

Abstract
Genetic factors that influence the transition from initial drinking to dependence remain enigmatic. Recent studies have leveraged chronic intermittent ethanol (CIE) paradigms to measure changes in brain gene expression in a single strain at 0, 8, 72 h, and even 7 days following CIE. We extend these findings using LCM RNA-seq to profile expression in 11 brain regions in two inbred strains – C57BL/6J (B6) and DBA/2J (D2) – 72 h following multiple cycles of ethanol self-administration and CIE. Linear models identified differential expression based on treatment, region, strain, or interactions with treatment. Nearly 40% of genes showed a robust effect (FDR < 0.01) of region, and hippocampus CA1, cortex, bed nucleus stria terminalis, and nucleus accumbens core had the highest number of differentially expressed genes after treatment. Another 8% of differentially expressed genes demonstrated a robust effect of strain. As expected, based on similar studies in B6, treatment had a much smaller impact on expression; only 72 genes (p < 0.01) are modulated by treatment (independent of region or strain). Strikingly, many more genes (415) show a strain-specific and largely opposite response to treatment and are enriched in processes related to RNA metabolism, transcription factor activity, and mitochondrial function. Over 3 times as many changes in gene expression were detected in D2 compared to B6, and weighted gene co-expression network analysis (WGCNA) module comparison identified more modules enriched for treatment effects in D2. Substantial strain differences exist in the temporal pattern of transcriptional neuroadaptation to CIE, and these may drive individual differences in risk of addiction following excessive alcohol consumption.
Keywords: C57BL/6J, DBA/2J, CIE, alcohol, addiction, RNA-seq
Introduction
Chronic alcohol intake can lead to the development of tolerance and dependence, which are risk factors for sustained excessive alcohol consumption. There are likely many genetic, environmental, and neuroadaptive factors that influence the transition from initial drinking to dependence, possibly through the complex interplay between brain reward and stress mechanisms (Koob & Volkow, 2010). Recent studies have leveraged genome-wide gene expression profiles (microarray and RNA sequencing platforms) to identify altered pathways in post mortem tissue from human alcoholics (Farris & Mayfield, 2014; Lewohl et al., 2000; Liu et al., 2006; Mayfield, Harris, & Schuckit, 2008; Ponomarev, Wang, Zhang, Harris, & Mayfield, 2012) and provide insight into the biological processes underlying the transition from chronic alcohol exposure to excessive alcohol intake in rodent models (Melendez, McGinty, Kalivas, & Becker, 2012; Osterndorff-Kahanek et al., 2015; Smith et al., 2016). Factors influencing excessive drinking are not yet well characterized, but recent studies have revealed some common themes: 1) chronic alcohol consumption leads to long-lasting changes in brain gene expression, 2) some brain regions, such as prefrontal cortex and hippocampus, seem to be exquisitely sensitive to chronic alcohol exposure, 3) only a handful of genes overlap across studies, and 4) at least in animal models, gene expression changes follow a clear time course following a prolonged period of chronic intake. However, few studies have chronicled the impact of genetic background on the chronic alcohol engram. To address this gap we have compared transcriptional profiles following chronic alcohol administration between two highly divergent inbred mouse strains – C57BL/6J (B6) and DBA/2J (D2).
Over 5 million loci are polymorphic between B6 and D2, and these strains have been shown to differ in a number of alcohol-related traits, including alcohol consumption (Yoneyama, Crabbe, Ford, Murillo, & Finn, 2008), initial sensitivity (Crabbe, 1983), withdrawal, and tolerance (Metten & Crabbe, 2005). These strains and their recombinant progeny are often used to explore individual differences that might influence stages of alcohol addiction. In particular, the B6 strain, compared to the D2 strain and most other inbred strains of mice, will voluntarily consume large quantities of ethanol mixed only with water. B6 mice also demonstrate increased alcohol intake, compared to baseline, after exposure to four cycles of continuous ethanol vapor (16 h) and withdrawal (8 h) using a chronic intermittent ethanol paradigm (CIE) (Becker & Lopez, 2004). For this reason, the B6 strain has been used to explore neurobiological adaptations that influence the transition between chronic alcohol exposure and excessive alcohol consumption. The CIE paradigm is also severely stressful and leads to long-lasting alterations in hypothalamic-pituitary-adrenal (HPA) axis signaling and function, which impacts the behavioral response to subsequent alcohol and stress exposure and may drive relapse drinking in susceptible individuals (Becker, 2012; Heilig, Egli, Crabbe, & Becker, 2010; Maldonado-Devincci et al., 2016). Several recent studies explored the temporal pattern of gene expression changes after multiple cycles of CIE in B6, and found a number of transcriptional changes immediately following CIE (time 0) or during withdrawal (8 h later); however, very little change was detected at 120 h (Melendez et al., 2012; Osterndorff-Kahanek et al., 2015). A more recent study identified a small but significant number of long-lasting alterations up to a week following the last CIE cycle (Smith et al., 2016). Here we extend these findings by characterizing transcriptional differences across 11 brain subregions at an intermediate 72-h time point following five cycles of CIE in both B6 and D2. Our data suggest substantial strain differences in the temporal pattern of transcriptional neuroadaptation in response to treatment and support differential brain region sensitivity and the existence of a long-lasting engram for chronic alcohol exposure.
Materials and methods
Design
Gene expression in response to chronic alcohol treatment was measured in 13 matched brain subregions from two inbred strains of mice (C57BL/6J and DBA/2J). The balanced study design included two males of each strain assigned to either the air control or chronic intermittent ethanol treatment group. The total number of samples was 104 (13 samples for each individual per condition). Tissue was collected by laser capture microdissection (LCM) followed by RNA extraction and sequencing (RNA-seq). Following quality control measures to detect and remove poorly performing samples (see Quality Control section below), the final data set included 11 brain regions and 87 samples.
Animals
Male C57BL/6J and DBA/2J mice (10 weeks old upon arrival) were purchased from the Jackson Labs and assigned to either the air control or CIE group. Mice were individually housed with free access to food (Harland Teklad, Madison, WI) and water throughout all phases of the experiments. Body weights were recorded weekly during ethanol-drinking weeks or daily during chronic intermittent ethanol (CIE) or air exposure (detailed below). Mice were housed in a temperature- and humidity-controlled animal facility under a reversed 12-h light/dark cycle (lights on at 0200 h). Mice were not food- or water-deprived at any time during the study. All procedures were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina (MUSC) and followed the NIH Guide for the Care and Use of Laboratory Animals (8th edition, National Research Council, 2011). Brain tissue was removed at MUSC and shipped to UTHSC for laser capture microdissection (LCM).
Chronic ethanol exposure
Mice were allowed to self-administer alcohol (15% v/v vs. water) for 2 h a day (5 days a week) 6 weeks prior to treatment in order to establish baseline consumption. Access to 15% alcohol versus water started 30 min prior to the start of the dark cycle. Following establishment of baseline drinking, two male mice representative of each strain were separated into two groups to be exposed to either weekly cycles of CIE exposure (CIE group) or air control (AIR group) exposure. There was no difference in baseline consumption or preference within strain prior to assignment to AIR or CIE treatment groups (Supplemental Fig. 1). As expected, there was a significant difference in alcohol consumption and preference between strains at baseline (B6 > D2, p < 0.01, Supplemental Fig. 1).
Mice assigned to the CIE treatment group were exposed to alcohol vapor for 16 h a day followed by 8 h of withdrawal for 4 days. Following the fourth vapor exposure, mice were given a 72-h abstinence period before resuming ethanol intake in the home cage for 5 days. Mice in the AIR control treatment group were similarly treated but exposed only to air in the inhalation chambers. This pattern of CIE or air control exposure followed by 5 days of ethanol self-administration was repeated for four cycles. A fifth cycle of CIE (or air) exposure followed the fourth ethanol intake evaluation period, and brain tissue was collected 72 h after the last cycle ended.
Pyrazole (1 mmol/kg) was used to stabilize blood ethanol levels (BEC) and was administered to both CIE and AIR groups. Average BEC across cycles was 212.11 ± 6.68 mg/dL for B6 and 269.96 ± 13.15 mg/dL for D2. There was a significant difference between strains in ethanol preference during AIR and CIE exposure (B6 > D2, p < 0.01, Supplemental Fig. 1). Although the number of individual replicates here is small, the variation in consumption of alcohol between strains and treatment groups is reflective of the larger population, and all individuals were included for LCM followed by RNA-seq.
LCM
Whole brain tissue was sectioned at 10 μm using a Leica cryostat and mounted in series with 6–8 sections per slide onto uncharged and uncoated glass slides. Mounted sections were dehydrated in 100% methanol (90 sec), 70% ethanol (1 min), 95% ethanol (1 min), 100% ethanol (1 min × 2), xylene (5 min). Slides were then allowed to air dry for 10 min under a fume hood.
