

Abstract
Rationale: Idiopathic pulmonary fibrosis (IPF) is a devastating disease characterized by epithelial phenotypic changes and fibroblast activation. Based on the temporal heterogeneity of IPF, we hypothesized that hyperplastic alveolar epithelial cells regulate the fibrotic response.
Objectives: To identify novel mediators of fibrosis comparing the transcriptional signature of hyperplastic epithelial cells and conserved epithelial cells in the same lung.
Methods: Laser capture microscope and microarrays analysis were used to identify differentially expressed genes in IPF lungs. Bleomycin-induced lung fibrosis was evaluated in Mmp19-deficient and wild-type (WT) mice. The role of matrix metalloproteinase (MMP)-19 was additionally studied by transfecting the human MMP19 in alveolar epithelial cells.
Measurements and Main Results: Laser capture microscope followed by microarray analysis revealed a novel mediator, MMP-19, in hyperplastic epithelial cells adjacent to fibrotic regions. Mmp19−/− mice showed a significantly increased lung fibrotic response to bleomycin compared with WT mice. A549 epithelial cells transfected with human MMP19 stimulated wound healing and cell migration, whereas silencing MMP19 had the opposite effect. Gene expression microarray of transfected A549 cells showed that PTGS2 (prostaglandin–endoperoxide synthase 2) was one of the highly induced genes. PTGS2 was overexpressed in IPF lungs and colocalized with MMP-19 in hyperplastic epithelial cells. In WT mice, PTGS2 was significantly increased in bronchoalveolar lavage and lung tissues after bleomycin-induced fibrosis, but not in Mmp19−/− mice. Inhibition of Mmp-19 by siRNA resulted in inhibition of Ptgs2 at mRNA and protein levels.
Conclusions: Up-regulation of MMP19 induced by lung injury may play a protective role in the development of fibrosis through the induction of PTGS2.
Keywords: MMPs, alveolar epithelial cells, PTGS2, idiopathic pulmonary fibrosis, laser capture microdissection
At a Glance Commentary
Scientific Knowledge on the Subject
Idiopathic pulmonary fibrosis (IPF) is a devastating and usually lethal disease of unknown etiology. Few matrix metalloproteinases (MMPs) have been studied in this disease, and the putative role of MMP-19, a relatively less-studied MMP, is unknown.
What This Study Adds to the Field
In this study, we discovered that MMP-19 is significantly increased in hyperplastic epithelial cells adjacent to fibrotic regions in IPF lungs. MMP-19 was required for normal cellular wound healing in vitro, and the loss of MMP-19 worsened bleomycin-induced fibrosis in vivo, likely by inducing the expression of cyclooxygenase-2. These data indicate that MMP-19 is a critical regulator of alveolar epithelial cell response to injury.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and fatal interstitial lung disorder with a median survival of 3 to 4 years after diagnosis, currently lacking effective therapy (1). Although there has been significant progress in understanding general mechanisms of lung fibrosis, the pathogenesis of IPF is still poorly understood. Currently, the disease is believed to be the result of injury to the alveolar epithelium that sets up a cascade of dysregulated epithelial–fibroblast crosstalk analogous to abnormal wound healing (2–4). Alveolar epithelial injury and activation, through the release of growth factors, cytokines, and matrix metalloproteinases (MMPs), leads to mesenchymal cell migration, activation/proliferation, formation of fibroblastic foci, differentiation of fibroblasts to myofibroblasts, and extracellular matrix (ECM) deposition, culminating in parenchymal destruction (5). The precise process by which alveolar epithelial cells (AECs) become aberrantly activated and communicate with fibroblast leading to unremitting fibrosis in IPF is unclear but may be associated with the recapitulation of developmental pathways and/or epigenetic changes (4).
In recent years, gene expression arrays have been increasingly used to profile the tissues of patients with IPF and to better understand the disease in humans. We and others applied gene expression microarrays and identified multiple novel targets, including MMP-7, osteopontin (SPP1), caveolin 1, MMP-1, adenosine-2b, CD133, Twist1, pigment epithelium-derived factor, and advanced glycation end products, as potential regulators of lung fibrosis (6–12). Additionally, we and others have identified gene expression patterns that distinguish IPF from hypersensitivity pneumonitis, variants of disease that characterize distinct patterns of progression and characterize patients with pulmonary hypertension (9, 13–16). A common limitation to all of these studies was the starting material used for analysis, since the IPF lung is known for its variable involvement and temporal heterogeneity (1). Thus, in the current study we applied laser capture microdissection (LCM) and gene expression microarray to study regional gene expression patterns in IPF lungs. We identified previously unrecognized overexpression of MMP-19 in the hyperplastic alveolar epithelium. After necessary validation steps that included the fibrotic response to bleomycin of the Mmp19-deficient mice, we established that up-regulated MMP-19 was a reactive response to lung injury and repair, attenuating pulmonary fibrosis through induction of PTGS2 (prostaglandin–endoperoxide synthase 2) expression.
Methods
See online supplement for detailed methods.
Laser Capture Microdissection
Lungs were from the University of Pittsburgh Health Sciences Tissue Bank (17, 18). A staining protocol for hematoxylin and eosin (http://cgap-mf.nih.gov/Protocols/) was used (19). Three regions were selected: Fibrotic (F), F-adjacent hyperplastic epithelial cells (C), and normal-looking alveolar epithelial cells (N). Careful examination was done to avoid contamination with inflammatory cells. Diagnosis of hyperplastic or reactive type II alveolar epithelial cells is based on the finding of (1) clusters of these cells, instead of cells occurring singly; and (2) qualitative morphologic alterations, including cuboidal shapes, increased nucleocytoplasmic ratio, enlarged nuclei, and prominent nucleoli (20). RNA was purified, including an additional amplification step (Sense Amp-Plus Protocol; Genisphere, Hatfield, PA). cRNA was hybridized to Codelink 55K Human Arrays and analyzed with CodeLink Expression Analysis Software (Tempe, AZ). Microarray data were submitted to the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov./geo/).
Cell Transfection
MMP19 cDNA with vector pCMV-sport6 (Open Biosystems, Lafayette, CO) were transfected into A549 cells (Lipofectamine 2000; Invitrogen, Carlsbad, CA). MMP19 expression and mock cell lines were generated with G418 selection. MMP19 siRNA was from Santa Cruz Biotechnology (Santa Cruz, CA).
