

SUMMARY
Segmental duplications contribute to human evolution, adaptation and genomic instability but are often poorly characterized. We investigate the evolution, genetic variation and coding potential of human-specific segmental duplications (HSDs). We identify 218 HSDs based on analysis of 322 deeply sequenced archaic and contemporary hominid genomes. We sequence 550 human and nonhuman primate genomic clones to reconstruct the evolution of the largest, most complex regions with protein-coding potential (n=80 genes/33 gene families). We show that HSDs are non-randomly organized, associate preferentially with ancestral ape duplications termed “core duplicons”, and evolved primarily in an interspersed inverted orientation. In addition to Homo sapiens-specific gene expansions (e.g., TCAF1/2), we highlight ten gene families (e.g., ARHGAP11B and SRGAP2C) where copy number never returns to the ancestral state, there is evidence of mRNA splicing, and no common gene-disruptive mutations are observed in the general population. Such duplicates are candidates for the evolution of human-specific adaptive traits.
Genetic mutations have shaped the unique adaptation and evolution of the human lineage, but their characterization has been a slow and difficult endeavor. Despite a few potential success stories over the years with various degrees of support1, the genetic basis of most of the unique aspects of human adaptation await discovery. As sequencing technologies have improved, more systematic efforts have been directed to discover regulatory differences among the great apes2–6. One potential source of genetic variation, which has been difficult to explore due to missing or erroneous sequences within reference genomes, are genes embedded within recently (<25 million years ago (mya)) duplicated regions also called segmental duplications (SDs)7. Unlike the focus on regulatory mutations or gene loss, which typically modify the expression of ancestral genes mapping to unique regions, duplicated regions have long been recognized as a potential source for the rapid evolution of new genes with novel functions8. Recent functional studies have emphasized the potential importance of SDs with respect to unique features of synaptogenesis, neuronal migration, and neocortical expansion in the human lineage9–12.
The genomes of apes are enriched in SDs having experienced a burst of interspersed duplications over the last 10 million years of evolution13,14. The mosaic and interspersed architecture of ape SDs offers tremendous potential for transcript innovation because duplicate paralogs may be truncated, combined with other transcripts to create fusion genes, or acquire alternate promoters directing the differential expression of novel transcripts15. Previous investigations have been limited to microarray studies16,17 and whole-genome sequencing read-depth comparisons14,18,19 between humans and great apes. None of these methods provide information regarding the structure and sequence of the duplicated segments, limiting gene annotation and the understanding of the functional potential of the duplicated genes.
In this study, we focus on understanding the sequence structure, genetic variation, and transcriptional potential of the largest human-specific segmental duplications (HSDs). HSDs are particularly problematic because they are highly identical (~99%), among the most copy number polymorphic parts of the genome, and frequently embedded within larger blocks of shared ape duplications. Not surprisingly, the genome assembly builds of these regions are highly enriched for euchromatic gaps and misassembly errors even within the most recent versions of the human reference20,21. We specifically target 33 human-specific gene families contained within these HSDs for high-quality sequence assembly by selecting large-insert bacterial artificial chromosome (BAC) clones from a library (CH17) generated from a well-characterized complete hydatidiform mole cell line (CHM1tert). The mole derives from the fertilization of an enucleated human oocyte with a single spermatozoon22,23 or from postzygotic loss of a complete parental genome24. The end result is a haploid as opposed to a diploid equivalent of the human genome where the absence of allelic variation allows high-identity paralogous regions of the genome to be rapidly resolved11. We apply the resulting high-quality sequence to more systematically investigate copy number variation, transcriptional potential, and human genetic variation in an effort to understand their evolutionary history as well as discover regions that have become fixed and potentially functional in the human species.
RESULTS
Refining regions of HSDs
With the wealth of deep-coverage Illumina sequence data from both humans and great apes, we began by first redefining the map location of HSDs. We mapped a genetically diverse panel of 236 human and 86 chimpanzee, gorilla and orangutan genomes to the human reference (GRCh37) to identify regions uniquely duplicated in humans (Figure 1, Supplementary Figure 1). Our approach identified 218 autosomal regions ranging in size from 5 kbp (our size threshold) to 362 kbp with HSDs dispersed non-randomly near each other (empirical median distance to nearest HSD 440 kbp, P < 1×10−7; Supplementary Figure 2; see Methods). Of these regions, 85 included entire or parts of RefSeq annotated genes (Supplementary Table 1). We orthogonally validated 87% (190/218) of our events as HSDs.
Figure 1. Identification of human-specific segmental duplications or HSDs.
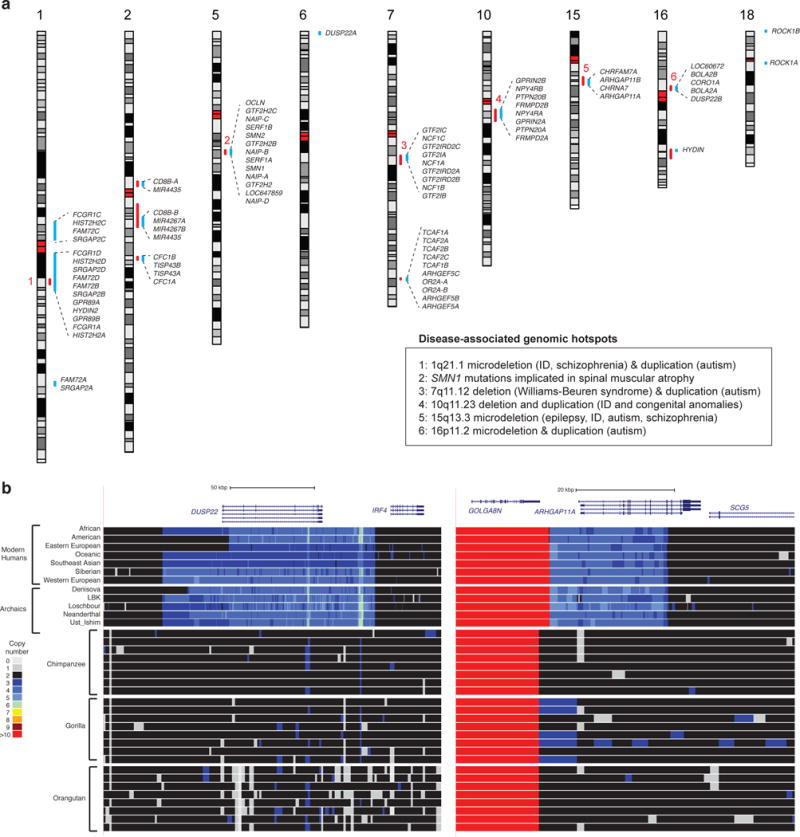
(a) The locations of large, gene-containing HSDs are highlighted (blue lines) with 80 individual gene paralogs from 33 gene families listed across 9 different human autosomes. Included in this set are paralogs of GPR89, which duplicated in other great apes but experienced human-specific expansions. Many of these HSDs overlap known disease-implicated genomic hotspots (red lines) prone to recurrent copy number variation associated with developmental delay. The genomic hotspots labeled with numbers (1 to 6) have significant associations with specific disorders including epilepsy, autism, schizophrenia and intellectual disability (ID). (b) Duplicated regions were detected based on read-depth analysis of Illumina reads mapped to the human reference genome (GRC37). The set included a diversity panel of humans (Human Genome Diversity Project or HGDP (N = 236)30) and nonhuman primates or NHPs (gorillas (N = 32), chimpanzees (N = 23), bonobos (N = 14), and orangutans (N = 17)34). Overall copy number (CN) was averaged across 500 bp sliding windows and depicted as colored heatmaps (see pictured index). Also pictured are heatmaps for Neanderthal, Denisovan, and archaic human (LBK, Loschbour, and Ust Ishim) individuals. Any genomic region >5 kbp shown to have diploid CN ≥ 3 in 90% of humans tested compared to all NHPs was considered an HSD.
