

SUMMARY
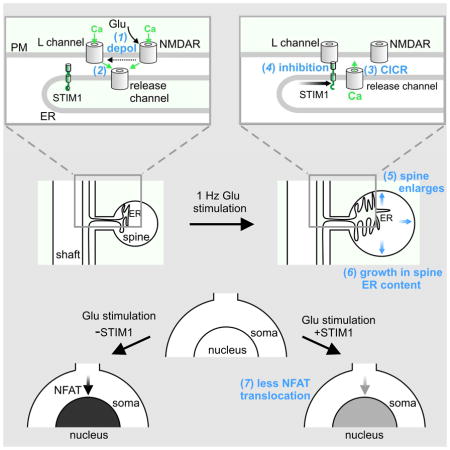
Potentiation of synaptic strength relies on postsynaptic Ca2+ signals, modification of dendritic spine structure and changes in gene expression. One Ca2+ signaling pathway supporting these processes routes through L-type Ca2+ channels (LTCC), whose activity is subject to tuning by multiple mechanisms. Here we show in hippocampal neurons that LTCC inhibition by the endoplasmic reticulum (ER) Ca2+ sensor, stromal interaction molecule 1 (STIM1), is engaged by the neurotransmitter glutamate, resulting in regulation of spine ER structure and nuclear signaling by the NFATc3 transcription factor. In this mechanism, depolarization by glutamate activates LTCC Ca2+ influx, releases Ca2+ from the ER and consequently drives STIM1 aggregation and an inhibitory interaction with LTCCs that increases spine ER content but decreases NFATc3 nuclear translocation. These findings of negative feedback control of LTCC signaling by STIM1 reveal interplay between Ca2+ influx and release from stores that controls both postsynaptic structural plasticity and downstream nuclear signaling.
Keywords: Voltage-gated Ca2+ channel, L-type Ca2+ channel, endoplasmic reticulum, stromal interaction molecule 1, N-methyl-D-aspartate receptor, dendritic spine, glutamate, structural plasticity, nuclear factor of activated T cells, cytoplasmic Ca2+
eTOC Blurb
Dittmer et al. show that postsynaptic activation of voltage-gated L-type Ca2+ channels triggers Ca2+ release from stores, activating feedback inhibition of L channels by the STIM1 Ca2+ sensor. Activated STIM1 also promotes L channel-dependent growth in ER content of dendritic spines and attenuates nuclear translocation of the NFAT transcription factor.
INTRODUCTION
At glutamatergic synapses in the brain, most notably hippocampal CA3-CA1 synapses, activity-dependent changes in synaptic strength that underlie certain forms of learning and memory formation commonly rely upon Ca2+ signaling initiated by NMDA receptors (NMDAR) (Bliss and Collingridge, 1993). High-frequency stimulation of this kind of synapse generates intracellular Ca2+ signals in the postsynaptic dendritic spine that recruit glutamate receptors to the synapse (Huganir and Nicoll, 2013) and cause enlargement of the spine (Lang et al., 2004; Matsuzaki et al., 2004; Yang et al., 2008), cellular responses which together support long-term potentiation (LTP) of synaptic transmission that can last up to ~2 hours (Colgan and Yasuda, 2014). This early phase of LTP is followed by a late phase of LTP (L-LTP) that may persist for several days or longer and relies upon synthesis of new mRNAs and proteins.
Stimulation of these synapses using theta burst or 200 Hz tetani is effective at initiating another form of LTP as well, one that is slower in onset and depends upon Ca2+ influx through postsynaptic voltage-gated L-type Ca2+ channels (LTCCs) (Bayazitov et al., 2007; Grover and Teyler, 1990; Moosmang et al., 2005; Pigott and Garthwaite, 2016; Zakharenko et al., 2001). It is now thought that slow LTCC-dependent and fast NMDAR-dependent LTP may occur in concert at the same synapse (Blundon and Zakharenko, 2008; Padamsey and Emptage, 2014) and it has been proposed that these distinct processes combine to support more flexible and reliable learning (Costa et al., 2015). In hippocampal pyramidal neurons, LTCCs are found most prominently in dendrites, dendritic spines (Davare et al., 2001; Hoogland and Saggau, 2004) and neuronal somata. An important function of LTCCs in neurons is to couple electrical activity, such as excitatory postsynaptic potentials (Mermelstein et al., 2000), to changes in gene transcription that help maintain changes in synaptic strength (Bading et al., 1993; Deisseroth et al., 1996; Dolmetsch et al., 2001; Graef et al., 1999; Moosmang et al., 2005; Murphy et al., 2014; Murphy et al., 1991; Oliveria et al., 2007).
In response to plasticity-inducing stimuli, cytoplasmic Ca2+ signals are also produced by release of Ca2+ from stores in the endoplasmic reticulum (ER) (Emptage et al., 1999; Holbro et al., 2009), which courses throughout the branches of the dendritic tree and also projects into some of the spines dotting the surface of hippocampal pyramidal neuron dendrites (Cooney et al., 2002; Spacek and Harris, 1997). Occupancy of spines by ER projections is dynamic, and controlled in part by NMDARs (Ng et al., 2014). Ca2+ is released from stores by Ca2+ itself, in a process referred to as Ca2+-induced Ca2+ release (CICR) (Rose and Konnerth, 2001). In this process, Ca2+ in the cytosol, arriving for example via Ca2+-permeable channels such as NMDARs or LTCCs, acts as an agonist to open two classes of Ca2+ channels present in the ER membrane: ryanodine receptors or inositol-1,4,5-trisphosphate receptors. Recently, it has been found that a sensor of ER luminal Ca2+, stromal interaction molecule 1 (STIM1), can bind to and modulate the activity of LTCCs (Park et al., 2010; Wang et al., 2010). Based on extensive work in immune system cells and other non-excitable cells, STIM1 is known to span the ER membrane, with a Ca2+-sensing EF hand located in the ER lumen and a large effector domain in the cytosol (Liou et al., 2005; Prakriya and Lewis, 2015; Roos et al., 2005; Zhang et al., 2005). Upon depletion of ER Ca2+, STIM1 aggregates within junctions formed between the plasma membrane (PM) and ER, and there executes it’s defining task: physical contact with and hence activation of Ca2+-permeable, plasma membrane-localized Orai channels, which provides Ca2+ that both acts as an important cytosolic Ca2+ signal and refills ER stores. In addition to being expressed in non-excitable cells, STIM1 is expressed in cardiac myocytes, smooth muscle and skeletal muscle cells, and many kinds of neurons (Soboloff et al., 2012); in hippocampal pyramidal neurons, STIM1 is found in the cell body and dendrites (Klejman et al., 2009). The surprise that STIM1 interacts with LTCCs (Park et al., 2010; Wang et al., 2010) was compounded by the finding that this interaction inhibits LTCC activity, which is opposite to the effect of STIM1 on Orai channels. At this point, two aspects of STIM1 control of LTCCs in neurons remain unknown: what physiologically-relevant stimuli engage STIM1 action on LTCCs, and what impact does this process have on neuronal function?