Series were created from distinct coronal sections (bregma positions based on a C57BL/6J reference brain atlas) and individual regions were matched across section and harvested by LCM (Supplemental Fig. 2). Prelimbic (PrL) and infralimbic (ILC) cortex included a series spanning from bregma 1.98 to 1.54 mm. The accumbens core (NAc) and shell (NAs) series were collected from bregma 1.54 to 0.98 mm, and dorsolateral (DLS) and dorsomedial (DMS) striatum and bed nucleus stria terminalis (BST) were collected from bregma 0.38 to −0.10 mm. Basolateral (BLA) and central nucleus (CeA) of the amygdala series were collected from bregma −0.58 to −1.22 mm and hippocampus (CA1 and CA3) was collected from bregma −1.46 to −2.46 mm. Finally, the ventral tegmental area (VTA) and primary visual cortex (VCX) series were collected from bregma −3.28 to −3.80 mm.
Arcturus XT (Life Technologies) was used to capture 13 brain areas. The infrared laser was then used to capture the tissue onto CapSure LCM caps (Life Technologies, laser spot power set to 70 mV with a duration of 25 msec).
RNA extraction
RNA from tissue trapped in the CapSure LCM caps was extracted using the PicoPure RNA isolation kit (Life Technologies) according to the manufacturer’s instructions (RNA was eluted from provided capture columns in 13.5-μL nuclease-free water). RNA quality was analyzed using a Bioanalyzer (Model 100, Agilent, Foster City, CA). Samples with an associated RNA integrity number (RIN) greater than 6 were subsequently used for RNA sequencing.
RNA sequencing
Poly-A enriched mRNA was sequenced on two platforms, ABI SOLID 550XL Wildfire (65 samples) and Ion Proton (39 samples). Read length was 50 nt for the SOLID system and the average read length for the Proton system was ~180 nt. Reads generated on the SOLID system were aligned to the mm10 reference genome using the LifeScope aligner, and BAM files were subsequently generated using custom scripts for third-party downstream analysis. For the Proton system, reads were also aligned to the mm10 (Ensemble GRCm38) reference genome using TopHat2. Settings for TopHat2 are as follows: “-p 15 -N 4 --read-gap-length 6 --read-edit-dist 8 --max-insertion-length 6 --max-deletion-length 6 --max-intron-length 300000 --b2-very-sensitive”. Alignments on both platforms are splice-aware. RSeQC-2.6.1 (RPKM_count.py) was used to generate count data based on mm10 GENECODE Basic transcript annotation (43,320 transcripts detected). We selected this annotation for greater reproducibility with existing microarray data sets and to simplify downstream analysis by limiting the number of transcript models for each gene. On average, 2.5 million and 7.8 million reads uniquely aligned to transcript models on the SOLID and Proton platforms, respectively. Data were further filtered to remove transcripts that had less than 1 count in 90% or more samples. After filtering, 24,597 transcripts representing 12,011 unique genes remained (Supplemental Table 1). The variance stabilizing transform (R package DESeq2, FitType = local) was applied to the count data, and transformed data were corrected by dividing by transcript length to generate log2 reads per kilobase gene model (RPK) values. The use of two different sequencing platforms was corrected using batch correction (ComBat, Supplemental Fig. 3). All data filtering, transformations, and batch correction were performed using custom R scripts. Batch-corrected and transformed log2 RPK and log2 count values are available in Supplemental Tables 2 & 3, and log2 RPK data are also available at GeneNetwork (www.genenetwork.org; Group = Chronic Intermittent Ethanol, Type = LCM Brain Regions mRNA, Data set = INIA LCM [11 Regions] CIE/AIR RNA-seq Transcript Level [Dec15]).
Quality control
Principal component analysis (PCA), a data-reduction technique, was used to identify sample outliers, visualize the data, and explore the main sources of variation driving expression. PCA analysis was performed in R using the prcomp function. Several tissues showed poor sample clustering and were excluded from analysis (BLA and DLS). The PrL sample from B6 mouse 34 (CIE treatment) was an extreme outlier in the PCA and was also excluded. All cortical samples (collected from ILC, PrL, and VCX) were considered as one sample type (CTX) due to the high degree of similarity in expression. The resulting data set included 9 tissue types (BST, CA1, CA3, CeA, CTX, DMS, NAc, NAs, VTA) and 87 samples.
Estimating effects on expression using Generalized Linear Models (GLM)
Differential gene expression was determined using the glm function in R where gene expression (Y) is dependent on treatment, strain, region, or treatment interactions with strain and region: Y ~ Treatment + Strain + Region + Treatment*Strain + Treatment*Region.
Treatment was coded as 0 for AIR and 1 for CIE, 0 for B6 and 1 for D2. Regions were collapsed into three groups based on the PCA clustering and coded as 0 for hippocampus (CA1 and CA3 regions), 1 for cortex (Prl, ILC, and VCX), and 2 for the remaining subcortical and limbic and mesolimbic tissue (VTA, NAc, NAs, DMS, CeA, and BST).
The local false discovery rate (q value) for each effect on expression in the model was estimated from the p value distribution in R using the q-value package (Storey & Tibshirani, 2003).
In many cases, the same gene is represented by multiple transcript models; however, the gene level analysis performed here cannot discriminate between transcript isoforms. For this reason, in the vast majority of cases, expression is similar for transcript models representing the same gene. In a few instances, a significant effect of treatment, region, or strain is detected for one isoform and a significant interaction is detected for another. In these cases, all transcript models for the gene are assigned to the interaction effect.
Confirmation of expression differences
Expression differences due to strain and region were confirmed using a bioinformatics approach since excess RNA for QPCR was not available. For regional expression differences, in situ expression data from the Allen Brain Atlas (mouse.brain-map.org) were compared with the top 27 genes with robust effects of region on expression. Log2 raw expression values based on quantified values for each structure at a grid voxel level were obtained for isocortex, hippocampal formation (HPF), striatum (STR), and midbrain (MB) for each gene and compared with average Prl, ILC and VCX; CA1 and CA3; DMS, NAc and NAs; and VTA log2 RPK expression data. Correlation was determined using the Pearson correlation coefficient. For strain differences, a list of genes modulated by locally acting (cis) polymorphisms was computed using resources available at GeneNetwork. Affymetrix M430 microarray data sets generated from hippocampus (Hippocampus Consortium M430v2 [Jun06] RMA), cortex (VCU BXD PFC Sal M430 2.0 [Dec06] RMA), striatum (HBP Rosen Striatum M430V2 [Apr05] RMA Clean), and nucleus accumbens (VCU BXD NAc Sal M430 2.0 [Oct07] RMA) of BXD strains were mined for cis-expression quantitative trait loci (cis eQTLs) using the GenomeGraph tool. Summarized eQTL data for each data set were downloaded and probe sets that had: 1) a maximum QTL (defined as the peak marker position) within (±) 2 Mb of the gene locus, and 2) a log of the odds (LOD) score greater than 2 (equivalent to a likelihood ratio statistic or LRS greater than 10) were nominated as cis eQTLs. A list consisting of unique Affymetrix M430 probe set IDs representing a transcript with a significant cis eQTL in one or more brain regions was generated. Ensembl BioMart was used to extract associated M430 probe set information for each Ensembl transcript ID associated with a significant strain effect in the RNA-seq data set, and the resulting list was overlapped with the significant brain cis-eQTL list. Some false positives are expected using this approach due to SNP effects on probe set expression (Ciobanu et al., 2010).
Enrichment analysis
WebGestalt was used for Gene Ontology (GO), KEGG pathway, miRNA binding site, and protein-protein interaction module enrichment (Zhang, Kirov, & Snoddy, 2005). Enrichment was determined based on the hypergeometric distribution, and FDR was estimated using Benjamni and Hochberg’s method (adjP < 0.05). Gene sets were selected after glm based on a nominal p-value threshold (p < 0.01). Gene sets and background gene lists contain unique genes only, and the identifier used was Ensemble transcript ID.
CIE time course overlap
A comparison of gene expression changes was performed between our study and a similar study from Smith and colleagues (Smith et al., 2016). The studies differ in strains included, expression platform used, number of CIE cycles, and whether or not subjects were allowed to self-administer alcohol. In the study from Smith et al., B6 mice underwent four cycles of CIE by vapor chamber. After the fourth CIE cycle, 12 mice (6 CIE and 6 air-control) were sacrificed, and brains were harvested at each of four time points: 0 h, 8 hours, 72 h, and 7 days. RNA was extracted from the prefrontal cortex, nucleus accumbens, hippocampus, bed nucleus of the stria terminalis, and central nucleus of the amygdala, and hybridized to Affymetrix Mouse Gene Chip 430, type 2 microarrays. Microarrays were processed, and differential gene expression was quantified using linear models for microarrays (LIMMA) as previously described (Smith et al., 2016). B6 time course genes were significantly differentially expressed between CIE and AIR mice at each time point and brain region, at a false discovery rate of 0.1 from Smith and colleagues. The B6 time course genes were used for comparison to RNA-seq genes that were significantly differentially expressed by treatment, or treatment-region interaction, in B6 and/or D2 mice in the current study. Significance of overlap was determined using Fisher’s Exact Test through the ‘GeneOverlap’ package for R. Overlaps with a false discovery rate of ≤0.05 were considered significant.
