

Abstract
RNA-protein interactions play a crucial role in every aspect of RNA metabolism, and also plays a major role in post-transcriptional gene regulation. RNA-binding proteins have been implicated in viral gene expression (Ray and Das, 2002) and microRNA-mediated gene regulation ( Poria et al., 2016 ). Here we have described the protocol which (1) covalently links transiently interacting RNA-protein complexes by UV crosslinking, (2) removes the unprotected RNA by RNase digestion and (3) detects the RNA-protein complexes by SDS-PAGE analysis. This protocol provides a rapid and reliable means to directly assay RNA-protein interactions and their kinetics using purified proteins and also help in identifying novel RNA-protein interactions
Keywords: RNA-protein interaction, UV-crosslinking, RNA-binding proteins
Background
RNA-protein interactions are mediated by transient non-covalent interactions such as electrostatic interactions and hydrogen bonds between specific residues in RNA and protein molecules. Short wave UV radiation can induce covalent bond formation between two closely placed aromatic rings. Aromatic ring structures are found in several amino acids in proteins and in nitrogenous bases in nucleic acids. Therefore, UV irradiation is used to covalently link RNA and interacting proteins, whereby the RNA-protein complex can be further analysed by SDS-Polyacrylamide gel electrophoresis. This protocol describes a simple and rapid assay system that can assay RNA-protein interactions and their binding kinetics in vitro. Also, mass spectrometric analysis of the fluorescently-labeled RNA-protein complexes obtained by this method can lead to identification of novel RNA-protein interactions.
Materials and Reagents
1.5 ml RNase, DNase free microcentrifuge tube (Corning, Axygen®, catalog number: MCT-150-C or equivalent)
96-well round-bottomed plate (Greiner Bio one International, catalog number: 650101)
Agarose (Lonza, catalog number: 50004)
Transcription Kit (MAXIscript® Kit) (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM1312 or any equivalent)
10 µCi/µl α-P32 UTP (BRIT, catalog number: PLC 108 or PerkinElmer, catalog number: BLU007H250UC) (Alternatively, Cy5-UTP can be used to generate fluorescently labelled RNA [GE Healthcare, catalog number: PA55026])
DNase I (optional) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EN0521)
100% ethanol (EMD Millipore, catalog number: 100983)
Nuclease free water
Ammonium acetate (Thermo Fisher Scientific, Affymetrix, catalog number: 75901)
Glycerol (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15514)
Urea-polyacrylamide gel
Yeast tRNA (Sigma-Aldrich, catalog number: R8759)
RNase inhibitor (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EO0381)
RNase A (Sigma-Aldrich, catalog number: R6513)
10% SOD-PAGE
Pre-stained protein markers or radiolabeled protein markers
HEPES (pH 7.4) (Thermo Fisher Scientific, Affymetrix, catalog number: 16926)
Potassium chloride (KCl) (AMRESCO, catalog number: 0395)
Magnesium chloride hexahydrate (MgCl2.6H2O) (AMRESCO, catalog number: 0288)
Dithiothreitol (DTT) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0861)
EDTA (AMRESCO, catalog number: 0105)
ATP (Sigma-Aldrich, catalog number: A8937)
SDS
Tris-Cl (pH 6.8)
Bromophenol blue
2x RNA binding buffer (see Recipes)
2x denaturing protein loading buffer (see Recipes)
Equipment
Refrigerated centrifuge (Eppendorf, model: 5418 R)
UV cross-linker or UV torch with 254 nm wavelength UV (UVP, model: CL1000)
Vertical gel electrophoretic system (Bio-Rad Laboratories, model: Mini-PROTEAN Tetra Cell, catalog number: 1658000EDU)
Scintillation counter (Hidex, model: Triathler or any equivalent model)
Phosphorimager (GE Healthcare, model: Typhoon Trio or any equivalent model)
Procedure
-
Template preparation for in vitro transcription
Clone the DNA template encoding the RNA of interest derived by a T7 promoter in a plasmid vector and linearize the plasmid at a site downstream of the DNA template using specific restriction enzyme.
Run the linearized plasmid DNA on a 0.8% agarose gel, excise the band and extract the linearized DNA for in vitro transcription.
Alternatively use a T7 promoter-adapter containing forward primer and gene specific reverse primer to amplify target DNA incorporating T7 promoter for in vitro transcription (Figure 1).
For oligo driven transcription of short RNAs, commercially synthesized template DNA oligo with T7 promoter site is annealed with a T7 promoter-adapter oligo to generate the template for in vitro transcription (Figure 2).
-
P32UTP body-labelled RNA preparation (proper biosafety protocol should be followed)
-
Setup the following in vitro transcription reaction using α-P32UTP as radiolabel
10x transcription buffer 2 μl
10 mM ATP 1 μl
10 mM CTP 1 μl
10 mM GTP 1 μl
100 μM UTP 1 μl
α-P32 UTP 2 μl
Linearized DNA template 10 μl (~1 µg)
T7 RNA polymerase 2 μl
Incubate for 1 h 30 min at 37 °C.
Add 1 μl DNase I (optional) to remove the DNA template. Incubate at 37 °C for 15 min.
Remove unincorporated nucleotides either by ethanol precipitation or column purification. For ethanol precipitation, add 29 μl nuclease free water to the transcription reaction to bring the volume to 50 μl. Add 5 μl 5 M ammonium acetate, 2 μg glycogen and 2 volumes of 100% ethanol, and precipitate at -80 °C for at least 2 h. Spin for 20 min at maximum speed in a 4 °C centrifuge and wash the pellet once with cold 70% ethanol before drying.
Dissolve the dried RNA pellet in 20 μl nuclease free water. Take 1 μl of labelled RNA and count the specific activity with a scintillation counter or run on an 8 M urea-polyacrylamide gel at the percentage appropriate for the size of the RNA.
-
-
RNA-protein binding and UV crosslinking
-
Setup the following binding reaction for each sample in 1.5 ml microcentrifuge tubes on ice
2x RNA binding buffer (with 3 mM ATP) 6 μl
10 mg/ml yeast tRNA 1 μl
Radiolabeled RNA x μl (~100,000 cpm)
RNase inhibitor (40 U/μl) 0.5 μl
Purified protein/cell lysate x μl (minimum 50 ng)
Nuclease free water to make up the volume to 12 µl
Incubate on ice for 30 min.
Carefully transfer to a precooled 96-well round-bottomed plate placed on ice.
Place the plate containing the reaction mixture under the UV light source of UV crosslinker or UV torch and apply 500 mJ/cm2 radiation for 10 min on ice.
Transfer the reaction to 1.5 ml tubes.
Add 2 µl RNase A (10 μg/μl) and incubate the tubes for 30 min at 37 °C.
-
-
Separation of RNA-protein complex and visualization (Figure 3)
Add 13 μl of 2x denaturing protein loading buffer.
Boil the samples for 5 min at 100 °C and resolve in 10% SOD-PAGE with pre-stained protein markers or radiolabeled protein markers.
Dry the gel and expose overnight to a phosphorimager screen.
Scan the screen in a phosphorimager.
Alternatively expose the dried gel to X-ray film for 24 h and develop the film.
Figure 1. Preparation of template DNA for transcription using T7 promoter.