The set included 38 previously unreported HSDs mapping to genic regions. Among these, we included HSDs where there was evidence of independent or distinct duplications in great apes (i.e., homoplasy; N = 21) and duplications corresponding to introns (N = 12). For example, the 3′ portion of MST1L (macrophage stimulating 1 like) on chromosome 1p36.13 is partially duplicated in chimpanzee and gorilla, but a complete duplication of the gene (>36 kbp) has risen to high copy uniquely in humans (diploid copy number (CN) > 8; Supplementary Figure 3). Similarly, we identified a 6.6 kbp duplication corresponding the third intron of CACNA1B (calcium voltage-gated channel subunit alpha1 B)—a pore-forming subunit of an N-type voltage-dependent calcium channel that controls neurotransmitter release from neurons (Supplementary Figures 3 and 4). We also identified a novel duplication of SCGB1C1 (secretoglobin family 1C member 1), a gene family whose products are secreted at large concentrations in the lung, lacrimal and salivary glands (Supplementary Figure 3).
Next, we focused on the largest gene-containing HSD regions (>20 kbp; Supplementary Figure 5). These HSDs and their ancestral counterparts reside on 16 autosomal regions with many appearing to cluster with other smaller HSDs and at “genomic hotspots”—regions prone to recurrent large-scale microdeletions and microduplications associated with neurodevelopmental disorders (Figure 1, Supplementary Table 2)25. Using the haploid BAC library (CH17), we generated alternate sequence assemblies (Supplementary Tables 3 and 4) of which 18.2 Mbp have now been incorporated into the most recent human reference build (GRCh38), allowing us to close 24 euchromatic gaps and correct large-scale errors in the human reference genome. The new sequence allowed us to distinguish 28 HSD events ranging in size from 11 kbp to 677 kbp corresponding to 33 HSD gene families accounting for 80 paralogous genes (Supplementary Table 5).
The majority of events (N = 24 events or 3.2 Mbp) were primary duplications—defined here as the initiating SD from the ancestral locus shared between human and chimpanzee (Figure 2). Compared to a random null distribution, we found that these primary HSDs map closer to each other than by chance (empirical median distance to nearest HSD 377 kbp, P = 1×10−7; Supplementary Figure 2). Consistent with previous observations26–28, we also found our primary HSDs to be significantly enriched near core duplicons—high-copy ancestral ape duplications significantly linked with the accumulation of SDs and breakpoints of rearrangement (empirical median distance to a core 250 kbp, P < 1×10−7; Supplementary Figure 6). We identified four secondary HSDs—additional duplications derived from a human-specific duplicate paralog. These secondary events account for 35% (1.7 Mbp) of HSD base pairs because the events are larger when compared to primary duplications (minimum median sizes: 497 kbp vs. 95 kbp, P = 0.041, Wilcoxon-Mann-Whitney test). The majority of HSDs are intrachromosomal and arranged in inverted orientation with respect to their ancestral paralogs (18/26, P = 0.014, binomial test), including all secondary duplications (4/4). HSD clustering is most pronounced on chromosome 1p12 to 1q32.1, which contains the greatest number of gene-containing HSDs (at least 6 independent HSD events including ~2 Mbp (0.8%) of human chromosome 1; Supplementary Note; Supplementary Figure 7). We find that 85% of this 8 Mbp region (chr1:119,989,248–121,395,939 and chr1:143,311,826–149,876,379, GRCh38) has been duplicated in humans and great apes with only 1.15 Mbp remaining unique in humans.
Figure 2. Timing of HSDs.
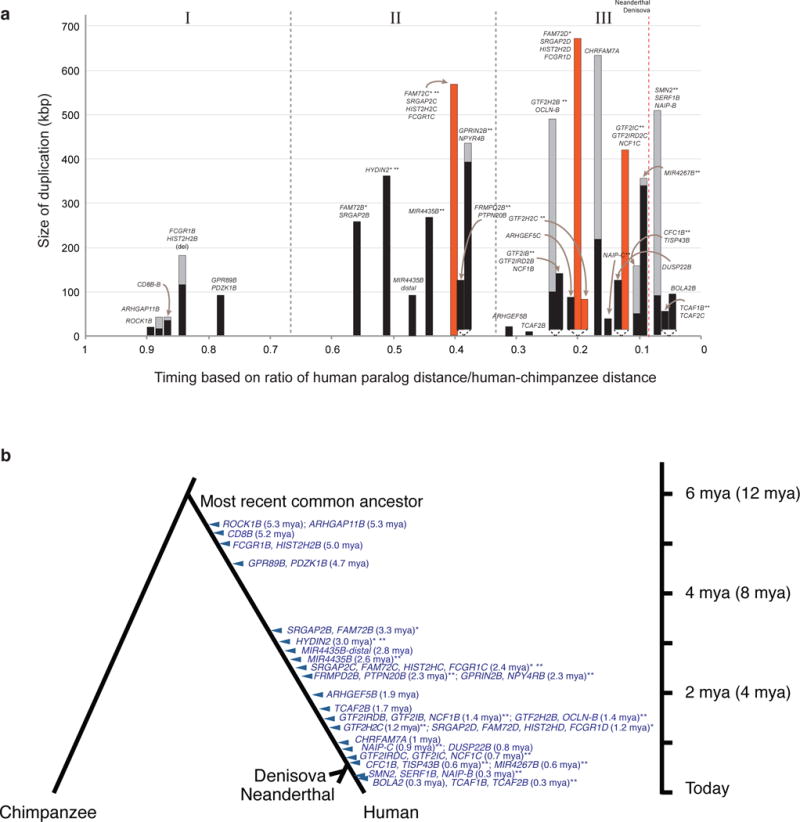
(a) Duplication timing estimates are plotted as a ratio of human–chimpanzee divergence (x-axis). The total estimated size of primary (black) and secondary (orange) duplications is shown for each event. Uncertainty in the size of each event is due to breakpoints mapping in high-identity flanking duplications (gray). (b) Generally accepted phylogeny indicating timing of each event assuming chimpanzee and human lineage divergence of 6 million years ago (mya)76–78. Recent estimates based on recalibrated substitution rates suggest an earlier divergence time of 12 mya, with this maximum scale indicated in parentheses. The analysis is based on high-quality sequencing, assembly and alignment of large-insert clones (human N = 224; NHP N = 269). Asterisks indicate adjusted timing estimates because of failed Tajima’s D relative rate (*) and genes with evidence of interlocus gene conversion (**).