WGCNA
We used the Weighted Gene Co-expression Network Analysis (WGCNA) package in R (version 1.49) to define correlated networks of transcripts from the gene expression data (Langfelder & Horvath, 2008). All 9 tissue types and 87 samples used for differential expression were included in WGCNA. For this, the transcripts were further filtered such that each gene had only a unique transcript. Specifically, for a gene with multiple transcripts, we retained the transcript with the highest variance across the samples as the representative expression trait, which resulted in a non-redundant list of 12,590 genes. WGCNA is a dimension-reduction procedure that first generates a gene-by-gene similarity matrix by computing pairwise correlations for each transcript. The similarity matrix is transformed into an adjacency matrix using soft thresholding power, β. The β is chosen so that the transformed data will fit a scale-free network topology. Finally, the connectivity between each node (i.e., transcript) is estimated from the topological overlap matrix, and modules that represent networks of tightly correlated transcripts are constructed. For more detail on the computation procedure for WGCNA please see Zhang & Horvath, 2005. Modules for B6 and D2 were constructed independently but using the same β of 6. In B6, this has a mean connectivity (k) = 56.8 and maximum k = 445 and fits a scale-free topology by R2 = 0.86. In D2, this has a mean connectivity (k) = 57 and maximum k = 429 and fits a scale-free topology by R2 = 0.83. Using this parameter, co-expression adjacencies and the topological overlap matrix were constructed, and hierarchical clustering of genes was done using the ‘hclust’ function using the average linkage method. Modules were defined using the dynamic cut-tree method with default cut height of 0.99 and a minimum module size of 35 with deepSplit set to 2. Highly correlated modules were merged at a cut height of 0.2 and transcripts with low overall connectivity were filtered into the grey module. Following module construction, transcripts with low overall connectivity are filtered into the grey module. Once modules are defined, the expression pattern of each module is summarized by the module eigengene (ME, the first principal component), and this can be used to examine inter-module correlations and association with other variables such as brain region, strain, and treatment. This allows the identification of factors that are associated with the summarized variance of a network of transcripts. We used linear regression analysis to test association between the MEs and Treatment + Region + Treatment*Region for the B6 and D2 modules. To examine overlap of transcript members between B6 and D2 modules, we performed a contingency analysis and counted how many transcript members in a particular D2 module are in a B6 module. Significance of overlap was derived from Fisher’s exact test.
Results
Differential expression results from region, strain, and treatment effects
The main source of variation in expression in the data set as a whole is tissue type. The first four principal components (PCs) reflect brain region and explain ~55% of the cumulative proportion of variance. Each PC captures approximately 27, 15, 8, and 5% of the variance in expression, respectively. Samples collected from hippocampus (CA1 and CA3), cortex, and ventral tegmental area (VTA) cluster together well after principal component analysis (PCA), regardless of strain or treatment group (Supplemental Fig. 3). Few samples fall outside the normal distribution probability ellipse for each region. Overlap in expression profiles is evident for samples from similar tissues such as DMS and NAc, NAs and CeA, and CeA and BST.
To capture expression patterns due to region, strain, and treatment or their interactions, we applied a linear model to the expression data: y ~ Treatment + Strain + Tissue + Treatment*Strain + Treatment*Region (Supplemental Table 4). At a nominal p value (p < 0.01), most expression differences are due to the effect of region (5464 unique genes) or strain (1111 unique genes), but there are many transcripts that demonstrate differential expression due to the effect of treatment – treatment alone (70 unique genes), treatment by strain (415 unique genes), or a treatment by region interaction (60 unique genes) (Fig. 1, Supplemental Table 5).
Fig. 1. Frequency of significant p values by model fit.

The number of genes associated with a main effect of strain, treatment, region, or a treatment by strain or treatment by region interaction is shown at an FDR of 10%. Values of approximately 100 or less are indicated on the chart for clarity. Only unique genes were included. Region and Strain have the strongest effect on gene expression, but the expression of a small number of genes shows either a main effect of treatment or an interaction 3 days following the fifth and final cycle of CIE exposure.
Robust detection and validation of regional and strain differences in gene expression
Over 40% of the unique genes surveyed show robust (FDR < 0.1) expression variation by region (Supplemental Table 6). In general, the effect of region is small – the average fold change is 1.35 (average log2 RPK estimate of 0.44). Within this gene set, 27 genes demonstrate a large effect of region with an estimated effect on expression of more than 2 log2 RPK units, which amounts to a greater than 4-fold difference (Fig. 2A–D). Expression patterns were confirmed for 21 of these genes (Fig. 2E) using in situ hybridization data available for the B6 strain (mouse.brain-map.org) from the Allen Brain Atlas project (Lein et al., 2007).
Fig. 2. Regional differences in expression.

Genes with a significant main effect of region on expression are shown in panels A through D with average log2 RPK values shown on the Y-axis. Only the top genes with a greater than 4-fold change in expression are shown. Genes with higher expression in HIP and cortical regions are shown in panel A and genes with higher expression in limbic and mesolimbic tissues are shown in panels B and C. Note that the genes in panel B have comparatively lower expression in HIP, cortex, and DMS and that Foxp2 in panel C has less expression in CeA and BST compared to DMS, NAc, NAs, and VTA. Genes in panel D have similar expression profiles that are opposite in HIP, cortical tissue, and VTA compared to limbic and mesolimbic tissue. Expression values and the correlation between the top 27 genes with regional expression (left matrix) and in situ expression (B6) from Allen Brain Atlas (right matrix) are shown in panel E. Expression strength (orange shading) is indicated by increasing hue intensity. Correlation strength is indicated by more intense hues and direction is indicated by color (yellow shading for positive correlations and blue shading for negative correlations). Genes not found in the Allan Brain Atlas have no corresponding expression value.
Over 8% of the unique genes surveyed demonstrate robust (FDR < 0.1) expression differences driven by strain. Over half of these genes are regulated by a strong cis eQTL (logarithm of the odds or LOD score > 2) in one or more brain tissues from the BXD recombinant inbred panel derived from B6 and D2 (Supplemental Table 6).
Small but significant strain differences in response to CIE treatment
At a false positive rate of 10% (q < 0.1), no significant main effect was detected for treatment or the interaction between region and treatment. However, 20 unique genes were significantly differentially expressed due to a treatment by strain interaction (Table 1, Supplemental Table 6). The top genes with treatment by strain interaction effects (q < 0.1) fell into two categories based on pattern of expression (Fig. 3). Some genes, such as mt-Rnr1 and Yme1l1, demonstrate differences in expression between AIR or CIE treatment groups in one strain (usually D2) only. Others demonstrate a transcriptional response to treatment that is opposite in each strain. For example, levels of Atp5a1, H1f0, Timm10, Denr, and Parn are increased after CIE compared to AIR exposure in B6, but decreased in D2. In contrast, levels of Creb1 are decreased after CIE treatment in B6 and increased in D2. No enrichment of biological function was detected for this small class of genes.
Table 1.
