

Abstract
Protein geranylgeranylation reactions are dependent on the availability of geranylgeranyl diphosphate (GGDP), which serves as the isoprenoid donor. Inhibition of GGDP synthase (GGDPS) is of interest from a drug development perspective as GGDPS inhibition results in impaired protein geranylgeranylation, which in multiple myeloma, disrupts monoclonal protein trafficking and induces apoptosis. We have recently reported on a series of isoprenoid triazole bisphosphonates and have demonstrated that a 3:1 mixture of homogeranyl and homoneryl isomers potently, and in a synergistic manner, inhibits GGDPS. We now present the synthesis and biological evaluation of a novel series of bishomoisoprenoid triazoles which furthers our understanding of the structure-function relationship of this class. These studies demonstrate the importance of chain length and olefin stereochemistry on inhibitory activity.
Keywords: GGDP synthase, inhibition, isoprenoid biosynthesis, bishomoisoprenoids, triazole, bisphosphonate
Graphical Abstract
1. Introduction
In animals, geranylgeranyl diphosphate synthase (GGDPS) is a key enzyme in the mevalonate pathway. This enzyme catalyzes the formation of the C20 geranylgeranyl diphosphate (GGDP) from the C15 compound farnesyl diphosphate (FDP) and the C5 isopentyl diphosphate (IPP). Both FDP and GGDP are utilized in protein prenylation reactions. This post-translational modification is required for the proper membrane localization and function of numerous proteins, including many members of the Ras small GTPase superfamily. Protein prenylation has emerged as a potential drug target for both malignant and non-malignant conditions1,2 and there has been interest in the development of agents which target either the prenyltransferases responsible for the prenylation reaction (FTase, GGTase I, GGTase II)3,4 or the prenyl synthases which produce the isoprenoid donor (FDPS or GGDPS).5 We have been focused on the disruption of Rab GTPase geranylgeranylation as a novel therapeutic strategy for multiple myeloma. Our past studies have demonstrated that agents which impair Rab geranylgeranylation induce ER stress and apoptosis as a consequence of interference with monoclonal protein trafficking.6,7
Our recent efforts to disrupt Rab geranylgeranylation have focused on the development of potent and selective GGDPS inhibitors based on triazole bisphosphonates (Figure 1). A mixture (2) of geranyl and neryl triazole bisphosphonates was noted to inhibit GGDPS and it was determined that the neryl isomer (3) was approximately 40-fold more potent than the geranyl isomer (1, IC50 375 nM vs 17 μM). 8,9 Subsequently, a homogeranyl/homoneryl triazole bisphosphonate mixture (5, Figure 1) was found to be significantly more potent, with an IC50 of 45 nM.10 Synthesis of the pure isomers allowed assessment of the biological activities of the individual compounds.11 These studies revealed that while the homoneryl isomer (6) was more potent than the homogeranyl isomer (4), when used in combination the two isomers exhibited synergistic inhibitory effects on the target enzyme.12 Modeling studies suggested that the homoneryl isomer (6) preferentially binds to the FDP site while the homogeranyl isomer (4) preferentially binds to the GGDP site, thus establishing a new paradigm for the development of GGDPS inhibitors.12 We were therefore interested in determining whether further extension of the tether between the triazole and the internal olefin would affect GGDPS inhibitory activity. To begin to address this question we report the synthesis and biological activity of three new compounds that can be viewed as the bishomoprenyl, bishomogeranyl, and bishomoneryl triazoles.
Figure 1.

Triazole bisphosphonates and their activity in enzyme (GGDPS) and cellular assays.
2. Synthesis
The synthesis of this series began with alkylation of dimethyl malonate (7) with prenyl (8a), geranyl (8b), or neryl bromide (8c) to afford the desired alkylated products 9a,13 9b,14 and 9c (Scheme 1), respectively, in moderate to good yield. The monoalkylated malonate was the main product of each reaction, but purification was necessary to separate unreacted allylic bromide and a minor component assumed to be the dialkylated malonate. The methyl esters 9a–c were hydrolyzed upon treatment with potassium hydroxide and then acidified with HCl15 to afford dicarboxylic acids 10a,16 10b, and 10c in good yield. Decarboxylation of an alkylated dimethyl malonate has been reported by Krapcho17 to occur in water, DMSO, and LiCl. The presence of a salt such as LiCl is necessary to increase the rate of decarboxylation because dimethyl malonate derivatives otherwise are known to undergo only slow decarboxylation.17 However when Krapcho’s conditions17 were employed with the malonate derivative 9a, only starting material was observed after the reaction was heated at reflux for 5 hours. Fortunately, other conditions were found for the hydrolysis, and the decarboxylation of the malonic acid derivatives 10a–c proceeded smoothly when each was allowed to react with pyridine followed by HCl16 to give the desired monocarboxylic acids 11a,16 11b,18 and 11c. The 1H and 13C NMR spectra confirmed that only the desired products had been formed through the hydrolysis and decarboxylation steps, and chromatographic purification was unnecessary.
Scheme 1.

Synthesis of bromides 13a–c.
The carboxylic acids 11a–c then were reduced upon treatment with LiAlH4 to the bishomoallylic alcohols 12a,19 12b,20 and 12c21 (Scheme 1). A 21:1 ratio of Z:E isomers was observed in the 13C NMR spectrum of bishomonerol (12c). The ratio was determined by integration of the resonances at 32.1 and 39.9 ppm, which correspond to the methylene carbon at C-6.11,22 Because the E-isomer was such a minor component, the mixture was carried forward. The primary alcohols then were allowed to react with MsCl and Et3N followed by LiBr to afford the corresponding bishomoallylic bromides 13a,23 13b,24 and 13c in low to moderate yields. The low yield of bromide 13a may result from losses due to its relatively high volatility.