Evolutionary timing of HSDs
We sequenced large-insert clones from nonhuman primate (NHP) genomic BAC libraries and applied standard phylogenetic methods under a model of gene conversion29 to understand the evolutionary timing of each large HSD (Supplementary Tables 6–8, Supplementary Figure 8). The results reveal differences in number and size of HSDs when we compare across three equal time periods during the evolution of the human lineage (P = 0.017; Figure 2). The first was a period of relative quiescence, which occurred after the human–chimpanzee divergence. This included five smaller primary duplications corresponding to seven genes with a median minimum size of 36 kbp for a total of 285 kbp. This was followed temporally by a set of larger primary (N = 6) and secondary (N = 1) duplications containing 12 HSD genes (median minimum size of primary events 262 kbp for a total of 1.5 Mbp, P = 0.026). The final set of duplications involved more secondary (N = 3) and primary (N = 13) duplications and are estimated to be the most recent. Although primary duplication lengths were not significantly different in size compared to either of the other two time periods (median minimum size 93 kbp for a total of 1.4 Mbp), they resulted in many more HSD genes (28 gene paralogs).
Human copy number diversity
We undertook three different approaches to assess the potential functional significance of HSDs—namely, copy number constraint, transcriptional potential and protein-coding mutations. We first assessed copy number in contemporary and archaic hominin (N= 2,384)30,31,32,33 as well as 86 NHP genomes34 in order to distinguish fixed duplications from those that are highly stratified among humans (Figure 3, Supplementary Tables 9–11). Thirteen HSD genes were among the most copy number polymorphic, including genes at chromosomes 7q35 (three units: ARHGEF5 and OR2A, TCAF1, and TCAF2), 5q13.1 (four units: SMN1 and SERF1, GTF2H2, OCLN, and NAIP), 16p11.2 (two units: BOLA2 and DUSP22), and 10q11.23 (one unit: GPRIN2 and NPY4R). Conversely, eight HSD genes were largely fixed for copy number, showing the lowest variance among contemporary human populations (six units: HYDIN, GPR89 and PDZK1, CFC1 and TISP43, CD8B, ROCK1, and ARHGAP11; Figure 3, Supplementary Figures 9 and 10, Supplementary Tables 12 and 13). As expected, higher copy number HSD genes are generally more copy number polymorphic (Supplementary Note). We identified 11/23 duplicated units with at least one normal individual identified who carried the ancestral state copy number (diploid CN of two) suggesting the HSD paralogs are missing in these individuals (e.g., DUSP22 and ROCK1; Figure 3B). Population differentiation (as measured by Vst35) generally correlated with copy number variance (R2 = 0.32; ρ = 0.54, Pearson correlation) but not copy number (R2 = 0.01; ρ = −0.01, Pearson correlation; Supplementary Figure 11).
Figure 3. Human copy number diversity.
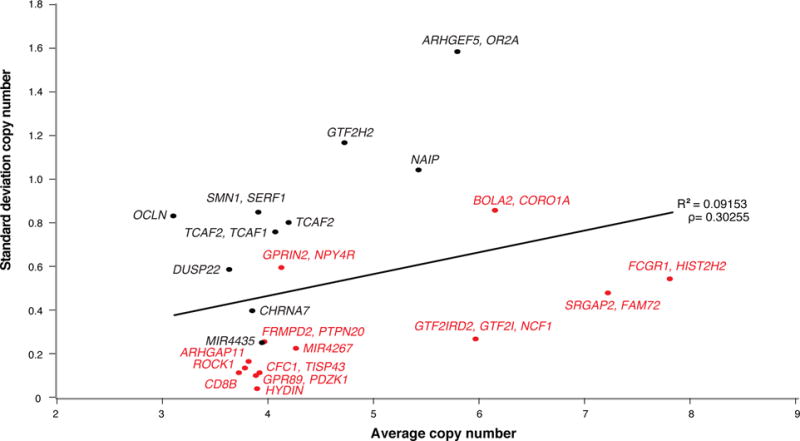
Overall average CN was calculated per individual from read depth produced from Illumina mappings across a set region defining each duplication (Supplementary Table 9) in human populations, including the HGDP (N = 236; GRCh38) and 1000 Genomes Project (1KG, N = 2,143; GRCh37) cohorts, NHPs, archaic humans, a Denisovan and a Neanderthal. From these results, the mean, standard deviation (s.d.), Vst, and number of individuals with CN = 2 indicating no duplicate paralogs exist were calculated for average CN of each duplicated gene family (Supplementary Table 11). For each gene family, plots are shown for the CN average vs. s.d. across all HGDP individuals with duplicate gene family names indicated next to each data point. Red data points indicate genes with no homozygous deletions in any human tested. Genes with higher s.d. are considered CN polymorphic and tend to have higher CN (R2 = 0.09; ρ = 0.30, Pearson correlation) and average Vst (R2 = 0.32; ρ = 0.54, Pearson correlation; Supplementary Figure 11).
We also identified three genes expanded uniquely in Homo sapiens when compared to two sequenced archaic hominins, a Neanderthal and a Denisovan. This included the previously reported BOLA2 on chromosome 1632,36 (Supplementary Figure 12) and two novel genes, TRPM8-associated TCAF1 and TCAF2 (formerly FAM115A and FAM115C), on chromosome 7 (Figure 4, Supplementary Table 14, Supplementary Note)37. In the case of TCAF1 and TCAF2, the timing estimate (0.048 ± 0.008 human–chimpanzee distance) is consistent with its absence in archaic hominins. The fact that we observe high copy number in two archaic humans (Loschbour and Ust Ishim individuals with CN ≥ 6) suggests these HSDs spread rapidly in the population. The TCAF1/TCAF2 HSD is differentiated (HGDP mean Vst = 0.11) between human populations with the highest copy number observed for African and European populations (in particular, Gambian and Esan from Nigeria where multiple individuals with diploid CN = 7 are observed) and the lowest copy number observed for Asian and Amerindian populations.
Figure 4. Copy number polymorphism across diverse populations of TCAF1 and TCAF2 HSDs.
