptic connectivity. These changes are accompanied by reduced frequency and increased amplitude of GABAergic synaptic events. Acute genetic disruption in early development, during the integration of post-embryonic-born GABAergic neurons into the circuit, produces irreversible effects on GABAergic synaptic connectivity that mimic those produced by chronic manipulations. In contrast, acute genetic disruption of cholinergic signaling in the adult circuit does not reproduce these effects. Our findings reveal that GABAergic signaling is regulated by cholinergic neuronal activity, probably through distinct mechanisms in the developing and mature nervous system.
KEY WORDS: GABA synapse, Neural circuit, Neural development, E/I balance
Highlighted Article: A non-cell-autonomous mechanism regulates inhibitory neuron synapse formation at the neuromuscular junction.
INTRODUCTION
Neurons in the brain are organized into circuits, and the activity of these circuits is regulated by the integration of excitatory and inhibitory synaptic inputs. Thus, appropriate numbers and proper positioning of excitatory and inhibitory synaptic connections are key factors governing circuit performance. Indeed, disruptions in the balance of excitatory and inhibitory synaptic activity are associated with debilitating brain diseases. For instance, impaired maturation of inhibitory signaling can produce circuit hyperexcitability and a predisposition to seizure activity that is thought to be important in epilepsy (Cossart et al., 2001; Powell et al., 2003; Briggs and Galanopoulou, 2011).
Though extensive studies of excitatory synapses have produced advances in our understanding of their development and regulation, far less is known about the processes that govern inhibitory synaptogenesis and patterning. Gamma-aminobutyric acid (GABA) is the principal inhibitory neurotransmitter in the mature mammalian brain. GABAergic inhibition controls spatiotemporal patterns of activity throughout many brain areas, and robust local GABAergic innervation of excitatory neurons is central for this role. Recent work in mammals has suggested that inhibitory connectivity is shaped by sensory experience, and identified activity-dependent transcriptional pathways important for this process (Morales et al., 2002; Chattopadhyaya et al., 2004; Jiao et al., 2006; Bloodgood et al., 2013; Pieraut et al., 2014). Despite these important advances, many aspects of inhibitory network development remain inadequately understood.
Caenorhabditis elegans provides a useful model in which to study neural circuit development and GABAergic function in vivo. As in mammals, inhibitory GABAergic signaling shapes C. elegans neural circuit activity and behavior. The identity, location and connectivity of all GABAergic neurons have been defined experimentally, and a wealth of genetic tools is available for manipulating GABAergic activity (Schuske et al., 2004). Informative genetic studies have previously identified genes involved in GABAergic synapse formation in C. elegans; however, the extent to which these genes identify mechanisms specific to GABAergic synapses versus more generalized roles remains unclear (Zhen and Jin, 1999; Schaefer et al., 2000; Zhen et al., 2000; Hallam et al., 2002; Liao et al., 2004; Ackley et al., 2005; Dai et al., 2006; Grill et al., 2007; Najarro et al., 2012). Here, we investigate the role of neuronal activity in the establishment and maintenance of inhibitory synaptic connections using the C. elegans motor circuit as a model. Prior work has provided evidence that activity-dependent processes regulate the timing of GABAergic developmental remodeling in C. elegans (Thompson-Peer et al., 2012; Han et al., 2015). Our studies here provide some of the first evidence that activity-dependent processes help to shape the mature pattern of synaptic connectivity in post-embryonic-born GABAergic neurons.
RESULTS
UNC-3 transcriptional regulation coordinates cholinergic synaptic connectivity with body wall muscles
The adult pattern of synaptic connectivity in the motor circuit has been well defined (White, 1976). Body wall muscles receive synaptic inputs from both excitatory (acetylcholine, ACh) and inhibitory (GABA) motor neurons and these inputs shape muscle activity and sinusoidal movement (Fig. 1A). Cholinergic motor neurons also provide excitatory synaptic drive onto inhibitory GABA motor neurons. GABA motor neurons then make inhibitory synaptic connections onto opposing musculature. The ventral D (VD) class GABAergic motor neurons that are the focus of our studies are integrated into the motor circuit post-embryonically following the first larval stage, and mediate inhibitory transmission onto ventral muscles in adults (Sulston, 1976; Han et al., 2015).
Fig. 1.

Cholinergic neuromuscular transmission is impaired in unc-3(e151) mutants. (A) The C. elegans motor circuit. Excitatory cholinergic motor neurons (gray) synapse onto inhibitory GABAergic motor neurons (blue) and onto body wall muscles (brown). (B) Representative recordings of endogenous EPSCs from body wall muscles of wild-type and unc-3(e151) mutant animals. Red traces are expanded views of time segments under the red bars in the upper traces. (C) The average frequency of endogenous EPSCs recorded from wild type and unc-3(e151) mutants. Each bar represents the mean±s.e.m., and numbers in bars indicate the n for each genotype in this figure and for all subsequent figures. ***P<0.0001; Student's t-test. (D) Confocal images of one wild-type (top) and two unc-3(e151) (middle and bottom) animals expressing GFP-tagged AChRs in body wall muscles (myo3p::ACR-16::GFP). Scale bar: 5 µm. (E) Normalized receptor cluster number per 75 µm of ventral nerve cord. *P<0.05; Student's t-test.
To address whether excitatory motor neurons are important for shaping functional connectivity in this circuit, we first examined unc-3 mutants. unc-3 encodes the sole C. elegans homolog of the COE family of transcription factors; in unc-3 mutants, most ventral cord motor neurons completely lack expression of the genes required for cholinergic function (e.g. cha-1 and unc-17) and exhibit additional defects in their organization (e.g. variable and/or disorganized process extension) (Prasad et al., 1998, 2008; Kratsios et al., 2012). Thus, cholinergic transmission onto body wall muscles should be severely disrupted in these animals. To confirm this, we made whole-cell patch clamp electrophysiology recordings of synaptic events from ventral body wall muscles of adult unc-3 mutants (1 mM Ca2+) and found significant defects in excitatory transmission (Fig. 1B,C). Specifically, the frequency of endogenous excitatory postsynaptic currents (EPSCs) is significantly reduced compared with wild type (79±6% reduction, P<0.001). The remaining cholinergic transmission might reflect activity of cholinergic VC motor neurons, which do not require unc-3 for expression of cholinergic markers (Kratsios et al., 2012). Prior work identified a requirement for unc-3 in the clustering of postsynaptic ionotropic (nicotinic) acetylcholine receptors (iAChRs) in muscles (Kratsios et al., 2015). Confirming these observations, we find that the distribution of muscle iAChRs (visualized with ACR-16::GFP) is disrupted by mutation of unc-3 (Fig. 1D,E). Although the total number of ACR-16::GFP clusters is only modestly reduced compared with wild type, large accumulations of receptor are often present in the shafts of muscle arms in unc-3 mutants, suggesting a re-distribution to non-synaptic locations. Thus, impaired terminal differentiation of cholinergic motor neurons leads to predicted defects in iAChR clustering and excitatory transmission at the cholinergic neuromuscular junction (NMJ).
Mutation of unc-3 impairs postsynaptic iAChR clustering in GABA neurons and reduces inhibitory transmission
Given that cholinergic motor neurons also make extensive synaptic contacts with inhibitory motor neurons, we next sought to determine whether the disruption of cholinergic innervation associated with mutation of unc-3 affected inhibitory activity. Using conditions that isolated inhibitory transmission, we measured endogenous inhibitory postsynaptic currents (IPSCs) from unc-3 mutants. The rate of endogenous IPSCs (5 mM Ca2+) is significantly lower in unc-3 mutants compared with wild type (63% reduction, P<0.0001) (Fig. 2A,B). Interestingly, mutation of unc-3 also produces a shift towards larger amplitude endogenous IPSCs (Fig. 2C), suggesting either altered muscle responsiveness to GABA release or increased GABA content in synaptic vesicles. Our analysis supports the notion that cholinergic neurons provide direct excitatory drive onto GABAergic motor neurons and perhaps also regulate functional attributes of GABA synaptic outputs onto muscles.
Fig. 2.

