

Abstract

Hydrophobins are surface-active proteins that form a hydrophobic, water-repelling film around aerial fungal structures. They have a compact, particle-like structure, in which hydrophilic and hydrophobic regions are spatially separated. This surface property renders them amphiphilic and is reminiscent of synthetic Janus particles. Here we report surface-specific chiral and nonchiral vibrational sum-frequency generation spectroscopy (VSFG) measurements of hydrophobins adsorbed to their natural place of action, the air–water interface. We observe that hydrophobin molecules undergo a reversible change in orientation (tilt) at the interface when the pH is varied. We explain this local orientation toggle from the modification of the interprotein interactions and the interaction of hydrophobin with the water solvent, following the pH-induced change of the charge state of particular amino acids.
Hydrophobins are the most surface-active proteins known and are exclusively produced by filamentous fungi.1 In nature, hydrophobin assemblies act to reduce the surface tension of the aqueous environment, which otherwise can be a barrier to the growth of hyphae into the air and subsequent spore production.2,3 Hydrophobin monolayers further provide a water-repellant coating on aerial hyphae, fruiting bodies, fungal spores, and gas cavities in lichens.4 The success of hydrophobins is witnessed by their wide distribution among fungi and their use in various technological applications such as stabilization of foams, dispersal of hydrophobic substances, and purification of recombinant proteins.5,6 At the air–water interface, class II hydrophobins form stable, highly ordered assemblies that show an exceptionally high surface elasticity (∼1000 mN/m), which is much larger than that reported for other proteins (typical values of ∼100 mN/m).7,8 Much effort has been devoted to understanding the unique surface properties of hydrophobins, and progress has been made in resolving their microscopic film structures, in particular, by transferring the interfacial film on solid support substrates.7,9 However, surface-specific information on hydrophobins in aqueous solution is lacking, and thus, the relation between the interfacial hydrophobin structure and the properties of hydrophobin films is still poorly understood.5 A hydrophobin protein is characterized by a pattern of eight cysteine residues that form intramolecular disulfide bonds and stabilize a rigid fold, as illustrated in Figure 1a. The overall structure of the protein is compact, with a central β-barrel accompanied by an α-helical structure and an unusually large solvent-exposed hydrophobic region.10,11 This hydrophobic patch comprises ∼18% of the total surface area and renders hydrophobins amphiphilic.11
Figure 1.

Crystal structure of HFBI and VSFG spectra at the air–water interface of a 14 μM aqueous solution (pH 7) of HFBI (black) and HFBII (blue) at room temperature. (a). Hydrophobin three-dimensional structure that consists of a β-barrel core, a small α-helix, and a distinguishable hydrophobic patch (colored in green). Basic and acidic residues are annotated and highlighted in blue and red, respectively. (b,c) VSFG spectra of HFBI and HFBII in the SSP polarization (s-SFG, s-VIS, p-IR) contain several signals associated with the protein. (d,e) VSFG spectra of HFBI and HFBII in PSP polarization (p-SFG, s-VIS, p-IR) show signals centered at ∼1640 and ∼1660 cm–1 that are associated with the central β-barrel of hydrophobins.
Here, we investigate the surface behavior of two class II hydrophobins from the fungi Trichoderma reesei (HFBI, HFBII) using a combination of surface-specific sum-frequency generation techniques and spectral calculations. Liquid surfaces can be probed with high selectivity using vibrational surface sum-frequency generation spectroscopy (VSFG). In this technique, an infrared light pulse and a visible pulse are combined at a surface to generate light at their sum-frequency.12 The sum-frequency generation is enhanced in case the infrared light is resonant with a molecular vibration at the surface. The technique is bulk-forbidden due to symmetry, and only ordered interfacial molecules generate a detectable signal, thus making VSFG a highly surface specific technique.