Experimental Model
C57BL/6/129O1 Mmp19-deficient (−/−) mice were generated as described (21). MMP-19−/− and wild-type (WT) (+/+) littermates were treated with an intratracheal injection of 25 μl of phosphate-buffered saline (PBS) or bleomycin (0.05 U/10 g) and killed at 7 and 21 days.
Bronchoalveolar lavage (BAL) was performed with 1.0 ml of PBS at 7 and 21 days, and lung hydroxyproline analysis was performed at 21 days, as described (22, 23). Data are expressed as μg/hydroxyproline/lung.
Scratch Assay
Scratches were made as described (24) on A549 cells transfected with mock, MMP19 siRNA, and MMP19 cDNA. Images were taken from 0 to 60 hours under light microscope (40×).
Cell Migration
Transmigration analysis through Matrigel (Becton Dickinson, Woburn, MA) was performed as described (25).
Quantitative Reverse Transcriptase–Polymerase Chain Reaction
MMP19, PTGS2, and β-GUS gene expression were assessed by reverse transcriptase–polymerase chain reaction (TaqMan; Applied Biosystems, Foster City, CA) as described (26).
Western Blot
Western blotting was performed as described (26). Proteins were visualized with Chemiluminescence Reagent Plus Kit (Perkin-Elmer Life Sciences, Boston, MA). Bands were semiquantified using the software ImageJ (National Institutes of Health, Bethesda, MD).
ELISA
An ELISA assay using recombinant and anti-MMP19 (Abcam, Cambridge, MA) was developed (see online methods). PGE2 from mice BAL was measured with ELISA kit from Enzo Life Sciences (Farmingdale, NY) following the manufacturer’s instructions.
Confocal Microscopy
Immunofluorescence of frozen lungs was performed as described (18). After primary antibodies for MMP19 and PTGS, samples were labeled with fluorescein isothiocyanate and Texas Red, respectively. Nuclei were counterstained with bisBenzimide Hoechst-33258.
Immunohistochemistry
MMP19 lung immunostaining was performed as described using 3-amino-9-ethyl-carbazole as substrate (27). For PTGS2, the antigen-antibody complex was visualized by diaminobenzidine.
Cell Microarray
Lysed A549-transfected cells were labeled with the Agilent Low RNA input linear amplification Kit PLUS (5184–3523) and hybridized to Agilent Whole Human Genome 4×44 arrays (G4112F; Agilent Technologies, Santa Clara, CA). Probes with annotations for Entrez-Gene ID were extracted (Agilent Feature Extraction 9.5.3 Software), and gene expression signals were normalized with cyclic LOESS. Microarray data were submitted to the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov./geo/).
Statistical Methods
For statistical analysis of microarrays, Genespring and Scoregene were used as described (18). P value was addressed for multiple comparisons using the false discovery method (6, 18, 28). For LCM, exact stratified permutation tests were applied. Expression ratios (F:N, F:C, C:N) were computed for each subject and gene, then combined across subjects using a median log-expression ratio. A P value was computed by comparing the observed median log-expression ratio to the permutation distribution of median log-expression ratios (29).
Results
MMP19 Is a Differentially Expressed Gene in IPF Microenvironments
To address the temporal and regional heterogeneity that characterize IPF lung, we applied LCM combined with microarray analysis to examine distinct microenvironments within the same lung. At least 100 to 1,000 cells were collected from regions of dense fibrotic foci (F), hyperplastic epithelial cells adjacent to such areas (C), and normal-looking alveolar epithelial cells (N) from each lung (Figure 1A). Out of the multiple sections, four IPF lungs had a complete set of samples from all three regions that passed quality control. Samples were hybridized to Codelink 55K Whole Genome Array. We implemented exact stratified permutation tests to determine statistical significance, because comparisons of interest involved a within-subject comparison (F, C, or N). Remarkably, 638 significantly different genes were identified that clearly distinguished the different IPF microenvironments (Figure 1B). Among them, MMP19 was revealed as one of the most significantly up-regulated genes that distinguished normal-looking epithelial cells from hyperplastic epithelial cells (Figure 1C). The up-regulation of MMP-19 was observed in only one of the two probes for the genes, an issue often observed in microarray experiments.
Figure 1.
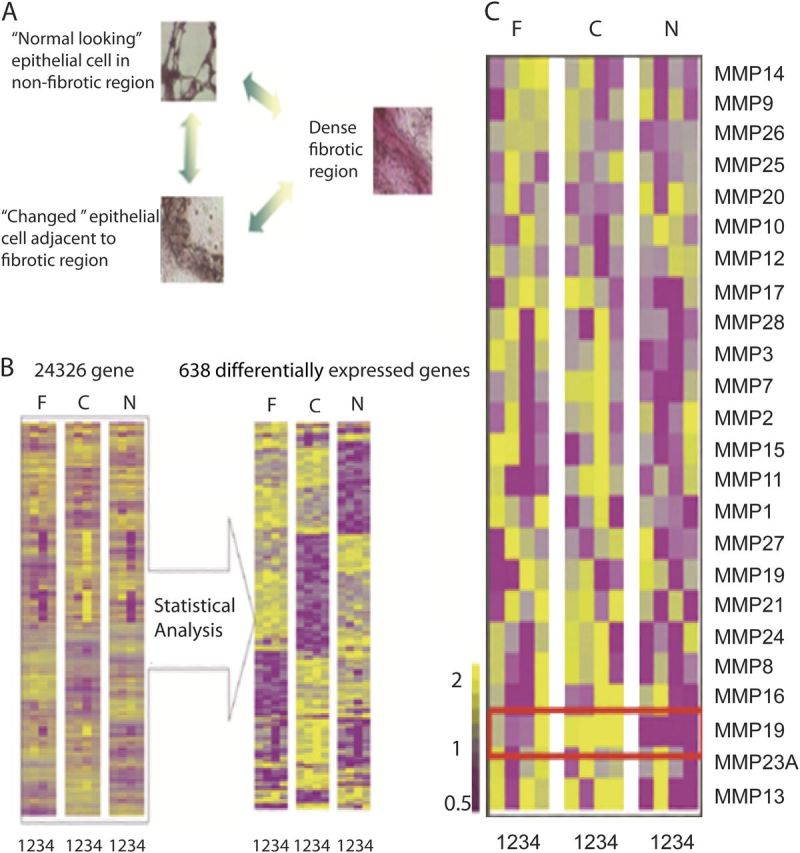
Laser capture microdissection (LCM) reveals matrix metalloproteinase (MMP)19 overexpression in hyperplastic epithelial cells of idiopathic pulmonary fibrosis (IPF) lungs. (A) Representative histological structures for LCM. Dense fibrotic regions, “Changed” epithelial cells adjacent to fibrotic regions, and “Normal looking” alveolar epithelial cells in nonfibrotic region. (B) Gene expression patterns in different regions with the same patient. The number denotes sample number and indicates samples from all three regions from four patients. (C) MMP gene expression patterns in different regions within the same patient. The number denotes sample number and indicates samples from all three regions from all four patients. Yellow indicates up-regulated genes, purple indicates down-regulated genes, gray indicates unchanged. C = hyperplastic epithelial cells adjacent to fibrotic regions; F = dense fibrotic regions; N = normal-looking alveolar epithelial cells in nonfibrotic regions.