unc-3 mutation causes defects in GABAergic transmission and iAChR clustering in GABAergic neurons. (A) Representative recordings of endogenous IPSCs from body wall muscles of wild-type animals and unc-3(e151) mutants. Red traces are expanded views of time segments under the red bars in the upper traces. (B) The average frequency of endogenous IPSCs recorded from wild type and unc-3(e151) mutants. ***P<0.0001, Student's t-test. (C) Cumulative distribution of amplitudes of endogenous IPSCs recorded from wild type and unc-3(e151) mutants. ***P<0.001; Kolmogorov–Smirnov test. (D) Confocal images showing the dorsal nerve cord of eight animals expressing ACR-12::GFP in GABA neurons for the genotypes indicated. Scale bars: 5 µm. (E) Quantification of the average number of receptor clusters per 85 µm of dorsal nerve cord for the genotypes indicated, normalized to wild type. *P<0.05, ****P<0.0001; ANOVA with Dunnett's multiple comparisons test.
Given these findings, we next investigated whether unc-3 is required for establishing proper synaptic connectivity between cholinergic and GABAergic motor neurons. The nicotinic AChR subunit ACR-12 is a constituent of GABA neuron iAChRs. ACR-12::GFP clusters localize opposite ACh release sites and relocate appropriately during developmental remodeling, suggesting that these clusters report mature postsynaptic structures (Petrash et al., 2013; He et al., 2015). We examined the distribution of ACR-12::GFP in the dorsal nerve cord where the majority of chemical synaptic inputs to VD neurons occur (VD synaptic contacts with muscles are exclusively ventral) (White et al., 1986). We found that receptor clustering is severely disrupted in two independent unc-3 mutant strains (n3435 and e151) (Fig. 2D,E). ACR-12 clusters are unevenly distributed and significantly reduced in number compared with wild type [e.g. 51% decrease in unc-3(e151), P<0.001]. These effects are rescued with expression of the wild-type unc-3 cDNA under control of a 1 kb unc-3 promoter, but not by either GABA or muscle-specific expression (Fig. 2D,E). This unc-3 promoter region drives expression in cholinergic, but not GABAergic, motor neurons (Fig. S1A). Together, these observations suggest that unc-3 expression in cholinergic neurons is required for proper ACR-12 clustering in postsynaptic GABAergic neurons.
madd-4, the C. elegans homolog of mammalian punctin-1 and punctin-2 (also known as Adamtsl1 and Adamtsl3, respectively), encodes a synapse-associated extracellular scaffolding protein (Pinan-Lucarre et al., 2014). Secretion of MADD-4 from cholinergic motor neurons is required for proper iAChR localization at neuromuscular synapses (Pinan-Lucarre et al., 2014). Prior work showed that UNC-3 transcriptional regulation of madd-4 is essential for this process (Kratsios et al., 2015). To test whether MADD-4 is similarly required at synaptic connections between cholinergic and GABAergic motor neurons, we examined ACR-12 clustering in GABAergic neurons of madd-4 mutants. The madd-4(ok2854) allele is a 962 bp deletion that eliminates the two largest madd-4 exons common to all madd-4 isoforms, has been previously reported to be a null allele (Seetharaman et al., 2011; Maro et al., 2015), and produces significant defects in muscle AChR clustering similar to those previously observed after deletion of the entire madd-4 locus (Pinan-Lucarre et al., 2014) (Fig. S1B,C). Mutation of madd-4 produces a slight decrease in the number of ACR-12 clusters in the dorsal nerve cord compared with wild type (18% decrease, P<0.05) (Fig. 2D,E). Notably, this effect is much less severe than that observed for mutation of unc-3, indicating that the postsynaptic defects in GABAergic neurons of unc-3 mutants cannot be accounted for by decreased madd-4 expression. Taken together, our results indicate that UNC-3 transcriptional control of cholinergic neuron development is essential for proper iAChR clustering in GABAergic motor neurons, but MADD-4 has a comparatively minor role.
Morphological development of body wall muscles and GABA motor neurons is not dramatically affected in unc-3 mutants
To address whether the connectivity changes we observed might reflect alterations in the gross development or assembly of the motor circuit, we examined the cellular morphology of body wall muscles and GABA motor neurons. In wild-type animals, body wall muscles project membrane extensions, called muscle arms, to the nerve cords where muscle arm termini bifurcate and project lengthwise along the nerve cord, forming en passant synapses with motor neurons (Dixon and Roy, 2005). We visualized muscles by specific expression of membrane-bound mCD8::GFP (him-4p::mCD8::GFP) (Dixon and Roy, 2005; Collins and Koelle, 2013) (Fig. 3A). Variable minor morphological defects (e.g. overgrowth, membrane blebbing) are apparent in the body wall muscles of unc-3 mutants (Fig. 3B). However, consistent with a prior study (Kratsios et al., 2015), we found that overall muscle morphology is preserved – muscle arms extend normally to the nerve cord and the extent of muscle membrane contact with the nerve cord region is not appreciably altered (Fig. 3D).
Fig. 3.

Gross morphological development of GABA motor neurons and muscles does not require cholinergic innervation. (A-C′) Confocal images of the ventral nerve cord and body wall muscles posterior to the vulva of an adult wild-type (A,A′), unc-3(e151) mutant (B,B′) and acr-2(L/S) transgenic (C,C′) animals expressing a membrane-bound GFP in the distal row of body wall muscles (him4p::mCD8::GFP) (A-C) or mCherry in the GABA nervous system (unc-47p::mCherry) (A′-C′). Arrows indicate muscle arm shafts and arrowheads indicate muscle arm termini. Asterisks indicate GABA neuron commissures. Scale bars: 10 µm (A-C); 20 µm (A′-C′). (D) Quantification of the percentage of the ventral nerve cord region covered by muscle membrane. ns, not significant; ANOVA with Dunnett's multiple comparisons test.
We visualized the morphology of GABA motor neurons using cell-specific expression of mCherry (unc-47p::mCherry) (Fig. 3A′). Similar to the case for muscle cells, disruption of cholinergic innervation does not produce significant defects in GABA motor neuron number, positioning or gross morphology (Fig. 3B′; Fig. S2A-C). Although in some cases we noted regions of nerve cord defasciculation in unc-3 mutants (and sometimes one fewer commissure was visible), we did not observe disruptions in the continuity of the dorsal nerve cord processes. Thus, our findings provide evidence that gross morphological development of body wall muscles and GABA motor neurons proceeds to completion when cholinergic development is disrupted, arguing that the iAChR clustering and GABA transmission defects we observed in unc-3 mutants are unlikely to arise secondary to impaired morphological development of these cell types.
Cholinergic innervation refines GABA synaptic outputs
Our results described above support the idea that UNC-3 regulation of cholinergic neuron differentiation is important for the establishment of synaptic connectivity with GABAergic neurons. We next investigated whether cholinergic neurons might similarly contribute to the development of GABA synaptic outputs onto muscles. We examined GABA synapses in a transgenic strain expressing the synaptic vesicle marker mCherry::RAB-3 in GABA motor neurons together with the GFP-tagged GABAA-like receptor UNC-49 in muscles (Bamber et al., 1999; Klassen and Shen, 2007) (Fig. 4A). In wild-type animals, GABA synapses are evenly spaced along the ventral nerve cord, with presynaptic vesicles and postsynaptic GABA receptor clusters in close apposition (Fig. 4B; Fig. S3A,D). Mutation of unc-3 produces striking changes in GABA synapses (Fig. 4C,D,G,H), a phenotype consistent across independent unc-3 mutant strains (n3435 and e151). Specifically, the number of pre- and postsynaptic clusters is significantly decreased, and the average size of postsynaptic UNC-49::GFP puncta is increased [e.g. by 95% in unc-3(e151)]. Furthermore, we noted significant regions of the ventral nerve cord lacking GABA synaptic vesicle clusters in unc-3 mutants [e.g. 56±4% in unc-3(e151), compared with 20±2% in the wild type, P<0.01]. A prior study reported diffusely distributed and enlarged clusters of the active zone marker SYD-2/α-liprin in GABA neurons of unc-3 mutants (Yeh et al., 2005), consistent with our findings.
Fig. 4.