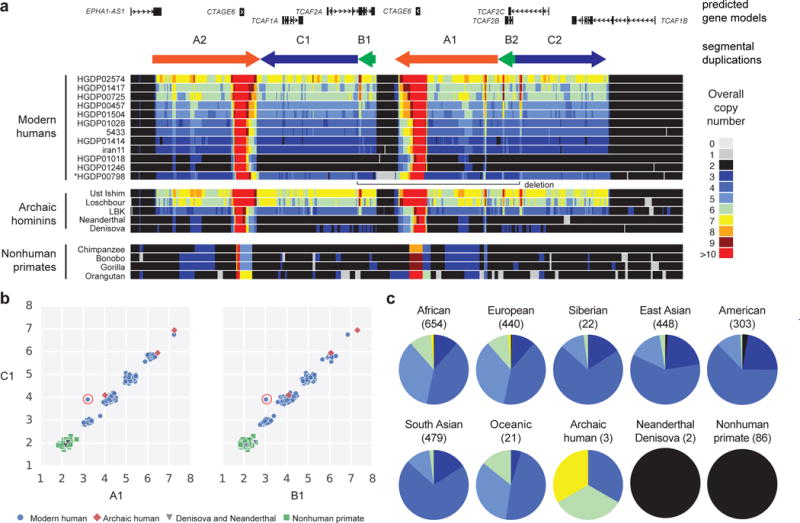
(a) Heatmap of overall CN of TCAF1 and TCAF2 HSD region on human chromosome 7 with predicted gene models and segmental duplications (SDs; depicted as colored arrows) pictured above. Representative modern humans are shown for each genotyped CN across the locus with a single person (*HGDP00798) showing deletion of the region, likely due to non-allelic homologous recombination between directly oriented SDs B1 and B2 (Supplementary Note). (b) A scatter plot of TCAF1 and TCAF2 SDs (A1, B1, and C1), overall CN of individuals from modern human (HGDP cohort), archaic humans, a Denisova and a Neanderthal, and NHPs (chimpanzee, bonobo, gorilla, and orangutan) plotted on each axis. The one Western European individual circled in red that deviates from the rest of the individuals’ copy numbers is the deletion carrier pictured in a. (c) CN predictions across modern humans from the 1KG and HGDP (N = 2,379), archaic hominins, and NHPs were made across a representative region (chr7:143,533,137-143,571,789; GRCh37). Overall CNs in the pie charts per population are represented as colors depicted in the legend shown panel a.
Patterns of HSD mRNA expression
To understand expression differences, we specifically examined RNA-seq data from GTEx38 and mapped the distribution of reads to HSDs in 45 different tissues across multiple individuals (Supplementary Tables 15 and 16, Supplementary Figures 13 and 14). Of the 26 comparisons that could be made between known ancestral and duplicate paralogs (Supplementary Figure 13), 65% (17/26) of duplicate paralogs showed significantly lower expression levels compared to their ancestral paralog (versus 19% (5/26) showing significantly greater expression and 15% (4/26) showing no difference in expression). In contrast, human-specific FRMPD2B and CHRFAM7A each show increased expression in specific tissues compared to their ancestral paralogs (FRMPD2A and CHRNA7). Both of the derived duplicates are incomplete, lacking the 5′ portion when compared to the ancestral gene. CHRFAM7A, for example, is the product of a gene fusion of FAM7A and CHRNA7 duplications and shows increased expression in the aorta, liver, lung, testis, and thyroid. FRMPD2B shows increased expression compared with FRMPD2A in several regions of the brain cortex as well as reproductive organs, including fallopian tubes and uterus.
Discovery of likely gene-disruptive events
Since pseudogenization is the most likely fate of duplicated genes, we tested whether paralogs had accumulated likely gene-disrupting (LGD) mutations by targeted sequencing of canonical protein-coding exons of 30 gene families using molecular inversion probes (MIPs)39. In 658 individuals from the 1000 Genomes Project, we identified 96 LGD variants (25 gene families) of which 33 could be definitively assigned to a copy (Supplementary Tables 10, 17–19). Ten duplicate paralogs and no ancestral paralogs harbored common loss-of-function mutations (population frequency >5%) suggesting potential functional constraint. The remaining LGD variants (N = 63) could not be unambiguously assigned but typically fell into gene families with more than one duplicate paralog. Overall, we identified no LGD variants in five gene families and discovered only rare LGD variants (<5% population frequency) in an additional 15 gene families (Supplementary Table 10).
Next, we sequenced and compared the LGD frequency in 3,444 children with autism and 2,617 unaffected siblings. From this set, we identified 4,069 total coding and splice variants, of which 247 were LGD, in both cases and controls for 30 genes (Supplementary Tables 20 and 21). The majority of LGD variants (N = 231) were considered rare and collectively found in equal proportions of cases and controls (24% of individuals). Examining burden of rare LGD variants of individual genes, we identified a nominal enrichment in cases versus controls of GPR89 with seven variants exhibiting an overall frequency of 19/3,430 cases versus 5/2,605 controls (puncorr = 0.02, Supplementary Figure 15), though this result did not pass Bonferroni multiple-testing correction. Common LGD variants existed in 11/30 genes, with six genes (FCGR1, GTF2I, GTF2IRD2, HIST2H2BF, HYDIN, and ROCK1) carrying fixed LGD variants in nearly all individuals tested. Notably, five of these genes represent partial duplications where we might expect a greater likelihood of disruptive mutations if the paralogs represent pseudogenes. Combining these results with our assessment of 1000 Genomes Project individuals, 16/30 gene families showed an absence of common protein-disrupting variants in the 6,719 humans tested (Supplementary Table 10).
The complexity of HSD evolutionary history
To highlight the complex evolutionary history associated with such regions, we selected three loci for further investigation (see Supplementary Note for details of genomic hotspot chromosome 10q11.23). Large deletions ~1.8 Mbp in size of the chromosome 7q11.23 region lead to Williams-Beuren syndrome (OMIM #194050) and reciprocal duplications are associated with autism and intellectual disability40. The directly oriented flanking HSDs (labeled B; Figure 5A) contain three genes: GTF2I, GTF2IRD2, and NCF1. Our analysis predicts that the most common human haplotype arose through a three-step evolutionary process. The first two events occurred within the distal duplication cluster of the region (Supplementary Figure 16). They involved an inverted duplication of a ~116 kbp SD (termed A, containing paralogs of the high-copy duplicon (SPDYE)) and a possible 90 kbp inversion (0.318 ± 0.028 human–chimpanzee distance) followed by a separate ~106 kbp inverted duplication of B (0.229 ± 0.019 human–chimpanzee distance). These events created truncated paralogs GTF2IB and GTF2IRD2B and a full-length version of NCF1B. A third large-scale inverted duplication transposed an ~395 kbp region comprised of SDs A, B, and C (containing POM121L) from the distal to proximal breakpoints of the disease-associated region (0.122 ± 0.014 human–chimpanzee distance). This tertiary duplication established “granddaughter” truncated copies of GTF2IRDC, GTF2IC, as well as a full-length paralog NCF1C, likely overwriting the 3′ end of the ancestral POM121 with POM121L. This final event created directly oriented SDs A and B, providing a substrate for non-allelic homologous recombination leading to disease-associated copy number variants. The great ape sequence (Supplementary Figure 17) matched nearly perfectly the deduced genomic configuration hypothesized previously41, with the exception of a large-scale inversion of the region proximal to BP1 in orangutan.
Figure 5. Complex models of HSD evolutionary history.