MMP19 Protein Is Up-regulated in IPF Lungs
To verify our gene expression microarray data, we performed immunohistochemical staining of IPF and control lungs for MMP-19. As shown in Figure 2, strong cytoplasmic immunostaining was observed in hyperplastic alveolar epithelial cells overlaying fibrotic areas (Figures 2A and 2B) as well as in endothelial cells from capillaries. A weak staining was occasionally observed in normal lungs.
Figure 2.

Up-regulation of matrix metalloproteinase (MMP)19 in idiopathic pulmonary fibrosis (IPF) lungs. (A, B) Intense intracytoplasmic MMP19 labeling is observed in alveolar epithelial cells in IPF lungs (A, 40×; B, 10×). (C) Normal histology lungs showing no staining (40×). (D) IPF lung negative control omitting the primary antibody (40×). (E) ELISA assay of MMP19 protein in bronchoalveolar lavage of IPF lungs and normal volunteer lungs. (F) Western blot analysis of MMP19 in lung homogenates from IPF and normal lungs. (G) Densitometry of Western blot of MMP19 relative to β-actin. Data in E and G represent the mean ± SEM. NL = normal lungs.
ELISA performed in BAL fluids and Western blot analysis in lung homogenates confirmed an increase in MMP-19 protein in IPF lungs compared with that of normal control lungs (Figures 2E and 2F). The densitometric analysis of the two immunoblotted bands (likely representing the pro- and activated forms) normalized to β-actin showed a twofold increase of MMP-19 in the IPF lungs (Figure 2G).
Further analysis revealed that Mmp19 expression was also increased in the murine bleomycin-induced lung fibrosis model. C57BL/6 mice were subjected to bleomycin or PBS intratracheally and killed at 7 and 28 days. We found increased Mmp19 mRNA and protein levels (see Figure E1 in the online supplement) in lungs harvested from animals treated with bleomycin but not control animals.
MMP19 Plays a Role in Wound Healing in Epithelial Cells
To address the role of MMP-19 in wound healing, we assessed the effect of silencing or overexpressing MMP19 on epithelial cell migration in the A549 cell line. Transfection of cells with MMP19 SiRNA caused significant silencing, both at the gene and protein levels (Figures 3A–3D). Scratch assay demonstrated that this silencing of MMP19 significantly reduced wound closure after 48 hours (Figure 3E). To determine the effect of adding MMP19 into the cells, we transfected A549 cells with full-length cDNA of MMP19 and performed transmigration assay with Matrigel. As shown in Figure 3F, cell migration and invasion through Matrigel were significantly higher in MMP19-transfected cells compared with control groups. Taken together, these results suggest that MMP-19 plays an important role in regulating epithelial cell migration.
Figure 3.

Effect of matrix metalloproteinase (MMP)19 on A549 cell migration. (A) Reverse transcriptase–polymerase chain reaction (qRT-PCR) from normal A549 epithelial cells (N), control SiRNA (C), and cells transfected with MMP19 SiRNA (M). Cells were harvested at 24 hours after transfection, and RNA was extracted and reverse transcribed. (B) Western blot data from parallel experiment as in A. (C, D) Quantification of RT-PCR and Western blot data. *P < 0.05. (E) Photomicrograph of scratch assay. Cells were seeded in triplicate in collagen IV–coated culture dishes at 1 × 105 cells/dish. A scratch through the center axis of the plate was gently made using pipette tip. Migration of the cells into the scratch was observed at time point 0 (upper panels) and 48 hours (bottom panels) after scratch. (F) Induction of A549 cell migration/invasion with MMP19 expression in Matrigel. Five high-power fields were counted for each treatment. Assays were performed in triplicate. Data represent mean ± SEM of five experiments.
Mmp19 Knockout Mice Develop Less Inflammation and More Severe Pulmonary Fibrosis in Response to Bleomycin
To obtain insight into the potential role of MMP19 in the development of lung fibrosis in vivo, we evaluated the response to bleomycin in the Mmp19-deficient or WT mice. Mmp19−/− mice are viable, reproduce normally, and develop without any gross phenotypic abnormalities. Also, histological evaluation does not demonstrate any difference between Mmp19−/− and Mmp19+/+ littermates.
At 7 days after bleomycin instillation, total cell count in BAL fluid was significantly higher in Mmp19+/+ mice compared with Mmp19−/− mice (Figure 4A). The analysis of the BAL cell profile revealed a significant decrease in the percent of neutrophils in Mmp19−/− mice (Figure 4B), which was corroborated by lower levels of myeloperoxidase activity in the BAL fluid of knockout mice (Figure 4C). No differences were found at 21 days.
Figure 4.

Total and differential cell count and myeloperoxidase activity in bronchoalveolar lavage (BAL). (A) Total cell counts from wild-type (WT) and matrix metalloproteinase (MMP)19-deficient mice of control and bleomycin instilled at 7 days (n = 7). (B) BAL cell profile. E = eosinophils; L = lymphocytes; M = macrophages; N = neutrophils. (n = 7). (C) BAL myeloperoxidase activity of Mmp19−/− and WT mice (n = 4). Results are shown as mean ± SD of two independent experiments. *P < 0.01; **P < 0.02.
Mmp19−/− mice developed significantly more extensive fibrosis 21 days after bleomycin instillation compared with Mmp19+/+ mice (Figure 5). Areas of epithelialization or regional variability usually characteristic of bleomycin-induced fibrosis were hardly observed in Mmp19−/−, suggesting a repair failure. Representative images are provided in Figures 5A–5G. The lungs also displayed extensive formation of fibroblast/myofibroblast foci and α-smooth muscle actin staining, suggesting that most of these cells were myofibroblasts in Mmp19−/− mice (Figure 5H). The levels of fibrosis assessed by hydroxyproline content paralleled the morphological results. As shown in Figure 5, bottom, lungs from Mmp19−/− mice displayed significant higher levels of hydroxyproline compared with Mmp19+/+ littermates (P < 0.05).