These primary bromides subsequently were treated with NaN3 in DMF to supply the azides 15a,25 15b,26 and 15c (Scheme 3). Once the bishomoallylic azides were formed they were immediately used in click reactions with acetylene bisphosphonate 14. These reactions proceeded smoothly under standard conditions27 to give the desired bishomoprenyl, bishomogeranyl, and bishomoneryl triazole bisphosphonates 16a–c as expected. The tetraethyl esters were hydrolyzed under standard conditions28 to provide the tetra sodium salts 17a–c. The neryl derivative 17c was a 96:4 Z:E mixture of olefin isomers as observed in the 13C NMR spectrum. In this case the ratio of olefin isomers was determined from the average integration of resonances corresponding to the sp2 olefin carbons (137.7 and 133.7 ppm for the Z-isomer and 137.6 and 133.5 ppm for the E-isomer) and the adjacent methylene group (31.1 ppm for the Z-isomer and 38.9 ppm for the E-isomer) in the 13C NMR spectrum.11 The 13C NMR spectrum of the geranyl-derived triazole 17b indicated that this analogue was a single isomer within the limits of detection.
3. Biological results and discussion
The ability of these three novel triazoles to disrupt protein geranylgeranylation in myeloma cells was determined via three independent methods: 1) immunoblot analysis for unmodified Rap1a (a substrate of GGTase I) (Figure 2A); 2) ELISA for intracellular lambda light chain which is a marker for disruption of Rab GTPase geranylgeranylation6 (Figure 2B); and 3) immunoblot analysis of Rab6 found in the detergent fraction (representing membrane-bound Rab6) from Triton X-114 lysis (Figure 2C). We already have demonstrated that the Rap1a and ELISA assays give concordant results for GGDPS inhibitors.9,10,12,29 The HMG-CoA reductase inhibitor lovastatin, which inhibits the synthesis of mevalonate and therefore depletes cells of all isoprenoids downstream of mevalonate, was used as a positive control in these studies.6 Of the three compounds examined, the bishomoneryl compound 17c most potently disrupted protein geranylgeranylation, with activity seen at a concentration as low as 0.5 μM. When ranked from most potent to least potent with respect to disruption of Rap1a geranylgeranylation, the order is 17c>17b>17a, indicating that the shorter isoprenoid chain length of 17a is less active than that of 17b/c, and that the olefin stereochemistry impacts activity. With respect to the olefin stereochemistry, these results are consistent with the previously described geranyl/neryl and homogeranyl/homoneryl sets of triazole bisphosphonates.9,12 Interestingly, however, while the results of all three assays were concordant for 17a and 17c, 17b appeared to more potently disrupt Rap1a geranylgeranylation (Figure 3A) than Rab geranylgeranylation (Figure 2B–C).
Figure 2. Effects of the bishomoisoprenoid triazole bisphosphonates on protein geranylgeranylation.

RPMI-8226 cells were incubated for 48 hours in the presence or absence of lovastatin (Lov, 10 μM) or varying concentrations of the test compounds. A) Immunoblot analysis of Rap1a (antibody detects only unmodified protein) and β-tubulin (as a loading control). B) Intracellular lambda light chain concentrations were determined via ELISA. Data are expressed as a percentage of control (mean + SD, n=3). The * denotes p < 0.05 per unpaired two-tailed t-test. C) Immunoblot analysis of the detergent fraction from Triton X-114 lysis of Rab6 and calnexin (loading control). Densitometric analysis of Rab6 (normalized to calnexin) for the treated cells normalized to untreated (control) cells is shown (relative intensity, Rel Int).
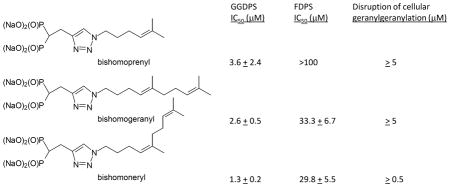
Next, the bishomoisoprenoids were tested for their ability to inhibit the prenyl synthases or prenyl transferases in in vitro enzyme assays. As shown in Table 1, all three inhibit GGDPS with a range of 1.3–3.6 μM for IC50 values. The compounds did not inhibit either FTase or GGTase I and displayed at least thirteen-fold selectivity for GGDPS over FDPS. For the most potent GGDPS inhibitor 17c, there was 23-fold selectivity for GGDPS over FDPS.
Table 1.
Inhibitory effects of the novel triazole bisphosphonates on prenyl synthases and prenyl transferases.
| GGDPS IC50 (μM) | FDPS IC50 (μM) | FTase IC50 (μM) | GGTase I IC50 (μM) | |
|---|---|---|---|---|
| 17a (bishomoprenyl) | 3.6 ± 2.4 | >100 | >100 | >100 |
| 17b (bishomogeranyl) | 2.6 ± 0.5 | 33.3 ± 6.7 | >100 | >100 |
| 17c (bishomoneryl) | 1.3 ± 0.2 | 29.8 ± 5.5 | >100 | >100 |
Because we had previously demonstrated that the combination of the homogeranyl and homoneryl triazole bisphosphonates synergistically inhibited GGDPS,12 we investigated whether the bishomo isomers behaved in a similar manner. The combination of the two isomers, either in a 1:1 or 3:1 (bishomogeranyl:bishomoneryl) mixture, did enhance GGDPS inhibition compared with either isomer alone (Supplementary Figure 1), but was not as strongly synergistic as the homoisomers.12 Similar studies performed in cell culture revealed that the combination of the two isomers did not result in a greater accumulation of unmodified Rap1a (Supplementary Figure 2). Interestingly, there was an antagonistic interaction with respect to intracellular light chain accumulation, particularly with higher concentrations of the isomers (Supplementary Figure 2).
4. Conclusions
In conclusion, we have developed a synthetic route to generate bishomoisoprenoid triazole bisphosphonates. These compounds have been evaluated in a series of enzyme and cell-based assays and found to be selective inhibitors of GGDPS, with the most potent inhibitor (17c) displaying submicromolar activity. This series expands our understanding of the structure-function relationship of isoprenoid triazole bisphosphonates as GGDPS inhibitors. Both the chain length and the olefin stereochemistry are important factors determining potency. With respect to GGDPS inhibitory activity, the bishomogeranyl isomer is intermediate in potency to the previously reported geranyl9 and homogeranyl12 compounds: 17b is six-fold more potent than geranyl 1 and 15-fold less potent than homogeranyl 4. However, the bishomoneryl 17c is less potent than both the corresponding neryl (3) and homoneryl (6) analogues in the GGDPS enzyme assay. Interestingly, although combinations of the bishomogeranyl (17b) and bishomoneryl (17c) isomers yielded synergistic/additive effects in the in vitro enzyme assay for GGDPS, antagonistic effects on protein geranylgeranylation were observed in cellular assays. These results are in contrast to those observed with the mixture of homogeranyl and homoneryl triazoles,12 suggesting unique properties of the bishomo derivatives. The mechanisms underlying these observations remain to be determined and future studies will explore these interactions.