Here we report that glutamatergic depolarization of neurons activates Ca2+ influx through NMDARs and LTCCs, causing CICR and activation of STIM1, which feeds back onto LTCCs to inhibit their activity. Moreover, STIM1 negative feedback onto LTCCs has profound consequences for key aspects of neuronal plasticity: it shapes LTCC cytoplasmic Ca2+ signals, controls LTCC-dependent ER content within dendritic spines, and potently tunes LTCC-dependent nuclear translocation of NFAT transcription factors.
RESULTS
LTCCs are inhibited by Ca2+ influx through NMDA receptors
We studied regulation of isolated L-type Ca2+ current by glutamate, using short-term cultured rat hippocampal neurons (Oliveria et al., 2012). The near absence of current following addition of the LTCC-selective antagonist nimodipine confirmed that inward Ca2+ current was carried almost entirely by LTCCs (Figure 1A). Using a train of 500-ms step depolarizations from −60 mV to +10 mV, with a step occurring every 15 s, we found that 15 s application via a multi-barrel fast perfusion system of a combination of 100 μM L-glutamate plus 1 μM glycine inhibited pharmacologically-isolated L-current by ~45% (Figure 1A). No effect of glutamate was evident on the kinetics of inactivation (500 ms steps) or on the shape and position of the current-voltage relationship, indicating that changes in neither inactivation, particularly Ca2+-dependent inactivation (CDI), nor the voltage-dependence of activation were involved in inhibition of L current by glutamate. Inhibition of L current by glutamate developed over an exponential time course (τ = 51 s), and a steady level of inhibition was reached within the 300 s duration of the experiments (Figure 1B). Reducing step depolarization duration to 50 ms and frequency to once every 30 s allowed recovery of current over the 300 s experimental period, suggesting that less intense Ca2+ signaling results in briefer inhibition of L current. Selective activation of NMDARs with a 15 s application of 100 μM NMDA plus 1 μM glycine in concert with membrane depolarization for 50 ms every 30 seconds also inhibited L current, with a time course similar that measured for glutamate plus glycine (Figure 1B). The relatively rapid reversibility of glutamate inhibition of L current suggests that channel internalization is not involved in the inhibition process, a conclusion supported by the failure of the endocytosis inhibitors Dyngo 4a and Pitstop-2 (McCluskey et al., 2013; von Kleist et al., 2011) to prevent glutamate inhibition of L current (Figure S1). In subsequent electrophysiological experiments, step depolarizations of 500 ms duration and at a frequency of once every 15 s were employed in order to allow inhibition by glutamate to reach a steady level.
Figure 1. Inhibition of L-Type Ca2+ current by NMDARs relies upon CICR and STIM1.

(A) Left, records of L currents (500-ms depolarization, −60 mV to +10 mV) before (black) and after (gray) glutamate application. Right, current-voltage relationship before (black) and after (gray) glutamate application and during block by 5 μM nimodipine (red).
(B) Time course of inhibition of normalized L currents by 15 s application of glutamate (100 μM) + glycine (1 μM), for trains of 500 ms steps at 1 every 15 s (○). Time course obtained with 500 ms step depolarizations was fit with an exponential function (0–360 s). Time course of isolated L current evoked by 50-ms tep-depolarization from −60 mV to +10 mV at 30 s intervals in response to perfusion of glutamate (100 μM, △) or NMDA (100 μM, ■), + 1 μM glycine. See also Figure S1.
(C,D) Pharmacological analysis of LTCC inhibition by 15 s glutamate application. Concentrations: 10 μM MK801, 25 μM DNQX, 25 μM MTEP, and 10 mM BAPTA substituted for 10 mM EGTA in the patch pipet. Percent inhibition (at time > 200 s) of L current by glutamate either alone or under conditions of BAPTA, MK801, DNQX or MTEP treatment. Glu control (n = 5), BAPTA (n = 6), MK801 (n = 5), MTEP (n = 6) and DNQX (n = 6).
(E) Time course of glutamate inhibition of normalized L current in neurons transfected with a short hairpin RNAi that suppressed endogenous STIM1 expression (STIM1RNAi; green), as compared to control (black) and rescue with human STIM1 (hSTIM1 rescue; gray). Data providing estimate of percent knockdown of STIM1 by RNAi presented in Figure S2.
(F) For the same conditions as in (E), steady inhibition (t > 200 s) of peak Ca2+ current density (pA/pF), calculated by dividing peak Ca2+ current (pA) by a measure of membrane surface area, cell capacitance (pF). rSTIM1 (n = 6), RNAi (n = 6) and hSTIM1 (n = 5).
(G) Neuron transfected with D1ER.
(H) Effects of nimodipine (5 μM) and MK801 (10 μM) on glutamate-initiated depletion of ER Ca2+ stores in neurons expressing D1ER. 100 μM glutamate + 1 μM glycine for 15 s. Calibration in Figure S3.
Throughout: neurons 4–5 DIV. Step depolarizations: 500 ms, every 15 s, except where marked as 50 ms steps (1 every 30 s). Mean ± SEM; comparisons via ANOVA and Bonferroni post-hoc correction. Significance: ***p < 0.001.
The uncompetitive NMDAR pore-blocker, MK801, strongly attenuated glutamate inhibition of L current (Figure 1C,D). As well, glutamate inhibition of L current was strongly attenuated by equimolar substitution in the whole cell pipet of BAPTA (10 mM) for the 50x slower Ca2+ chelator EGTA. Although BAPTA is known to suppress CDI of LTCCs (Oliveria et al., 2012), the fact that L current exhibited strong and rapid inactivation in the absence or presence of glutamate (Figure 1A) indicated that glutamate did not inhibit L current by promoting CDI, but rather that Ca2+ flux into the cytosol via, at least in part, glutamate-activated NMDARs most likely led to LTCC inhibition. Indeed, DNQX inhibition of AMPA and kainate subtypes of ionotropic glutamate receptors had no effect on the ability of glutamate to inhibit L current, and nor did MTEP, a non-competitive inhibitor of group I metabotropic glutamate receptors (mGluR), prevent glutamate inhibition of L current. Together these data indicate that Ca2+ influx through NMDA receptors leads to inhibition of L-type Ca2+ current.
Inhibition of LTCCs by NMDARs requires STIM1
Based on the discovery that STIM1, a sensor of [Ca2+]ER, can inhibit LTCC activity (Park et al., 2010; Wang et al., 2010), we tested whether RNAi suppression of STIM1 expression in rat hippocampal neurons (STIM1 RNAi) might interfere with glutamate inhibition of L current. Compared to control neurons transfected with YFP and the empty pSilencer vector, neurons expressing STIM1RNAi exhibited ~55% reduction in STIM1 expression (Figure S2), reduced glutamate inhibition of L current (Figure 1E,F) and increased L current density (Figure 1F). The density of L current, and both the degree and speed of inhibition by glutamate, were restored in rat STIM1RNAi-expressing neurons by co-expression of RNAi-insensitive human STIM1 (hSTIM1).