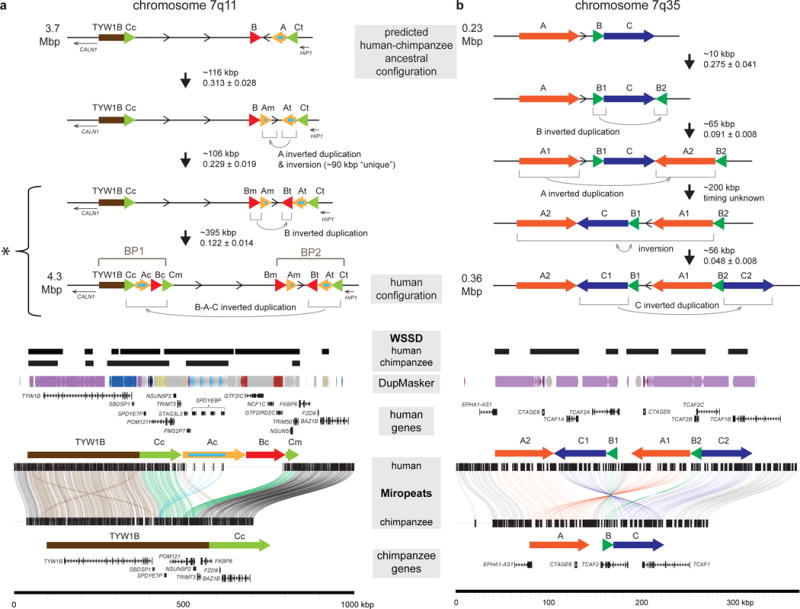
BACs tiling across human chromosome (a) 7q11 and (b) 7q35 regions were sequenced and assembled (representing human and additional great apes) and supercontigs were created. Estimates of sizes and evolutionary timing (human–chimpanzee distance; Supplementary Table 8) of events are denoted between each predicted intermediate genomic structure. SD organization is depicted as colored arrows across the 7q11 (SDs annotated with subscripts representing relative positions including centromeric (c), middle (m), and telomeric (t) as previously defined41) and 7q35 regions. The orientations of intervening regions are shown with arrows. Models of the predicted evolutionary histories of the HSDs at all loci are depicted starting with the predicted human–chimpanzee common ancestor to the most common haplotype present in modern humans. A Miropeats comparison of the human and chimpanzee contigs shows the pairwise differences between the orthologous regions. Lines connect stretches of homologous regions based on a chosen threshold (s), defined as the number of matching bases minus the number of mismatching bases (s = 500 for a, s = 1000 for b) and match the arrow colors when they connect SD blocks. Additional annotations include whole-genome shotgun sequence detection (WSSD) in human and chimpanzee, indicating duplicated regions identified by sequence read depth56, DupMasker79, and genes.
We also characterized one of the youngest HSD regions unique to modern humans on chromosome 7q35 containing TCAF1 and TCAF2 and primate-duplicated CTAGE642 (Figure 4, Supplementary Note, Supplementary Figure 18). We note that expansion of a CTAGE-paralog also occurred in the duplication of HSD gene ARHGEF5, located less than 500 kbp distal to this locus. Pairwise comparisons between human and chimpanzee suggest the possibility of three distinct duplication events (A: 65 kbp, B: 10 kbp, and C: 56 kbp) as well as a large-scale inversion (~200 kbp) (Figure 5B). We estimate an initial 10 kbp inverted duplication of HSD B containing the 3′ end of TCAF2A (0.275 ± 0.041 human–chimpanzee distance) creating a truncated TCAF2B. The subsequent events occurred very recently during human evolution potentially during or after the split from a common ancestor of Denisova and Neanderthal. These subsequent rearrangements created a new full-length paralog of CTAGE6 (contained in A; 0.091 ± 0.008 human–chimpanzee distance) and truncated paralogs TCAF1A (the putative ancestral paralog contained in C1; 0.048 ± 0.008 human–chimpanzee distance) and TCAF2C (contained in C2). Notably, we estimate that the full-length and functional TCAF1B and TCAF2A now reside on distinct SD paralogs that are separated by 130 kbp transcribed on opposite strands—as opposed to the ancestral configuration where the genes are tandem, adjacent, and transcribed on the same strand.
DISCUSSION
In this study we generated new reference sequence for some of the most complex and gap-ridden sequence of the human genome. Several important features emerge from our targeted sequencing (48.4 Mbp) and evolutionary reconstruction of HSD regions (Supplementary Discussion). The largest HSDs are significantly clustered near core duplicons, including at chromosomes 1q21, 5q13 and 7q11.3. Most regions have been subjected to multiple large structural variation events during human evolution with inverted duplications being the predominant mode of structural change (71.4% of the total predicted 28 intrachromosomal duplication events, P = 0.006; Supplementary Table 5). Inverted SDs have been noted before in complex structural rearrangements associated with genomic disorders, such as Pelizaeus-Merzbacher disease43,44 and Smith-Magenis syndrome45, and may be a product of replication-based mechanisms, such as fork-stalling and template switching (FoSTeS)43 and/or microhomology-mediated break-induced repair (MMBIR)46.
We enriched for potential functional HSD genes by applying three criteria: (1) all humans must carry the duplicate paralog; (2) no common truncating mutations are observed in the human population and (3) duplicates show evidence of spliced mRNA expression. Ten HSD gene families met all criteria, including two genes previously implicated in cortical development and neuronal spine density, ARHGAP11B12 and SRGAP2C10,11, as well as the gene families BOLA2, CD8B, CFC1, FAM72, GPR89, GPRIN2, NPY4R, and TISP43. GPRIN2 (G protein regulated inducer of neurite outgrowth 2) has been shown to interact directly with G-coupled proteins (GNAO1 and GNAZ)47 and has been implicated in the control of neurite outgrowth48. Our RNA-seq analysis points to localized expression in various regions of the brain, including the cerebellum and hypothalamus (Figure 5). Other genes of interest include CFC1 (cripto, FRL-1, cryptic family 1), which encodes a member of the epidermal growth factor important in patterning the left-right embryonic axis49, and NPY4R (neuropeptide Y receptor Y4)—a gene implicated in energy homeostasis. Large copy number variants of the region are associated with obesity50 (Supplementary Discussion; Supplementary Figure 20).