Figure 5.

Bleomycin induced severe pulmonary fibrosis in matrix metalloproteinase (Mmp)19-deficient mice. (A–F) Light micrographs in the lung sections from Mmp19−/− and WT mice. (A, B) Wild-type (WT) mice, 21 days after saline or bleomycin instillation, stained with hematoxylin and eosin (10×). (C) WT mice, 21 days after bleomycin (40×). (D, E) MMP19−/− mice, 21 days after saline or bleomycin instillation, stained with hematoxylin and eosin (10×). (F) Mmp19−/− mice, 21 days after bleomycin (40×). (G, H) Immunostaining of αSMA in the lung sections from Mmp19−/− and WT mice; samples were counterstained with hematoxylin (10×). Inset: positive fibroblasts in a fibroblast focus (40×). Bottom: lung hydroxyproline levels in Mmp19−/− and their WT littermates after bleomycin; results represent mean ± SD from six knockout and WT mice; *P < 0.05.
Because tenascin-C, a multimeric ECM glycoprotein expressed during tissue remodeling, is a substrate of MMP-19 (30), we explored whether tenascin-C is augmented in bleomycin-instilled Mmp19−/− mice compared with Mmp19+/+ littermates. As illustrated in Figure 6, tenascin C was increased at 21 days post bleomycin in both WT and deficient mice, but it was significantly higher in Mmp19−/− mice as examined by Western blot (Figures 6A and 6B) and by immunohistochemistry (Figures 6C and 6D). The protein accumulated mostly in the thickened fibrotic alveolar walls.
Figure 6.

Tenascin C is increased in bleomycin-exposed matrix metalloproteinase (Mmp)19-deficient mice. (A) Lungs from control mice and 21-day bleomycin-instilled mice were homogenized in RIPA lysis buffer, and tenascin C was analyzed by Western blot. (B) Densitometric analysis of the bands of tenascin C normalized with β-tubulin. Results are presented as mean ± SD from two independent experiments; *P < 0.01, **P < 0.02. (C, D) Immunohistochemical evaluation of tenascin C in Mmp19+/+ (C) and Mmp19−/− (D) mice. Pictures are representative of three WT and three MMP-19–deficient mice.
MMP19 Regulates PTGS2 Expression in A549 Cells
To identify target genes that may mediate the effects of MMP19, we profiled MMP19-transfected A549 cells using Agilent gene expression microarrays using mock-transfected A549 cells as controls. A total of 768 genes were significantly differentially expressed in the MMP19-overexpressing cells (P < 0.0001). Table E1 shows the 50 most markedly up-regulated genes. PTGS2, which was increased 3.11-fold (P = 1.12 × 10−7) after MMP19 transfection, was chosen for further experiments because of the known antifibrotic potential of PGE2 and the similarity of the response of PTGS2-deficient mice to bleomycin to that of Mmp-19–deficient mice (31). The induction of PTGS2 mRNA was verified by quantitative reverse transcriptase–polymerase chain reaction (Figure E2). Further experiments corroborated significant increase of PTGS2 expression both at mRNA and at the protein level (Figures 7A–7C). Inhibition of MMP19 by siRNA led to a concomitant decrease in PTGS2 mRNA expression (Figure 7A), but not protein, at the time point we assessed (Figures 7B and 7C). In addition, the induction of PTGS2 by MMP-19 was inhibited not only by MMP19 siRNA but also by the GM6001, an MMP general inhibitor, and MMP inhibitor IV, suggesting that it is dependent on its proteolytic activity. However, it is important to consider that these inhibitors are nonspecific and also inhibit many other MMP or ADAM members that may be involved in cell migration (Figure 7D). We examined the microarrays database (Figure 1) to evaluate if there was a correlation between MMP19 and PTGS2 in hyperplastic epithelial cells. We found a trend toward positive correlation, although it was not statistically significant, potentially because of the small number of samples.
Figure 7.

Matrix metalloproteinase (MMP)19 regulation of PTGS2 (prostaglandin–endoperoxide synthase 2) expression in A549 cells. A549 cells were transfected with full-length cDNA or siRNA of MMP19 and harvested 24 hours after the treatment. (A) MMP19 and PTGS2 mRNA levels were measured by quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR) (MMP19: MMP19+ vs. control, *P = 6.23 × 10−6; MMP19− vs. control, *P = 0.0004; PTGS2: MMP19+ vs. control, *P = 0.0002; MMP19− vs. control, P = 0.04). (B) MMP19 and PTGS2 protein by Western blot analysis in homogenates of A549 cells. (C) Densitometric analysis of the Western blot shown in B. MMP19 versus scramble, *P = 0.03; MMP19+ versus mock, *P = 0.004; PTGS2 (MMP19+) versus mock, *P < 0.007. (D) PTGS2 mRNA levels quantified by qRT-PCR. RNA was extracted from A549 cells 24 hours after transiently transfected with MMP19 that were pretreated with PBS, MMP inhibitors (GM6001 and MMP inhibitors IV), and MMP19 siRNA transfection for 6 hours. Y-axis: fold expression over mock transfection. Data represent mean ± SEM of three experiments. (E) A549 cell migration was attenuated by the MMP inhibitor GM6001 as well as the specific PTGS2 inhibitor, COX-2 inhibitor II, a cell-permeable isoxazolyl-benzenesulfonamide compound; *P < 0.05; **P < 0.01. DMSO = dimethyl sulfoxide.
Because MMP19 affects epithelial cell migration, we wondered if PTGS2 would have a similar effect. As shown in Figure 7E, transmigration of A549 cells on Matrigel was significantly inhibited by both the MMP inhibitor GM6001 and by the cell-permeable isoxazolyl-benzenesulfonamide compound that acts as a potent and highly selective inhibitor of COX-2.