Because depletion of ER Ca2+ stores is the natural trigger for STIM1 association with the PM, we examined the effect of glutamate on [Ca2+]ER by transfecting hippocampal neurons with the genetically-encoded, ER lumen-targeted, ratiometric Ca2+ sensor, D1ER (Palmer et al., 2004) (Figure 1G). At the end of each experiment with the D1ER sensor, measured FRET values were converted to [Ca2+]ER using the calibration protocol illustrated in Figure S3. Bath application of glutamate decreased [Ca2+]ER by ~50% within 2 min, and [Ca2+]ER then returned to its initial level within 4–5 min after application of glutamate (Figure 1H). Treatment with the LTCC antagonist nimodipine prevented much of the release of ER Ca2+, but left intact an early phase of ER Ca2+ release that peaked at ~45 s post-glutamate. In contrast, treatment with the NMDAR blocker MK801 prevented glutamate-triggered release of Ca2+ from the ER.
The results in Figure 1 can be accounted for by a model in which NMDARs initiate CICR (CICRNMDA) and via depolarization (in the physiologically-relevant situation of a non-clamped neuron), also activate LTCCs; in turn, LTCCs further deplete [Ca2+]ER via additional CICR (CICRLTCC), activating STIM1, which partially inhibits subsequent LTCC activity. Because activation of LTCCs by voltage-clamp steps did not result in obvious inhibition of L current prior to glutamate application, CICRLTCC alone may be unable to initiate strong STIM1-dependent inhibition of LTCCs; CICRNMDA is required for this process. Apparently, ionotropic, non-NMDARs such as AMPA receptors were ineffective in providing either the depolarizing drive needed to activate LTCCs or in initiating CICR (CICRAMPA) equivalent to that produced by Ca2+-permeable NMDARs.
CICR promotes STIM1 clustering and association of STIM1 with CaV1.2
Following depletion of ER Ca2+ stores in non-neuronal cells, STIM1 is known to aggregate within junctions formed between the plasma membrane (PM) and ER (Liou et al., 2005; Prakriya and Lewis, 2015; Roos et al., 2005; Zhang et al., 2005), prompting us to test whether activation of NMDARs and LTCC activity might also promote clustering of STIM1 in neurons. Using total internal reflection fluorescence (TIRF) microscopy to image PM-proximal regions of neurons transfected with STIM1-YFP revealed that application of glutamate for 15 s indeed caused clustering of STIM1-YFP in dendrites (Figure 2A). The constitutively active mutant STIM1 (D76A)-YFP (Liou et al., 2005) appeared to be chronically clustered, even without glutamate treatment. In contrast, block of NMDARs (MK801) or LTCCs (nimodipine) prevented glutamate-triggered clustering of STIM1-YFP (Figure 2B). We examined the ability of locally restricted application of glutamate to cluster STIM1 by using photolysis of a caged glutamate compound, 4-methoxy-7-nitroindolinyl-caged-L-glutamate (MNI-glutamate), in a 1 μm by 1 μm region adjacent to a dendritic spine imaged using spinning-disc confocal microscopy (Figure 2C). As shown in the example dendrite, STIM1-YFP was relatively widely distributed prior to glutamate uncaging (−40 s), and an uncaging train of 60 laser flashes at 1 s−1 caused STIM1-YFP to condense into bright clusters by the end of the train (60 s). By 200 s after the first laser pulse (140 s after the end of stimulation), the spatial distribution of STIM1-YFP had become more diffuse, presumably reflecting disaggregation and deactivation of STIM1-YFP. The decrease in overall STIM1-YFP fluorescence intensity by the end of the experiment (200 s) may be attributable to bleach of the fluorophore. Block of NMDARs (MK801) or LTCCs (nimodipine) prevented the clustering of STIM-YFP caused by uncaging of glutamate (Figure S4).
Figure 2. Activation of NMDARs and LTCCs promotes clustering of STIM1-YFP and FRET with CFP-CaV1.2 LTCCs.

(A) TIRF images of STIM1-YFP clustering in response to bath application of glutamate (100 μM + 1 μM glycine, 15 s) compared to clustering of STIM1(D76A)-YFP. Results similar to those illustrated were obtained from: STIM1-YFP (n = 10), STIM1-YFP + Glu (n = 17) and STIM1(D76A)-YFP (n = 17) neurons.
(B) MK801 (10 μM) or nimodipine (5 μM) prevented STIM1-YFP clustering in response to glutamate. Results similar to those illustrated were obtained for n = 7 MK801-treated and n = 5 nimodipine-treated neurons.
(C) YFP intensity plots derived from confocal images illustrate clustering of STIM1-YFP in response to glutamate uncaging, followed by de-clustering. Uncaging protocol: 2 ms laser pulses once per second, for 1 min starting at time = 0 s. White box indicates uncaging region. Bath: 2 mM MNI-glutamate, 1 μM glycine, 3 mM CaCl2 and 0 Mg2+. For each condition, observations were obtained from 5–6 neurons, with results similar to those illustrated: no blockers (6), nimodipine (5) and MK801 (5). Figure S4 presents exemplar images illustrating prevention by MK801 or by nimodipine of glutamate uncaging-driven increases in STIM1-YFP.
(D) Two-dimensional, summed-intensity projection images of STIM1-YFP and CFP-CaV1.2 expression in a dendrite.
(E) Based on a differential interference contrast image, outline of the section of dendrite analyzed in (D).
(F) Same dendrite as in (D–E), showing STIM1-YFP/CFP-CaV1.2 FRET images corrected for spectral bleed-through and cross-excitation (FRETC). Shown are images collected 40 s prior to onset of the 1 Hz uncaging train (−40 s), at the end of the 60 s train (60 s) and 200 s after the onset of the 60 s train (200 s). Uncaging: 1 μm x 1 μm box.
(G) Time course of glutamate uncaging-driven changes in FRET between STIM1-YFP and CFP-CaV1.2. FRET ratio (R) normalized to values measured 60 s prior to the train of uncaging pulses (R0).
(H) FRET ratio prior to glutamate uncaging (R0).
(I) For single spines (position: 0 μm) stimulated by 1 Hz uncaging of glutamate for 60 s, average spatial extent along the adjacent dendritic shaft of CaV1.2:STIM1 FRET ratio and of ER Ca2+ depletion (D1ER signal) from experiments presented in Figure 4.
Throughout: 12–14 DIV neurons. Means presented ± SEM. *p < 0.05.
As biochemical experiments have indicated that activated STIM1 can bind to CaV1.2 channels (Park et al., 2010; Wang et al., 2010), we tested in live neurons whether glutamate might induce association of STIM1 with LTCCs. To this end, we co-expressed STIM1-YFP and CFP-CaV1.2 in neurons (Figure 2D) and measured Förster resonance energy transfer (FRET) between these fluorescent partners as a gauge of their proximity to one another. For the dendrite illustrated in Figure 2E, uncaging was carried out within the box outlined in black, and FRET ratios were measured in the gray-shaded region of interest. For this dendrite, we found that 60 s of 1 Hz glutamate uncaging increased FRET between STIM1-YFP and CFP-CaV1.2 by the end of the train (60 s), and FRET relaxed to the pre-uncaging level by 140 s after the end (200 s after the onset) of the uncaging train (Figure 2F). The time course of the rise and decay of the FRET signal between STIM1-YFP and CFP-CaV1.2, illustrated with greater temporal resolution in Figure 2G, was comparable to that for STIM1-YFP clustering, indicating that STIM1 clustering and translocation to regions of the ER that are near the PM was followed closely by STIM1-CaV1.2 interaction and inhibition of the channel’s activity. The increase in FRET between STIM1-YFP and CFP-CaV1.2 initiated by glutamate was strongly reduced by block of LTCCs with nimodipine, and was virtually absent when NMDARs were blocked by MK801. In response to glutamate uncaging, the constitutively active mutant STIM1(D76A)-YFP failed to show any change in its chronically elevated level of FRET with CFP-CaV1.2 (Figure 2H), as though these two FRET partners were already, and persistently, co-associated.