5. Experimental procedures and methods
5.1 General experimental conditions
Tetrahydrofuran was freshly distilled from sodium/benzophenone, while methylene chloride was distilled from calcium hydride prior to use. All other reagents and solvents were purchased from commercial sources and used without further purification. All reactions in nonaqueous solvents were conducted in flame-dried glassware under a positive pressure of argon and with magnetic stirring. All NMR spectra were obtained at 300 or 500 MHz for 1H, and 75 or 125 MHz for 13C, with internal standards of (CH3)4Si (1H, 0.00) or CDCl3 (1H, 7.27; 13C, 77.2 ppm) for non-aqueous samples or D2O (1H, 4.80) and 1,4-dioxane (13C, 66.7 ppm) for aqueous samples. The 31P chemical shifts were reported in ppm relative to 85% H3PO4 (external standard). High resolution mass spectra were obtained at the University of Iowa Mass Spectrometry Facility. Silica gel (60 Å, 0.040–0.063 mm) was used for flash chromatography.
5.2 Representative procedure for alkylation of dimethyl malonate: Dimethyl 2–(3–methylbut–2–enyl)malonic acid dimethyl ester (9a)
According to the published procedure,13 solid NaH (60% dispersion in mineral oil, 477 mg, 11.9 mmol) was added to a stirred solution of dimethyl malonate (1.35 mL, 11.5 mmol) in THF (25 mL) at 0 °C and the reaction was allowed to stir for 30 minutes. Prenyl bromide (1.09 mL, 9.44 mmol) then was added dropwise to the reaction. The resulting mixture was allowed to stir overnight while slowly warming to rt and subsequently was quenched by addition of saturated NH4Cl. The resulting mixture was extracted with EtOAc (3x), dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (3% EtOAc in hexanes) afforded diester 9a (1.53 g, 81%) as a yellow oil, with 1H NMR data that is consistent with the literature data.13
5.3 2–(3,7–Diemthylocta–2E,6–dienyl)malonic acid dimethyl ester (9b)
According to the procedure described for preparation of dimethyl ester 9a, dimethyl malonate (4.2 mL, 35.6 mmol) in THF (79 mL) was treated with NaH (60% dispersion in mineral oil, 1.57 g, 39.2 mmol), followed by geranyl bromide (6.31 g, 29.1 mmol). A parallel work-up and purification afforded dimethyl ester 9b (4.86 g, 62%) as a clear oil. The 1H and 13C NMR data were consistent with literature data.14
5.4 2–(3,7–Diemthylocta–2Z,6–dienyl)malonic acid dimethyl ester (9c)
According to the procedure described for preparation of dimethyl ester 9a, dimethyl malonate (4.11 mL, 34.9 mmol) in THF (76 mL) was treated with NaH (60% dispersion in mineral oil, 1.52 g, 37.9 mmol), followed by neryl bromide (6.07 g, 27.9 mmol). A parallel work-up and purification afforded dimethyl ester 9c (4.17 g, 56%) as a pale yellow oil. The 1H and 13C NMR data were consistent with literature data.30
5.5 Representative procedure for hydrolysis of alkylated malonic acid dimethyl esters: Dimethyl 2–(3–methylbut–2–enyl)malonic acid (10a).16
According to the published procedure,31,15 to a stirred solution of dimethyl ester 9a (1.27 g, 6.36 mmol) in MeOH (24 mL), 5N KOH (3.8 mL, 19.0 mmol) was added. The resulting solution was heated at reflux for 1 hour, and the solvent then was removed in vacuo. The resulting residue was dissolved in H2O, acidified with 2N HCl to pH 2, and extracted with Et2O (4x). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to provide dicarboxylic acid 10a (905 mg, 83%) as a pale yellow solid, which was used without further purification: 1H NMR (300 MHz, CDCl3) δ 11.59 (br s, 2H), 5.11 (m, 1H), 3.46 (t, J = 7.8 Hz, 1H), 2.65 (t, J = 7.5 Hz, 2H), 1.71 (s, 3H), 1.65 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 175.3 (2C), 136.2, 118.9, 52.1, 27.6, 26.0, 18.0.
5.6 2–(3,7–Diemthylocta–2E,6–dienyl)malonic acid (10b).32
According to the procedure described for preparation of alkylated malonic acid 10a, dimethyl ester 9b (3.05 g, 11.4 mmol) in MeOH (43 mL) was treated with 5N KOH (11.4 mL, 57.0 mmol) and heated at reflux for 1 hour. A parallel work-up, except 1N HCl was used for acidification, provided dicarboxylic acid 10b (2.17 g, 79%) as a white solid which was used without further purification: 1H NMR (300 MHz, CDCl3) δ 10.93 (br s, 2H), 5.17–5.02 (m, 2H), 3.46 (t, J = 7.5 Hz, 1H), 2.66 (dd, J = 7.5, 7.5 Hz, 2H), 2.12–1.96 (m, 4H), 1.67 (s, 3H), 1.65 (s, 3H), 1.60 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 175.1 (2C), 139.7, 131.8, 124.1, 118.9, 52.0, 39.9, 27.6, 26.7, 25.8, 17.9, 16.3; HRMS (ES+) m/z calcd for C13H20O4Na (M + Na)+ 263.1259, found 263.1248.