Although our analysis provides a framework for the evolution of new human-specific genes, there are a number of limitations. First, additional mutation events, such as interlocus gene conversion (IGC), frequently occur between high-identity paralogs51–53 (Supplementary Figure 19). We identified 2.9% of the sequence showing signatures of IGC, consistent with previous estimates51 (Supplementary Table 7). Though such duplications will make HSDs appear evolutionarily “younger”, excluding these regions increases our timing estimates only by a small degree (on average an increase of 0.008 human–chimpanzee distance across 17 HSDs; Supplementary Table 8). The second caveat is that the full extent of HSDs is often difficult to assess because they frequently occur in duplication blocks where there have been multiple rounds of structural variation over the last 15 million years. Breakpoints and boundaries become challenging to delineate due to a series of overlapping complex rearrangements (e.g., SMN1 region on chromosome 5q13.3; Supplementary Discussion; Supplementary Figure 21). Third, we assume that any individual with two copies of a gene family represents the ancestral non-duplicated state. It is possible that the alternative scenario of duplication followed by subsequent deletion of the ancestral paralog may have occurred (Supplementary Discussion; Supplementary Figures 22 and 23; Supplementary Tables 22 and 23). Fourth, this study focuses on protein-encoding gene models and does not consider the possibility of functional noncoding RNA. Notably, three of the annotated genes (MIR4435, MIR4267, and OR2A) mapping to HSDs are identified as noncoding RNA (Ensemble Variant Effect Finder). Moreover, the canonical gene model being investigated in our analysis is heavily weighted by the ancestral intron-exon structure (Supplementary Discussion; Supplementary Figure 24). Thus, novel fusion genes and transcripts not previously annotated that have gained alternate promoters would not have been considered36. It is likely that long-read genome and transcriptome data will be required to explore such gene innovations20.
Finally, although we focused on HSDs that had become fixed in the human population, it may be that some of the most copy number polymorphic loci (or, additionally, loci that exist at <90% frequency in the population) are candidates for more recent adaptations between populations30. In this regard, duplications of TCAF1/TCAF2 are particularly intriguing. The genes encode TRP channel-associated factors that bind to TRPM8—the primary detector of environmental cold54,55 expressed in 10–15% of somatosensory neurons. The two TCAF proteins are thought to exert opposing effects in TRPM8 gating and insertion into the plasma membrane37. Our copy number analysis agrees with our evolutionary finding that duplications of this locus are Homo sapiens-specific—not existing in Neanderthal and Denisova but at high copy in archaic humans. In modern humans, African and European populations show the greatest copy numbers while Asians show the lowest with some humans showing no duplication of the region (Figure 4). The model suggests that a single full-length paralog of TCAF1B (predicted HSD duplicate paralog) and TCAF2A (predicted ancestral paralog) exist at the locus, respectively, while additional TCAF1/TCAF2 copies appear to be truncated or incomplete. It is interesting to note that the conserved function of full-length TCAF2 may have been co-opted by a duplicate paralog after truncation of the ancestral paralog, a mechanism we also suggest occurred for duplicate family PTPN20 (Supplementary Figures 25 and 26). Although the function of the truncated duplicates awaits further characterization, it is clear that this locus has been radically restructured in most humans resulting in the ancestral functional loci being separated by hundreds of kilobase pairs and being transcribed in opposite orientations with the potential effect of altering regulation of these genes important in cold sensation.
METHODS
Characterization of HSD regions
HSD regions >5 kbp in length were initially identified by read-depth analysis of 236 human30 and 86 NHP34 whole-genome Illumina sequence data sets mapped against the human reference genome (GRCh37/hg19). We defined HSDs as regions with evidence of copy number gain in >90% of all humans (>2.5 copies) but where >90% of all great apes did not harbor duplications of the locus (<2.8 copies). Previously uncharacterized HSDs were validated by WSSD56 and whole-genome analysis comparison (WGAC)57 methods (Supplementary Table 1) and comparison to a genome assembly of the CHM1 haploid hydatidiform mole (NCBI Assembly PacBioCHM1_r2_GenBank_08312015) using BLASR. A combination of BLAST58, BLAT59, and WGAC57 methods were used to annotate HSD paralogous regions and identify duplication breakpoints. HSD clustering simulations were performed 10 million times using BEDTools shuffle (v2.23.0)60, median midpoint distances to specific genomic features for each iteration of the simulation were calculated, and comparisons of these distribution were made to the empirical values.
BAC clone sequencing
Large-insert clones from primate BAC libraries (CH17, CH251, CH276 and CH277) were sequenced using either capillary-based methods or single-molecule, real-time (SMRT) sequencing using Pacific Biosciences (PacBio) RSII P4C2 or P6C4 chemistry. Inserts were assembled using Quiver and HGAP (Hierarchical Genome Assembly Process) as described previously61 (Supplementary Tables 3 and 6). Tiling paths of human BAC clones (CH17) were subjected to capillary (N = 205) or PacBio (N = 76) sequence and assembly, resulting in 47.2 Mbp of high-quality sequence from 224 BACs. Of these, 85 BACs were included in the most recent human reference assembly (GRCh38). Contig sequences not included in the human reference may be found in Supplementary Dataset 1. All NHP clones were subjected to SMRT sequence and assembly (N = 269 clones).
Evolutionary analyses
Multiple sequence alignments were generated using MAFFT62 (Supplementary Dataset 1), visualized for manual editing using Jalview63, and phylogenetic analyses were performed using MEGA664 (Supplementary Dataset 2). Evolutionary timing of HSDs was estimated as a fraction of the human–chimpanzee branch length. IGC regions were identified by GENECONV29, masked using BEDTools, and timing estimates repeated with masked alignments. Duplication mechanisms were predicted using a combined approach of defining ancestral paralogs/configurations using genomic synteny taken from chimpanzee and/or orangutan and evolutionary timing estimates to predict the order of rearrangements.
Copy number genotyping
Copy number genotyping was performed from genome sequence data from 2,379 humans from the HGDP30 and Phase 3 of the 1000 Genomes Project31, 86 NHP individuals from the Great Ape Genome Project [including bonobo (N = 14), chimpanzee (N = 23), gorilla (N = 32), and orangutan (N = 17)]34, a Denisovan individual33, a Neanderthal individual32, and three archaic hominids65,66. Copy number variant genotypes were determined based on mrsFAST sequence alignment67 and paralog-specific read-depth (SUNK) mapping19. We used the Vst statistic35 (custom python script available at https://github.com/EichlerLab/vst_calc) to measure copy number stratification between populations. In some cases, gene copy numbers were validated via fluorescence in situ hybridization (FISH) using fosmid clones performed on lymphoblast cell lines (Coriell Cell Repository, Camden, NJ) as described previously68 (Supplementary Tables 12 and 13).
RNA-seq
GTEx RNA-seq data from different subtissues (dbGaP version phs000424.v3.p1) were used to analyze the expression of a set of representative transcripts from hg38 RefSeq annotation. We quantified relative levels of expression using an adjusted version of RPKM (reads per kilobase of transcript per million mapped reads) with reads intersecting unique genomic 30-mers of a canonical isoform (RefSeq) corresponding to each gene paralog. Alternatively, we also applied the Sailfish69 method version 0.63 with the default parameters and k = 20.
MIP sequencing
Single-molecule MIPs (N = 1,105 capturing 415 exonic regions of 30 gene families) designed using MIPgen70 were phosphorylated, captured, barcoded and sequenced as previously described71. Variants were identified using FreeBayes (https://github.com/ekg/freebayes) with relaxed constraints allowing for reduced allele ratios (0.07) and annotated with the Ensembl Variant Effect Predictor72 based on the canonical transcript for each gene. We sequenced a total of 1,096 MIPs from 6,719 individuals, including population controls from the 1000 Genomes Project and cases and controls from the Simons Simplex Collection (SSC)73, Autism Genetic Resource Exchange (AGRE)74, and The Autism Simplex Collection (TASC)75 cohorts.