Figure 1b,c shows VSFG spectra of 14 μM solutions of the class II hydrophobins HFBI and HFBII in water (pH 7, 20 °C) at the air–water interface in the frequency regions from 1400 to 1800 cm–1, measured in the achiral SSP (s-SFG, s-VIS, p-IR) polarization configuration. In this region, we identify several bands that are associated with the interfacial hydrophobin films. We assign the band centered at ∼1410 cm–1 to the symmetric stretch vibration of carboxylate groups and the band at ∼1450 cm–1 to C–H bending vibrations.13 Signals at around 1530 cm–1 are associated with the amide II mode and consist mostly of the out-of-phase combination of the N–H in-plane bend vibration and the C–N stretching vibration. Signals near ∼1650 cm–1 are assigned to amide I vibrations and arise primarily from the C=O stretching vibration of the protein backbone. The amide I region is well-known to be sensitive to the secondary structure of a protein.14 For both hydrophobins, we observe strong narrow signals centered at ∼1675 cm–1. We assign the central peak at ∼1675 cm–1 to a combination of the B1 mode of the antiparallel β-sheets (typically centered at 1685 cm–1) and β-turn elements (typically centered at 1665 cm–1). We observe a weaker band at ∼1635 cm–1 that we assign to the B2 vibrational mode of antiparallel β-sheets.15 This assignment of the observed bands to β-turn and B1 and B2 modes of antiparallel β-sheets agrees very well with previous work,16−18 and the signals find their origin in the central β-barrel of the protein.18 We measured cVSFG spectra (cVSFG) in the amide I frequency region using the PSP (p-SFG, s-VIS, p-IR) polarization configuration (Figure 1c,d). cVSFG spectra have as an advantage that the achiral resonances, including the water vibrations, are not observed. Hence, cVSFG spectra form an ideal means to identify chiral secondary structure elements of proteins at interfaces.19−21 The cVSFG signals observed in the amide I region can only result from β-sheet structures.20,22 The measured cVSFG spectra show a peak at ∼1640 cm–1 and a shoulder at ∼1660 cm–1. We assign the signal at ∼1640 cm–1 to the B2 mode of antiparallel β-sheets and the signal at ∼1660 cm–1 to β-turn elements, as we did for the SSP spectrum shown in Figure 1c. This assignment agrees with the results of previous studies. The difference in the signal strength of vibrational modes between the SSP and PSP spectra can be explained by their different selection rules.15,22 Overall, both proteins show similar VSFG spectra with a dominant peak at 1675 cm–1, as expected for proteins with similar three-dimensional structures (Supporting Information Figure S1).
Both the conventional (achiral) and the cVSFG data show the presence of a highly ordered interfacial protein structure that is rich in β-sheets. The observation of cVSFG bands further confirms that hydrophobins remain in a well-ordered fold upon interface adsorption as the unfolding of the VSFG active secondary structure elements would reduce chirality and hence the cVSFG signals.
We examined the effects of a change in environmental conditions on the structure of the hydrophobin films. Hydrophobins are known to be extremely stable and to withstand high temperatures, a broad range of pH values, and high concentrations of common denaturants.23 Interestingly, changing the pH of the solution does have a strong effect on the observed VSFG spectra. In Figure 2, we present achiral VSFG spectra of HFBI that were recorded at acidic and alkaline pH values. In the SSP spectra, the main signal observed at ∼1676 cm–1 at pH = 8 shifts to higher frequencies (∼1680 cm–1) when the pH is decreased to pH = 4.5, while the signal centered at 1635 cm–1 shifts to lower frequencies. The dip at ∼1650 cm–1 becomes more pronounced when the pH is decreased. A complete pH series showing these trends is presented in the Supporting Information Figure S2. In the corresponding PSP spectra (Figure 2b), we observe bands at 1640 and 1660 cm–1. The signal intensity of the band at 1660 cm–1 decreases when the pH is decreased. Similar spectral changes were observed for HFBII (Figure S3).
Figure 2.

VSFG spectra of a 14 μM HFBI solution at acidic (red) and alkaline (black) pH values. (a) At acidic pH values, signals are observed at ∼1630, ∼1680, and ∼1725 cm–1 in the SSP spectra. At alkaline pH, the signal at 1630 cm–1 has shifted to higher frequencies while the signal at 1680 cm–1 has shifted to lower frequencies. (b) In the PSP spectra, the signal at 1660 cm–1 increases relative to the signal at 1630 cm–1 when the pH is increased.