Impaired cholinergic innervation alters the distribution and size of GABA synapses. (A) Diagram depicting the location of fluorescent reporters used to label GABA pre- (unc-47p::mCherry::RAB-3) and postsynaptic (UNC-49::GFP) structures. (B-F) Confocal images of the ventral nerve cord in adult wild type (B), unc-3(n3435) mutants (C), unc-3(e151) mutants (D), unc-3(e151) mutants expressing an unc-3 cDNA rescuing array (unc-3p::unc-3 cDNA) (E), and transgenic acr-2(L/S) animals (F) co-expressing unc-47p::mCherry::RAB-3 in GABA motor neurons with UNC-49::GFP in body wall muscles. Note areas devoid of synaptic clusters (brackets) and enlarged clusters in unc-3 mutants and acr-2(L/S) transgenic animals. Motor neuron cell bodies are outlined in this figure and all subsequent figures. Scale bars: 20 µm. (G) Average number of pre- and postsynaptic clusters per 50 µm, normalized to wild type. (H) Average size of pre- and postsynaptic clusters at GABA synapses. *P<0.05, **P<0.01, ****P<0.0001 compared with either control or unc-3 mutant as indicated; ANOVA with Dunnett's multiple comparisons test. (I,J) Confocal images of the ventral cord in wild type or unc-3(e151) mutants expressing the GFP-tagged synaptic vesicle marker synaptobrevin (SNB-1::GFP) (I), or the Rim1 homolog (UNC-10::mCherry) (J). Scale bars: 20 µm. (K) Quantification of synaptic cluster number per 50 μm for each marker, normalized to wild type. (L) Quantification of presynaptic cluster area for genotypes indicated. **P<0.01, ***P<0.0001; Student's t-test.
The localization of other synaptic markers, including the synaptic vesicle marker SNB-1/synaptobrevin (Nonet, 1999) and the active zone marker UNC-10/RIM, are similarly altered by mutation of unc-3 (Fig. 4I-L). The size and distribution of SNB-1 clusters are both affected similarly to RAB-3. The number of UNC-10 clusters is decreased significantly but we do not observe a change in their average area, suggesting that synaptic vesicle clustering is more dramatically affected than the size of the active zone itself. Despite the altered distribution of synaptic markers, we did not observe obvious changes in the apposition of presynaptic mCherry::RAB-3 and postsynaptic UNC-49::GFP fluorescent signals (Fig. S3D) [wild type: 88±3%; unc-3(e151): 82±4% in apposition], suggesting that trans-synaptic signaling important for establishing proper registration between pre- and postsynaptic specializations is maintained.
Most aspects of these GABA synapse defects are reversed with expression of the wild-type unc-3 cDNA in cholinergic neurons (Fig. 4E,G,H). In particular, the decreased number of GABAergic presynaptic RAB-3 clusters and the increases in synapse size are each rescued with unc-3p::unc-3 expression, but not by GABA-specific expression. Although decreases in the number of GABA receptor clusters are not significantly rescued by unc-3p::unc-3 expression, GABA- or muscle-specific expression enhances the severity of this phenotype, arguing against a requirement for unc-3 expression in these cells. Finally, we noted that muscle-specific expression of unc-3 appears to partially reverse the increases in synapse size; however, decreases in synapse number are not rescued. Together, our transgenic rescue experiments provide additional support for the idea that unc-3 expression in cholinergic neurons is required for establishing proper GABAergic connectivity.
Surprisingly, unc-3p::unc-3 cDNA expression is not sufficient to restore movement to unc-3 mutants. In contrast, expression of an unc-3 minigene containing the first three unc-3 introns under control of the same promoter improves movement and restores synaptic connectivity (Fig. S4), suggesting that additional aspects of unc-3 regulation are important for motor function and that unc-3 intronic regions are crucial in this process.
To further test the idea that cholinergic innervation influences the number and positioning of GABAergic synapses, we ablated ACh motor neurons by expressing a putative gain-of-function variant of the ACh receptor subunit acr-2, acr-2(L/S), that produces cell-autonomous death of cholinergic motor neurons during late embryogenesis (Barbagallo et al., 2010). Similar to unc-3, we did not observe gross morphological changes in GABA neurons or reduced muscle membrane contact with the nerve cord (Barbagallo et al., 2010) (Fig. 3C,C′,D). Nonetheless, genetic ablation of ACh motor neurons produces striking defects in inhibitory synapses, similar to those observed for unc-3 mutants, including a significant decrease in the number of both presynaptic mCherry::RAB-3 and postsynaptic UNC-49::GFP clusters, as well as significant increases in their size (Fig. 4F-H; Fig. S3C). Thus, impaired differentiation or genetic ablation of ACh motor neurons does not affect the gross morphology of GABA neurons but instead alters the density and size of GABA synaptic outputs. Our analysis of inhibitory synapses in unc-3 mutants and acr-2(L/S) transgenic animals supports the hypothesis that presynaptic cholinergic neurons play important roles in both clustering iAChRs in GABA neurons and in shaping GABA synaptic outputs.
Vesicular release from cholinergic neurons shapes GABA synapses
We next investigated whether vesicular release from cholinergic neurons is required to properly establish GABA neuron connectivity. We disrupted vesicular release by specific expression of tetanus toxin light chain (TetTx) (Fig. 5; Fig. S5). TetTx expression in cholinergic motor neurons does not affect morphology or outgrowth, but produces changes in sensitivity to the cholinesterase inhibitor aldicarb consistent with decreased transmission (Fig. S5). Cholinergic expression of TetTx does not alter ACR-12 clustering in GABA neurons (Fig. 5A,B). We did, however, observe significant abnormalities in GABA synaptic outputs (Fig. 5C-F). These include enlargement of both presynaptic mCherry::RAB-3 and postsynaptic receptor clusters, as well as gaps in fluorescence that are similar to those observed for unc-3 mutants and acr-2(L/S) transgenic animals. Thus, synaptic structures between cholinergic and GABAergic neurons appear to be properly established when vesicular release from cholinergic motor neurons is impaired. Despite this, the distribution of GABAergic synaptic outputs is disrupted, implicating vesicular signals from ACh motor neurons in contributing to this process.
Fig. 5.
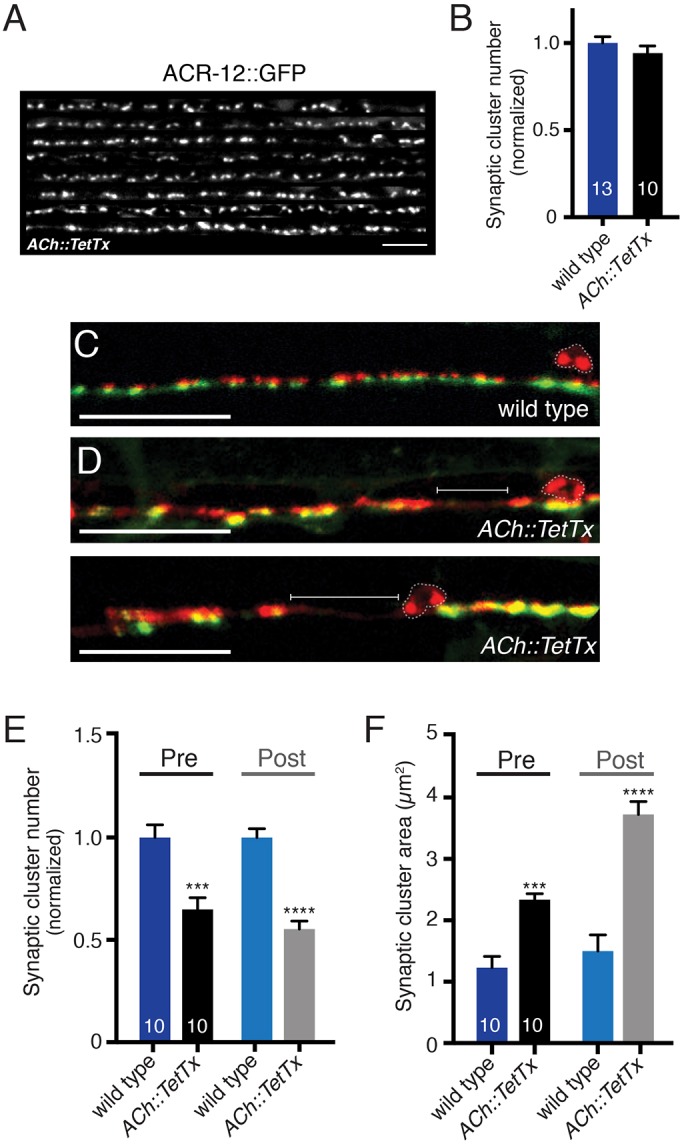
Vesicular release from cholinergic motor neurons shapes GABA synaptic outputs. (A) Confocal images showing the dorsal nerve cord of eight animals co-expressing ACR-12::GFP in GABA neurons and tetanus toxin in cholinergic neurons. Scale bar: 5 µm. (B) Quantification of the average number of receptor clusters per 85 µm of dorsal nerve cord for the genotypes indicated, normalized to wild type. (C,D) Merged confocal images of unc-47p::mCherry::RAB-3 and UNC-49::GFP labeling in the ventral nerve cord of adult wild-type (C) and transgenic animals with specific TetTx expression in cholinergic neurons (D). Brackets indicate areas devoid of synaptic clusters. Scale bars: 20 µm. (E) Average number of pre- and postsynaptic clusters per 50 µm for each genotype, normalized to wild type. (F) Average area of pre- and postsynaptic clusters at GABA synapses for each genotype as indicated. ***P<0.001, ****P<0.0001; Student's t-test.