Evidence that STMI1-YFP:CFP-CaV1.2 interaction (FRET) spreads outside the spine and into the dendritic shaft is visible in Figure 2F. Relative to the position of the dendritic spine stimulated by glutamate uncaging, interaction between STIM1 and CaV1.2 extends about ±10 μm along the dendritic shaft (Figure 2I). The spatial extent of ER Ca2+ depletion is slightly larger, about ±15 μm along the shaft. Thus in response to 1 Hz uncaging near a spine, STIM1:CaV1.2 interaction and ER Ca2+ release occur in both the spine and adjacent shaft, but these signals do not spread beyond the immediate vicinity of the stimulated spine.
[Ca2+]ER and [Ca2+]cyto in dendritic spines and shafts
We next examined the effects of glutamate uncaging on cytosolic Ca2+ ([Ca2+]cyto) and [Ca2+]ER in dendritic spines and shafts. Measurement of [Ca2+]cyto was made using the genetically-encoded Ca2+ indicator, RGECO1 (Zhao et al., 2011), and simultaneously, measurement of [Ca2+]ER was made with D1ER (Figure 3A). Photo-uncaging of glutamate near an individual spine at 1 Hz for 60 s generated a rapid elevation of [Ca2+]cyto in both the dendritic spine and the adjoining dendritic shaft (Figure 3B), and a much slower, progressive decrease in [Ca2+]ER (Figure 3D,E). Blocking NMDARs with MK801 eliminated both elevation of [Ca2+]cyto and subsequent CICR from ER (Figure 3C,E), highlighting the reliance upon NMDARs for initiation of signaling.
Figure 3. Simultaneous measurement of changes in [Ca2+]cyto and [Ca2+]ER following uncaging of glutamate near a dendritic spine.

Neurons (DIV 12–14) expressing both RGECO1 and D1ER were used to image [Ca2+]cyto and [Ca2+]ER. MNI-glutamate was uncaged at 1 Hz for 60 s.
(A) Left, fluorescence image of neuron shows RGECO1 fills spines and dendrites. High magnification shows presence of RGECO1 (middle) and D1ER (right) in the same spine. Uncaging of MNI-glutamate was carried out in the region marked by white box.
(B) In the absence (left) or presence of MK801 (right), kymographic displays of RGECO1 fluorescence (related to [Ca2+]cyto) along each of a series of line scans acquired as marked by the yellow line passing through a spine and dendritic shaft in A. Position is represented on the vertical axis (spine near top, shaft toward bottom), and time on the horizontal axis. Yellow hash marks inset at top of each kymograph indicate the 60-s period of 1 Hz uncaging.
(C) For RGECO1, time course of average ΔF/F0 along the line scan in the absence (black, n = 10) of MK801 superimposed on that in the presence of MK801 (gray, n = 5). Mean ± SEM.
(D) In the absence (left) or presence of MK801 (right), FRET imaging of D1ER (related to [Ca2+]ER) was carried out in the same neuron as in (A), and along the same line.
(E) Time course of average total D1ER response (R/R0) along the line scan in the absence (black, n = 10) and presence of MK801 (gray, n = 5). Mean ± SEM.
Varying glutamate uncaging frequency during a 1 minute period of stimulation provided insight into the dependence upon “synaptic drive” (uncaging) of both [Ca2+]cyto and [Ca2+]ER. Stimulation at 1 Hz for 60 s triggered a rapid rise in [Ca2+]cyto that was partially sensitive to the LTCC blocker nimodipine (Figure 4A). Depletion of ER Ca2+ was slower, and a larger fraction of release was blocked by nimodipine (Figure 4B). Defining the LTCC-dependent component of change in cytosolic or ER Ca2+ as the difference in area between the normalized time course with and without nimodipine showed that for both the cytosol and ER, the LTCC component became progressively smaller as stimulus frequency was decreased over the 1 min stimulus period (Figure 4A–F; Figure S5), as summarized in Figure S6. The correlation between the magnitudes of LTCC-dependent Ca2+ influx and ER release of Ca2+ supports the idea that Ca2+-induced Ca2+ release drives stores depletion. Intriguingly, the relatively small contribution of LTCCs to the total cytosolic Ca2+ signal was nonetheless dominant in releasing Ca2+ from ER stores within dendrites.
Figure 4. Frequency dependence of LTCC-driven Ca2+ signals.

For 12–14 DIV neurons expressing RGECO1 and D1ER, simultaneous imaging of [Ca2+]cyto and [Ca2+]ER was carried out for three different frequencies of glutamate uncaging: 1 Hz, 0.333 Hz and 0.167 Hz. Black hash marks at top of each time course indicate period of glutamate uncaging. Bath contained 1 μM glycine, 3 mM CaCl2 and 0 Mg2+; MNI-glutamate = 2 mM. Measurement of the time course of change in [Ca2+] was carried out in the dendritic spine or adjoining shaft. Throughout, mean ± SEM.
(A) Left, black and white images of RGECO1 in a dendritic shaft with spines, collected before, during and after 1 Hz uncaging. Right, in spines or adjoining shafts, average time course of RGECO1 responses (ΔF/F0) to uncaging in the absence (black) or presence of 5 μM nimodipine (red). Yellow area between the red and black time courses represents the nimodipine-sensitive component of the [Ca2+]cyto response to uncaging. No response detected in the absence of MNI-glutamate (gray).
(B) Left, pseudo color-coded FRET image of the same neuron as in A, for D1ER imaging of [Ca2+]ER. Right, average time course of [Ca2+]ER depletion from the dendritic spine and adjoining shaft, normalized to initial D1ER signal (R/R0), in the absence (black) or presence of nimodipine (red). Yellow area between the black and red time courses represents the nimodipine-sensitive component of Ca2+ release from the ER. No ER depletion detected in the absence of MNI-glutamate (gray).
(C–F) Reducing uncaging frequency reduced the size of LTCC-dependent components of [Ca2+]cyto and [Ca2+]ER responses. Figure S5 presents images of dendritic spines studied using 0.333 Hz and 0.167 Hz uncaging. See also Figure S6.
(G) Frequency dependence of the LTCC-dependent component of [Ca2+]cyto and [Ca2+]ER responses. To preserve fixed total stimulation, varying only frequency of stimulation, the R/R0 signals were integrated over the first 10 uncaging flashes, with the flashes presented at 0.167, 0.333 or 1 Hz (integration periods of 60, 30 or 10s).