Mmp19-Deficient Mice Exhibit Decreased Expression of Ptgs2 after Bleomycin
To determine whether Mmp19 expression was associated with Ptgs2 expression in response to lung injury in vivo, we examined the Ptgs2 protein in the BAL and whole lung homogenates of Mmp19 WT and Mmp19−/− mice after bleomycin treatment (Figures 8A and 8B). Western blot analysis of Ptgs2 protein showed a threefold increase in BAL Ptgs2 protein in Mmp19+/+ compared with Mmp19−/− mice (1.04 ± 0.03 vs. 0.34 ± 0.05; P < 0.0001) after bleomycin (Figure 8C). Similarly, the increase in PTGS2 after bleomycin was significantly higher in Mmp19+/+ mouse lungs compared with Mmp19−/− mouse lung. Ptgs2 expression was not detected in both Mmp19 WT and Mmp19−/− mouse control lungs.
Figure 8.
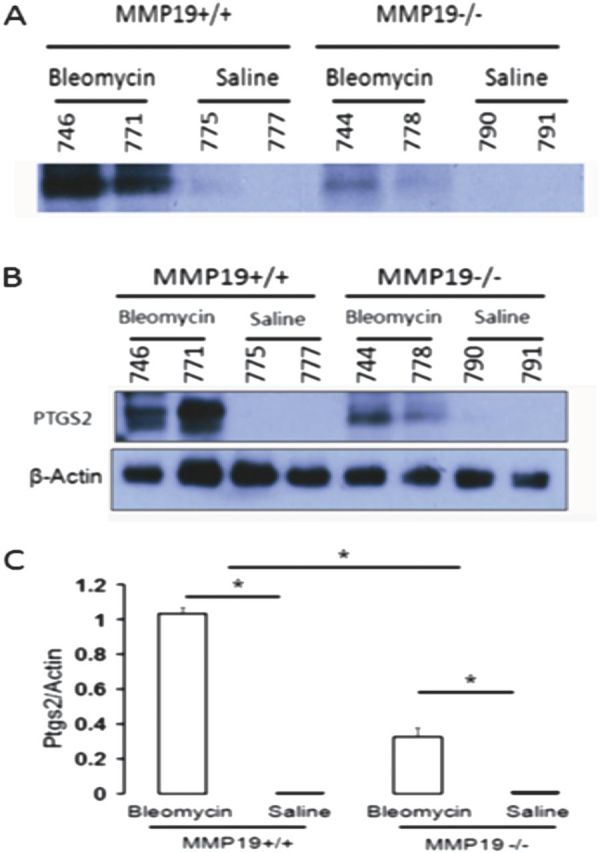
Matrix metalloproteinase (Mmp)19 is required for PTGS2 (prostaglandin–endoperoxide synthase 2) expression in mice. (A) Western blot analysis of PTGS2 protein in bronchoalveolar lavage from the lungs of Mmp19+/+ and Mmp19−/− mice treated with bleomycin or saline control. (B) Western blot analysis of PTGS2 protein in the lungs of Mmp19+/+ and Mmp19−/− mice treated with bleomycin or saline control. (C) Densitometry analysis of the Western blot of PTGS2 protein in the lungs of Mmp19+/+ and Mmp19−/− mice treated with bleomycin or saline control. Y-axis: the ratio of PTGS2 to β-actin. Data represent mean of three experiments with SEM (P < 0.01).
Elevated PTGS2 Was Present and Colocalized with MMP19 in IPF
Given that MMP19 was up-regulated in IPF and MMP19 can induce PTGS2 in epithelial cells, we analyzed the expression of PTGS2 in patients with IPF. Immunoblot analysis revealed a threefold increase of PTGS2 protein in IPF lungs compared with normal histology control lungs (P = 0.0014) (Figures 9B and 9C). A similar increase was found in BAL samples from IPF lungs compared with control lungs (Figure 9A). Immunohistochemistry revealed that PTGS2 expression was higher in IPF lungs compared with normal histology controls. PTGS2 immunostaining localized to hyperplastic alveolar epithelial cells in IPF lungs (Figures 9D–9F). This finding suggests that the PTGS2 protein detected in BAL fluids probably originated from alveolar epithelial cells, although some contribution of alveolar macrophages cannot be ruled out. To localize PTGS2 expression, we performed a confocal microscope analysis and confirmed that PTGS2 was expressed in the hyperplastic alveolar epithelial cells and bronchial epithelial cells, where MMP19 protein was also strongly stained. Thus, PTGS2 and MMP19 colocalized in hyperplastic epithelial cells (Figure 9G).
Figure 9.

PTGS2 (prostaglandin–endoperoxide synthase 2) is increased in patients with idiopathic pulmonary fibrosis (IPF). (A) Western blot analysis of PTGS2 protein in bronchoalveolar lavage from normal volunteers and patients with IPF. (B) Western blot analysis of PTGS2 protein in the lungs from normal volunteers and patients with IPF. (C) Densitometric analysis of the Western blot shown in E. Data represent mean ± SEM of three experiments (P < 0.01). (D, E) Immunostaining of PTGS2 showing strong staining in epithelial cells of IPF lungs (D, 10×; E, 40×). (F) Control lung. (G) Representative images of colocalization of PTGS2 with matrix metalloproteinase (MMP)19 in IPF lungs. Red indicates PTGS2, green indicates MMP19, blue indicates nucleus.
Discussion
IPF is characterized by an aberrant activation of alveolar epithelial cells followed by mesenchymal cell migration/proliferation and differentiation to myofibroblasts and excessive matrix accumulation. The fibrotic lung reaction is likely the final result of a complex interplay between growth factors, cytokines, and chemokines. In this study, we discovered that MMP19 is significantly increased in hyperplastic epithelial cells adjacent to fibrotic regions in IPF lungs. Contrary to our initial hypothesis that the increased expression of MMP19 in hyperplastic epithelial cells surrounding fibrotic areas may have a profibrotic effect, we discovered that MMP-19 is important for normal cellular wound healing in vitro and for limiting bleomycin-induced lung fibrosis in vivo. In fact, MMP19-deficient mice demonstrated a more severe fibrotic response, without a concurrent increase in inflammatory response. Finally, perturbations of MMP-19 levels in vivo and in vitro were associated with changes in PTGS2. Taken together, our findings indicate that up-regulation of MMP-19 induced by lung injury may play a pivotal protective role in the progression of IPF and other fibrotic lung disorders.