To investigate the possibility that ACh itself is an important signal, we examined GABA synapses in mutant strains with specific defects in cholinergic transmission. unc-17 mutants are defective in a vesicular transporter (vAChT) that loads ACh into synaptic vesicles (Brenner, 1974; Rand and Russell, 1984; Rand, 1989). The unc-17(e113) mutation disrupts the UNC-3 binding site in the unc-17 promoter, disrupting UNC-17 and CHA-1 expression in cholinergic motor neurons (J. Rand, personal communication), and these mutants are presumably null for ACh release from cholinergic motor neurons.
ACR-12::GFP clustering in the dorsal nerve cord is not significantly altered in unc-17(e113) mutants or in madd-4;unc-17 double mutants (Fig. 6A,B), consistent with the results described above for TetTx expression. In contrast, GABAergic pre- and postsynaptic clusters are significantly decreased in number in unc-17(e113) mutants (by 65% and 64% respectively, P<0.001) (Fig. 6D,G). A second strongly hypomorphic unc-17 allele (e245) produced similar effects. The e245 mutation replaces a glycine with an arginine (G347R) in the ninth transmembrane domain of the vAChT protein (Alfonso et al., 1993) and decreases UNC-17 protein levels (Sandoval et al., 2006). ACR-12::GFP clustering is unchanged in unc-17(e245) compared with wild type (Fig. 6B), but significant defects in GABA synaptic outputs are apparent. These include gaps in the distribution of synapses as well as enlarged pre- and postsynaptic clusters (Fig. 6E,G,H). The increased size of GABAergic synapses in unc-17(e245) mutants, but not unc-17(e113) mutants, might indicate that partial loss of ACh release triggers changes in GABA synapse size. Alternatively, ACh release from cholinergic neurons in which unc-17 transcriptional regulation occurs independently of unc-3 (e.g. VC neurons) might influence GABA synapse size. Together, our analysis of unc-17 mutants provides evidence for the involvement of cholinergic transmission in directing the development of GABA synaptic outputs. Nonetheless, these effects are less pronounced than those observed after either TetTx expression or mutation of unc-3, suggesting participation of additional ACh-independent signals.
Fig. 6.

Reduced cholinergic transmission alters GABA synapse density and localization. (A) Confocal images showing the dorsal nerve cord of eight unc-17(e113) and madd-4(ok2854);unc-17(e113) animals expressing ACR-12::GFP in GABA neurons. Scale bars: 5 µm. (B) Quantification of the average number of receptor clusters per 85 µm of dorsal nerve cord for the genotypes indicated, normalized to wild type. (C-F) Merged confocal images of unc-47p::mCherry::RAB-3 and UNC-49::GFP labeling in the ventral nerve cord region of adult wild-type animals (C), unc-17(e113)/vAChT mutants (D), unc-17(e245)/vAChT mutants (E) or unc-29(x29);acr-16(ok789) double mutants, which lack functional muscle iAChRs (F). Scale bars: 20 µm. (G) Average number of GABA pre- and postsynaptic clusters per 50 µm for each genotype, normalized to wild type. (H) Average size of GABA pre- and postsynaptic clusters. *P<0.05, **P<0.01, ****P<0.0001; ANOVA with Dunnett's multiple comparisons test.
To address whether the effects of decreased ACh release reflect a requirement for ACh signaling onto muscles, we examined GABA synapses in animals lacking functional iAChRs at the NMJ (unc-29;acr-16 double mutants) (Francis et al., 2005). Eliminating synaptic activation of muscles causes a slight reduction in the number of postsynaptic GABA receptor clusters (Fig. 6F-H) but does not alter the density or positioning of presynaptic structures, suggesting that cholinergic activation of muscles is not essential for shaping GABAergic neuromuscular connectivity.
Acute reductions in cholinergic activity in adults lead to decreased GABA synapse size
Our previous genetic analysis indicated that cholinergic transmission helps to shape the distribution of GABA synaptic outputs, but did not distinguish whether these effects occur in the mature or developing nervous system. To address this question, we used a temperature-sensitive allele (y226) of cha-1/choline acetyltransferase (ChAT) (Rand and Russell, 1984). When maintained at a permissive temperature (15°C), cha-1(y226) mutants develop similarly to wild type, generating the full complement of cholinergic motor neurons (Fig. S6A). When switched to a restrictive temperature (25°C), cha-1(y226) mutants become uncoordinated within minutes, consistent with rapid impairment of cholinergic transmission, and these behavioral effects are reversible upon shifting back to the permissive temperature (Zhang et al., 2008) (Fig. S6B).
We first investigated the effects of reduced cholinergic transmission in adult animals. We shifted cha-1(y226) mutants to 25°C for a 4 h period in early adulthood, and visualized GABA synapses immediately following this shift (Fig. 7A). Interestingly, this transient reduction in cholinergic transmission does not produce appreciable changes in the distribution or number of GABA synapses. Instead, we noted a significant decrease in the size of both pre- and postsynaptic clusters (Fig. 7B-G), whereas GABA synapses in wild-type animals remain unaffected. This temperature-shift paradigm also produces a significant decrease in movement velocity in cha-1(y226) mutants, and these effects are reversible within a 4 h recovery period at 15°C (Fig. S6B). GABA synaptic clusters in cha-1 mutants are also restored to their normal size following a 4 h recovery period (Fig. 6D-G). Increasing the duration of shifts to the restrictive temperature (8 h) produces further decreases in the size of GABA pre- and postsynaptic clusters. Our findings indicate that acute changes in cholinergic activity during adulthood are sufficient to alter the size of GABA synapses, without producing obvious effects on GABA synapse positioning or density. The reversible nature of the synaptic alterations in adults provides evidence for active mechanisms that regulate the size of GABA synapses in response to changing levels of cholinergic transmission.
Fig. 7.

Acute reduction of cholinergic transmission during adulthood produces reversible decreases in GABA synapse size. (A) Diagram indicating predicted timeline of wild-type C. elegans development at 15°C and temperature shifts employed. Approximate time of hatch is indicated. Temperature shifts to 25°C (red) and time of imaging (green) are indicated. E, embryo. (B,C) Merged confocal images of unc-47p::mCherry::RAB-3 and UNC-49::GFP labeling in the ventral nerve cord region of adult wild-type animals (B) or cha-1(y226) mutants (C) following a shift to 25°C for the durations indicated. Scale bar: 20 µm. (D,E) Average number of GABA presynaptic (D) and postsynaptic (E) clusters per 50 µm for wild type (black) and cha-1(y226) mutants (gray). (F,G) Average size of GABA presynaptic (F) and postsynaptic (G) clusters for wild type (black) and cha-1(y226) mutants (gray). *P<0.05, **P<0.01, ***P<0.0001; ANOVA with Dunnett's multiple comparisons test.