| ID | Name | Description | Chr | Transcript Start | Transcript Length | BioType | Estimate | Std.Error | t value | P-value | q value | Effect |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ENSMUST00000162890 | 0610040B10Rik | RIKEN cDNA 0610040B10 gene | 5 | 143329327 | 600 | antisense | −0.99 | 0.24 | −4.14 | 8.60E-05 | 8.80E-02 | Treatment*Strain |
| ENSMUST00000177533 | AA465934 | expressed sequence AA465934 | 11 | 83291742 | 308 | antisense | −1.16 | 0.28 | −4.17 | 7.68E-05 | 8.80E-02 | Treatment*Strain |
| ENSMUST00000026495 | Atp5a1 | ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1 | 18 | 77773729 | 2471 | protein_coding | −0.41 | 0.10 | −4.21 | 6.50E-05 | 8.65E-02 | Treatment*Strain |
| ENSMUST00000088307 | BC003965 | cDNA sequence BC003965 | 17 | 25184561 | 3022 | protein_coding | −0.58 | 0.13 | −4.36 | 3.74E-05 | 7.17E-02 | Treatment*Strain |
| ENSMUST00000049932 | Creb1 | cAMP responsive element binding protein 1 | 1 | 64532809 | 5009 | protein_coding | 0.59 | 0.11 | 5.26 | 1.16E-06 | 6.70E-03 | Treatment*Strain |
| ENSMUST00000023869 | Denr | density-regulated protein | 5 | 123907175 | 2339 | protein_coding | −0.70 | 0.17 | −4.11 | 9.38E-05 | 9.00E-02 | Treatment*Strain |
| ENSMUST00000068791 | Eif2d | eukaryotic translation initiation factor 2D | 1 | 131153181 | 3316 | protein_coding | 0.61 | 0.14 | 4.27 | 5.33E-05 | 8.18E-02 | Treatment*Strain |
| ENSMUST00000003123 | Fam32a | family with sequence similarity 32, member A | 8 | 72219730 | 1910 | protein_coding | −0.81 | 0.14 | −5.94 | 6.81E-08 | 5.22E-04 | Treatment*Strain |
| ENSMUST00000180086 | H1f0 | H1 histone family, member 0 | 15 | 79028212 | 2287 | protein_coding | −0.88 | 0.13 | −6.56 | 4.72E-09 | 1.09E-04 | Treatment*Strain |
| ENSMUST00000015800 | Hspa8 | heat shock protein 8 | 9 | 40800984 | 2394 | protein_coding | 1.65 | 0.40 | 4.13 | 8.79E-05 | 8.80E-02 | Treatment*Strain |
| ENSMUST00000056732 | Mfap1b | microfibrillar-associated protein 1B | 2 | 121460235 | 3362 | protein_coding | −1.00 | 0.16 | −6.23 | 2.00E-08 | 2.30E-04 | Treatment*Strain |
| ENSMUST00000082388 | mt-Rnr1 | mitochondrially encoded 12S rRNA | MT | 70 | 955 | Mt_rRNA | 0.59 | 0.12 | 4.82 | 6.58E-06 | 2.16E-02 | Treatment*Strain |
| ENSMUST00000046951 | Pak1ip1 | PAK1 interacting protein 1 | 13 | 41001023 | 1722 | protein_coding | −0.60 | 0.13 | −4.43 | 2.95E-05 | 6.69E-02 | Treatment*Strain |
| ENSMUST00000058884 | Parn | poly(A)-specific ribonuclease (deadenylation nuclease) | 16 | 13537964 | 2902 | protein_coding | −0.71 | 0.17 | −4.30 | 4.73E-05 | 7.78E-02 | Treatment*Strain |
| ENSMUST00000086519 | Rplp0 | ribosomal protein, large, P0 | 5 | 115559467 | 1358 | protein_coding | −0.56 | 0.14 | −4.13 | 8.71E-05 | 8.80E-02 | Treatment*Strain |
| ENSMUST00000040487 | Rsad1 | radical S-adenosyl methionine domain containing 1 | 11 | 94539798 | 4143 | protein_coding | −1.75 | 0.40 | −4.41 | 3.20E-05 | 6.69E-02 | Treatment*Strain |
| ENSMUST00000185815 | Snhg14 | small nucleolar RNA host gene 14 | 7 | 59676393 | 670 | antisense | 0.96 | 0.21 | 4.66 | 1.23E-05 | 3.14E-02 | Treatment*Strain |
| ENSMUST00000111631 | Timm10 | translocase of inner mitochondrial membrane 10 | 2 | 84827384 | 564 | protein_coding | −0.75 | 0.15 | −4.92 | 4.40E-06 | 2.02E-02 | Treatment*Strain |
| ENSMUST00000116556 | Wash1 | WAS protein family homolog 1 | 17 | 66111645 | 2887 | protein_coding | −0.61 | 0.15 | −4.21 | 6.53E-05 | 8.65E-02 | Treatment*Strain |
| ENSMUST00000028117 | Yme1l1 | YME1-like 1 (S. cerevisiae) | 2 | 23156369 | 4707 | protein_coding | −0.50 | 0.12 | −4.30 | 4.69E-05 | 7.78E-02 | Treatment*Strain |
Fig. 3. Differential strain response to treatment.

At a 10% FDR level, 20 genes are associated with a treatment by strain effect on expression. Genes that show a significant effect of treatment on expression primarily in a single strain are shown in the top panel and genes that show a divergent expression response between strains following treatment are shown in the bottom panel. Significance levels based on pair-wise comparisons as follows: * = p < 0.05, ** = p < 0.01, *** = p < 0.005, **** = p < 0.001. Average log2 RPK values are shown on the X-axis for each group.
The lower number of significant results for treatment effects at FDR < 0.1 compared with region and strain effects are expected given: 1) a high level of variation associated with gene expression measurements, 2) the low number of biological replicates (n = 2), and 3) a much smaller effect size for treatment compared to region or strain. The use of LCM precludes validation by quantitative PCR; however, these data are sufficient for an exploratory analysis using nominal p values in order to identify biological pathways that are exquisitely sensitive to chronic alcohol exposure and may mediate long-lasting changes involved in the development of tolerance, dependence, and addiction.
Long-lived transcriptional changes after CIE treatment reflect mainly divergent biological processes between strains
Selecting a nominal p-value threshold of p < 0.01 results in 72 and 415 unique genes with differential expression due to a main effect of Treatment and a Treatment by Strain interaction, respectively (Supplemental Table 5). For treatment (independent of strain or region), 42 and 30 unique genes were up- or down-regulated, respectively. No GO categories were significantly enriched for either the combined or up- or down-regulated list at an FDR < 0.05 (Benjamini & Hochberg method), probably due to the small size of the gene set.
Genes with a significant Treatment by Strain interaction effect on expression display differential regulation in each strain and were split into two categories: 1) general down-regulation in B6 and up-regulation in D2 due to treatment (140 unique genes) and 2) general up-regulation in B6 and down-regulation in D2 due to treatment (262 unique genes). Genes in category 1 were significantly enriched (FDR < 0.05) for a number of GO categories related to transcription, including regulation of gene expression, regulation of RNA biosynthetic process, regulation of transcription, DNA-dependent, SMAD binding, and sequence-specific DNA binding transcription factor activity (Supplemental Fig. 4, Supplemental Table 7). The gene set also included significant enrichment for the KEGG TGF-beta signaling pathway and binding sites for MIR-142-5P, MIR-505, and MIR-29 (A, B, & C) (Supplemental Table 7). Although no protein-protein interaction (PPI) networks were significantly enriched in the gene set, there was a suggestive enrichment (FDR = 6.3%) for a PPI network module related to transcription in which several genes with altered expression in category 1 are positioned as hub genes or highly connected genes. These genes include Ube2d3, Ep300, Ercc6, Cul1, Taf5, Pou2f1, Myobp1a, Papola, Cpsf2, and Fos (Supplemental Fig. 5, Supplemental Table 7).
Category 2 genes show differential strain expression (up-regulation in B6 and down-regulation in D2) after CIE treatment and are significantly enriched for many GO categories related to mitochondria, oxidation-reduction processes, and energy production, including: electron transport chain; oxidative phosphorylation; mitochondrial ATP synthesis coupled-electron transport; iron-sulfur cluster binding, oxidoreductase activity; and localization to mitochondrion (Supplemental Fig. 6, Supplemental Table 8). Many overlapping KEGG pathways are also significantly enriched in this gene set, including Parkinson’s, Huntington’s, and Alzheimer’s disease, oxidative phosphorylation, drug metabolism-cytochrome P450, metabolism of xenobiotics by cytochrome P450, cardiac muscle contraction, metabolic pathways, and tyrosine and glutathione metabolism pathways (Supplemental Table 8). PPI modules related to mitochondrial proton-transporting ATP synthase complex (Supplemental Fig. 7), unfolded protein binding (Supplemental Fig. 8), proteasome (Supplemental Fig. 9), and microtubule-based movement (Supplemental Fig. 10) were also enriched with several category 2 genes positioned as hub genes.
Markedly different transcriptional responses between B6 and D2 were observed due to CIE treatment using our regression analysis. To further explore this difference, linear models were used in a post hoc analysis to evaluate the effect of treatment and region (y ~ treatment*region) on gene expression within each strain. The results of this analysis are striking. At a nominal significance threshold (p < 0.05), 1516 unique genes are differentially expressed due to a treatment or treatment by region interaction in D2 compared to 369 in B6 (Fig. 4, Supplemental Table 9). Only 59 genes overlap and the direction is often inconsistent. No significantly enriched GO categories were detected for the B6 gene set after correction for multiple testing. For the D2 gene set, localization to mitochondrial respiratory chain complex 1 (n = 16, adjP = 1.66E−05) was significantly enriched.
Fig. 4. Higher number of significant expression differences due to treatment in D2.

There is a striking difference in the number of significant differences (p < 0.05) detected following within-strain regression analysis (y ~ treatment + region + treatment*region). Counts of differentially expressed genes are shown on the X-axis and are based on unique genes only. A greater number of genes are differentially expressed due to treatment or treatment*region (interaction) effects in D2 compared to B6.
Marked regional differences in response to treatment
Sixty genes show significant (p < 0.01) expression differences due to an interaction between treatment and region (Treatment*Region). The effect of region on expression is quite small for this subset; average estimated effect of the interaction on expression is 0.32 (1.24-fold) with a maximum value of 0.5 log2 RPK units (1.4-fold). To further investigate the effect of treatment within each region, regression analysis was performed for all 11 brain areas (y ~ Treatment*Strain). PrL, VCX, CA1, BST, and NAc show the largest number of treatment and treatment by strain interaction effects (p < 0.01) 72 h following CIE treatment (Fig. 5). In addition, BST (n = 78), CA1 (n = 60), and PrL (n = 65) have the highest number of nominally significant changes in expression due to the effect of treatment.
Fig. 5. Differential response to CIE treatment across brain regions.