(H) For single spines (at 0 μm) stimulated by 1 Hz uncaging for 60 s: extent along the adjacent dendritic shaft of total (black) and nimodipine-insensitive (red) time-integrated RGECO1 Ca2+ signals (reflecting [Ca2+]cyto)
(I) Subtraction of nimodipine-insensitive signal (red) from total (black) in H, converted to percent inhibition of the nimodipine-sensitive component (reflecting region of LTCC inhibition), is plotted as a function of distance along the dendritic shaft from the stimulated spine.
The relationship between LTCC-dependent [Ca2+]cyto and stimulus frequency can be extracted from these data by integrating the fluorescence signal over a fixed number of uncaging flashes (first 10), where the frequency of the flashes varied from 0.167 Hz to 1 Hz (Figure 4G). At this fixed drive, the slight decrease in size of the LTCC-dependent [Ca2+]cyto at 1 Hz compared to 0.333 Hz may be attributable to increased feedback inhibition of LTCCs by STIM1 at higher stimulation frequency.
For 1Hz, 60 s uncaging at a spine, elevation of [Ca2+]cyto in the adjacent shaft dropped off with distance along the shaft (Figure 4H). Subtracting the nimodipine-insensitive component of the time-integrated RGECO1 signal (red) from the total signal (black) yielded the LTCC-dependent component, which is displayed in Figure 4I as percent inhibition of total by nimodipine. The inhibition of LTCC-dependent [Ca2+]cyto extends about ±5 μm along the dendrite, consistent with the lateral extent of the region in which ER Ca2+ is depleted and CaV1.2 and STIM1 interact (Figure 2I).
RNAi knockdown and rescue experiments revealed that STIM1 limits LTCC-dependent Ca2+ signals in the cytosol of both dendritic spines and shafts and in the ER of these regions (Figure 5). Compared to control neurons transfected with YFP and the empty pSilencer vector, knockdown neurons expressing STIM1RNAi exhibited a two-fold increase in dendritic shaft and spine [Ca2+]cyto, along with greater Ca2+ release from the ER (Figure 5A,C). Responses to glutamate uncaging—increases in dendritic shaft and spine [Ca2+]cyto and decreases in [Ca2+]ER—were significantly blunted in STIM1 knockdown neurons co-expressing the constitutively active mutant, human STIM1(D76A). Figure 5B,D summarizes these results, illustrating the limits STIM1 imposes on postsynaptic [Ca2+]cyto and [Ca2+]ER signals. In STIM1 knockdown neurons, uncaging at 1 Hz for 60 s near a spine generated total and nimodipine-insensitive Ca2+ profiles along the adjacent dendritic shaft as shown (Figure 5E). Subtracting nimodipine-insensitive from total signal yields the LTCC-dependent component: in contrast to the case for intact STIM1, no inhibition was detected near spines in STIM1 knockdown neurons (Figure 5F), supporting the idea that STIM1 mediates inhibition of LTCCs in dendrites.
Figure 5. STIM1 impacts L channel-dependent changes in both cytosolic and ER Ca2+.

For RNAi knockdown of STIM1 (green), RNAi knockdown of STIM1 plus rescue with hSTIM1 (black), and RNAi knockdown of STIM1 plus replacement with hSTIM1 (D76A) (gray), the time courses of changes in [Ca2+] in response to 1 Hz uncaging for 60 s are displayed as mean ± SEM.
(A) For STIM1 knockdown, rescue and replacement conditions, average time courses for [Ca2+]cyto responses in the dendritic spine and shaft.
(B) Nimodipine-sensitive component of the peak response in dendritic spine and shaft [Ca2+]cyto calculated by subtracting Ca2+ transients after nimodipine from Ca2+ transients before nimodipine in the neurons from A. Fluorescent protein bleach prevented measurement of Ca2+ both before and after nimodipine in some neurons.
(C) For STIM1 knockdown, rescue and replacement, average time courses for depletion of ER Ca2+.
(D) Nimodipine-sensitive component of the uncaging-initiated release of Ca2+ from the ER, for STIM1 knockdown, rescue and replacement in neurons from C.
(E) In neurons expressing STIM1 RNAi, 1 Hz uncaging for 60 s near a spine (0 μm) generated total (green) and nimodipine-insensitive (red) cytoplasmic Ca2+ signals (RGECO1) with the illustrated profiles along the dendritic shaft.
(F) Subtraction of nimodipine-insensitive signal (red) from total (green) in E, converted to percent inhibition of the nimodipine-sensitive LTCC component, is plotted versus distance along the adjacent shaft (green). Data from Figure 4I (STIM1 intact) reproduced in gray.
STIM1 and LTCCs in dendritic spine structural plasticity
What is the impact on neuronal function of STIM1 inhibition of LTCCs? The 1 Hz glutamate uncaging paradigm we used to study STIM1 clustering and interaction with LTCCs is known to promote long-term potentiation of synaptic transmission, incorporation of additional synaptic glutamate receptors, and spine enlargement (Colgan and Yasuda, 2014; Matsuzaki et al., 2004), hence we re-analyzed our uncaging data with the goal of determining whether STIM1 regulation of LTCC activity might be involved in structural plasticity of dendritic spines. Imaging cytosolic RGECO1 revealed that 1 Hz glutamate uncaging for 60 s increased spine cross-sectional area, as expected (Figure 6A,C). Spines are known to possess ER (Spacek and Harris, 1997), and imaging of D1ER in the same spines that were enlarged by glutamate revealed a corresponding increase in the cross-sectional area of the spine ER domain (Figure 6B,C). Laser illumination in the absence of MNI-glutamate had no effect on either spine or ER cross-sectional area (Figure 6A–C). MK801 block of NMDARs fully prevented uncaging-induced growth in spine cross-sectional area and ER content (Figure 6D–F). In contrast, block of LTCCs with nimodipine did not prevent spine enlargement, but nonetheless did prevent growth in ER content of enlarged spines.
Figure 6. STIM1- and LTCC-driven growth in spine ER content.

In neurons (12–14 DIV) expressing both RGECO1 and D1ER, 1 Hz uncaging of glutamate increased cross-sectional areas of spines and their ER compartment. (A) RGECO1 imaging of spine cross-sectional area.
(B) D1ER imaging of spine ER cross-sectional area.
(C) Quantitation of spine enlargement and growth in spine ER content in response to 1 Hz uncaging.
(D–F) Effects of antagonists of NMDARs (10 μM MK801) or of LTCCs (5 μM nimodipine) on uncaging-driven spine enlargement and growth in spine ER content. In F, the lines connecting filled and open symbols indicates that these measurements of spine and ER cross-sectional area were carried out in the same spine. Mean values in red.
(G–I) Effect on spine enlargement and growth in ER content of STIM1 knockdown, knockdown and rescue with hSTIM1, and knockdown and replacement with constitutively active hSTIM1(D76A).
Experimental data describing the growth in neighboring spine size is presented in Figure S7.