Gene and protein studies in whole IPF lungs have shown that some MMPs, including MMP-7, MMP-1, MMP-2, MMP-9, and MMP-3, are strongly up-regulated (6, 27, 28, 32). In a previous study also using LCM, in which only few MMPs were included, it was found that MMP-9 and MMP-7 were up-regulated in the epithelium adjacent to fibroblastic foci (33). MMP-1 and MMP-7 have been proposed as potential peripheral blood biomarkers in IPF (17), and MMP-7 increases in the plasma are predictive of significantly worse prognosis in IPF (34). Interestingly, these enzymes have shown epithelial localization in diverse areas of the IPF lungs, suggesting that different events are occurring in the epithelial regions and in the surrounding connective tissue. MMP-19, a relatively less-studied matrix metalloproteinase, exhibits a number of unique structural features and a distinct expression pattern and functional characteristics. Human MMP19 differs from other MMPs by a C-terminal threonine-rich sequence of 36 residues downstream of the conserved cysteine generally found at the carboxy terminus (35, 36). MMP-19 is able to hydrolyze diverse components of the ECM, such as aggrecan, cartilage oligomeric matrix protein (COMP), fibronectin, and the basement membrane proteins laminin-5γ2, nidogen, and collagen type IV (30, 37), all found in fibrotic matrix. MMP-19 has been shown to cleave IGFBP3 (38), a molecule increased in IPF as well as other fibrotic disorders (39), as well as to cleave tenascin-C, an ECM component required for lung fibrosis in the bleomycin model (40). Similar to our observation in MMP19−/− mice exposed to bleomycin, exaggerated tenascin C accumulation was observed in MMP-19–deficient mice exposed to a murine model of asthma. Interestingly, the MMP-19–deficient mice demonstrated exaggerated allergen-induced airway eosinophilic inflammation and a shift toward Th2-driven inflammation (41), which is consistent with the effects of PTGS2 on dendritic cells (42). The up-regulation and localization of MMP19 to the regressing keloid center in skin scarring lesions (43) also support a putative antifibrotic role of MMP-19. Our findings that Mmp19-deficient mice show an increased lung fibrotic response to bleomycin are consistent with these observations.
A novel finding in our study is the observation that MMP-19 induces PTGS2 (also known as COX2). PTGS2 is a key regulatory enzyme in the synthesis of the prostaglandins E2 (PGE2) and D2 (PGD2) (44). PGE2 has long been considered an antifibrotic molecule because it inhibits fibroblast proliferation, collagen synthesis, migration, and differentiation to myofibroblasts and induces fibroblasts apoptosis (45–47). Fibroblasts derived from IPF lungs exhibit a relative deficiency of PGE2 production as well as reduced responsiveness to PGE2 (48–50). Several studies suggested that bleomycin-induced fibrosis is enhanced when PTGS2-derived PGE2 is low or absent (51–53), whereas others did not observe the effects to the same extent (54) or suggested alternative pathways to PGE2 production (55). Important to our findings, which demonstrated that silencing MMP-19 in alveolar epithelial cells reduced wound healing and transfection of MMP-19 increased cell migration, is the evidence that inhibiting PTGS2 results in a dose-dependent inhibition of wound closure in human and feline epithelial cells (56). A similar result was obtained in our study, wherein inhibition of PTGS2 decreased epithelial cell transmigration through Matrigel. By contrast, the exogenous application of PGE2 stimulated wound closure in a dose-dependent manner. Notably, inhibition of PTGS2 only at initial time points resulted in a sustained inhibition of wound closure, indicating that this mediator is involved in early wound repair processes such as spreading and migration (56). Although less studied in lung disease, PGD2 also seems to have antifibrotic effects in the bleomycin-induced lung injury model (57). Thus, it seems that PTGS2 exerts its antifibrotic effects through its two major products, PGE2 and PGD2.
Although at this stage we have not determined the mechanism by which MMP-19 induces PTGS2, our results in vitro and in vivo demonstrate that changes in PTGS2 expression are dependent on MMP-19 expression and that these changes are associated with the antifibrotic effect of MMP-19. Interestingly enough, it has been recently reported that PGE2 induces MMP-9 expression in dendritic cells and that PGE2-induced MMP-9 is required for dendritic cells migration in vivo and in vitro (58). All together, these findings indicate the existence of a close bidirectional association between some MMPs and PTGS2.
Our findings are consistent with the dramatic shift in the perception of the role of MMPs in IPF that happened over the last decade (59). Although traditionally only considered antifibrotic because of their ECM-degrading properties, MMPs have emerged in recent years as regulators of the complex interaction between secreted molecules (growth factors, cytokines, chemokines), matrix molecules (laminins and collagens), and membrane-anchored proteins (60). It is now well accepted that MMPs regulate fibrosis through their effects on immune and inflammatory processes, cell migration and activation, wound healing, angiogenesis, and apoptosis (32, 59, 60). In this context, the discovery of MMP-19 as a potential antifibrotic MMP through its effects on PTGS2 highlights the complexity of the role of MMPs in lung as well as the potential difficulties inherent in modifying MMP activity for therapeutic interventions in IPF.
IPF is believed to result from aberrant repair of chronic or recurrent alveolar epithelial injury. Alveolar epithelial cells overlying fibroblast foci with presence of altered phenotypes are the most distinctive feature in IPF lung. In IPF, the reestablishing of the intact epithelium after injury, a key component of normal wound healing, is impaired. Our results suggest that expression of MMP-19 by the epithelium is necessary for recovery of the injured epithelium and suppression of mesenchymal cell activation. Thus, the increased expression of MMP-19 (and PTGS2) in the hyperplastic epithelial cells suggests a normal but potentially insufficient response to continuous epithelial injury in IPF lungs. The reasons for this failure are unclear; a downstream cellular resistance may be possible, as would be an ongoing cellular stress or an overwhelming profibrotic balance. Although the reasons for this failure are unclear, the observations highlight the diverse repertoire of responses alveolar epithelial cells display in response to injury and suggest that the balance of these responses may play a role in determining the lung phenotype in IPF. In this context, it is important to note that although we observed and highlighted the up-regulation of MMP-19 in alveolar epithelial cells, we cannot rule out that worsened fibrosis observed in MMP-19–deficient mice may also be attributed to the loss of MMP-19 in other cell types. Additional experiments will be required to address this issue.
In summary, we have discovered that MMP19 is significantly up-regulated in hyperplastic epithelial cells overlying fibroblast foci in IPF lung, that MMP19 is required for normal wound healing in vitro, and that mice deficient in MMP19 present a much more severe pulmonary fibrosis. Increased MMP19 can induce PTGS2 expression in alveolar epithelial cells, enhancing epithelial cell migration and promoting wound healing, whereas mice deficient in MMP19 are also deficient in PTGS2 induction in response to bleomycin. Together, our results suggest that MMP19 is a critical regulator of alveolar epithelial cell response to injury. Understanding why this response is not effective in IPF may be key to our understanding of the sustained phenotype of the lung in IPF and thus to development of novel and effective therapeutic interventions.