Acetylcholine release is required during a highly synaptogenic period for proper GABA synapse localization
We next investigated whether cholinergic activity during development might help to shape the density or distribution of GABA synapses. To address this question, we again used cha-1(y226) mutants, shifting animals to 25°C for 8 h periods during the first larval (L1) stage, then imaging GABA synapses later in adulthood (Fig. 8A). We noted a time window beginning approximately 20 h after hatch (late L1 stage in wild type at 15°C) during which an 8 h shift to the restrictive temperature produces significant defects by adulthood. Specifically, we found the number of pre- and postsynaptic clusters is decreased by 40% and 27%, respectively, in cha-1 mutants subjected to temperature shift (Fig. 8B-D). We also noted a significant increase in the size of presynaptic clusters (Fig. 8E), although the size of postsynaptic clusters does not appear to be altered (Fig. 8F). These effects do not occur in control animals exposed to the same shift in temperature, or in cha-1 mutants grown continuously at 15°C. This temperature shift paradigm also produces a modest decrease in movement velocity in cha-1(y226) adult animals, suggesting that transient reduction of cholinergic transmission during development might be sufficient to produce behavioral defects that extend into adulthood (Fig. S6C). The timing of the 8 h temperature shift is predicted to overlap the L1/L2 transition for wild-type animals during which GABAergic VD neurons are initially integrated into the motor circuit, although the precise timing of these events can vary in mutant and transgenic strains. Thus, reduced cholinergic activity early in the development of GABAergic VD neurons affects their synaptic distribution, implicating cholinergic signaling in defining the mature pattern of inhibitory connectivity.
Fig. 8.

Reduced cholinergic neurotransmission during a period of GABAergic synaptogenesis alters GABAergic synaptic connectivity. (A) Diagram indicating predicted timeline of wild-type C. elegans development at 15°C and temperature shifts employed. Approximate time of hatch is indicated. (B,C) Confocal images of the ventral nerve cord in adult wild-type (B) and cha-1(y226) (C) temperature-sensitive mutant animals co-expressing GABA pre- and postsynaptic markers (unc-47p::mCherry::RAB-3 and UNC-49::GFP) grown at the permissive temperature and after an 8 h shift to the non-permissive temperature during the first larval stage. Note that the representative image for the cha-1 no shift control group shown in C is the same as that displayed in Fig. 7C. Scale bar: 20 µm. (D) Average number of GABA presynaptic and postsynaptic clusters per 50 µm for wild type (black) and cha-1(y226) mutants (gray). (E,F) Average size of GABA presynaptic (E) and postsynaptic (F) clusters for wild type (black) and cha-1(y226) mutants (gray). *P<0.05, **P<0.01, ***P<0.0001; Student's t-test.
DISCUSSION
Establishing the appropriate pattern of GABAergic innervation is crucial for proper inhibitory regulation of mature neural circuits. In this study, we explored the role of cholinergic motor neurons in sculpting the synaptic connectivity of GABAergic neurons in the C. elegans motor circuit. Our results show that integration of post-embryonic-born GABAergic VD neurons into the motor circuit is extensively regulated by cholinergic motor neurons, complementing prior studies that provided evidence for activity-dependent synaptic remodeling of embryonic-born GABAergic DD neurons (Thompson-Peer et al., 2012; Han et al., 2015). First, mutation of the COE transcription factor unc-3, which is required for terminal differentiation of cholinergic motor neurons, disrupts postsynaptic development (iAChR clustering) in GABAergic neurons. These effects are not strongly dependent on either cholinergic vesicular release or expression of the synaptic organizer madd-4, suggesting involvement of a novel molecular pathway. Second, mutation of unc-3 produces defects in GABAergic VD neuron synaptic connectivity with muscles. Third, genetic ablation of cholinergic motor neurons or specific TetTx expression in cholinergic neurons similarly alters the size and distribution of GABA synaptic outputs to muscles. Mutation of the cholinergic vesicular transporter unc-17 produces similar effects on GABAergic connectivity with muscles. Finally, the density of GABAergic synaptic outputs is decreased by acute reduction of cholinergic transmission during a developmental period that corresponds with the time of initial integration of GABAergic VD neurons into the circuit in wild type. Our findings suggest a model in which vesicle-borne signals from cholinergic motor neurons, both ACh and additional signals, are important for shaping GABAergic connectivity.
Cholinergic motor neurons innervate both GABAergic motor neurons and body wall muscles. Disruption of cholinergic innervation did not impair gross morphological development or outgrowth in either of these cell types, indicating that cell-intrinsic genetic programs are sufficient to guide normal morphological development. In contrast, additional activity-dependent processes appear to be required for establishing the mature pattern of GABAergic synaptic connectivity. These results could be indicative of a functional segregation between processes underlying activity-dependent regulation of synaptic connectivity and intrinsic genetic regulation of gross morphological features. Prior work showed that elimination of glutamatergic synaptic inputs has little effect on the morphological development of mouse hippocampal neurons (Lu et al., 2013), providing support for a similar segregation between these processes in mammals.
Mutation of unc-3 produced the most striking effects on synaptic connectivity in our experiments, both impairing postsynaptic ACR-12 iAChR clustering in VD neurons and decreasing their synaptic outputs. The requirement for UNC-3 in ACR-12 iAChR clustering is unlikely to indicate involvement of cholinergic activity-dependent regulation because neither TetTx expression nor mutation of unc-17 altered ACR-12 clustering. Similarly, mutation of the synaptic organizer madd-4 did not strongly affect ACR-12 iAChR clustering in VD neurons, either alone or in combination with mutation of unc-17, suggesting that a novel, as yet unidentified, transcriptional target of UNC-3 might be important for this process.
Our studies suggest that cholinergic activity might have a more prominent role in directing the distribution of GABAergic synaptic outputs. Our electrophysiological analysis of unc-3 mutants showed that GABAergic VD motor neurons are capable of sustaining inhibitory transmission. Notably, however, the frequency of GABAergic synaptic events is dramatically reduced compared with wild type, and their average amplitude increased significantly, mirroring the changes we observed in GABAergic outputs using our synaptic markers. We noted similar structural changes in GABAergic synaptic outputs when cholinergic neurons were genetically ablated [acr-2(L/S)], and when cholinergic vesicular release was disrupted (ACh::TetTx). For all of these, presynaptic alterations were paralleled by similar effects on the clustering of postsynaptic GABA receptors, suggesting that these effects reflect a change in synapse patterning/maturation rather than direct impairment of synapse formation per se.
Although synapse dynamics continue throughout the life of an animal, there is often increased plasticity earlier in development (Hensch and Fagiolini, 2005). The effects of cholinergic activity we observed might reflect an ongoing requirement for cholinergic transmission or a more specific requirement during a developmental period of increased plasticity. Our analysis of temperature-sensitive cha-1 mutants provides support for the latter possibility. A temporally defined reduction in cholinergic transmission that overlaps with the predicted timing of the highly synaptogenic L1/L2 transition in wild type produced irreversible defects in the patterning and size of GABAergic synaptic outputs. At the L1/L2 transition, 13 GABAergic VD neurons are added to the motor circuit, forming synapses onto ventral muscles. Our findings suggest that cholinergic activity during this developmental period influences the patterning of GABA synapses, and that deficits in transmission during this time period cannot be compensated for in later developmental stages. How might reduced cholinergic activity in early development alter GABAergic synaptic outputs in the adult? Synaptic inputs to the VDs primarily come from the dorsal A and B (DA/DB) classes of cholinergic motor neurons, which are present from hatch and are re-wired to innervate VD neurons after the L1 stage. One interesting possibility is that a reduction in cholinergic activity impairs re-wiring and DA/DB innervation of VD GABAergic neurons. According to this model, defects in GABA synapse patterning would then arise due to impaired DA/DB activation of GABAergic neurons. In contrast, acute reduction of cholinergic activity in the adult produces effects that are distinct from those during development, including changes in synapse size without decreases in synapse number, raising the possibility that homeostatic mechanisms are established after synapse patterning is completed. Similar scaling mechanisms have been reported at excitatory glutamatergic synapses in worms, flies and mammals (Grunwald et al., 2004; Turrigiano, 2008; Davis, 2013).