5.7 2–(3,7–Diemthylocta–2Z,6–dienyl)malonic acid (10c)
According to the procedure described for preparation of alkylated malonic acid 10a, dimethyl ester 9c (2.00 g, 7.44 mmol) in MeOH (28 mL) was treated with 5N KOH (4.46 mL, 22.3 mmol) and heated at reflux for 1 hour. A parallel work-up, provided dicarboxylic acid 10c (1.54 g, 86%) as a white solid that was used without further purification: 1H NMR (300 MHz, CDCl3) δ 10.84 (br s, 2H), 5.17–5.06 (m, 2H), 3.44 (t, J = 7.5 Hz, 1H), 2.65 (dd, J = 7.2, 7.2 Hz, 2H), 2.10–2.04 (m, 4H), 1.71 (s, 3H), 1.69 (s, 3H), 1.61 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 175.2 (2C), 139.8, 132.3, 124.1, 119.6, 52.3, 32.1, 27.4, 26.6, 25.9, 23.6, 17.9; HRMS (ES+) m/z calcd for C13H20O4Na (M + Na)+ 263.1259, found 263.1255.
5.8 Representative procedure for decarboxylation of alkylated malonic acid: 5–Methyl–4–hexenoic acid (11a)
According to the published protocol,16 a stirred solution of dicarboxylic acid 10a (1.46 g, 8.47 mmol) in pyridine (3.4 mL) and H2O (0.15 mL) was heated at reflux for 2 hours. The reaction mixture then was allowed to cool to rt, diluted with H2O, acidified with 2N HCl to pH 2, and extracted with CH2Cl2 (5x). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to afford the desired monocarboxylic acid 11a (883 mg, 81%) as an orange oil that was used without further purification. The 1H and 13C NMR data were consistent with literature data.33
5.9 (4E)–5,9–Dimethyl–4, 8–decadienoic acid (11b)
According to the procedure used for formation of the carboxylic acid 11a, dicarboxylic acid 10b (2.15 g, 8.94 mmol) in pyridine (3.6 mL) and H2O (0.16 mL) was heated at reflux for 1 hour. A parallel work-up provided the desired monocarboxylic acid 11b (1.48 g, 84%) as an orange oil, which was used without further purification. The 1H and 13C NMR data were consistent with literature data.33
5.10 (4Z)–5,9–Dimethyl–4, 8–decadienoic acid (11c).34
According to the procedure used for formation of monocarboxylic acid 11a, dicarboxylic acid 10c (2.07 g, 8.63 mmol) in pyridine (3.5 mL) and H2O (0.15 mL) was heated at reflux for 2 hours. A parallel work-up provided the desired monocarboxylic acid 11c (1.37 g, 81%) as an orange oil, which was used without further purification: 1H NMR (300 MHz, CDCl3) δ 5.16–5.08 (m, 2H), 2.43–2.27 (m, 4H), 2.09–2.04 (m, 4H), 1.70 (s, 6H), 1.62 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 179.7, 137.3, 132.0, 124.3, 123.0, 34.6, 32.1, 26.7, 25.9, 23.6, 23.3, 17.9.
5.11 Representative procedure for carboxylic acid reduction to form bishomoallylic alcohols: Bishomoprenol (12a)
According to the published protocol,35 to a stirred suspension of LiAlH4 (95%, 504 mg, 13.3 mmol) in Et2O (35 mL) at 0 °C, carboxylic acid 11a (852 mg, 6.65 mmol) was added as a solution in Et2O (10 mL) over 10 minutes. The reaction flask then was removed from the ice bath, and the mixture was allowed to stir overnight while warming to room temperature. The reaction then was cooled to 0 °C, quenched by slow addition of 1N HCl followed by H2O, and extracted with Et2O (3x). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to afford the desired alcohol 12a (543 mg, 72%) as a clear oil that was used without further purification. The 1H and 13C NMR data were consistent with literature data.19
5.12 Bishomogeraniol (12b)
According to the procedure used for the production of alcohol 12a, to a suspension of LiAlH4 (95%, 596 mg, 14.9 mmol) in Et2O (65 mL) at 0 °C was added carboxylic acid 11b (1.46 g, 7.46 mmol) in Et2O (10 mL) over 7 minutes. The resulting mixture was allowed to stir for 2.5 hours while warming to room temperature. A parallel work-up gave the desired alcohol 12b (1.19 g, 88%) as a pale yellow oil that was used without further purification. Both the 1H and 13C NMR data were consistent with literature data.20
5.13 Bishomonerol (12c)
According to the procedure used for production of alcohol 12a, to a suspension of LiAlH4 (95%, 305 mg, 7.64 mmol) in Et2O (30 mL) at 0 °C was added carboxylic acid 11c (748 mg, 3.81 mmol) in Et2O (8 mL) over 9 minutes. The resulting mixture was allowed to stir overnight while warming to room temperature. A parallel work-up gave the desired alcohol 12c (502 mg, 72%) as a clear oil that was used without further purification. The 13C NMR spectrum shows a 95:5 mixture of Z and E olefin isomers. Both the 1H and 13C NMR data were consistent with literature data.21
5.14 Representative preparation of bishomoallylic bromides: Bishomogeranyl bromide (13b)
According to the published protocol,36 a stirred solution of alcohol 12b (1.18 g, 6.46 mmol) in CH2Cl2 (60 mL) at 0 °C, was treated with NEt3 (1.4 mL, 9.96 mmol), followed by methanesulfonyl chloride (0.65 mL, 8.40 mmol). The reaction mixture was allowed to stir at 0 °C for 1 hour and then was quenched by addition of H2O. The organic extract was washed with 1N HCl (2x), brine, and saturated NaHCO3 (2x), dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. The resulting mesylate then was added to a stirred solution of LiBr (1.69 g, 19.5 mmol) in anhydrous acetone (23 mL) and was heated at reflux. After 1 hour, the heat was turned off, and the reaction was allowed to stir overnight while it cooled to room temperature. After the solvent was removed in vacuo, the resulting residue was dissolved in H2O and extracted with Et2O (4x). The combined organic extracts were dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (100% hexanes) afforded the desired bromide 13b (966 mg, 61%) as a clear oil. The 1H NMR data was consistent with literature data.37 13C NMR (75 MHz, CDCl3) δ 137.0, 131.7, 124.4, 122.6, 39.9, 33.7, 33.0, 26.8, 26.5, 25.9, 17.9, 16.3.