The number of significant differences (p < 0.01) based on within-region regression analysis (y ~ treatment + strain + treatment*strain) is shown by region. The number of differentially expressed genes with a main effect of treatment or an interaction between strain and treatment is shown for each region and includes unique genes only. Prefrontal cortex (and visual cortex), hippocampus (CA3), BS, and NAc show the largest number of expression differences due to treatment effects.
Overlap with previous studies
Smith and colleagues recently performed a comprehensive time course of expression changes in multiple brain regions following CIE treatment in B6. Top significant genes at each time point and region were compared with nominally significant expression changes detected in B6 and D2 following CIE in our study (Fig. 6, Supplemental Tables 10 & 11). There was significant overlap at an FDR of 0.05, detected primarily at 0 and 8-h time points in prefrontal cortex (PFC), nucleus accumbens, BNST, and hippocampus, potentially indicating a persistent molecular signal, or engram, following chronic alcohol treatment. The pattern of overlap across brain regions likely reflects the larger number of significant treatment differences detected in CA3, BNST, NAc, and cortex detected in our study. Remarkably, no significant overlap was detected for the 72-h time point across studies, perhaps due to the small number of significant changes in B6 detected in both the Smith et al. study and our own.
Fig. 6. Overlap between CIE paradigms.
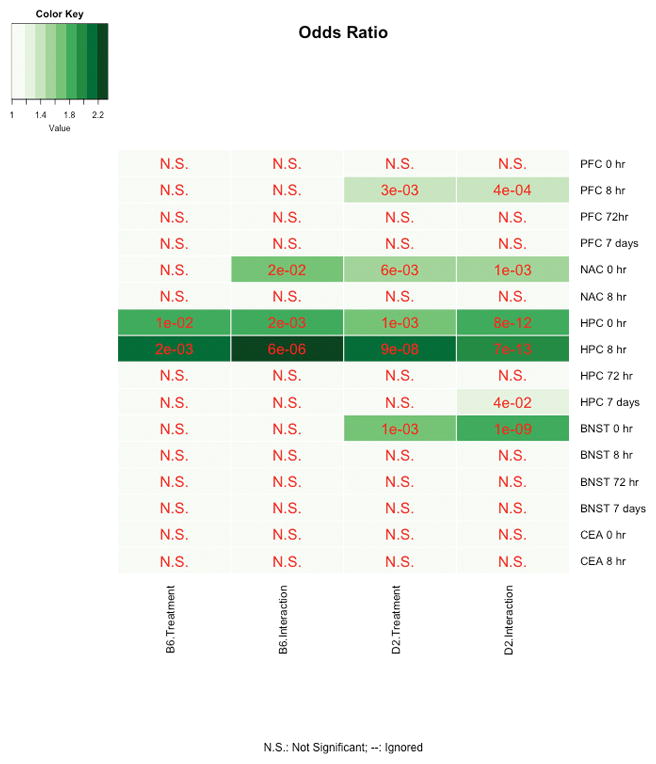
Smith and colleagues (2016) measured expression differences between AIR control and CIE-treated B6 mice in multiple brain regions at several time points following four cycles of CIE. We compared expression changes due to treatment or treatment by region interactions within strain generated from our study (tissue collected 72 h after five cycles of CIE combined with voluntary alcohol self-administration). Heatmaps show overlap for each study category (the current study as columns and Smith et al., 2016 as rows) and associated FDR-adjusted p value. Criterion for overlap is FDR < 0.05. Despite the lack of overlap at 72 h (probably due to the low number of expression differences due to treatment detected in B6 in both studies), overlap across brain regions and at 0- and 8-h time points suggests that some of the changes we detect at the 72-h time point may also be altered at earlier time points following CIE treatment.
WGCNA reveals a more robust effect of treatment on network organization of the D2 transcriptome
Statistical analysis of gene expression patterns in the data set indicated that D2 was more sensitive to CIE treatment in terms of gene expression changes at 72 h post-exposure. To evaluate global patterns of organization within each strain, co-expression networks were constructed independently for B6 and D2. This organized the D2 transcriptome into 25 modules ranging in size from 42 to 1949 members (Supplemental Table 12). The B6 transcriptome was organized into 24 modules that range in size from 48 to 2923 members (Supplemental Table 13). Each module is a network of correlated transcripts that have similar expression patterns across samples. To test if treatment has an influence on the correlated network of transcripts, we extracted the first principal component for each module (i.e., the module eigengene or ME) and used this in a multiple regression analysis to test association with treatment, region, or treatment by region interactions. As expected, brain region had the most pervasive influence on the global organization of expression traits and most modules show significant association with brain region in both strains. This indicates that the co-expression of these transcripts is strongly driven by brain region-specific expression patterns. By comparing the network members in the D2 and B6 modules, we found that for the most part, modules constructed in D2 overlap extensively with B6 modules. However, 6 D2 modules have limited overlap with B6 modules, and we consider these to be D2 strain-specific modules (Supplemental Table 14).
In B6, only 1 ME (B6darkolivegreen) showed a modest association with a treatment by region interaction effect at the 72-h post-exposure time point. However, this did not reach significance following multiple test correction (Bonferroni p < 0.05). None of the B6 modules was enriched for transcripts with altered expression due to treatment or treatment by region interaction effects (Supplemental Table 15). In contrast, the ME of 9 D2 modules was associated with a treatment or treatment by region interaction effect (Supplemental Table 16). Even after setting a Bonferroni-corrected threshold, 4 D2 MEs (D2blue, D2darkgreen, D2darkorgange, and D2salmon) had a highly significant association with treatment or treatment by region interaction. When we examined the members of these modules, we found that several are enriched for transcripts differentially expressed due to a treatment or interaction effect in D2. In particular, we highlight three modules: D2blue, D2darkorange, and D2salmon. These represent tightly correlated networks of transcripts that are responsive to treatment. Taken together, the network analysis shows that while CIE has a strong influence on the global organization of gene expression in the D2 brain, the B6 brain is less responsive to CIE.
Discussion
In this study, we generated an extensive set of brain-region RNA-seq profiles for two inbred strains at a single time point following chronic exposure to alcohol. We identified and validated regional changes in expression using in situ profiles available at the Allen Brain Atlas. We also identified strain differences in expression and leveraged cis-eQTL data from the BXD panel to determine that over half of these differences may result from variants segregating between B6 and D2. Despite a low number of biological replicates and the potential for sampling bias, the detection and replication of brain region and strain differences from our study using independent data sets from the Allen Brain Atlas and GeneNetwork indicate that our data set is robust. We leveraged these data to identify differential expression due to the effect of treatment using linear models and WGCNA and found a marked difference in the transcriptional response between B6 and D2, both at the level of single gene expression and at networks of genes. Previous studies using microarray platforms have examined expression changes across brain regions at different time points following the last cycle of CIE in a single strain, B6 (Melendez et al., 2012; Osterndoff et al., 2015; Smith et al., 2016). Our study is the first to explore the lasting impact of CIE on gene expression 72 h following treatment in multiple brain regions in both B6 and D2 using the RNA-seq platform. Although the number of individuals profiled here is small, the depth of sampling across tissues is broad. The data presented here provide a rich snapshot of strain-dependent transcriptional changes following CIE, and provide the impetus for future focused studies in a larger number of individuals.
As expected based on findings from previous studies, the effect of treatment on gene expression 72 h after multiple cycles of CIE is small compared to regional and strain effects. We identified only a handful of common transcriptional responses to treatment. Studies using B6 have reported a smaller number of significant changes in gene expression 120 h following repeated cycles of CIE (Smith et al., 2016). This is in contrast to gene expression changes at 0 and 8 h following chronic exposure, time points thought to represent response to and withdrawal from alcohol (Melendez et al., 2012; Osterndorff-Kahanek et al., 2015; Smith et al., 2016). Interestingly, a cross-study comparison of gene expression changes (Smith et al., 2016) revealed significant overlap for both B6 and D2 at earlier time points following exposure (0 h and 8 h) in several brain regions that are particularly sensitive to chronic alcohol treatment, including hippocampus and cortex. Genes altered by CIE in both B6 and D2 over an expanded time course may serve as biomarkers or even drive dependence-induced escalation of drinking. Interestingly, there was no significant overlap detected between data sets for the matched time point 3 days after the last CIE cycle. The lack of overlap at the 72-h time point between the Smith study and our own may reflect differences in expression platform or experimental paradigm. For example, microarrays can produce a broader signal range, whereas coverage of low abundance transcripts is sparse in our study. In addition, the mice included in our study consumed alcohol between vapor exposures whereas the mice in the Smith study did not. It is also highly likely that the lack of overlap at matched time points reflects the paucity of transcriptional changes detected in B6. Similar to previous studies, we found fewer than 400 genes with differential expression due to treatment (air control versus CIE treatment) in B6, even at a nominal p value (p < 0.05). Smith and colleagues found only 3 and 238 genes altered by CIE in B6 hippocampus and prefrontal cortex at the 72-h time point at a stringent 1% FDR. However sparse, changes in expression in B6 have been reported at 72 h, 120 h, and even a week following CIE (Smith et al., 2016), suggesting the presence of a lasting transcriptional imprint or engram. Here we report an even larger long-lasting imprint in D2. This signature may reflect long-lasting alterations that underlie susceptibility or resistance to addiction.