Amide I frequency shifts often arise from changes in the folding state of a protein.24 We exclude this explanation because hydrophobins are known to be extremely stable over a wide pH range.23 Furthermore, unfolding would lead to a disappearance of the chiral signals of VSFG active secondary structural elements, which is not observed.18 Another explanation might be that the density of molecules adsorbed at the interface changes with pH. We also exclude this explanation as it was shown that the adsorption of hydrophobins to the interface is broadly independent of solution pH; similar film thicknesses are observed over a wide range of pH values.25 A third possibility is that the observed reversible spectral changes (Figure S4) result from pH-dependent interference effects of the amide vibrations and the interfacial water bending mode, which is typically centered at 1670 cm–1.26 In fact, differently charged protein surfaces are known to be able to flip the orientation of water molecules in their vicinity, which can have a severe impact on the appearance of the signals of C–H stretch vibrations in the frequency region of 2800–3100 cm–1.27 To study this effect, we performed control experiments in heavy water (D2O), for which the solvent bending mode is centered at 1250 cm–1. This exchange of water for D2O did not lead to a change of the pH dependence of the measured spectra (Figure S5), which demonstrates that the strong dependence of the VSFG spectra on the pH value does not result from interference of the protein signals with the water background. We also observed pH-dependent spectral changes in the PSP polarization configuration, where the influence of water is negligible. We further rule out significant contributions from the nonresonant background because hydrophobins generate large VSFG signals at the interface.
We explain the observed pH-dependent spectral changes of the amide I VSFG spectra with a reorientation of hydrophobin molecules at the interface. To quantify the pH-induced orientational changes, we compare the measured spectra in the amide I region with spectral calculations as shown in Figure 3. Our calculations show that the experimentally observed changes can be reproduced well with different protein orientations for the different pH values. The calculated VSFG response is found to be very sensitive to the protein’s orientation because the orientation determines how the different amide I vibrational modes of the protein interfere to give rise to the total VSFG signal. By performing a global fit of the calculated VSFG response to the SSP and PSP data, we find that for neutral pH the hydrophobicity vector of the protein (as defined in the Supporting Information) is not far from the surface normal (θ = 26.8 ± 2.3° and ψ = 213.6 ± 5.3°). We integrate φ from 0 to 360° to account for the azimuthal symmetry of the surface. When the pH is lowered, the hydrophobicity vector protein tilts away farther from the surface normal, to θ = 43.6 ± 10.4° and ψ = 271.0 ± 10.1°.
Figure 3.
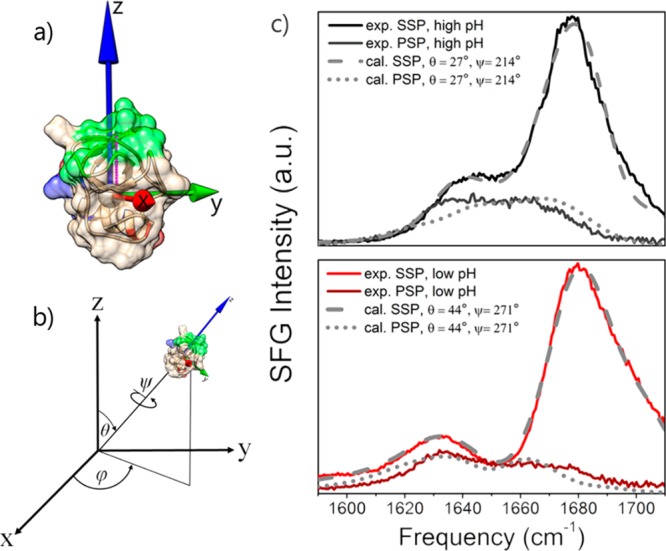
Calculated VSFG spectra of HFBI. (a) Definition of the molecular axes of hydrophobin with the Z axis overlapping with the hydrophobic moment vector. (b) Euler angles (θ, φ, ψ) that transform the atom coordinates from the molecular to the lab frame and define the orientation of the hydrophobic vector (shown in magenta) of the protein. (c) Calculated VSFG spectra that best resemble the experimental data. The experimental acidic (red) and basic (black) pH spectra are reproduced best with (θ, ψ) = (26.8 ± 2.3°, 213.6 ± 5.3°) and (43.6 ± 10.4° and 271.0 ± 10.1°), respectively.
From comparison of the experimental and calculated spectra, we conclude that the orientation angle θ of the protein changes by ∼20° (from ∼25 to ∼45°) when the pH is decreased from alkaline to acidic values. At the same time, the rotation angle ψ around the long axis of the protein changes by ∼60° (from ∼210 to ∼270°). As a result of this change in protein orientation, the contribution of the α-helix at ∼1650 cm–1 to the overall signal in the amide I region increases, thus explaining the observed spectral changes with pH. The differences in the ∼1650 cm–1 region between HFBI and HFBII that were shown in Figure 1 can now also be explained from small differences in the helical segment of these proteins. Even small differences in this segment cause significant differences in the VSFG response in the 1650 cm–1 region.