5.15 Bishomoprenyl bromide (13a)
According to the procedure used for formation of bromide 13b, alcohol 12a (487 mg, 4.27 mmol) in CH2Cl2 (25 mL) was treated with NEt3 (0.90 mL, 6.4 mmol) and methanesulfonyl chloride (0.43 mL, 5.6 mmol) in succession, and the resulting mixture was allowed to stir for 40 minutes. A parallel work-up provided the desired mesylate. To a stirred solution of flame dried LiBr (947 mg) in anhydrous acetone (10 mL), the mesylate (574 mg, 2.98 mmol) in anhydrous acetone (7 mL) was added. The resulting mixture was heated at reflux for 40 minutes. A parallel work-up provided the desired bromide 13a (203 mg, 27%) as a yellow oil that was used without further purification. The 1H NMR data was consistent with literature data.23
5.16 Bishomoneryl bromide (13c)
According to the procedure used for formation of bromide 13b, alcohol 12c (502 mg, 2.76 mmol) in CH2Cl2 (26 mL) was treated with NEt3 (0.60 mL, 4.1 mmol) and methanesulfonyl chloride (0.28 mL, 3.6 mmol) in succession, and the resulting mixture was allowed to stir for 2 days. A parallel work-up provided the desired mesylate. To a stirred solution of flame dried LiBr (612 mg) in anhydrous acetone (8 mL), the mesylate (284 mg, 1.09 mmol) in anhydrous acetone (3 mL) was added. The resulting mixture was heated at reflux for 2.5 hours. A parallel work-up provided the desired bromide 13c (281 mg, 42%) as an orange oil that was used without further purification: 1H NMR (300 MHz, CDCl3) δ 5.13–5.09 (m, 2H), 3.43–3.39 (t, J = 6.5 Hz, 2H), 2.19–2.11 (m, 2H), 2.07–2.06 (m, 4H), 1.94–1.85 (m, 2H), 1.70 (m, 6H), 1.62 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 137.0, 131.9, 124.3, 123.4, 33.8, 33.3, 32.1, 26.8, 26.5, 25.9, 23.6, 17.8.
5.17 Tetraethyl (2–(1–(5–methylhexa–4–enyl)–1H–1,2,3–triazol–4–yl)ethane–1,1–diyl)bis(phosphonate) (16a)
According to the procedures described for preparation of triazoles,11 solid NaN3 (120 mg, 1.85 mmol) was added to a solution of bishomoprenyl bromide (13a, 202 mg, 1.14 mmol) in DMF (5.5 mL). A parallel work-up using Et2O provided the desired azide 15a (48%). The azide intermediate then was dissolved in a solution of t-BuOH and H2O (4:1, 3.4 mL), followed by addition of acetylene 14 (136 mg, 0.42 mmol), saturated CuSO4 (0.01 mL), and sodium ascorbate (25 mg, 0.13 mmol) in sequence. Standard work-up and purification afforded the desired triazole 16a (118 mg, 61%) as a yellow oil: 1H NMR (300 MHz, CDCl3) δ 7.47 (s, 1H), 5.10–5.04 (m, 1H), 4.27 (t, J = 7.3 Hz, 2H), 4.20–4.06 (m, 8H), 3.32 (td, JHP = 16.2 Hz, J = 6.7 Hz, 2H), 2.95 (tt, JHP = 23.4 Hz, J = 6.7 Hz, 1H), 2.06–1.97 (m, 2H), 1.96–1.87 (m, 2H), 1.69 (s, 3H), 1.57 (s, 3H), 1.32–1.24 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 145.1, 133.6, 122.5, 122.4, 63.0 (d, JCP = 6.5 Hz, 2C), 62.7 (d, JCP = 6.8 Hz, 2C), 49.8, 36.8 (t, JCP = 133.0 Hz), 30.6, 25.9, 25.0, 22.3 (br), 18.0, 16.6 (d, JCP = 3.6 Hz, 2C), 16.5 (d, JCP = 3.4 Hz, 2C); 31P NMR δ 22.5; HRMS (ES+) m/z calcd for C19H38N3O6P2 (M + H)+ 466.2236, found 466.2234.
5.18 Tetraethyl (4E)–(2–(1–(5,9–dimethyldeca–4,8–dienyl)–1H–1,2,3–triazol–4–yl)ethane–1,1–diyl)bis(phosphonate) (16b)
According to the general procedure for preparation of triazoles,11 solid NaN3 (128 mg, 1.97 mmol) was added to a solution of bishomogeranyl bromide (13b, 315 mg, 1.29 mmol) in DMF (6.2 mL). Standard work-up using Et2O provided the desired azide 15b (73%). The azide intermediate then was dissolved in a solution of t-BuOH and H2O (4:1, 5.09 mL), followed by addition of acetylene 14 (235 mg, 0.72 mmol), saturated CuSO4 (0.01 mL), and sodium ascorbate (43 mg, 0.22 mmol) in sequence. Standard work-up and purification afforded the desired triazole 16b (202 mg, 53%) as a yellow oil: 1H NMR (300 MHz, CDCl3) δ 7.47 (s, 1H), 5.14–5.05 (m, 2H), 4.29 (t, J = 7.2 Hz, 2H), 4.24–4.06 (m, 8H), 3.33 (td, JHP = 16.1 Hz, J = 6.3 Hz, 2H), 2.97 (tt, JHP = 23.4 Hz, J = 6.0 Hz, 1H), 2.13–1.88 (m, 8H), 1.68 (s, 3H), 1.60 (s, 3H), 1.58 (s, 3H), 1.30 (t, J = 6.9 Hz, 6H), 1.28 (t, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 145.1, 137.3, 131.7, 124.3, 122.4 (2C), 63.0 (d, JCP = 6.5 Hz, 2C), 62.7 (d, JCP = 6.6 Hz, 2C), 49.8, 39.9, 36.9 (t, JCP = 132.1 Hz), 30.6, 26.8, 25.9, 24.9, 22.3 (t, JCP = 5.0 Hz), 17.9, 16.5 (d, JCP = 6.3 Hz, 2C), 16.5 (d, JCP = 6.1 Hz, 2C), 16.3; 31P NMR δ 22.5; HRMS (ES+) m/z calcd for C24H45N3O6P2Na (M + Na)+ 556.2681, found 556.2690.