Strikingly, we observed a large number of strain-dependent (and often divergent) responses to CIE treatment, which is known to produce both metabolic stress and dysregulation of the HPA axis. Nearly four times as many nominally significant changes due to treatment were detected in D2 compared to B6, and many of the genes significantly modified by a main effect of treatment (independent of strain) after FDR correction were either unchanged in B6 or reflected a divergent response in both strains. These genes may serve as biomarkers for disruptions in mitochondrial function, protein metabolism, epigenetic modification, and telomerase maintenance. For example, Parn is required for telomere function, and mutations in this gene are part of a collection of Mendelian disorders that result in telomere shortening (Bertuch, 2015; Moon et al., 2015; Tseng et al., 2015). Interestingly, H1f0 has been identified as a potential biomarker of acetylation based on response to pharmacological application of histone deacetylase inhibitors (Reardon et al., 2015). Yme1l1 is an oxidative stress-sensitive mitochondrial inner membrane protein that is up-regulated in response to the mitochondrial unfolded protein response (Aldridge, Horibe, & Hoogenraad, 2007; Rainbolt, Saunders, & Wiseman, 2015; Stiburek et al., 2012), and Atp5a1 functions as a component in mitochondrial complex V and is involved in energy production (Hejzlarová et al., 2014; Lucas et al., 2014). mt-Rnr1 encodes the catalytic subunit of the ribonucleotide reductase complex that is the rate-limiting step in dNTP production – vital for a number of biological processes, including mitochondrial replication and DNA repair. Rplp0 is a member of the ribosomal P complex involved in protein synthesis; down-regulation of this complex leads to autophagy, ER stress, and the unfolded protein response (Artero-Castro et al., 2015). Hspa8 is also involved in chaperone-mediated autophagy (Chiang, Terlecky, Plant, & Dice, 1989; Kaushik & Cuervo, 2012). Eif2d is an initiation factor that plays a key role in delivering initiator tRNA to the ribosome (Holcik, 2015) and may be involved in ribosome recycling (Dever & Green, 2012). Similarly, Denr is involved in ribosome recycling and reinitiation at uORFs, and has been recently linked to circadian periodicity in protein levels (Janich, Arpat, Castelo-Szekely, Lopes, & Gatfield, 2015; Schleich et al., 2014). Finally, ORF19 (Wash1) is involved in endosome-to-Golgi retrieval of membrane proteins (Harbour et al., 2010; Wang et al., 2014).
Changes in mitochondrial function, DNA levels, and telomere length have been reported as biomarkers of chronic stress and depression (Cai et al., 2015), and changes in protein function and epigenetic modifications have long been posited as mechanisms driving neuroadaptation and response to environment. Our strain comparison based on linear models suggests that these pathways diverge between strains following chronic alcohol treatment. These results are mirrored by global WGCNA. Despite a high degree of module overlap between B6 and D2 primarily driven by brain region differences, following multiple test correction, no modules were associated with treatment effects on expression in B6 compared to four in D2. Only the darkorange module is unique to D2 with relatively poor overlap with B6 modules. This relatively small module (164 members) is both significantly associated with treatment and treatment by region interaction effects and is enriched for transcripts that are altered in expression by treatment. The D2 darkorange module represents an expression signature unique to D2 that is responsive to CIE treatment. The remaining three treatment-associated modules (D2blue, D2darkgreen, D2salmon) are larger (346 to 1394 members) and have significant overlap with B6 modules. These modules represent genes that show a differential strain response to treatment.
The large and generally opposite response to treatment between strains observed in this study was unanticipated. Most recent experiments have been performed in a single strain – B6 – due to the robust enhancement in alcohol consumption and behavior observed in this strain following CIE (Lopez, Becker, & Chandler, 2014; Lopez, Griffin, Melendez, & Becker, 2012) and following CIE and exposure to additional environmental stressors (Anderson, Lopez, & Becker, 2016a). These studies demonstrate concomitant changes in neurotransmitters (Griffin, Ramachandra, Knackstedt, & Becker, 2015) and neuropeptides (Maldonado-Devincci et al., 2014) following CIE, and suggest alterations in specific circuits and regions (Anderson, Lopez, & Becker, 2016b; Lopez, Moorman, Aston-Jones, & Becker, 2016; Samantaray et al., 2015). Whether or not these changes are unique to B6 or represent common mechanisms in the addiction process remains an open question. However, accumulating evidence suggests that the D2 strain also exhibits behavior associated with addiction. This includes observations that alcohol dependence can elicit a high rate of self-administration in D2 using methods that bypass the innate alcohol avoidance demonstrated in this strain, such as intragastric delivery (Cunningham, Fidler, Murphy, Mulgrew, & Smitasin, 2013; Fidler et al., 2012) and the clever use of tastant-substitution procedures (McCool & Chappell, 2014). Indeed, McCool and colleagues found a robust increase in alcohol self-administration in both B6 and D2 following multiple cycles of CIE using an umami-fading procedure that produces high levels of baseline alcohol consumption in both strains (McCool & Chappell, 2015). B6 and D2 also show profound differences in the behavioral and physiological response to acute (Belzung, El Hage, Moindrot, & Griebel, 2001; Jacobson & Cryan, 2007; Jones, Sarrieau, Reed, Azar, & Mormède, 1998; Millstein & Holmes, 2007) and chronic (Mozhui et al., 2010; Pleil et al., 2012; Pothion, Bizot, Trovero, & Belzung, 2004; Shea et al., 2015) stress. This differential response to stress could account for some of the transcriptional differences between strains following CIE. Our results, combined with recent studies, provide evidence that the D2 strain is a unique and genetically distinct model for investigating the contribution of stress and alcohol interactions to addiction and alcohol-use disorders. However, it is also possible that the gene expression patterns we observed, especially with regard to the enrichment of genes localized to mitochondria in D2, may represent increased toxicity and metabolic stress to the alcohol inhalation paradigm in this strain. Most CIE paradigms involve the use of pyrazole to increase blood alcohol levels and reduce ethanol elimination through inhibition of alcohol dehydrogenase. Pyrazole is a known hepatotoxin that increases oxidative stress through upregulation of cytochrome P450 2E1 (Cyp2e1) – which is also up-regulated by alcohol (Song, 1996). Interaction between chronic alcohol exposure and pyrazole, combined with strain differences in mitochondrial supercomplexes (Buck, Walter, & Denmark, 2014), could result in a divergent, and perhaps more profound and long-lasting, metabolic response to CIE treatment in D2. Indeed, D2 typically have elevated BEC compared to B6 (212.11 ± 6.68 mg/dL for B6 and 269.96 ± 13.15 mg/dL for D2 in this study), and this may account for some of the strain differences in treatment effects on gene expression. Disentangling the effects of treatment and BEC on expression is difficult and will require studies with a larger number of individuals that either match across BECs or identify correlated expression signatures. Future studies in both strains that include additional time points following CIE without the use of pyrazole will be also be required to elucidate the influence of pyrazole, chronic alcohol exposure, and strain on gene expression.
Similar to previous human and mouse studies, we observed a differential impact of treatment across brain regions with cortical, hippocampal region CA1, BNST, and NAc showing the most pronounced response to treatment. This suggests that the chronic alcohol engram may be unique across structures and especially pronounced in areas important for cognition and reward. Surprisingly perhaps, given the role of the amygdala in stress and addiction pathways, we did not detect a large number of changes in this structure (at least in the central nucleus). Previous work has demonstrated reduced baseline alcohol consumption following lesions of the lateral posterior bed nucleus of the stria terminalis and central nucleus of the amygdala, but these lesions did not block dependence-induced drinking in B6 (Dhaher, Finn, Snelling, & Hitzemann, 2008), suggesting that other regions may contribute more to this behavior. A caveat to this analysis is that tissue dissection and heterogeneity can contribute to the ability to detect significant changes within a tissue. In addition, neuroadaptation is expected to occur partly at the level of circuits and protein function, neither of which can be reliably inferred from gene expression studies. Nevertheless, our findings agree well with recent work in mice and suggest long-lasting alterations in cortex and hippocampus that are conserved between two different inbred strains and also overlap with critically impacted brain areas in human alcoholics.
In summary, we have generated transcriptional profiles for two inbred strains across 11 brain regions and identified genes whose expression is altered by CIE treatment as well as interactions between treatment and brain region or strain. Although changes in expression due to the effect of chronic ethanol inhalation are small 72 h post-exposure, these alterations may represent a chronic alcohol engram. This transcriptional signal is highly divergent between strains, and may be related to individual differences in the response to metabolic and physiological stress, development of tolerance, dependence, and excessive alcohol consumption.