Footnotes
Supported by National Institutes of Health grants R01HL073745, R01HL095397, R01LM009657, and T32HL007563; Consejo Nacional de Ciencia y Tecnología grant CONACYT 80473; and the Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease.
Author Contributions: N.K., A. Pardo, and M.S. conceived and designed experiments; N.K. and A. Pardo supervised research; G.Y., E.K-N., A. Pardo, D.K., V.R., P.J., S.C., L.J.C., and R.R. performed the experiments, including quality control; C.L.-O. developed the MMP-19–deficient mice and supervised research. N.K. and T.J.R. performed statistical analysis/analyzed data; I.O.R., K.F.G., A. Parwani, and S.A.Y. contributed to cohort enrollment/data collection/pathology. All authors contributed to the writing of this manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201202-0302OC on August 7, 2012
References
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. ; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. Idiopathic pulmonary fibrosis: evidence based guidelines for diagnosis and management. A Joint Statement by the American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Asociación Latinoamericana de Tórax. Am J Respir Crit Care Med 2011;183:788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001;134:136–151. [DOI] [PubMed] [Google Scholar]
- 3.Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res 2002;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selman M, Pardo A, Kaminski N. Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs? PLoS Med 2008;5:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King TE, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011;378:1949–1961. [DOI] [PubMed] [Google Scholar]
- 6.Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, Lollini L, Morris D, Kim Y, DeLustro B, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA 2002;99:6292–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, Yousem S, Herrera I, Ruiz V, Selman M, et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med 2005;2:e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, Aziz N, Kaminski N, Zlotnik A. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med 2006;173:188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selman M, Carrillo G, Estrada A, Mejia M, Becerril C, Cisneros J, Gaxiola M, Pérez-Padilla R, Navarro C, Richards T, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS ONE 2007;2:e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, Fattman CL, Ramsgaard L, Richards TJ, Loutaev I, Nawroth PP, et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol 2008;172:583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bridges RS, Kass D, Loh K, Glackin C, Borczuk AC, Greenberg S. Gene expression profiling of pulmonary fibrosis identifies Twist1 as an antiapoptotic molecular “rectifier” of growth factor signaling. Am J Pathol 2009;175:2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cosgrove GP, Brown KK, Schiemann WP, Serls AE, Parr JE, Geraci MW, Schwarz MI, Cool CD, Worthen GS. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: a role in aberrant angiogenesis. Am J Respir Crit Care Med 2004;170:242–251. [DOI] [PubMed] [Google Scholar]
- 13.Kaminski N, Rosas IO. Gene expression profiling as a window into idiopathic pulmonary fibrosis pathogenesis: can we identify the right target genes? Proc Am Thorac Soc 2006;3:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum 2011;63:783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, Ahmad F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol 2010;298:H1235–H1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, Brown KK, Schwarz MI, Schwartz DA. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS ONE 2009;4:e5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, Lindell KO, Cisneros J, Macdonald SD, Pardo A, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med 2008;5:e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konishi K, Gibson KF, Lindell KO, Richards TJ, Zhang Y, Dhir R, Bisceglia M, Gilbert S, Yousem SA, Song JW, et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009;180:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards RA. Laser capture microdissection of mammalian tissue. J Vis Exp 2007;(8):309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsui K, Riemenschneider W, Hilbert SL, Yu ZX, Takeda K, Travis WD, Moss J, Ferrans VJ. Hyperplasia of type II pneumocytes in pulmonary lymphangioleiomyomatosis. Arch Pathol Lab Med 2000;124:1642–1648. [DOI] [PubMed] [Google Scholar]
- 21.Pendás AM, Folgueras AR, Llano E, Caterina J, Frerard F, Rodríguez F, Astudillo A, Noël A, Birkedal-Hansen H, López-Otín C. Diet-induced obesity and reduced skin cancer susceptibility in matrix metalloproteinase 19-deficient mice. Mol Cell Biol 2004;24:5304–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardo A, Ruiz V, Arreola JL, Ramírez R, Cisneros-Lira J, Gaxiola M, Barrios R, Kala SV, Lieberman MW, Selman M. Bleomycin-induced pulmonary fibrosis is attenuated in gamma-glutamyl transpeptidase-deficient mice. Am J Respir Cell Mol Biol 2003;167:925–932. [DOI] [PubMed] [Google Scholar]
- 23.Woessner JF. The determination of hydroxyproline in tissue and protein samples containing small proportions of this amino acid. Arch Biochem Biophys 1961;93:440–447. [DOI] [PubMed] [Google Scholar]
- 24.Akagi T, Shishido T, Murata K, Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc Natl Acad Sci USA 2000;97:7290–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu G, Tseng GC, Yu YP, Gavel T, Nelson J, Wells A, Michalopoulos G, Kokkinakis D, Luo JH. CSR1 suppresses tumor growth and metastasis of prostate cancer. Am J Pathol 2006;168:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh M, Handley D, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2010;182:220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selman M, Ruiz V, Cabrera S, Segura L, Ramírez R, Barrios R, Pardo A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis: a prevailing nondegradative lung microenvironment? Am J Physiol Lung Cell Mol Physiol 2000;279:L562–L574. [DOI] [PubMed] [Google Scholar]
- 28.Yamashita CM, Dolgonos L, Zemans RL, Young SK, Robertson J, Briones N, Suzuki T, Campbell MN, Gauldie J, Radisky DC, et al. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol 2011;179:1733–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Good P, editor. Permutation, parametric, and bootstrap tests of hypotheses. New York: Springer. 2005. pp. 57–279. [Google Scholar]
- 30.Stracke JO, Hutton M, Stewart M, Pendás AM, Smith B, López-Otin C, Murphy G, Knäuper V. Biochemical characterization of the catalytic domain of human matrix metalloproteinase 19: Evidence for a role as potent basement membrane degradation enzyme. J Biol Chem 2000;275:14809–14816. [DOI] [PubMed] [Google Scholar]