5.19 Tetraethyl (4Z)–(2–(1–(5,9–dimethyldeca–4,8–dienyl)–1H–1,2,3–triazol–4–yl)ethane–1,1–diyl)bis(phosphonate) (16c)
According to the general procedure for preparation of triazoles,11 solid NaN3 (116 mg, 1.78 mmol) was added to a solution of bishomoneryl bromide (13c, 281 mg, 1.15 mmol) in DMF (5.5 mL). Standard work-up using Et2O provided the desired azide 15c (75%). This azide intermediate then was dissolved in a solution of t-BuOH and H2O (4:1, 5.25 mL), followed by addition of acetylene 14 (216 mg, 0.66 mmol), saturated CuSO4 (0.01 mL), and sodium ascorbate (202 mg, 1.02 mmol) in sequence. Standard work-up and purification afforded the desired triazole 16c (169 mg, 48%) as a yellow oil: 1H NMR (300 MHz, CDCl3) δ 7.48 (s, 1H), 5.14–5.05 (m, 2H), 4.29 (t, J = 7.5 Hz, 2H), 4.23–4.07 (m, 8H), 3.33 (td, JHP = 16.2 Hz, J = 7.0 Hz, 2H), 2.97 (tt, JHP = 23.4 Hz, J = 6.4, 1H), 2.06–1.99 (m, 6H), 1.96–1.89 (m, 2H), 1.69–1.67 (m, 3H), 1.68 (s, 3H), 1.61 (s, 3H), 1.33–1.26 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 145.2, 137.3, 132.0, 124.2, 123.2, 122.3, 63.0 (d, JCP = 6.7 Hz, 2C), 62.7 (d, JCP = 6.7 Hz, 2C), 49.9, 36.9 (t, JCP = 133.0 Hz), 32.1, 30.9, 26.7, 25.9, 24.9, 23.6, 22.3 (t, JCP = 4.5 Hz), 17.8, 16.5 (d, JCP = 6.0 Hz, 2C), 16.5 (d, JCP = 6.3 Hz, 2C); 31P NMR δ 22.5; HRMS (ES+) m/z calcd for C24H46N3O6P2 (M + H)+ 534.2862, found 534.2874.
5.20 Sodium (2–(1–(5–methylhexa–4–enyl)–1H–1,2,3–triazol–4–yl)ethane–1,1–diyl)bis(phosphonate) (17a)
According to the general procedure described for formation of sodium salt 4,11 triazole 16a (116 mg, 0.25 mmol) in CH2Cl2 (7 mL) was treated with collidine (0.34 mL, 2.5 mmol) and TMSBr (97%, 0.40 mL, 3.0 mmol). The resulting residue was washed with toluene (10 mL, 3x) and then treated with 1N NaOH (20 mL, 20 mmol). Precipitation and removal of water on a lyophilizer afforded the initial salt. This process was repeated to remove excess NaOH. Purification by column chromatography using Dianion HP-20 resin (100% H2O) provided the desired salt 17a (23 mg, 21%) as a white powder. (Resonances observed at 7.90–7.84 ppm and 7.59–7.44 ppm in the 1H NMR spectrum and at 134.3, 131.2, 128.9, and 128.3 ppm in the 13C NMR spectrum correspond to styrene from the Dianion HP-20 resin): 1H NMR (500 MHz, D2O) δ 7.86 (s, 1H), 5.22–5.13 (m, 1H), 4.40–4.32 (m, 2H), 3.18 (td, JHP = 16.0 Hz, J = 7.2 Hz, 2H), 2.24–2.08 (m, 1H), 2.05–1.91 (m, 4H), 1.69 (s, 3H), 1.56 (s, 3H); 13C NMR (125 MHz) δ 165.6, 148.3, 136.4, 124.0, 122.8, 49.8, 40.1 (t, JCP = 111.9 Hz), 29.4, 24.9, 24.3, 22.3 (br), 17.0; 31P NMR δ 18.7; HRMS (ES+) m/z calcd for C11H19N3O6P2Na3 (M + Na)+ 420.0442, found 420.0443.
5.21 Sodium (4E)–(2–(1–(5,9–dimethyldeca–4,8–dienyl)–1H–1,2,3–triazol–4–yl)ethane–1,1–diyl)bis(phosphonate) (17b)
According to the general procedure described for formation of sodium salt 4,11 triazole 16b (90 mg, 0.17 mmol) in CH2Cl2 (4.7 mL) was treated with collidine (0.23 mL, 1.7 mmol) and TMSBr (97%, 0.27 mL, 2.0 mmol). The resulting residue was washed with toluene (10 mL, 3x) and then treated with 1N NaOH (0.90 mL, 0.90 mmol) under Ar (to exclude carbonate formation) for 2 hours. A parallel work-up, precipitation, and removal of water on a lyophilizer afforded the initial salt. The salt then was dissolved in H2O/D2O and allowed to stir in 1N NaOH (0.07 mL, 0.07 mmol) under Ar for 1 hour to remove residual collidine observed in the 1H NMR spectrum. The salt then was precipitated by addition of acetone, filtered, and lyophilized to remove water and provide the desired salt 17b (58 mg, 67%) as a white powder: 1H NMR (500 MHz, D2O) δ 7.86 (s, 1H), 5.23–5.16 (m, 2H), 4.37 (t, J = 7.0 Hz, 2H), 3.20 (td, JHP = 15.3 Hz, J = 7.0 Hz, 2H), 2.27 (tt, JHP = 21.0 Hz, J = 7.0 Hz, 1H), 2.16–2.09 (m, 2H), 2.06–2.00 (m, 4H), 2.00–1.93 (m, 2H), 1.69 (s, 3H), 1.63 (s, 3H), 1.57 (s, 3H); 13C NMR (125 MHz, D2O) δ 147.6 (t, JCP = 9.0 Hz), 137.6, 133.5, 124.5, 124.0, 123.0, 49.7, 39.8 (t, JCP = 114.1 Hz), 38.8, 29.3, 25.8, 24.9, 24.1, 21.9 (t, JCP = 4.1 Hz), 17.0, 15.2; 31P NMR δ 18.7; HRMS (ES−) m/z calcd for C16H28N3O6P2 (M – H)− 420.1453, found 420.1461.