GABA release itself has been suggested to be key for proper inhibitory synapse maturation in mammals (Chattopadhyaya, 2011). Notably, disruption of GABA biosynthesis (unc-25/GAD mutants) does not impair GABA synapse formation in worms (Jin et al., 1999; Gally and Bessereau, 2003). Likewise, mutations that produce global reductions in neurotransmission (e.g. mutations in unc-13/Munc13 or unc-64/syntaxin) do not dramatically alter inhibitory synaptogenesis (Saifee et al., 1998; Richmond et al., 1999). For our studies, we used genetic tools that specifically affect cholinergic neurons. Under these conditions, inhibitory synapse formation appears to be properly initiated. Presynaptic specializations and postsynaptic receptor clusters are in close apposition; however, the size and density of these synapses are altered, implicating vesicular signals from cholinergic neurons in the refinement or patterning of GABAergic synaptic outputs. Although the mechanism underlying this process remains to be determined, one possibility is that vesicular signals from cholinergic neurons may regulate expression of synapse-organizing molecules in GABA neurons. Several recent studies have identified genetic pathways important for GABA synapse development and regulation in worms (Maro et al., 2015; Tong et al., 2015; Tu et al., 2015). It will be interesting to investigate whether cholinergic neurons influence expression levels of these and other potential synaptic organizers. Notably, activity-dependent transcriptional cascades shape the development and placement of GABA synapses in mammals (Lin et al., 2008; Bloodgood et al., 2013). Our findings lead us to propose that similar processes might be important for integrating post-embryonic-born GABAergic VD neurons into the motor circuit and patterning their synaptic outputs.
MATERIALS AND METHODS
Strains
C. elegans strains were grown at room temperature (22-24°C) on nematode growth media (NGM) plates seeded with the Escherichia coli strain OP50 unless otherwise noted. Wild type represents the N2 Bristol strain. Transgenic strains were obtained by microinjection to achieve germline transformation (Mello et al., 1991). Multiple independent lines were obtained for each transgenic strain, and data presented are from a single representative transgenic line. Transgenic strains were identified either by co-injection of lin-15(+) (pL15Ek) into lin-15(n765ts) mutants or by co-injection of either lgc-11p::GFP or lgc-11p::mCherry (restricted expression to the pharynx). As required, transgenes were integrated using x-ray bombardment and outcrossed to wild type. A complete list of strains is available in the Table S1.
Molecular biology
Plasmids were constructed using the two-slot Gateway Cloning system (Invitrogen) as described previously (Bhattacharya et al., 2014).
him-4p::mCD8::GFP
Entry vector: A him-4p promoter fragment (2156 bp) was amplified from pPRZ138.2 (a gift from Peter Roy, University of Toronto, ON, Canada) and cloned into pENTR-D-TOPO to create pENTR-3′-him4. Destination vector: mCD8::GFP (1566 bp) was amplified from pKMC72 and ligated into pDEST-16 to create pDEST-20. pENTR-3′-him4 and pDEST-16 were recombined to generate pBB102 (him-4p::mCD8::GFP).
Tetanus toxin construct
Entry Vector: An acr-2 promoter fragment (3386 bp) was amplified from pBB67 and subcloned into pENTR-D-TOPO to create pENTR-5′-acr2. Destination Vector: Tetanus toxin light chain (1374 bp) was amplified from TetTx-pWD157 to create pDEST-34. pENTR-5′-acr2 and pDEST-34 were recombined to generate pBB112 (acr-2p::TetTx).
unc-3 reporter and rescue constructs
Transcriptional reporter: An unc-3 promoter fragment (−1068 bp relative to start) was amplified from genomic DNA and cloned into pENTR-D-TOPO to generate pENTR-17. pENTR-17 was recombined with pDEST-94 to generate unc-3p::GFP (pAP192). cDNA rescue: Wild-type unc-3 cDNA was RT-PCR amplified from total N2 RNA and inserted into a destination vector to create pDEST-140. pDEST-140 was recombined with pENTR-17 (unc-3 promoter), pENTR-3′-myo3 (muscle-specific promoter) and pENTR-3′-unc47 (GABA-specific promoter) to generate pAP194 (unc-3p::unc-3 cDNA), pAP172 (myo-3p::unc-3 cDNA) and pAP176 (unc-47p::unc-3 cDNA), respectively. Minigene rescue: A 3154 bp fragment extending from the unc-3 start to exon 4 of the unc-3 genomic locus was amplified from N2 genomic DNA and ligated into pDEST-140 to create pDEST-142. pDEST-142 was recombined with pENTR-17 to create pAP196 (unc-3p::unc-3 minigene) containing the first three unc-3 introns [a 463 bp repetitive fragment within intron 1 (+2181 relative to start) was not included] and all of the unc-3 coding sequence.
Electrophysiology
Endogenous postsynaptic currents (PSCs) were recorded from body wall muscles as described previously (Petrash et al., 2013). The extracellular solution consisted of the following (in mM): 150 NaCl, 5 KCl, 4 MgCl2, 1 or 5 CaCl2 (as indicated), 15 HEPES and 10 glucose, pH 7.4, osmolarity adjusted with 20 sucrose. To facilitate analysis of unc-3 mutants that display very low rates of GABAergic activity, recordings of GABAergic currents were performed in 5 mM CaCl2. The intracellular fluid consisted of the following (in mM): 115 potassium gluconate, 25 KCl, 0.1 CaCl2, 50 HEPES, 5 Mg-ATP, 0.5 Na-GTP, 0.5 cGMP, 0.5 cAMP and 1 BAPTA, pH 7.4, osmolarity adjusted with 10 sucrose. At least 60-90 s of continuous data were used in the analysis. Data analysis was performed using Igor Pro (Wavemetrics) and Mini Analysis (Synaptosoft) software.
Aldicarb assay
Aldicarb assays were performed on adult animals (24 h post L4) at room temperature (22-24°C) with the researcher blind to genotype. Staged animals (ten per plate) were transferred to NGM plates containing 1 mM Aldicarb (ChemService) and assessed for movement every 15 min for 2 h.
Microscopy and image analysis
For all imaging, nematodes were immobilized with sodium azide (0.3 M) on a 5% agarose pad. Each n represents analysis of the nerve cord from an independent animal. Confocal microscopy of GABA NMJ synapses was performed using an LSM Pascal 5 confocal microscope (Zeiss). Images were analyzed using ImageJ software (open source). Images were obtained using a 63× oil lens and aligned using Photoshop (Adobe) or ImageJ.
Synapse density was calculated in a 50 µm section of the ventral nerve cord directly posterior to the vulva. A minimum fluorescence threshold was set to identify synaptic peaks (1500 a.u. for cholinergic synapses and 500 a.u. for GABAergic synapses). Peaks were identified as two or more consecutive data points above threshold and spaces were defined as two or more data points below threshold. Synapse area was calculated by separating particles using the ImageJ analyze particles function. Particles ≤0.2 µm2 were excluded as background.
To quantify orphan puncta at the GABAergic NMJ, the fluorescence intensities of both GFP and mCherry signals in an 85 µm region of the ventral nerve cord were analyzed. Intensity data were exported to Microsoft Excel and normalized to the corresponding highest value. Each peak of fluorescence was categorized as a colocalized puncta, presynaptic orphan puncta (mCherry signal without overlapping GFP signal) or postsynaptic orphan puncta (GFP signal without overlapping mCherry signal).
The extent of muscle membrane coverage of the ventral nerve cord region was calculated by measuring the length of the ventral midline of the posterior body wall muscles. The length of muscle membrane along the ventral midline (visualized by him-4p::mCD8::GFP) was quantified and used to calculate a percentage of total nerve cord length.
ACR-12::GFP (ufIs92), ACR-16::GFP (ufIs8) and UNC-29::GFP (ufIs2) images were acquired using a 3i (Intelligent Imaging Innovations) Everest spinning-disk confocal microscope and collected at 0.27 µm/steps using a 63× objective.
Confocal montages of ACR-12::GFP were assembled by imaging the dorsal nerve cord posterior to the vulva in adult hermaphrodites. Identical image and laser settings were used for each genotype. Straightened dorsal cords were extracted from these images using the ‘straighten to line’ function.