We explain the observed reorientation with a change of the charge state of the residues of the protein. At neutral pH, HFBI contains three negatively charged (Asp30, Asp40, Asp43) and three positively charged (Lys32, Lys50, Arg45) residues, as seen in Figure 1.7 Asp40, Asp43, Lys50, and Arg45 are located opposite to the hydrophobic patch, while Asp30 and Lys32 are located on the lateral side, as shown in Figure 1. In particular, the position of Asp30 is interesting as it lies in direct vicinity to the hydrophobic patch. As a result, there is a competition between the driving force to remove the hydrophobic patch from the aqueous environment and the driving force to form lateral interactions or to solvate Asp30. These lateral intermolecular interactions likely involve salt bridges as a recent computational modeling study of HFBI membranes reported the formation of salt bridges between D30 and K32 of adjacent hydrophobin molecules.9 A variation of the pH will change the protonation state of charged residues and the net charge of the protein. As a consequence, interprotein salt bridges can be disrupted and the solvation interactions will change, causing a modification of the orientation of the protein.
The solvation interactions with water will be strongest for carboxylate groups (COO–) as these groups can form strong hydrogen bonds with surrounding water molecules. Indeed, at high pH values, we observe a signal at ∼1410 cm–1, corresponding to the symmetric stretch vibration of COO–, which vanishes at acidic pH values due to protonation (Figure S6). At low pH, we also observe a shoulder in the spectrum at ∼1725 cm–1, which we assign to the C=O stretch vibration of carboxylic acid groups. The more upright position of hydrophobin at neutral and high pH can be explained from the fact that the Asp30, Asp40, and Asp43 residues are deprotonated at these pH values and that the resulting carboxylate groups are better hydrated when the proteins are in a more upright position. For a tilted configuration, the carboxylate groups are probably more shielded from the water solvent. At low pH, the Asp40 and Asp43 residues are in their neutral (carboxylic acid) state, which weakens the solvation interactions, thus explaining a more tilted orientation of the protein.
At low pH values, we observe little intensity in the part of the amide I spectrum that corresponds to α-helical structures (∼1650 cm–1). At alkaline pH, the intensity at this frequency increases, leading to a red shift of the main band to ∼1680 cm–1 and a blue shift of the weaker band to ∼1630 cm–1. Strikingly, we find similar spectral changes in the amide III region (Figure S7), where the α-helical signal at ∼1300 cm–1 is weak in amplitude at low pH and gains in intensity at higher pH values. Tsuboi et al. have shown that the amide I and III Raman tensors (defined with respect to the α-helix axis) have approximately the same principal components apart from an overall scaling factor.28 Hence, the increase in intensity of VSFG signals associated with α-helical structures (∼1650 cm–1 amide I, ∼1300 cm–1 amide III) with increasing pH likely find a common origin in a change of the orientation of the hydrophobin molecules.
A special role in the interprotein interactions may be played by intermolecular salt bridges between negatively charged carboxylate groups of the Asp residues and the positively charged side groups of the lysine and arginine residues.7,9 These salt bridges can best form at near-neutral pH values and can contribute to the intermolecular connectivity of hydrophobins. At low pH, the carboxylate groups are neutralized to carboxylic acid groups, which implies that the salt bridges would be disrupted, which would lead to a decrease of the strength of the interprotein interactions. In the fungal aerial structures, where liquid water is absent, salt bridges could be strong and could fulfill an important role in maintaining a strong water-repellent film. As single hydrophobins often have multiple biological roles (e.g., lowering the surface tension and coating different fungal structures), it would be beneficial if the film properties can be fine-tuned to suit diverse biological functions. In this respect, it is interesting to note that changes in the pH of the fungal surroundings elicited by cell metabolism alter hydrophobin film structure and elasticity8 and that this mechanism may play an important role in fungal development.
Acknowledgments
This work is part of the research program of the Stichting voor Fundamenteel Onderzoek der Materie (FOM), which is financially supported by the Nederlandse organisatie voor Wetenschappelijk Onderzoek (NWO). K.M. gratefully acknowledges the European Commission for funding through the award of a Marie Curie fellowship. A.P. and G.R.S. acknowledges support by the Academy of Finland through its Centres of Excellence Programme (2014-2019). Riitta Suihkonen is thanked for technical assistance with hydrophobins. S.J.R. and S.W. acknowledge the European Research Council (ERC) for funding through Grant 210999 and The Netherlands Organization for Scientific Research (NWO) for financially supporting this research.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.7b00394.