STIM1 was also critical for control of ER content of spines during their enlargement: RNAi knockdown of STIM1 allowed normal enlargement of spines in response to 1 Hz glutamate uncaging (Figure 6G,I), but fully prevented concomitant growth in spine ER content (Figure 6H,I). Uncaging-induced growth in ER content in STIM1 knockdown neurons was fully rescued by co-expression of hSTIM1 or hSTIM1 (D76A). Neighboring spines on the same shaft showed a lesser degree of enlargement and increased ER content, and only for neighbors within 10 μm (Figure S7). This range is similar to that for elevation of [Ca2+]cyto, depletion of [Ca2+]ER, and interaction of STIM1 with CaV1.2, which may in part reflect the extent of lateral diffusion of glutamate during the 1 Hz, 60 s uncaging train. The finding that 1 Hz, plasticity-inducing stimuli elicit STIM1- and LTCC-dependent growth of spine ER content raises the possibility that a coordinated change in the ER content of a spine may subserve functional plasticity of an individual synapse.
STIM1 attenuates LTCC-dependent nuclear translocation of NFATc3
NFAT transcription factors are involved in LTCC-dependent excitation-transcription coupling in neurons (Graef et al., 1999), prompting us to ask whether NFATs are regulated, via LTCC modulation, by STIM1. We therefore monitored the coupling between LTCCs and NFATc3 translocation (Murphy et al., 2014; Oliveria et al., 2007) under conditions designed to impact STIM1 signaling. At 5–120 min after stimulation with glutamate (Figure 7A), subcellular distribution of GFP-NFATc3 was analyzed by immunostaining. Nuclear GFP-NFATc3 staining was increased within 5 min following glutamate application, reached a maximum level by 15 min, and elevated nuclear GFP-NFATc3 persisted for ~90 min before returning to the pre-stimulation level by 120 min post-stimulation (Figure 7B,C). Knockdown of endogenous STIM1 with STIM1RNAi strongly enhanced the degree of nuclear translocation of GFP-NFATc3. In STIM1RNAi-expressing neurons, glutamate-stimulated GFP-NFATc3 translocation was restored to the wild-type level by co-expression of hSTIM1. In STIM1RNAi neurons co-expressing constitutively active hSTIM1 (D76A), glutamate-stimulated nuclear translocation of GFP-NFATc3 was reduced below wild-type levels. Glutamate-stimulated GFP-NFATc3 translocation was fully prevented by nimodipine block of LTCCs (Figure 7D); translocation was strongly suppressed but not fully prevented by the NMDAR blocker MK801 (Figure 7D). The small fraction of LTCC-dependent NFATc3 translocation that could not be blocked by MK801 may reveal a small, glutamatergic but non-NMDAR-mediated activation of LTCCs.
Figure 7. Attenuation of nuclear translocation of NFATc3 following STIM1 inhibition of L channels.

(A) Following 30 min pre-incubation in TTX to silence spontaneous synaptic activity in the cultures, neurons (4–5 DIV) were stimulated by 15 s application of 100 μM glutamate + 1 μM glycine. Neurons were returned to TTX-containing solution until fixed at times indicated in the schematic.
(B) Two-dimensional, summed-intensity projection images of neurons expressing GFP-NFATc3 and stained with anti-GFP (black) and, to visualize the nucleus, DAPI (blue). Top row of images illustrates glutamate-triggered GFP-NFATc3 translocation in neurons with endogenous STIM1. Second row: STIM1RNAi neurons transfected with a short hairpin construct to suppress endogenous (rat) STIM1 expression. Third row: neurons co-expressed anti-rat STIM1RNAi and human STIM1 (hSTIM1); fourth row: neurons co-expressed anti-rat STIM1RNAi and constitutively active human STIM1 (D76A).
(C) Average time courses (± SEM) of GFP-NFATc3 nuclear translocation following application of glutamate. GFP-NFATc3 distribution was measured as the ratio between nuclear and cytoplasmic fluorescence intensity. Inset: same data, expanded time scale.
(D) Effects of MK801 and nimodipine on translocation of GFP-NFATc3 induced by 15 s bath application of glutamate.
(E) Left, black and white image of an RGECO1a-expressing neuron before glutamate uncaging. NFAT signal indicated in black. Yellow arrow indicates site of uncaging and blue arrow indicates region where NFAT translocation was measured (neuronal soma).
(F) Black and white images of somatic sGFP2-NFATc3 fluorescence intensity at three time points: before uncaging (NG = no glutamate) and at 5 min and 40 min after uncaging. Black marks NFAT signal. Left column, STIM1RNAi neurons. Right column, STIM1 wt neurons transfected with empty pSilencer vector.
(G) Average time courses (± SEM) of sGFP2-NFATc3 nuclear translocation following glutamate uncaging. sGFP2-NFATc3 distribution measured as the ratio between nuclear and cytoplasmic fluorescence intensity, normalized to pre-uncaging values.
We also investigated nuclear translocation of NFATc3 in response to glutamate uncaging near a single dendritic spine. In these experiments, we imaged living neurons that expressed NFATc3 fused to sGFP2 (Kremers et al., 2007), a brighter version of GFP (Figure 7E). Glutamate was uncaged using 1 ms laser flashes applied at 1 Hz during three 20 s episodes, with episodes separated by 6 s inter-episode intervals (60 flashes total). sGFP2-NFATc3 fluorescence was monitored in the cytosolic compartment of the neuronal soma and in the neuronal nucleus (Figure 7F). In stimulated neurons, nuclear sGFP2-NFATc3 fluorescence reached maximum intensity 5 min post-uncaging and returned to a steady state level near the pre-stimulation level after 40 min (Figure 7F,G). As seen with 15 s global glutamate application, STIM1RNAi-expressing neurons showed strongly enhanced nuclear translocation of sGFP-NFATc3 following glutamate uncaging. STIM1RNAi neurons co-expressing constitutively-active hSTIM1(D76A) showed nuclear translocation of sGFP2-NFATc3 that was reduced below the wild-type level. sGFP2-NFATc3 translocation in response to glutamate uncaging was fully prevented by nimodipine block of LTCCs (Figure 7G). In sum, these results are consistent with the idea that STIM1 feedback inhibition of LTCCs limits LTCC-dependent translocation of NFATc3 to the nucleus.
DISCUSSION
STIM1’s defining role, activation of PM Orai channels and hence of stores-operated Ca2+ entry (SOCE), was discovered in non-excitable cells (Liou et al., 2005; Roos et al., 2005). In neurons, SOCE has been implicated in hippocampal synaptic potentiation (Baba et al., 2003), short-term presynaptic plasticity (Emptage et al., 2001), and long-term depression in hippocampal (Holbro et al., 2009) and cerebellar (Hartmann et al., 2014) synapses. Control of ER dynamics (Toresson and Grant, 2005) and of Ca2+ release (Emptage et al., 1999) within spines by NMDARs (Ng et al., 2014) has also each been reported, but roles for LTCCs and STIM1 in regulating these processes had not previously been identified. Thus, our findings that negative feedback control of LTCC activity by STIM1 tunes ER Ca2+ release, spine ER content, and synapse-to-nucleus signaling sheds new light on links between activity-dependent PM Ca2+ signaling and ER STIM1 signaling that are important for regulation of neuronal function.