Figure 2. Preparation of DNA template for oligo-driven transcription.

Figure 3. UV-crosslinking of 32P labelled PDCD4-3’UTR RNA with 500 ng of purified 6x His-tagged HuR protein (lane 1) and in the presence of 10x (lane 2) and 100x unlabelled PDCD4 3’UTR RNA.
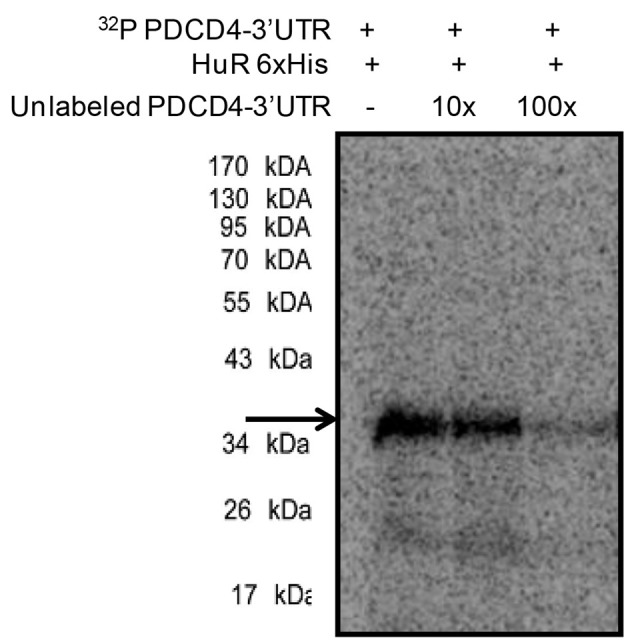
RNA-protein complexes were digested with RNase A and resolved in 10% SDS-PAGE and exposed for phosphorimaging. The HuR-32P PDCD4-3’UTR RNA complexes are indicated by the arrow.
Recipes
-
2x RNA binding buffer
10 mM HEPES (pH 7.4)
50 mM KCl
4 mM MgCl2
4 mM dithiothreitol (DTT)
0.2 mM EDTA
7.6% glycerol
3 mM ATP
-
2x denaturing protein loading buffer
4 ml 10% (w/v) SDS (final concentration 4%)
2 ml glycerol (final concentration 20%)
1.2 ml 1 M Tris-Cl (pH 6.8) (120 mM)
2.8 ml DEPC-H2O (to make volume upto 10 ml)
Add bromophenol blue to a final concentration of 0.02% (w/v)
Acknowledgments
We thank members of our laboratory for trying out and standardizing this protocol. Research which led to the development of these protocols was funded by a Wellcome Trust-DBT India Alliance Intermediate fellowship (WT500139/Z/09/Z) to PSR and a CSIR, India Senior Research Fellowship to DKP. This protocol was adapted from the protocol described in Ray and Das (2002).
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1.Poria D. K., Guha A., Nandi I. and Ray P. S.(2016). RNA-binding protein HuR sequesters microRNA-21 to prevent translation repression of proinflammatory tumor suppressor gene programmed cell death 4. Oncogene 35(13): 1703-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ray P. S. and Das S.(2002). La autoantigen is required for the internal ribosome entry site- mediated translation of Coxsackievirus B3 RNA. Nucleic Acids Res 30(20): 4500-4508. [DOI] [PMC free article] [PubMed] [Google Scholar]