Supporting materials and methods and several supporting figures showing structures, VSFG spectra and charge states, and sum of squares plots (PDF)
Author Contributions
# K.M. and S.J.R. contributed equally to the presented work.
The authors declare no competing financial interest.
Supplementary Material
References
- Wessels J.; De Vries O.; Asgeirsdottir S. A.; Schuren F. Hydrophobin Genes Involved in Formation of Aerial Hyphae and Fruit Bodies in Schizophyllum. Plant Cell 1991, 3, 793–799. 10.2307/3869273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wösten H. A. B.; van Wetter M.-A.; Lugones L. G.; van der Mei H. C.; Busscher H. J.; Wessels J. G. H. How a Fungus Escapes the Water to Grow into the Air. Curr. Biol. 1999, 9, 85–88. 10.1016/S0960-9822(99)80019-0. [DOI] [PubMed] [Google Scholar]
- Askolin S.; Penttilä M.; Wösten H. A. B.; Nakari-Setälä T. The Trichoderma Reesei Hydrophobin Genes Hfb1 and Hfb2 Have Diverse Functions in Fungal Development. FEMS Microbiol. Lett. 2005, 253, 281–288. 10.1016/j.femsle.2005.09.047. [DOI] [PubMed] [Google Scholar]
- Kwan A. H.; Winefield R. D.; Sunde M.; Matthews J. M.; Haverkamp R. G.; Templeton M. D.; Mackay J. P. Structural Basis for Rodlet Assembly in Fungal Hydrophobins. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3621–3626. 10.1073/pnas.0505704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder M. B. Hydrophobins: Proteins that Self-Assemble at Interfaces. Curr. Opin. Colloid Interface Sci. 2009, 14, 356–363. 10.1016/j.cocis.2009.04.001. [DOI] [Google Scholar]
- Wösten H. B.; Scholtmeijer K. Applications of Hydrophobins: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 1587–1597. 10.1007/s00253-014-6319-x. [DOI] [PubMed] [Google Scholar]
- Lienemann M.; Grunér M. S.; Paananen A.; Siika-aho M.; Linder M. B. Charge-Based Engineering of Hydrophobin HFBI: Effect on Interfacial Assembly and Interactions. Biomacromolecules 2015, 16, 1283–1292. 10.1021/acs.biomac.5b00073. [DOI] [PubMed] [Google Scholar]
- Cox A. R.; Cagnol F.; Russell A. B.; Izzard M. J. Surface Properties of Class II Hydrophobins from Trichoderma Reesei and Influence on Bubble Stability. Langmuir 2007, 23, 7995–8002. 10.1021/la700451g. [DOI] [PubMed] [Google Scholar]
- Magarkar A.; Mele N.; Abdel-Rahman N.; Butcher S.; Torkkeli M.; Serimaa R.; Paananen A.; Linder M.; Bunker A. Hydrophobin Film Structure for HFBI and HFBII and Mechanism for Accelerated Film Formation. PLoS Comput. Biol. 2014, 10, e1003745. 10.1371/journal.pcbi.1003745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakanpää J.; Paananen A.; Askolin S.; Nakari-Setälä T.; Parkkinen T.; Penttilä M.; Linder M. B.; Rouvinen J. Atomic Resolution Structure of the HFBII Hydrophobin, a Self-assembling Amphiphile. J. Biol. Chem. 2004, 279, 534–539. 10.1074/jbc.M309650200. [DOI] [PubMed] [Google Scholar]
- Hakanpää J.; Szilvay G. R.; Kaljunen H.; Maksimainen M.; Linder M.; Rouvinen J. Two Crystal Structures of Trichoderma Reesei Hydrophobin HFBI--the Structure of a Protein Amphiphile with and without Detergent Interaction. Protein Sci. 2006, 15, 2129–2140. 10.1110/ps.062326706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y. R. Surface Properties Probed by Second-Harmonic and Sum-Frequency Generation. Nature 1989, 337, 519–525. 10.1038/337519a0. [DOI] [Google Scholar]
- Engelhardt K.; Peukert W.; Braunschweig B. Vibrational Sum-Frequency Generation at Protein Modified Air–Water Interfaces: Effects of Molecular Structure and Surface Charging. Curr. Opin. Colloid Interface Sci. 2014, 19, 207–215. 10.1016/j.cocis.2014.03.008. [DOI] [Google Scholar]
- Barth A.; Zscherp C. What Vibrations Tell us about Proteins. Q. Rev. Biophys. 2002, 35, 369–430. 10.1017/S0033583502003815. [DOI] [PubMed] [Google Scholar]