LTCCs were much more strongly engaged by stronger stimulation that resulted from uncaging at higher frequency (Figure 4A–F, Figure S6), likely owing to summation of NMDAR-mediated depolarizations. This finding is in accord with evidence that LTCCs, with slow kinetics of activation combined with a relatively low threshold for activation, respond preferentially to longer-lasting, low-amplitude postsynaptic depolarizations as occurs with excitatory postsynaptic potentials but not action potentials (Mermelstein et al., 2000). The requirement of higher frequency stimulation for generation of substantial LTCC Ca2+ signals may also help account for past challenges in dissecting LTCC-dependent components of LTP from NMDAR-dependent ones, as synaptic activity is key to activating both of these processes (Blundon and Zakharenko, 2008; Padamsey and Emptage, 2014). The preferential engagement of LTCCs by higher frequency stimulation also raises the possibility that non-linear summation of synaptic potentials might result in frequency-dependent effects on signaling molecules downstream of LTCCS, such as NFAT. As STIM1, via negative feedback onto LTCCs, acts to scale down signaling through NFAT, the frequency-dependence of activation of LTCCs might function to non-linearly depress NFAT signaling which, speculatively, could result in resetting the mid-point of LTCC signaling to the nucleus and thereby maintain the dynamic range of this process. A natural question to pursue in future is whether remodeled spine structure persisting beyond the initial phase investigated here might involve lasting alteration in spine ER content, a concomitant increase in the fraction of LTCCs that interact with STIM1, and a consequent chronic scaling of downstream signaling through NFAT or other LTCC-regulated transcription factors that could support sustained change in synaptic transmission.
Our finding that activation of STIM1 attenuates nuclear translocation of NFAT in cultured hippocampal neurons contrasts strikingly with findings in various non-excitable cell types (Prakriya and Lewis, 2015), skeletal muscle (Stiber et al., 2008), cardiomyocytes (Hulot et al., 2011) and neural progenitor cells (Somasundaram et al., 2014), in which STIM1 promotes nuclear translocation and activity of NFAT. The basis for this difference is straightforward: STIM1 feedback inhibition of LTCC Ca2+ influx reduces LTCC-dependent activation of calcineurin and NFAT (Graef et al., 1999), whereas in cases in which STIM1 activates SOCE, including in neurons, Ca2+ entry via this mechanism activates calcineurin, which in turn promotes NFAT translocation and action. An intriguing question is how these two STIM1 signaling pathways are coordinated within a neuron in order to provide integrated regulation of downstream effectors such as NFAT.
What other roles in synaptic plasticity might growth in spine ER content play? Activation of NMDARs triggers large postsynaptic store-based Ca2+ transients (Emptage et al., 1999), and augmentation of synaptically-stimulated NMDAR activity enhances ER growth in spines (Ng et al., 2014), but little is known about long-lasting ER structural changes specific to high-frequency stimulation of an individual synapse. From a structural point of view, growth in ER content might help stabilize mushroom spines that have become enlarged during the course of LTP. Increased ER content would also increase the Ca2+ storage capacity of spines, which could simply match store capacity to the enlarged spine volume, or perhaps increase storage and Ca2+ buffering above spine volume-normalized pre-growth levels. Increased spine Ca2+ stores could support increased Ca2+ release in response to either synaptic NMDAR/LTCC activity or neuromodulatory Gq-coupled signals. Finally, increased ER content and Ca2+ capacity in spines could support increased SOCE via Orai channels.
STIM1-dependent growth in spine ER content might be anticipated to increase the spine complement of another important ER Ca2+ sensor, STIM2, a sensor which has been found to promote insertion of AMPA receptors into the neuronal PM (Garcia-Alvarez et al., 2015a) and impact long-term potentiation (Garcia-Alvarez et al., 2015b). In addition, it is now emerging that regions of close apposition between ER and PM are critical zones in the control of protein trafficking (Fox et al., 2013; Garcia-Alvarez et al., 2015a) and, in agreement with our current findings for NFAT regulation, gene expression (Lalonde et al., 2014; Somasundaram et al., 2014). STIM-based signaling in these regions may also be critical in the formation (Garcia-Alvarez et al., 2015a) and stabilization of dendritic spines (Sun et al., 2014), particularly through the action of STIM2, as reduced STIM2 signaling in neurons is related to cognitive deficits in aging neurons and in a mouse model of familial Alzheimer’s disease (Sun et al., 2014).
In conclusion, we have identified a mechanism that links activation of postsynaptic NMDARs and LTCCs to signaling by the ER Ca2+ sensor STIM1, thereby providing control of local LTCC signaling, dendritic spine ultrastructure and signaling from synapse to nucleus. What subtypes of LTCCs are involved in this process, and whether ryanodine receptors versus inositol-1,4,5-trisphosphate receptors are involved, remain to be determined. Other questions to pursue include identification of the molecular underpinnings of growth in spine ER content, investigation of potential roles of the spine apparatus and synaptopondin (Korkotian et al., 2014) in such growth, and how changes in spine ER content impact spine function.
EXPERIMENTAL PROCEDURES
Culture and transfection of primary hippocampal neurons
Primary hippocampal neurons were prepared from postnatal day 0–2 Sprague-Dawley rats of both sexes. All procedures with animals were approved by the Institutional Animal Care and Use Committee of the University of Colorado Anschutz Medical Campus. For all measurements in Figure 1, Figures S1–S3 and Figure 7A–D, neurons 4–5 days in vitro (DIV) were used. For experiments using photo-uncaging of MNI-glutamate (Figures 2–6, Figure 7E–G, and Figures S4–S7), we used 12–16 DIV neurons to allow development of dendritic spines.
Patch-clamp recording
To isolate L-type Ca2+ currents from other neuronal Ca2+ currents, 4–5 DIV neurons were treated for 30 min prior to recording with blockers of N- and P/Q-type channels, ω-CTx-GVIA (1 μM) and ω-CTx-MVIIC (5 μM). Neurons were used within 1 hr of blocker pre-treatment to minimize adulteration of L current as N- and P/Q-type channels became unblocked (Oliveria et al., 2007). The whole-cell pipet contained (mM): 120 CsMeSO4, 30 tetraethylammonium-Cl, 10 ethylene glycol-bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) or 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), 5 MgCl2, 5 Na2ATP and 10 HEPES; pH 7.2 with TEA-OH. The bath solution contained 1 μM tetrodotoxin (TTX) and (mM): 125 NaCl, 10 CaCl2, 5.85 KCl, 22.5 TEA-Cl, 1.2 MgCl2, 10 HEPES(Na), 11 D-glucose; pH 7.4 with HCl. Glutamate and other agents were applied using continuous perfusion (~1 ml/min) via a 3-barrel system (SF-77B, Warner Instruments) that allowed rapid solution exchange between control solution, test (e.g., glutamate + glycine), and when needed, antagonist (e.g., glutamate + glycine + MK801). (+)-MK801 maleate, DNQX disodium salt (6,7-dinitroquinoxaline-2,3-dione), MTEP hydrochloride (3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride), and TTX citrate were obtained from Tocris Bioscience. Nimodipine was obtained from Sigma-Aldrich.