For quantifying ACR-12::GFP, ACR-16::GFP and UNC-29::GFP puncta along the nerve cord, background fluorescence was first subtracted by calculating the average intensity of each image in a region devoid of puncta. Puncta were quantified across 80 µm of the nerve cord region with ImageJ ‘analyze particles’, using an intensity threshold specific for each marker. Particles ≤0.04 µm2 were excluded as background.
unc-3 movement analysis
For locomotion assays, 1-day-old adults were allowed to acclimate for 1 min on NGM plates thinly seeded with a bacterial lawn. WormLab software (MBF Bioscience) was used to capture and analyze 30 s digital videos of individual worms.
cha-1 time course
Adult shifts and recovery
Wild-type and cha-1 animals were cultured at 15°C. Animals were synchronized at the L4 stage, and then placed at 15°C for an additional 24 h. Young adult animals were then shifted to 25°C for either 4 or 8 h and imaged immediately. A third group of animals were shifted to 25°C for 4 hours, then placed back at 15°C for 4 h (recovery) before imaging.
Developmental shift
Embryos were staged at the 21+ cell stage and allowed to develop at 15°C for 20 h. Animals were then shifted to 25°C for 8 h and returned to 15°C. Animals were synchronized at the L4 stage and allowed to grow for an additional 24 h at 15°C prior to imaging.
Behavior
Temperature shifts were performed as described above. For behavioral analysis, animals were transferred individually to plates without food for a period of 5 min. Movement was quantified using Wormlab software to track animals and measure their average forward velocity over a period of 5 min.
Acknowledgements
We would like to thank Claire Bénard and members of the Francis lab for critical reading of the manuscript, Will Joyce for technical assistance, Mei Zhen, Kevin Collins, Bruce Bamber and Peter Roy for sharing reagents. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR).
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.M.F.; Methodology: M.M.F., B.B., C.M.L.; Validation: M.M.F., A.P.; Formal analysis: M.M.F., B.B., A.P., D.T., D.O., N.B.; Investigation: M.M.F., B.B., A.P., D.T., D.O., N.B.; Resources: C.M.L.; Writing - original draft: M.M.F., B.B.; Writing - review & editing: M.M.F., A.P.; Supervision: M.M.F.
Funding
This work was funded by the National Institute of Neurological Disorders and Stroke (R01NS064263 to M.M.F. and F31NS076326 to B.B.) and the National Institute on Drug Abuse (F31DA038399 to A.P.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.141911.supplemental
References
- Ackley B. D., Harrington R. J., Hudson M. L., Williams L., Kenyon C. J., Chisholm A. D. and Jin Y. (2005). The two isoforms of the Caenorhabditis elegans leukocyte-common antigen related receptor tyrosine phosphatase PTP-3 function independently in axon guidance and synapse formation. J. Neurosci. 25, 7517-7528. 10.1523/JNEUROSCI.2010-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso A., Grundahl K., Duerr J. S., Han H. P. and Rand J. B. (1993). The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter. Science 261, 617-619. 10.1126/science.8342028 [DOI] [PubMed] [Google Scholar]
- Bamber B. A., Beg A. A., Twyman R. E. and Jorgensen E. M. (1999). The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J. Neurosci. 19, 5348-5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo B., Prescott H. A., Boyle P., Climer J. and Francis M. M. (2010). A dominant mutation in a neuronal acetylcholine receptor subunit leads to motor neuron degeneration in Caenorhabditis elegans. J. Neurosci. 30, 13932-13942. 10.1523/JNEUROSCI.1515-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R., Touroutine D., Barbagallo B., Climer J. Lambert C. M., Clark C. M., Alkema M. J. and Francis M. M. (2014). A conserved dopamine-cholecystokinin signaling pathway shapes context-dependent Caenorhabditis elegans behavior. PLoS Genet. 10, e1004584 https://doi.org/10.1371/journal.pgen.1004584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood B. L., Sharma N., Browne H. A., Trepman A. Z. and Greenberg M. E. (2013). The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503, 121-125. 10.1038/nature12743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs S. W. and Galanopoulou A. S. (2011). Altered GABA signaling in early life epilepsies. Neural Plast. 2011, 527605 10.1155/2011/527605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B. (2011). Molecular mechanisms underlying activity-dependent GABAergic synapse development and plasticity and its implications for neurodevelopmental disorders. Neural Plast. 2011, 734231 10.1155/2011/734231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B., Di Cristo G., Higashiyama H., Knott G. W., Kuhlman S. J., Welker E. and Huang Z. J. (2004). Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J. Neurosci. 24, 9598-9611. 10.1523/JNEUROSCI.1851-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K. M. and Koelle M. R. (2013). Postsynaptic ERG potassium channels limit muscle excitability to allow distinct egg-laying behavior states in Caenorhabditis elegans. J. Neurosci. 33, 761-775. 10.1523/JNEUROSCI.3896-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R., Dinocourt C., Hirsch J. C., Merchan-Perez A., De Felipe J., Ben-Ari Y., Esclapez M. and Bernard C. (2001). Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat. Neurosci. 4, 52-62. 10.1038/82900 [DOI] [PubMed] [Google Scholar]
- Dai Y., Taru H., Deken S. L., Grill B., Ackley B., Nonet M. L. and Jin Y. (2006). SYD-2 Liprin-alpha organizes presynaptic active zone formation through ELKS. Nat. Neurosci. 9, 1479-1487. 10.1038/nn1808 [DOI] [PubMed] [Google Scholar]
- Davis G. W. (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718-728. 10.1016/j.neuron.2013.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. J. and Roy P. J. (2005). Muscle arm development in Caenorhabditis elegans. Development 132, 3079-3092. 10.1242/dev.01883 [DOI] [PubMed] [Google Scholar]
- Francis M. M., Evans S. P., Jensen M., Madsen D. M., Mancuso J., Norman K. R. and Maricq A. V. (2005). The Ror receptor tyrosine kinase CAM-1 is required for ACR-16-mediated synaptic transmission at the C. elegans neuromuscular junction. Neuron 46, 581-594. 10.1016/j.neuron.2005.04.010 [DOI] [PubMed] [Google Scholar]
- Gally C. and Bessereau J. L. (2003). GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans. J. Neurosci. 23, 2591-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill B., Bienvenut W. V., Brown H. M., Ackley B. D., Quadroni M. and Jin Y. (2007). C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron 55, 587-601. 10.1016/j.neuron.2007.07.009 [DOI] [PubMed] [Google Scholar]
- Grunwald M. E., Mellem J. E., Strutz N., Maricq A. V. and Kaplan J. M. (2004). Clathrin-mediated endocytosis is required for compensatory regulation of GLR-1 glutamate receptors after activity blockade. Proc. Natl. Acad. Sci. USA 101, 3190-3195. 10.1073/pnas.0306156101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam S. J., Goncharov A., McEwen J., Baran R. and Jin Y. (2002). SYD-1, a presynaptic protein with PDZ, C2 and rhoGAP-like domains, specifies axon identity in C. elegans. Nat. Neurosci. 5, 1137-1146. 10.1038/nn959 [DOI] [PubMed] [Google Scholar]
- Han B., Bellemer A. and Koelle M. R. (2015). An evolutionarily conserved switch in response to GABA affects development and behavior of the locomotor circuit of Caenorhabditis elegans. Genetics 199, 1159-1172. 10.1534/genetics.114.173963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S., Philbrook A., McWhirter R., Gabel C. V., Taub D. G., Carter M. H., Hanna I. M., Francis M. M. and Miller D. M. (2015). Transcriptional control of synaptic remodeling through regulated expression of an immunoglobulin superfamily protein. Curr. Biol. 25, 2541-2548. 10.1016/j.cub.2015.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch T. K. and Fagiolini M. (2005). Excitatory-inhibitory balance and critical period plasticity in developing visual cortex. Prog. Brain Res. 147, 115-124. 10.1016/S0079-6123(04)47009-5 [DOI] [PubMed] [Google Scholar]