5.22 Sodium (4Z)–(2–(1–(5,9–dimethyldeca–4,8–dienyl)–1H–1,2,3–triazol–4–yl)ethane–1,1–diyl)bis(phosphonate) (17c)
According to the general procedure described for formation of sodium salt 4,11 triazole 16c (82 mg, 0.16 mmol) in CH2Cl2 (4.5 mL) was treated with collidine (0.22 mL, 1.6 mmol) and TMSBr (97%, 0.26 mL, 2.0 mmol). The resulting residue was washed with toluene (10 mL, 3x) and then treated with 1N NaOH (0.86 mL, 0.86 mmol) under Ar for 25 minutes. The salt then was dissolved in H2O/D2O and allowed to stir in 1N NaOH (0.11 mL, 0.11 mmol) for 35 minutes to remove residual collidine observed in the 1H NMR spectrum. The salt then was precipitated by addition of acetone, filtered, and lyophilized to remove water and provide the desired salt 17c (39 mg, 48%) as a white powder. The 13C NMR spectrum shows a 96:4 mixture of Z and E olefin isomers. For the Z–isomer: 1H NMR (500 MHz, D2O) δ 7.87 (s, 1H), 5.22–5.15 (m, 2H), 4.37 (t, J = 6.9 Hz, 2H), 3.19 (td, JHP = 15.0 Hz, J = 7.0 Hz, 2H), 2.24–2.13 (m, 1H), 2.11–2.00 (m, 6H), 1.98–1.91 (m, 2H), 1.70 (s, 3H), 1.67 (s, 3H), 1.60 (s, 3H); 13C NMR (125 MHz, D2O) δ 148.1, 137.3, 133.7, 124.5, 124.3, 123.8, 49.9, 40.1 (t, JCP = 111.0 Hz), 31.1, 29.7, 25.8, 24.9, 24.2, 22.5, 22.1 (br), 16.9; 31P NMR δ 18.7; HRMS (ES−) m/z calcd for C16H28N3O6P2 (M – H)− 420.1453, found 420.1460.
5.23 Immunoblot analysis
RPMI-8226 (ATCC, Manassas, VA) cells were incubated (37 °C and 5% CO2) with test compounds for 48 hrs in RPMI-1640 media containing 10% fetal bovine serum and penicillin-streptomycin. Whole cell lysate was obtained using RIPA buffer (0.15 M NaCl, 1% sodium deoxycholate, 0.1% SDS, 1% Triton (v/v) X-100, 0.05 M Tris HCl) containing protease and phosphatase inhibitors. For Rab6, cells were lysed with Triton X-114 to generate detergent (membrane) fractions.38 Protein content was determined using the bicinchoninic acid (BCA) method (Pierce Chemical, Rockford, IL). Equivalent amounts of cell lysate were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membrane, probed with the appropriate primary antibodies, and detected using HRP-linked secondary antibodies and Bio-Rad Clarity ECL Substrate Western blotting reagents per manufacturer’s protocols.
5.24 Lambda light chain ELISA
Human lambda light chain kit (Bethyl Laboratories, Montgomery, TX) was used to quantify intracellular monoclonal protein levels of whole cell lysate. Lambda light chain levels were normalized to total protein levels (as determined by BCA).
5.25 FDPS and GGDPS enzyme assays
Recombinant FDPS was kindly provided by Dr. Raymond Hohl, Penn State Cancer Institute. Recombinant GGDPS was obtained from MyBioSource (San Diego, CA). Enzyme assays were performed as previously described.10 Compounds were tested in duplicate at multiple concentrations and three independent experiments were performed.
5.26 FTase and GGTase I assays
FTase and GGTase I activity was determined using the method of Temple et al.,39 with some modifications. For FTase, reaction mixtures were initiated by the addition of 18 nM recombinant FTase (Jena Biosciences) to reaction buffer (50 mM HEPPSO, 5 mM TCEP-HCl, 5 mM MgCl2) containing 10 μM FDP (Sigma) and 5 μM dansyl-GCVLS (Biosynthesis, Inc.). For GGTase I, 24 nM of recombinant GGTase I (Jena Biosciences) was added to reaction buffer (50 mM HEPPSO, 5 mM TCEP-HCl,) containing 10 μM GGDP (Sigma) and 5 μM dansyl-GCVLS (Biosynthesis, Inc.). Reactions were carried out at 30 °C in the presence or absence of test compounds for 1 hour (FTase) or 2 hours (GGTase I). Prenylation of the peptide results in an increase in fluorescence intensity of the dansyl group (λex = 340 nm, λem = 520 nm). Changes in fluorescence over time were detected using a Molecular Devices Spectramax Gemini EM fluorescence microplate reader.
5.27 Statistics
Two-tailed t-testing was used to calculate statistical significance. An α of 0.05 was set as the level of significance. CompuSyn software (ComboSyn, Inc.,) was used to analyze the concentration response curves and determine the IC50 values as well as the combination indices for the enzyme assays. This software is based on the work of Chou and Talalay.40,41
Supplementary Material
Scheme 2.

Synthesis of the bishomoallylic compounds 17a–c.
Acknowledgments
Financial support from the Roy J. Carver Charitable Trust (01–224) and the National Institutes of Health (R01CA-172070) is gratefully acknowledged. We thank the UI Graduate College for a Dean’s Graduate Fellowship and an AGEP Fellowship (to V.S.W.) and the Center for Biocatalysis and Bioprocessing for a fellowship (to V.S.W.) through the predoctoral Training Program in Biotechnology (T32 GM008365).