Statistical analysis
We applied the Wilcoxon-Mann-Whitney test when comparing primary versus secondary HSD sizes and the Kruskal-Wallis rank sum test to assess size differences across three different evolutionary periods. We applied a Wilcoxon-Mann-Whitney test post hoc to identify the duplication period(s) with significant differences and adjusted for multiple comparisons using the Holm method. For paralogous gene expression comparisons median RPKM values of annotated RefSeq transcripts were compared across all tissue types using a Wilcoxon signed-rank test and a Bonferroni correction applied for multiple test comparisons. A one-tailed Fisher’s exact test was used to compare frequency of HSD-exonic mutations in autism cases versus unaffected sibling controls and Bonferroni-corrected for multiple testing comparisons.
Human subjects
The 1000 Genomes and SSC cohorts included in this study did not meet the U.S. federal definitions for human subjects research. All samples were publicly available or encoded, with no individual identifiers available to the study authors. The University of Washington institutional review board (IRB) approved the AGRE and TASC cohorts for human subjects research. All samples were collected at respective institutions after receiving informed consent and approval by the appropriate IRBs. There are no new health risks to participants.
Data availability
BAC sequencing data generated during the current study are available in GenBank with the primary accession numbers provided in Supplementary Tables 3 and 6. Targeted MIP sequencing data generated during this study are available from NCBI BioProject (1000 Genomes Project cohort, ID# PRJNA356308) and the National Database for Autism Research (autism cohorts, NDAR project number #431; doi to be assigned).
Supplementary Material
Acknowledgments
We would like to thank many individuals that contributed to the results described here. We thank B. Coe for assistance in statistical analyses, T. Brown for manuscript editing, and L. Vives, T. Wang, and B. Xiong for technical assistance with MIP sequencing. We would also like to thank B. Dumont, C. Campbell, K. Meltz Steinberg, S. Girirajan, C. Payan, C. Alkan, and E. Karakoc for helpful discussion and advice. We thank Milinn Kremitzki for technical support in generating sequence maps and contigs of BACs not currently included in the human reference sequence. For DNA samples used in MIP sequencing, we would like to thank the investigators and families participating in the 1000 Genomes Project, Autism Speaks, the Autism Simplex Collection, and Simons Simplex Collection (SSC). Additionally, we would like to thank the principal investigators involved in the SSC (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman). Approved researchers can obtain the SSC population dataset described in this study (https://sfari.org/resources/autism-cohorts/simons-simplex-collection) by applying at https://base.sfari.org. The BAC clones from the complete hydatidiform mole were derived from a cell line created by U. Surti. This work was supported, in part, by U.S. National Institutes of Health (NIH) grants from NINDS (R00NS083627, M.Y.D.), NIMH (R01MH101221 to E.E.E.), and NHGRI (R01HG002385 to E.E.E. and U41HG007635 to R.K.W. and E.E.E.) as well as The Paul G. Allen Family Foundation (11631 to E.E.E.). S.C. is supported by a National Health and Medical Research Council (NHMRC) CJ Martin Biomedical Fellowship (#1073726). E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
CONTRIBUTIONS
M.Y.D. and E.E.E. conceived and designed the experiments. M.Y.D., L.H., B.J.N., O.P., S.C., J.H., F.A., L.D., K.M., C.E. performed the experiments. M.Y.D., L.H., B.J.N., O.P., S.C., J.H. analyzed data. M.Y.D., K.P., A.R., C.B., M.M., N.J., K.H. provided technical support. H.A.F.S., X.N., T.A.L.G., R.K.W., E.E.E. provided materials or analyses tools. M.Y.D., L.H., B.J.N., O.P., S.C., E.E.E. wrote the paper.
COMPETING FINANCIAL INTERESTS
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc., is a consultant for Kunming University of Science and Technology (KUST) as part of the 1000 China Talent Program, and was an SAB member of Pacific Biosciences, Inc. (2009–2013).
References
- 1.O’Bleness M, Searles VB, Varki A, Gagneux P, Sikela JM. Evolution of genetic and genomic features unique to the human lineage. Nat Rev Genet. 2012;13:853–866. doi: 10.1038/nrg3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallego Romero I, et al. A panel of induced pluripotent stem cells from chimpanzees: a resource for comparative functional genomics. Elife. 2015;4:e07103. doi: 10.7554/eLife.07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan Z, et al. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science. 2013;342:1100–1104. doi: 10.1126/science.1242379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLean CY, et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 2011;471:216–219. doi: 10.1038/nature09774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prescott SL, et al. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell. 2015;163:68–83. doi: 10.1016/j.cell.2015.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vermunt MW, et al. Epigenomic annotation of gene regulatory alterations during evolution of the primate brain. Nat Neurosci. 2016;19:494–503. doi: 10.1038/nn.4229. [DOI] [PubMed] [Google Scholar]
- 7.Eichler EE, Clark RA, She X. An assessment of the sequence gaps: unfinished business in a finished human genome. Nat Rev Genet. 2004;5:345–354. doi: 10.1038/nrg1322. [DOI] [PubMed] [Google Scholar]
- 8.Ohno S. Evolution by gene duplication. Allen & Unwin; Springer-Verlag; 1970. [Google Scholar]
- 9.Boyd JL, et al. Human-chimpanzee differences in a FZD8 enhancer alter cell-cycle dynamics in the developing neocortex. Curr Biol. 2015;25:772–779. doi: 10.1016/j.cub.2015.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charrier C, et al. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell. 2012;149:923–935. doi: 10.1016/j.cell.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dennis MY, et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell. 2012;149:912–922. doi: 10.1016/j.cell.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Florio M, et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science. 2015;347:1465–1470. doi: 10.1126/science.aaa1975. [DOI] [PubMed] [Google Scholar]
- 13.Marques-Bonet T, et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457:877–881. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sudmant PH, et al. Evolution and diversity of copy number variation in the great ape lineage. Genome research. 2013;23:1373–1382. doi: 10.1101/gr.158543.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 16.Fortna A, et al. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2004;2:E207. doi: 10.1371/journal.pbio.0020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locke DP, et al. Large-scale variation among human and great ape genomes determined by array comparative genomic hybridization. Genome research. 2003;13:347–357. doi: 10.1101/gr.1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Z, et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature. 2005;437:88–93. doi: 10.1038/nature04000. [DOI] [PubMed] [Google Scholar]
- 19.Sudmant PH, et al. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaisson MJ, et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2015;517:608–611. doi: 10.1038/nature13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eichler EE. Segmental duplications: what’s missing, misassigned, and misassembled–and should we care? Genome research. 2001;11:653–656. doi: 10.1101/gr.188901. [DOI] [PubMed] [Google Scholar]
- 22.Fan JB, et al. Paternal origins of complete hydatidiform moles proven by whole genome single-nucleotide polymorphism haplotyping. Genomics. 2002;79:58–62. doi: 10.1006/geno.2001.6676. [DOI] [PubMed] [Google Scholar]
- 23.Kajii T, Ohama K. Androgenetic origin of hydatidiform mole. Nature. 1977;268:633–634. doi: 10.1038/268633a0. [DOI] [PubMed] [Google Scholar]