- Jiao Y., Zhang C., Yanagawa Y. and Sun Q. Q. (2006). Major effects of sensory experiences on the neocortical inhibitory circuits. J. Neurosci. 26, 8691-8701. 10.1523/JNEUROSCI.2478-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Jorgensen E., Hartwieg E. and Horvitz H. R. (1999). The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J. Neurosci. 19, 539-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen M. P. and Shen K. (2007). Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell 130, 704-716. 10.1016/j.cell.2007.06.046 [DOI] [PubMed] [Google Scholar]
- Kratsios P., Stolfi A., Levine M. and Hobert O. (2012). Coordinated regulation of cholinergic motor neuron traits through a conserved terminal selector gene. Nat. Neurosci. 15, 205-214. 10.1038/nn.2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratsios P., Pinan-Lucarré B., Kerk S. Y., Weinreb A., Bessereau J.-L. and Hobert O. (2015). Transcriptional coordination of synaptogenesis and neurotransmitter signaling. Curr. Biol. 25, 1282-1295. 10.1016/j.cub.2015.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao E. H., Hung W., Abrams B. and Zhen M. (2004). An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature 430, 345-350. 10.1038/nature02647 [DOI] [PubMed] [Google Scholar]
- Lin Y., Bloodgood B. L., Hauser J. L., Lapan A. D., Koon A. C., Kim T.-K., Hu L. S., Malik A. N. and Greenberg M. E. (2008). Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198-1204. 10.1038/nature07319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Bushong E. A., Shih T. P., Ellisman M. H. and Nicoll R. A. (2013). The cell-autonomous role of excitatory synaptic transmission in the regulation of neuronal structure and function. Neuron 78, 433-439. 10.1016/j.neuron.2013.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maro G. S., Gao S., Olechwier A. M., Hung W. L., Liu M., Özkan E., Zhen M. and Shen K. (2015). MADD-4/punctin and neurexin organize C. elegans GABAergic postsynapses through neuroligin. Neuron 86, 1420-1432. 10.1016/j.neuron.2015.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C. C., Kramer J. M., Stinchcomb D. and Ambros V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales B., Choi S. Y. and Kirkwood A. (2002). Dark rearing alters the development of GABAergic transmission in visual cortex. J. Neurosci. 22, 8084-8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najarro E. H., Wong L., Zhen M., Carpio E. P., Goncharov A., Garriga G., Lundquist E. A., Jin Y. and Ackley B. D. (2012). Caenorhabditis elegans flamingo cadherin fmi-1 regulates GABAergic neuronal development. J. Neurosci. 32, 4196-4211. 10.1523/JNEUROSCI.3094-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet M. L. (1999). Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J. Neurosci. Methods 89, 33-40. 10.1016/S0165-0270(99)00031-X [DOI] [PubMed] [Google Scholar]
- Petrash H. A., Philbrook A., Haburcak M., Barbagallo B. and Francis M. M. (2013). ACR-12 ionotropic acetylcholine receptor complexes regulate inhibitory motor neuron activity in Caenorhabditis elegans. J. Neurosci. 33, 5524-5532. 10.1523/JNEUROSCI.4384-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieraut S., Gounko N., Sando R., Dang W., Rebboah E., Panda S., Madisen L., Zeng H. and Maximov A. (2014). Experience-dependent remodeling of basket cell networks in the dentate gyrus. Neuron 84, 107-122. 10.1016/j.neuron.2014.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinan-Lucarre B., Tu H., Pierron M., Cruceyra P. I., Zhan H., Stigloher C., Richmond J. E. and Bessereau J.-L. (2014). C. elegans Punctin specifies cholinergic versus GABAergic identity of postsynaptic domains. Nature 511, 466-470. 10.1038/nature13313 [DOI] [PubMed] [Google Scholar]
- Powell E. M., Campbell D. B., Stanwood G. D., Davis C., Noebels J. L. and Levitt P. (2003). Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J. Neurosci. 23, 622-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B. C., Ye B., Zackhary R., Schrader K., Seydoux G. and Reed R. R. (1998). unc-3, a gene required for axonal guidance in Caenorhabditis elegans, encodes a member of the O/E family of transcription factors. Development 125, 1561-1568. [DOI] [PubMed] [Google Scholar]
- Prasad B., Karakuzu O., Reed R. R. and Cameron S. (2008). unc-3-dependent repression of specific motor neuron fates in Caenorhabditis elegans. Dev. Biol. 323, 207-215. 10.1016/j.ydbio.2008.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand J. B. (1989). Genetic analysis of the cha-1-unc-17 gene complex in Caenorhabditis. Genetics 122, 73-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand J. B. and Russell R. L. (1984). Choline acetyltransferase-deficient mutants of the nematode Caenorhabditis elegans. Genetics 106, 227-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond J. E., Davis W. S. and Jorgensen E. M. (1999). UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat. Neurosci. 2, 959-964. 10.1038/14755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifee O., Wei L. and Nonet M. L. (1998). The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol. Biol. Cell 9, 1235-1252. 10.1091/mbc.9.6.1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval G. M., Duerr J. S., Hodgkin J., Rand J. B. and Ruvkun G. (2006). A genetic interaction between the vesicular acetylcholine transporter VAChT/UNC-17 and synaptobrevin/SNB-1 in C. elegans. Nat. Neurosci. 9, 599-601. 10.1038/nn1685 [DOI] [PubMed] [Google Scholar]
- Schaefer A. M., Hadwiger G. D. and Nonet M. L. (2000). rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26, 345-356. 10.1016/S0896-6273(00)81168-X [DOI] [PubMed] [Google Scholar]
- Schuske K., Beg A. A. and Jorgensen E. M. (2004). The GABA nervous system in C. elegans. Trends Neurosci. 27, 407-414. 10.1016/j.tins.2004.05.005 [DOI] [PubMed] [Google Scholar]
- Seetharaman A., Selman G., Puckrin R., Barbier L., Wong E., D'Souza S. A. and Roy P. J. (2011). MADD-4 is a secreted cue required for midline-oriented guidance in Caenorhabditis elegans. Dev. Cell 21, 669-680. 10.1016/j.devcel.2011.07.020 [DOI] [PubMed] [Google Scholar]
- Sulston J. E. (1976). Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 275, 287-297. 10.1098/rstb.1976.0084 [DOI] [PubMed] [Google Scholar]
- Thompson-Peer K. L., Bai J., Hu Z. and Kaplan J. M. (2012). HBL-1 patterns synaptic remodeling in C. elegans. Neuron 73, 453-465. 10.1016/j.neuron.2011.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X. J., Hu Z., Liu Y., Anderson D. and Kaplan J. M. (2015). A network of autism linked genes stabilizes two pools of synaptic GABA(A) receptors. Elife 4, e09648 10.7554/eLife.09648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H., Pinan-Lucarré B., Ji T., Jospin M. and Bessereau J.-L. (2015). C. elegans punctin clusters GABA(A) receptors via neuroligin binding and UNC-40/DCC recruitment. Neuron 86, 1407-1419. 10.1016/j.neuron.2015.05.013 [DOI] [PubMed] [Google Scholar]
- Turrigiano G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422-435. 10.1016/j.cell.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. G., Southgate E., Thomson J. N. and Brenner S. (1976). The structure of the ventral nerve cord of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 275, 327-348. 10.1098/rstb.1976.0086 [DOI] [PubMed] [Google Scholar]
- White J. G., Southgate E., Thomson J. N. and Brenner S. (1986). The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1-340. 10.1098/rstb.1986.0056 [DOI] [PubMed] [Google Scholar]
- Yeh E., Kawano T., Weimer R. M., Bessereau J. L. and Zhen M. (2005). Identification of genes involved in synaptogenesis using a fluorescent active zone marker in Caenorhabditis elegans. J. Neurosci. 25, 3833-3841. 10.1523/JNEUROSCI.4978-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Chung S. H., Fang-Yen C., Craig C., Kerr R. A., Suzuki H., Samuel A. D., Mazur E. and Schafer W. R. (2008). A self-regulating feed-forward circuit controlling C. elegans egg-laying behavior. Curr. Biol. 18, 1445-1455. 10.1016/j.cub.2008.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M. and Jin Y. (1999). The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature 401, 371-375. 10.1038/43886 [DOI] [PubMed] [Google Scholar]
- Zhen M., Huang X., Bamber B. and Jin Y. (2000). Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26, 331-343. 10.1016/S0896-6273(00)81167-8 [DOI] [PubMed] [Google Scholar]