Supplementary Material
Panels A and B show individual level alcohol consumption and preference data for B6, respectively. Panels C and D show individual level consumption and preference data for D2, respectively. Average alcohol consumption (y-axis) is plotted across weeks for baseline (BASE) and treatment (TREAT) conditions and average preference data is shown on the y-axis for all 6 weeks of baseline drinking (BASE) and all 4 weeks of treatment (TREAT). B6 shows higher alcohol preference than D2 for all conditions (p = 1.29E−05). Although not significant, likely due to the low number of n and expected high variability in consumption between individuals of the same strain, both B6 mice in the CIE condition showed a trend toward elevated drinking from baseline compared to D2 (p = 0.17) and compared to the B6 AIR condition (p = 0.24). Note that statistical tests (e.g., t tests) are not recommended for n < 3.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module.
Sections are shown after removal of each region by LCM.
Panel on the left shows PCA biplot for PC1(X-axis) and PC2 (Y-axis) prior to batch correction. Tissue is identified by color and condition by shape. S (Solid) and P (Proton) labels indicate the RNA-seq platform used. Probability ellipses indicate grouping by batch as a major source of variation in the data set. Right panel shows PCA biplot following ComBat batch correction. Strain is indicated by B (B6) or D (D2), condition by geometric type, and tissue by color. Tissue origin is a major factor driving expression. CA1, CA3, CTX, and VTA samples have distinct and non-overlapping patterns of expression compared to samples from BNST, amygdala, dorsal striatum, and accumbens.
Directed acyclic graph tree (DAG) showing significantly (adjP < 0.05) enriched categories (red). Many enriched categories are related to transcription.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module. This module is enriched for terms related to transcription.
DAG tree showing significantly (adjP < 0.05) enriched categories (red). Many enriched categories are related to energy production, metabolism, and mitochondria.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module.
Highlights.
This is the first study to compare expression profiles across 11 brain regions and between two inbred strains following CIE.
We report long-lived alterations in expression – a putative chronic alcohol engram – 72 h following chronic alcohol exposure.
Several brain regions, including the cortex and hippocampus, show long-lasting enhanced sensitivity to CIE treatment.
Strikingly divergent transcriptional responses are observed between strains with 3 times more genes altered by CIE in D2.
Acknowledgments
Funding support was provided by INIA grant U01AA13499.
References
- Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PloS One. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RI, Lopez MF, Becker HC. Forced swim stress increases ethanol consumption in C57BL/6J mice with a history of chronic intermittent ethanol exposure. Psychopharmacology. 2016a;233:2035–2043. doi: 10.1007/s00213-016-4257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RI, Lopez MF, Becker HC. Stress-Induced Enhancement of Ethanol Intake in C57BL/6J Mice with a History of Chronic Ethanol Exposure: Involvement of Kappa Opioid Receptors. Frontiers in Cellular Neuroscience. 2016b;10:45. doi: 10.3389/fncel.2016.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artero-Castro A, Perez-Alea M, Feliciano A, Leal JA, Genestar M, Castellvi J, et al. Disruption of the ribosomal P complex leads to stress-induced autophagy. Autophagy. 2015;11:1499–1519. doi: 10.1080/15548627.2015.1063764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HC. Effects of alcohol dependence and withdrawal on stress responsiveness and alcohol consumption. Alcohol Research. 2012;34:448–458. [PMC free article] [PubMed] [Google Scholar]
- Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcoholism: Clinical and Experimental Research. 2004;28:1829–1838. doi: 10.1097/01.alc.0000149977.95306.3a. [DOI] [PubMed] [Google Scholar]
- Belzung C, El Hage W, Moindrot N, Griebel G. Behavioral and neurochemical changes following predatory stress in mice. Neuropharmacology. 2001;41:400–408. doi: 10.1016/s0028-3908(01)00072-7. [DOI] [PubMed] [Google Scholar]
- Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biology. 2015;13:696–706. doi: 10.1080/15476286.2015.1094596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck KJ, Walter NA, Denmark DL. Genetic variability of respiratory complex abundance, organization and activity in mouse brain. Genes, Brain, and Behavior. 2014;13:135–143. doi: 10.1111/gbb.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai N, Chang S, Li Y, Li Q, Hu J, Liang J, et al. Molecular signatures of major depression. Current Biology. 2015;25:1146–1156. doi: 10.1016/j.cub.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- Ciobanu DC, Lu L, Mozhui K, Wang X, Jagalur M, Morris JA, et al. Detection, validation, and downstream analysis of allelic variation in gene expression. Genetics. 2010;184:119–128. doi: 10.1534/genetics.109.107474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC. Sensitivity to ethanol in inbred mice: genotypic correlations among several behavioral responses. Behavioral Neuroscience. 1983;97:280–289. doi: 10.1037//0735-7044.97.2.280. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Fidler TL, Murphy KV, Mulgrew JA, Smitasin PJ. Time-dependent negative reinforcement of ethanol intake by alleviation of acute withdrawal. Biological Psychiatry. 2013;73:249–255. doi: 10.1016/j.biopsych.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Green R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harbor Perspectives in Biology. 2012;4:a013706. doi: 10.1101/cshperspect.a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaher R, Finn D, Snelling C, Hitzemann R. Lesions of the extended amygdala in C57BL/6J mice do not block the intermittent ethanol vapor-induced increase in ethanol consumption. Alcoholism: Clinical and Experimental Research. 2008;32:197–208. doi: 10.1111/j.1530-0277.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- Farris SP, Mayfield RD. RNA-Seq reveals novel transcriptional reorganization in human alcoholic brain. International Review of Neurobiology. 2014;116:275–300. doi: 10.1016/B978-0-12-801105-8.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler TL, Powers MS, Ramirez JJ, Crane A, Mulgrew J, Smitasin P, et al. Dependence induced increases in intragastric alcohol consumption in mice. Addiction Biology. 2012;17:13–32. doi: 10.1111/j.1369-1600.2011.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, Ramachandra VS, Knackstedt LA, Becker HC. Repeated cycles of chronic intermittent ethanol exposure increases basal glutamate in the nucleus accumbens of mice without affecting glutamate transport. Frontiers in Pharmacology. 2015;6:27. doi: 10.3389/fphar.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour ME, Breusegem SY, Antrobus R, Freeman C, Reid E, Seaman MN. The cargo-selective retromer complex is a recruiting hub for protein complexes that regulate endosomal tubule dynamics. Journal of Cell Science. 2010;123(Pt 21):3703–3717. doi: 10.1242/jcs.071472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Egli M, Crabbe JC, Becker HC. Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addiction Biology. 2010;15:169–184. doi: 10.1111/j.1369-1600.2009.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejzlarová K, Mráček T, Vrbacký M, Kaplanová V, Karbanová V, Nůsková H, et al. Nuclear genetic defects of mitochondrial ATP synthase. Physiological Research. 2014;63(Suppl 1):S57–71. doi: 10.33549/physiolres.932643. [DOI] [PubMed] [Google Scholar]
- Holcik M. Could the eIF2α-Independent Translation Be the Achilles Heel of Cancer? Frontiers in Oncology. 2015;5:264. doi: 10.3389/fonc.2015.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson LH, Cryan JF. Feeling strained? Influence of genetic background on depression-related behavior in mice: a review. Behavior Genetics. 2007;37:171–213. doi: 10.1007/s10519-006-9106-3. [DOI] [PubMed] [Google Scholar]
- Janich P, Arpat AB, Castelo-Szekely V, Lopes M, Gatfield D. Ribosome profiling reveals the rhythmic liver translatome and circadian clock regulation by upstream open reading frames. Genome Research. 2015;25:1848–1859. doi: 10.1101/gr.195404.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BC, Sarrieau A, Reed CL, Azar MR, Mormède P. Contribution of sex and genetics to neuroendocrine adaptation to stress in mice. Psychoneuroendocrinology. 1998;23:505–517. doi: 10.1016/s0306-4530(98)00014-6. [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends in Cell Biology. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Wang L, Miles MF, Zhang L, Dodd PR, Harris RA. Gene expression in human alcoholism: microarray analysis of frontal cortex. Alcoholism: Clinical and Experimental Research. 2000;24:1873–1882. [PubMed] [Google Scholar]