- 31.Hodges RJ, Jenkins RG, Wheeler-Jones CP, Copeman DM, Bottoms SE, Bellingan GJ, Nanthakumar CB, Laurent GJ, Hart SL, Foster ML, et al. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E(2) production. Am J Pathol 2004;165:1663–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pardo A, Selman M. Matrix metalloproteases in aberrant fibrotic tissue remodeling. Proc Am Thorac Soc 2006;3:383–388. [DOI] [PubMed] [Google Scholar]
- 33.Kelly MM, Leigh R, Gilpin SE, Cheng E, Martin GE, Radford K, Cox G, Gauldie J. Cell-specific gene expression in patients with usual interstitial pneumonia. Am J Respir Crit Care Med 2006;174:557–565. [DOI] [PubMed] [Google Scholar]
- 34.Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, Li K, Choi J, Vuga LJ, Lindell KO, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;185:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pendás AM, Knäuper V, Puente XS, Llano E, Mattei MG, Apte S, Murphy G, López-Otín C. Identification and characteristics of a novel human matrix metalloproteinase with unique structural characteristics, chromosomal location, and tissue distribution. J Biol Chem 1997;272:4281–4286. [DOI] [PubMed] [Google Scholar]
- 36.Mueller MS, Harnasch M, Kolb C, Kusch J, Sadowski T, Sedlacek R. The murine ortholog of matrix metalloproteinase 19: its cloning, gene organization, and expression. Gene 2000;256:101–111. [DOI] [PubMed] [Google Scholar]
- 37.Sadowski T, Dietrich S, Koschinsky F, Ludwig A, Proksch E, Titz B, Sedlacek R. Matrix metalloproteinase 19 processes the laminin 5 gamma 2 chain and induces epithelial cell migration. Cell Mol Life Sci 2005;62:870–880. [DOI] [PubMed] [Google Scholar]
- 38.Sadowski T, Dietrich S, Koschinsky F, Sedlacek R. Matrix metalloproteinase 19 regulates insulin-like growth factor-mediated proliferation, migration, and adhesion in human keratinocytes through proteolysis of insulin-like growth factor binding protein-3. Mol Biol Cell 2003;14:4569–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pilewski JM, Henry AC, Knauer AV, Feghali-Bostwick CA. Insulin-like growth factor binding proteins 3 and 5 are overexpressed in idiopathic pulmonary fibrosis and contribute to extracellular matrix deposition. Am J Pathol 2005;166:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carey WA, Taylor GD, Dean WB, Bristow JD. Tenascin-C deficiency attenuates TGF-beta-mediated fibrosis following murine lung injury. Am J Physiol Lung Cell Mol Physiol 2010;299:L785–L793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gueders MM, Hirst SJ, Quesada-Calvo F, Paulissen G, Hacha J, Gilles C, Gosset P, Louis R, Foidart JM, Lopez-Otin C, et al. Matrix metalloproteinase-19 deficiency promotes tenascin-C accumulation and allergen-induced airway inflammation. Am J Respir Cell Mol Biol 2010;43:286–295. [DOI] [PubMed] [Google Scholar]
- 42.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol 2012;188:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seifert O, Bayat A, Geffers R, Dienus K, Buer J, Löfgren S, Matussek A. Identification of unique gene expression patterns within different lesional sites of keloids. Wound Repair Regen 2008;16:254–265. [DOI] [PubMed] [Google Scholar]
- 44.Smith WL, Langenbach R. Why there are two cyclooxygenase isozymes. J Clin Invest 2001;107:1491–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White ES, Atrasz RG, Dickie EG, Aronoff DM, Stambolic V, Mak TW, Moore BB, Peters-Golden M. Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am J Respir Cell Mol Biol 2005;32:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang SK, White ES, Wettlaufer SH, Grifka H, Hogaboam CM, Thannickal VJ, Horowitz JC, Peters-Golden M. Prostaglandin E2 induces fibroblast apoptosis by modulating multiple survival pathway. FASEB J 2009;23:4317–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E-prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol 2003;29:537–544. [DOI] [PubMed] [Google Scholar]
- 48.Maher TM, Evans IC, Bottoms SE, Mercer PF, Thorley AJ, Nicholson AG, Laurent GJ, Tetley TD, Chambers RC, McAnulty RJ. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2010;182:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang SK, Fisher AS, Scruggs AM, White ES, Hogaboam CM, Richardson BC, Peters-Golden M. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol 2010;177:2245–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang SK, Wettlaufer SH, Hogaboam CM, Flaherty KR, Martinez FJ, Myers JL, Colby TV, Travis WD, Toews GB, Peters-Golden M. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am J Respir Crit Care Med 2008;177:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Striter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesis prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 1995;95:1861–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol 2010;190:693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hartney JM, Coggins KG, Tilley SL, Jania LA, Lovgren AK, Audoly LP, Koller BH. Prostaglandin E2 protects lower airways against bronchoconstriction. Am J Physiol Lung Cell Mol Physiol 2006;290:L105–L113. [DOI] [PubMed] [Google Scholar]
- 54.Card JW, Voltz JW, Carey MA, Bradbury JA, Degraff LM, Lih FB, Bonner JC, Morgan DL, Flake GP, Zeldin DC. Cyclooxygenase-2 deficiency exacerbates bleomycin-induced lung dysfunction but not fibrosis. Am J Respir Cell Mol Biol 2007;37:300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bauman KA, Wettlaufer SH, Okunishi K, Vannella KM, Stoolman JS, Huang SK, Courey AJ, White ES, Hogaboam CM, Simon RH, et al. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J Clin Invest 2010;120:1950–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savla U, Appel HJ, Sporn PH, Waters CM. Prostaglandin E(2) regulates wound closure in airway epithelium. Am J Physiol Lung Cell Mol Physiol 2001;280:L421–L431. [DOI] [PubMed] [Google Scholar]
- 57.Ando M, Murakami Y, Kojima F, Endo H, Kitasato H, Hashimoto A, Kobayashi H, Majima M, Inoue M, Kondo H, et al. Retrovirally introduced prostaglandin D2 synthase suppresses lung injury induced by bleomycin. Am J Respir Cell Mol Biol 2003;28:582–591. [DOI] [PubMed] [Google Scholar]
- 58.Yen JH, Kocieda VP, Jing H, Ganea D. Prostaglandin E2 induces matrix metalloproteinase 9 expression in dendritic cells through two independent signaling pathways leading to activator protein 1 (AP-1) activation. J Biol Chem 2011;286:38913–38923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pardo A, Selman M, Kaminski N. Approaching the degradome in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol 2008;40:1141–1155. [DOI] [PubMed] [Google Scholar]
- 60.Folgueras AR, Pendás AM, Sánchez LM, López-Otín C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol 2004;48:411–424. [DOI] [PubMed] [Google Scholar]