Footnotes
Supplementary data associated with this article, including NMR spectra and supplementary figures 1–2, can be found in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang M, Casey PJ. Nat Rev Mol Cell Biol. 2016;17:110. doi: 10.1038/nrm.2015.11. [DOI] [PubMed] [Google Scholar]
- 2.Gao S, Yu R, Zhou X. Mol Neurobiol. 2016;53:6925. doi: 10.1007/s12035-015-9594-3. [DOI] [PubMed] [Google Scholar]
- 3.Holstein SA, Hohl RJ. Curr Opin Pharmacol. 2012;12:704. doi: 10.1016/j.coph.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 4.Palsuledesai CC, Distefano MD. ACS Chem Biol. 2015;10:51. doi: 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiemer AJ, Hohl RJ, Wiemer DF. Anticancer Agents Med Chem. 2009;9:526. doi: 10.2174/187152009788451860. [DOI] [PubMed] [Google Scholar]
- 6.Holstein SA, Hohl RJ. Leuk Res. 2011;35:551. doi: 10.1016/j.leukres.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dykstra KM, Allen C, Born EJ, Tong H, Holstein SA. Oncotarget. 2015;6:41535. doi: 10.18632/oncotarget.6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou X, Hartman SV, Born EJ, Smits JP, Holstein SA, Wiemer DF. Bioorg Med Chem Lett. 2013;23:764. doi: 10.1016/j.bmcl.2012.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou X, Ferree SD, Wills VS, Born EJ, Tong H, Wiemer DF, Holstein SA. Bioorg Med Chem. 2014;22:2791. doi: 10.1016/j.bmc.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wills VS, Allen C, Holstein SA, Wiemer DF. ACS Med Chem Lett. 2015;6:1195. doi: 10.1021/acsmedchemlett.5b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthiesen RA, Wills VS, Metzger JI, Holstein SA, Wiemer DF. J Org Chem. 2016;81:9438. doi: 10.1021/acs.joc.6b01693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen C, Kortagere S, Tong H, Matthiesen RA, Metzger JI, Wiemer DF, Holstein SA. Mol Pharmacol. 2017;91:229. doi: 10.1124/mol.116.107326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nyfeler E, Renaud P. Org Lett. 2008;10:985. doi: 10.1021/ol702832x. [DOI] [PubMed] [Google Scholar]
- 14.Tripp JC, Schiesser CH, Curran DP. J Am Chem Soc. 2005;127:5518. doi: 10.1021/ja042595u. [DOI] [PubMed] [Google Scholar]
- 15.Yu JS. University of Iowa; 2007. [Google Scholar]
- 16.Brimioulle R, Bach T. Science. 2013;342:840. doi: 10.1126/science.1244809. [DOI] [PubMed] [Google Scholar]
- 17.Krapcho AP, Weimaster JF, Eldridge JM, Jahngen J, EGE, Lovey AJ, Stephens WP. J Org Chem. 1978;43:138. [Google Scholar]
- 18.Pereira AR, Strangman WK, Marion F, Feldberg L, Roll D, Mallon R, Hollander I, Andersen RJ. J Med Chem. 2010;53:8523. doi: 10.1021/jm100531u. [DOI] [PubMed] [Google Scholar]
- 19.Flachsbarth B, Fritzsche M, Weldon PJ, Schulz S. Chem Biodiversity. 2009;6:1. doi: 10.1002/cbdv.200800265. [DOI] [PubMed] [Google Scholar]
- 20.Cosgrove KL, McGeary RP. Synlett. 2009:1749. [Google Scholar]
- 21.Roe AN, McPhail AT, Porter NA. J Am Chem Soc. 1983;105:1199. [Google Scholar]
- 22.Bohlmann F, Zeisberg R, Klein E. Organic Magnetic Resonance. 1975;7:426. [Google Scholar]
- 23.Boyer FD, Hanna I. Org Lett. 2007;9:2293. doi: 10.1021/ol070708j. [DOI] [PubMed] [Google Scholar]
- 24.Zakharova S, Fulhorst M, Luczak L, Wessjohann L. ARKIVOC. 2004:79. [Google Scholar]
- 25.Chong WKM, Chu S, Duvadie RK, Li L, Na J, Schaffer L, Yang Y. Pfizer Inc; USA: 2004. p. 179. [Google Scholar]
- 26.Reddy PG, Varghese B, Baskaran S. Org Lett. 2003;5:583. doi: 10.1021/ol027563v. [DOI] [PubMed] [Google Scholar]
- 27.Skarpos H, Osipov SN, Vorob’eva DV, Odinets IL, Lork E, Roeschenthaler GV. Org Biomol Chem. 2007;5:2361. doi: 10.1039/b705510b. [DOI] [PubMed] [Google Scholar]
- 28.McKenna CE, Higa MT, Cheung NH, McKenna MC. Tetrahedron Lett. 1977;18:155. [Google Scholar]
- 29.Foust BJ, Allen C, Holstein SA, Wiemer DF. Bioorg Med Chem. 2016;24:3734. doi: 10.1016/j.bmc.2016.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trost BM, Verhoeven TR. J Am Chem Soc. 1980;102:4730. doi: 10.1021/ja00418a063. [DOI] [PubMed] [Google Scholar]
- 31.Yu J, Kleckley TS, Wiemer DF. Org Lett. 2005;7:4803. doi: 10.1021/ol0513239. [DOI] [PubMed] [Google Scholar]
- 32.Dicker DW, Whiting MC. J Chem Soc. 1958:1994. [Google Scholar]
- 33.Cermak DM, Wiemer DF, Lewis K, Hohl RJ. Bioorg Med Chem. 2000;8:2729. doi: 10.1016/s0968-0896(00)00212-1. [DOI] [PubMed] [Google Scholar]
- 34.Jaeger V, Kuhn W, Buddrus J. Tetrahedron Lett. 1986;27:2587. [Google Scholar]
- 35.Mori K, Sugai T, Maeda Y, Okazaki T, Noguchi T, Naito H. Tetrahedron. 1985;41:5307. [Google Scholar]
- 36.Gash RC, MacCorquodale F, Walton JC. Tetrahedron. 1989;45:5531. [Google Scholar]
- 37.Biller SA, Magnin DR. E. R. Squibb and Sons, Inc; USA: 1992. p. 52. [Google Scholar]
- 38.Wasko BM, Dudakovic A, Hohl RJ. J Pharmacol Exp Ther. 2011;337:540. doi: 10.1124/jpet.110.175521. [DOI] [PubMed] [Google Scholar]
- 39.Temple KJ, Wright EN, Fierke CA, Gibbs RA. Bioorg Med Chem Lett. 2016;26:3503. doi: 10.1016/j.bmcl.2016.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou TC. Pharmacol Rev. 2006;58:621. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 41.Chou TC, Talalay P. Adv Enzyme Regul. 1984;22:27. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.