- Liu J, Lewohl JM, Harris RA, Iyer VR, Dodd PR, Randall PK, et al. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology. 2006;31:1574–1582. doi: 10.1038/sj.npp.1300947. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Becker HC, Chandler LJ. Repeated episodes of chronic intermittent ethanol promote insensitivity to devaluation of the reinforcing effect of ethanol. Alcohol. 2014;48:639–645. doi: 10.1016/j.alcohol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MF, Griffin WC, 3rd, Melendez RI, Becker HC. Repeated cycles of chronic intermittent ethanol exposure leads to the development of tolerance to aversive effects of ethanol in C57BL/6J mice. Alcoholism: Clinical and Experimental Research. 2012;36:1180–1187. doi: 10.1111/j.1530-0277.2011.01717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MF, Moorman DE, Aston-Jones G, Becker HC. The highly selective orexin/hypocretin 1 receptor antagonist GSK1059865 potently reduces ethanol drinking in ethanol dependent mice. Brain Research. 2016;1636:74–80. doi: 10.1016/j.brainres.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, McMeekin LJ, Reid CS, Dobrunz LE, West AB, et al. PGC-1 α provides a transcriptional framework for synchronous neurotransmitter release from parvalbumin-positive interneurons. The Journal of Neuroscience. 2014;34:14375–14387. doi: 10.1523/JNEUROSCI.1222-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Devincci AM, Cook JB, O’Buckley TK, Morrow DH, McKinley RE, Lopez MF, et al. Chronic intermittent ethanol exposure and withdrawal alters (3α,5α)-3-hydroxy-pregnan-20-one immunostaining in cortical and limbic brain regions of C57BL/6J mice. Alcoholism: Clinical and Experimental Research. 2014;38:2561–2571. doi: 10.1111/acer.12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Devincci AM, Kampov-Polevoi A, McKinley RE, Morrow DH, O’Buckley TK, Morrow AL. Chronic Intermittent Ethanol Exposure Alters Stress Effects on (3α,5α)-3-hydroxy-pregnan-20-one (3α,5α-THP) Immunolabeling of Amygdala Neurons in C57BL/6J Mice. Frontiers in Cellular Neuroscience. 2016;10:40. doi: 10.3389/fncel.2016.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield RD, Harris RA, Schuckit MA. Genetic factors influencing alcohol dependence. British Journal of Pharmacology. 2008;154:275–287. doi: 10.1038/bjp.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, Chappell AM. Persistent enhancement of ethanol drinking following a monosodium glutamate-substitution procedure in C57BL6/J and DBA/2J mice. Alcohol. 2014;48:55–61. doi: 10.1016/j.alcohol.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, Chappell AM. Chronic intermittent ethanol inhalation increases ethanol self-administration in both C57BL/6J and DBA/2J mice. Alcohol. 2015;49:111–120. doi: 10.1016/j.alcohol.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI, McGinty JF, Kalivas PW, Becker HC. Brain region-specific gene expression changes after chronic intermittent ethanol exposure and early withdrawal in C57BL/6J mice. Addiction Biology. 2012;17:351–364. doi: 10.1111/j.1369-1600.2011.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metten P, Crabbe JC. Alcohol withdrawal severity in inbred mouse (Mus musculus) strains. Behavioral Neuroscience. 2005;119:911–925. doi: 10.1037/0735-7044.119.4.911. [DOI] [PubMed] [Google Scholar]
- Millstein RA, Holmes A. Effects of repeated maternal separation on anxiety- and depression-related phenotypes in different mouse strains. Neuroscience and Biobehavioral Reviews. 2007;31:3–17. doi: 10.1016/j.neubiorev.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, et al. Poly(A)-specific ribonuclease (PARN) mediates 3’-end maturation of the telomerase RNA component. Nature Genetics. 2015;47:1482–1488. doi: 10.1038/ng.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozhui K, Karlsson RM, Kash TL, Ihne J, Norcross M, Patel S, et al. Strain differences in stress responsivity are associated with divergent amygdala gene expression and glutamate-mediated neuronal excitability. The Journal of Neuroscience. 2010;30:5357–5367. doi: 10.1523/JNEUROSCI.5017-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterndorff-Kahanek EA, Becker HC, Lopez MF, Farris SP, Tiwari GR, Nunez YO, et al. Chronic ethanol exposure produces time- and brain region-dependent changes in gene coexpression networks. PloS One. 2015;10:e0121522. doi: 10.1371/journal.pone.0121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleil KE, Lopez A, McCall N, Jijon AM, Bravo JP, Kash TL. Chronic stress alters neuropeptide Y signaling in the bed nucleus of the stria terminalis in DBA/2J but not C57BL/6J mice. Neuropharmacology. 2012;62:1777–1786. doi: 10.1016/j.neuropharm.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. The Journal of Neuroscience. 2012;32:1884–1897. doi: 10.1523/JNEUROSCI.3136-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothion S, Bizot JC, Trovero F, Belzung C. Strain differences in sucrose preference and in the consequences of unpredictable chronic mild stress. Behavioural Brain Research. 2004;155:135–146. doi: 10.1016/j.bbr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Rainbolt TK, Saunders JM, Wiseman RL. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Reports. 2015;16:97–106. doi: 10.15252/embr.201438976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon B, Beliakova-Bethell N, Spina CA, Singhania A, Margolis DM, Richman DR, et al. Dose-responsive gene expression in suberoylanilide hydroxamic acid-treated resting CD4+ T cells. AIDS. 2015;29:2235–2244. doi: 10.1097/QAD.0000000000000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, Knaryan VH, Patel KS, Mulholland PJ, Becker HC, Banik NL. Chronic intermittent ethanol induced axon and myelin degeneration is attenuated by calpain inhibition. Brain Research. 2015;1622:7–21. doi: 10.1016/j.brainres.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleich S, Strassburger K, Janiesch PC, Koledachkina T, Miller KK, Haneke K, et al. DENR-MCT-1 promotes translation re-initiation downstream of uORFs to control tissue growth. Nature. 2014;512:208–212. doi: 10.1038/nature13401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea CJ, Carhuatanta KA, Wagner J, Bechmann N, Moore R, Herman JP, et al. Variable impact of chronic stress on spatial learning and memory in BXD mice. Physiology & Behavior. 2015;150:69–77. doi: 10.1016/j.physbeh.2015.06.022. [DOI] [PubMed] [Google Scholar]
- Smith ML, Lopez MF, Archer KJ, Wolen AR, Becker HC, Miles MF. Time-Course Analysis of Brain Regional Expression Network Responses to Chronic Intermittent Ethanol and Withdrawal: Implications for Mechanisms Underlying Excessive Ethanol Consumption. PloS One. 2016;11:e0146257. doi: 10.1371/journal.pone.0146257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song BJ. Ethanol-inducible cytochrome P450 (CYP2E1): biochemistry, molecular biology and clinical relevance: 1996 update. Alcoholism: Clinical and Experimental Research. 1996;20(8 Suppl):138A–146A. doi: 10.1111/j.1530-0277.1996.tb01764.x. [DOI] [PubMed] [Google Scholar]
- Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L, et al. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Molecular Biology of the Cell. 2012;23:1010–1023. doi: 10.1091/mbc.E11-08-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CK, Wang HF, Burns AM, Schroeder MR, Gaspari M, Baumann P. Human Telomerase RNA Processing and Quality Control. Cell Reports. 2015;13:2232–2243. doi: 10.1016/j.celrep.2015.10.075. [DOI] [PubMed] [Google Scholar]
- Wang F, Zhang L, Zhang GL, Wang ZB, Cui XS, Kim NH, et al. WASH complex regulates Arp2/3 complex for actin-based polar body extrusion in mouse oocytes. Scientific Reports. 2014;4:5596. doi: 10.1038/srep05596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama N, Crabbe JC, Ford MM, Murillo A, Finn DA. Voluntary ethanol consumption in 22 inbred mouse strains. Alcohol. 2008;42:149–160. doi: 10.1016/j.alcohol.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Statistical Applications in Genetics and Molecular Biology. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Research. 2005;33:W741–748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Panels A and B show individual level alcohol consumption and preference data for B6, respectively. Panels C and D show individual level consumption and preference data for D2, respectively. Average alcohol consumption (y-axis) is plotted across weeks for baseline (BASE) and treatment (TREAT) conditions and average preference data is shown on the y-axis for all 6 weeks of baseline drinking (BASE) and all 4 weeks of treatment (TREAT). B6 shows higher alcohol preference than D2 for all conditions (p = 1.29E−05). Although not significant, likely due to the low number of n and expected high variability in consumption between individuals of the same strain, both B6 mice in the CIE condition showed a trend toward elevated drinking from baseline compared to D2 (p = 0.17) and compared to the B6 AIR condition (p = 0.24). Note that statistical tests (e.g., t tests) are not recommended for n < 3.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module.
Sections are shown after removal of each region by LCM.
Panel on the left shows PCA biplot for PC1(X-axis) and PC2 (Y-axis) prior to batch correction. Tissue is identified by color and condition by shape. S (Solid) and P (Proton) labels indicate the RNA-seq platform used. Probability ellipses indicate grouping by batch as a major source of variation in the data set. Right panel shows PCA biplot following ComBat batch correction. Strain is indicated by B (B6) or D (D2), condition by geometric type, and tissue by color. Tissue origin is a major factor driving expression. CA1, CA3, CTX, and VTA samples have distinct and non-overlapping patterns of expression compared to samples from BNST, amygdala, dorsal striatum, and accumbens.
Directed acyclic graph tree (DAG) showing significantly (adjP < 0.05) enriched categories (red). Many enriched categories are related to transcription.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module. This module is enriched for terms related to transcription.
DAG tree showing significantly (adjP < 0.05) enriched categories (red). Many enriched categories are related to energy production, metabolism, and mitochondria.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module.
Genes highlighted in green show divergent expression differences between strains following chronic alcohol treatment. Many of these genes are well-connected hub genes in the PPI network module.