- 24.Destouni A, et al. Zygotes segregate entire parental genomes in distinct blastomere lineages causing cleavage-stage chimerism and mixoploidy. Genome research. 2016;26:567–578. doi: 10.1101/gr.200527.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itsara A, et al. Population analysis of large copy number variants and hotspots of human genetic disease. American journal of human genetics. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antonacci F, et al. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nature genetics. 2014;46:1293–1302. doi: 10.1038/ng.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang Z, et al. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nature genetics. 2007;39:1361–1368. doi: 10.1038/ng.2007.9. [DOI] [PubMed] [Google Scholar]
- 28.Steinberg KM, et al. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nature genetics. 2012;44:872–880. doi: 10.1038/ng.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawyer S. Statistical tests for detecting gene conversion. Molecular biology and evolution. 1989;6:526–538. doi: 10.1093/oxfordjournals.molbev.a040567. [DOI] [PubMed] [Google Scholar]
- 30.Sudmant PH, et al. Global diversity, population stratification, and selection of human copy-number variation. Science. 2015;349:aab3761. doi: 10.1126/science.aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sudmant PH, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prufer K, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43–49. doi: 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer M, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338:222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prado-Martinez J, et al. Great ape genetic diversity and population history. Nature. 2013;499:471–475. doi: 10.1038/nature12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nuttle X, et al. Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature. 2016 doi: 10.1038/nature19075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gkika D, et al. TRP channel-associated factors are a novel protein family that regulates TRPM8 trafficking and activity. J Cell Biol. 2015;208:89–107. doi: 10.1083/jcb.201402076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.GTEx_Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiatt JB, Pritchard CC, Salipante SJ, O’Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome research. 2013;23:843–854. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antonell A, de Luis O, Domingo-Roura X, Perez-Jurado LA. Evolutionary mechanisms shaping the genomic structure of the Williams-Beuren syndrome chromosomal region at human 7q11.23. Genome research. 2005;15:1179–1188. doi: 10.1101/gr.3944605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Q, Su B. Evolutionary origin and human-specific expansion of a cancer/testis antigen gene family. Molecular biology and evolution. 2014;31:2365–2375. doi: 10.1093/molbev/msu188. [DOI] [PubMed] [Google Scholar]
- 43.Lee JA, Carvalho CM, Lupski JRA. DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 44.Carvalho CM, et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nature genetics. 2011;43:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park SS, et al. Structure and evolution of the Smith-Magenis syndrome repeat gene clusters, SMS-REPs. Genome research. 2002;12:729–738. doi: 10.1101/gr.82802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iida N, Kozasa T. Identification and biochemical analysis of GRIN1 and GRIN2. Methods Enzymol. 2004;390:475–483. doi: 10.1016/S0076-6879(04)90029-8. [DOI] [PubMed] [Google Scholar]
- 48.Chen LT, Gilman AG, Kozasa T. A candidate target for G protein action in brain. J Biol Chem. 1999;274:26931–26938. doi: 10.1074/jbc.274.38.26931. [DOI] [PubMed] [Google Scholar]
- 49.Bamford RN, et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nature genetics. 2000;26:365–369. doi: 10.1038/81695. [DOI] [PubMed] [Google Scholar]
- 50.Sha BY, et al. Genome-wide association study suggested copy number variation may be associated with body mass index in the Chinese population. J Hum Genet. 2009;54:199–202. doi: 10.1038/jhg.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dumont BL. Interlocus gene conversion explains at least 2.7% of single nucleotide variants in human segmental duplications. BMC Genomics. 2015;16:456. doi: 10.1186/s12864-015-1681-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fawcett JA, Innan H. The role of gene conversion in preserving rearrangement hotspots in the human genome. Trends Genet. 2013;29:561–568. doi: 10.1016/j.tig.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 53.Nuttle X, et al. Rapid and accurate large-scale genotyping of duplicated genes and discovery of interlocus gene conversions. Nature methods. 2013;10:903–909. doi: 10.1038/nmeth.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Colburn RW, et al. Attenuated cold sensitivity in TRPM8 null mice. Neuron. 2007;54:379–386. doi: 10.1016/j.neuron.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 55.Bautista DM, et al. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature. 2007;448:204–208. doi: 10.1038/nature05910. [DOI] [PubMed] [Google Scholar]
- 56.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 57.Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome research. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 59.Kent WJ. BLAT–the BLAST-like alignment tool. Genome research. 2002;12:656–664. doi: 10.1101/gr.229202. Article published online before March 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan AR. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics. 2014;47(11–12):11–34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huddleston J, et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome research. 2014;24:688–696. doi: 10.1101/gr.168450.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular biology and evolution. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu Q, et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature. 2014;514:445–449. doi: 10.1038/nature13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lazaridis I, et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513:409–413. doi: 10.1038/nature13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hach F, et al. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nature methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antonacci F, et al. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nature genetics. 2010;42:745–750. doi: 10.1038/ng.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nature biotechnology. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boyle EA, O’Roak BJ, Martin BK, Kumar A, Shendure J. MIPgen: optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics. 2014;30:2670–2672. doi: 10.1093/bioinformatics/btu353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Roak BJ, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cunningham F, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 74.Geschwind DH, et al. The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. American journal of human genetics. 2001;69:463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buxbaum JD, et al. The Autism Simplex Collection: an international, expertly phenotyped autism sample for genetic and phenotypic analyses. Mol Autism. 2014;5:34. doi: 10.1186/2040-2392-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brunet M, et al. New material of the earliest hominid from the Upper Miocene of Chad. Nature. 2005;434:752–755. doi: 10.1038/nature03392. [DOI] [PubMed] [Google Scholar]
- 77.Brunet M, et al. A new hominid from the Upper Miocene of Chad, Central Africa. Nature. 2002;418:145–151. doi: 10.1038/nature00879. [DOI] [PubMed] [Google Scholar]
- 78.Vignaud P, et al. Geology and palaeontology of the Upper Miocene Toros-Menalla hominid locality, Chad. Nature. 2002;418:152–155. doi: 10.1038/nature00880. [DOI] [PubMed] [Google Scholar]
- 79.Jiang Z, Hubley R, Smit A, Eichler EE. DupMasker: a tool for annotating primate segmental duplications. Genome research. 2008;18:1362–1368. doi: 10.1101/gr.078477.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
BAC sequencing data generated during the current study are available in GenBank with the primary accession numbers provided in Supplementary Tables 3 and 6. Targeted MIP sequencing data generated during this study are available from NCBI BioProject (1000 Genomes Project cohort, ID# PRJNA356308) and the National Database for Autism Research (autism cohorts, NDAR project number #431; doi to be assigned).