- Nguyen K. T.; King J. T.; Chen Z. Orientation Determination of Interfacial β-Sheet Structures in Situ. J. Phys. Chem. B 2010, 114, 8291–8300. 10.1021/jp102343h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L.; et al. Structure and Orientation of Interfacial Proteins Determined by Sum Frequency Generation Vibrational Spectroscopy: Method and Application. Adv. Protein Chem. Struct. Biol. 2013, 93, 213–255. 10.1016/B978-0-12-416596-0.00007-5. [DOI] [PubMed] [Google Scholar]
- Wang Z.; et al. A Narrow Amide I Vibrational Band Observed by Sum Frequency Generation Spectroscopy Reveals Highly Ordered Structures of a Biofilm Protein at the Air/Water Interface. Chem. Commun. 2016, 52, 2956. 10.1039/C5CC05743D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister K.; Bäumer A.; Szilvay G. R.; Paananen A.; Bakker H. J. Self-Assembly and Conformational Changes of Hydrophobin Classes at the Air–Water Interface. J. Phys. Chem. Lett. 2016, 7, 4067–4071. 10.1021/acs.jpclett.6b01917. [DOI] [PubMed] [Google Scholar]
- Wang J.; Chen X.; Clarke M. L.; Chen Z. Detection of Chiral Sum Frequency Generation Vibrational Spectra of Proteins and Peptides at Interfaces in Situ. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4978–4983. 10.1073/pnas.0501206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L.; Liu J.; Yan E. C. Y. Chiral Sum Frequency Generation Spectroscopy for Characterizing Protein Secondary Structures at Interfaces. J. Am. Chem. Soc. 2011, 133, 8094–8097. 10.1021/ja201575e. [DOI] [PubMed] [Google Scholar]
- Yan E. C. Y.; Fu L.; Wang Z.; Liu W. Biological Macromolecules at Interfaces Probed by Chiral Vibrational Sum Frequency Generation Spectroscopy. Chem. Rev. 2014, 114, 8471–8498. 10.1021/cr4006044. [DOI] [PubMed] [Google Scholar]
- Fu L.; Xiao D.; Wang Z.; Batista V. S.; Yan E. C. Y. Chiral Sum Frequency Generation for In Situ Probing Proton Exchange in Antiparallel β-Sheets at Interfaces. J. Am. Chem. Soc. 2013, 135, 3592–3598. 10.1021/ja3119527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisko K.; Szilvay G. R.; Vainio U.; Linder M. B.; Serimaa R. Interactions of Hydrophobin Proteins in Solution Studied by Small-Angle X-ray Scattering. Biophys. J. 2008, 94, 198–206. 10.1529/biophysj.107.112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vandenAkker C. C.; Engel M. F. M.; Velikov K. P.; Bonn M.; Koenderink G. H. Morphology and Persistence Length of Amyloid Fibrils Are Correlated to Peptide Molecular Structure. J. Am. Chem. Soc. 2011, 133, 18030–18033. 10.1021/ja206513r. [DOI] [PubMed] [Google Scholar]
- Tucker I. M.; Petkov J. T.; Penfold J.; Thomas R. K.; Cox A. R.; Hedges N. Adsorption of Hydrophobin–Protein Mixtures at the Air–Water Interface: The Impact of pH and Electrolyte. Langmuir 2015, 31, 10008–10016. 10.1021/acs.langmuir.5b02403. [DOI] [PubMed] [Google Scholar]
- Nagata Y.; Hsieh C. S.; Hasegawa T.; Voll J.; Backus E. H.; Bonn M. Water Bending Mode at the Water-Vapor Interface Probed by Sum-Frequency Generation Spectroscopy: A Combined Molecular Dynamics Simulation and Experimental Study. J. Phys. Chem. Lett. 2013, 4, 1872–1877. 10.1021/jz400683v. [DOI] [PubMed] [Google Scholar]
- Strazdaite S.; Meister K.; Bakker H. J. Orientation of Polar Molecules near Charged Proteins Interfaces. Phys. Chem. Chem. Phys. 2016, 18, 7414–7418. 10.1039/C5CP06372H. [DOI] [PubMed] [Google Scholar]
- Tsuboi M.; Benevides J. M.; Thomas G. J. Jr. Raman Tensors and Their Application in Structural Studies of Biological Systems. Proc. Jpn. Acad., Ser. B 2009, 85, 83–97. 10.2183/pjab.85.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.