Measurement of [Ca2+]ER with D1ER
For D1ER measurements in Figure 1H and Figure S3, fluorescence images were acquired from transfected cultured neurons (4–5 DIV) using a Zeiss Axiovert 200M inverted microscope. The bath solution was identical to that used for patch-clamp recording, but omitted TTX. Neurons on glass coverslips were mounted in a sealed-top, diamond-shaped chamber (RC-21BDW, Warner Instruments). Gravity-driven continuous laminar flow (~1 ml/min) through the chamber allowed rapid exchange between control, glutamate- and blocker-containing solutions.
Photo-uncaging MNI-glutamate and simultaneous imaging of [Ca2+]cyto and [Ca2+]ER
DIV 12–14 neurons transfected with D1ER and RGECO1 were imaged in bath solution containing 1 μM TTX and (mM): 135 NaCl, 3 CaCl2, 5 KCl, 25 HEPES, 10 D-glucose; pH 7.4 with NaOH. Fluorescence images were acquired using a Zeiss Observer.Z1 inverted microscope equipped with α Plan-Apochromat 40x (1.30 NA) oil-immersion objective lens; 405 nm/50 mW, 445 nm/40 mW and 561 nm/50 mW lasers; an mSAC unit for correction of spherical ablation (Intelligent Imaging Innovations); a Yokogawa CSU-X1 spinning disk unit; a Photometrics Evolve EMCCD camera with 16-bit dynamic range; a Vector scanner (Intelligent Imaging Innovation) for positioning the photo-uncaging laser; and an mSwitcher ms optical switching unit (Intelligent Imaging Innovation) that enabled simultaneous use of the CSU-X1 and Vector units. Instrumentation was controlled by SlideBook 6.0 software (Intelligent Imaging Innovation). A dichroic mirror (445/515/640; Semrock) and three different filter sets (RFP, 561 nm excitation, 617/73 nm emission; CFP, 445 nm excitation, 445/45 nm emission; and FRET (rawFRET), 445 nm excitation, 525/50 nm emission) were used to serially capture a set of three images using a 100 ms exposure from a fixed image plane. Without MNI-glutamate in the bath, a 1 μm x 1 μm region was selected near a dendritic spine of interest. Glutamate was photo-uncaged from MNI-glutamate in this region using 2 ms pulses of light from a 405 nm/50 mW laser. MNI-caged L-glutamate was obtained from Tocris Bioscience.
FRET between STIM1-YFP and CFP-CaV1.2
For sensitized Förster resonance energy transfer (FRET) measurements [FRETC; three filter (3F)] in Figure 2F–H, a dichroic mirror (445/515/640; Semrock) and three different filter sets were used to serially capture a set of three images using a 100 ms exposure from a fixed image plane: YFP, 515 nm excitation, 525/50 nm emission; CFP, 445 nm excitation; 482/35 nm emission; and FRET (rawFRET), 445 nm excitation, 525/50 nm emission.
Measurement of spine and ER cross-sectional areas
Cross-sectional areas of the cytosolic compartment of dendritic spines were measured using RGECO1 as a reporter (561 nm excitation; 617/73 nm emission). Cross-sectional areas of spine ER compartments were measured using emission from the citrine moiety of D1ER as a reporter (515 nm excitation; 525/50 nm emission).
NFATc3 translocation measurements
For experiments with bath-applied glutamate, cultured neurons were transfected with GFP-NFATc3 and studied at 4–5 DIV. Prior to stimulation with glutamate, coverslips bearing neurons were incubated in 1 μM TTX for 30 min at 37°C. Neurons were then stimulated fo r 15 s in TTX-free solution using glutamate (100 μM) + glycine (1 μM), and finally returned to the TTX solution until fixation at the indicated time points. For experiments with glutamate uncaging near spines, neurons were transfected with sGFP2-NFATc3 and, to provide a reporter of spine stimulation, with RGECO1a. Prior to glutamate uncaging, these neurons (14–16 DIV) were incubated with 1 μM TTX in growth medium, at 37°C. After incubation i n the TTX solution, coverslips were placed in an imaging chamber with bath solution containing: 1.5 mM MNI-caged L-glutamate, 10 μM glycine and (mM): 135 NaCl, 3 CaCl2, 5 KCl, 25 HEPES, 10 D-glucose; pH 7.4 with NaOH.
Statistical analysis
Values reported are mean ± standard error of the mean. Statistical analyses were carried out using SigmaPlot version 11.0 (Systat). Comparisons were made using one-way analysis of variance and a Bonferroni correction for multiple comparisons; the data were normally distributed as judged by a Shapiro-Wilk test.
Supplementary Material
Highlights.
NMDA receptor activation of L-type Ca2+ channels triggers Ca2+ release from ER
ER Ca2+ depletion activates STIM1, which feeds back onto L channels to inhibit them
Activated STIM1 promotes L channel-dependent growth in dendritic spine ER content
Activated STIM1 attenuates L channel-dependent nuclear translocation of NFAT
Acknowledgments
This work was supported by NIH grants T32-HL007822, R01-MH102338 and R01-HL088548. Core facility imaging was supported by NIH grant P30-NS048154 and NIH/NCATS Colorado CTSI grant UL1 TR001082.
Footnotes
AUTHOR CONTRIBUTIONS
PJD, ARW, MLD and WAS designed experiments; PJD and ARW carried out experiments; PJD, MLD and WAS wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baba A, Yasui T, Fujisawa S, Yamada RX, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J Neurosci. 2003;23:7737–7741. doi: 10.1523/JNEUROSCI.23-21-07737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Bayazitov IT, Richardson RJ, Fricke RG, Zakharenko SS. Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J Neurosci. 2007;27:11510–11521. doi: 10.1523/JNEUROSCI.3077-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Blundon JA, Zakharenko SS. Dissecting the components of long-term potentiation. Neuroscientist. 2008;14:598–608. doi: 10.1177/1073858408320643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan LA, Yasuda R. Plasticity of dendritic spines: subcompartmentalization of signaling. Annu Rev Physiol. 2014;76:365–385. doi: 10.1146/annurev-physiol-021113-170400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney JR, Hurlburt JL, Selig DK, Harris KM, Fiala JC. Endosomal compartments serve multiple hippocampal dendritic spines from a widespread rather than a local store of recycling membrane. J Neurosci. 2002;22:2215–2224. doi: 10.1523/JNEUROSCI.22-06-02215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RP, Froemke RC, Sjostrom PJ, van Rossum MC. Unified pre- and postsynaptic long-term plasticity enables reliable and flexible learning. eLife. 2015:4. doi: 10.7554/eLife.09457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel CaV1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Fox PD, Haberkorn CJ, Weigel AV, Higgins JL, Akin EJ, Kennedy MJ, Krapf D, Tamkun MM. Plasma membrane domains enriched in cortical endoplasmic reticulum function as membrane protein trafficking hubs. Mol Biol Cell. 2013;24:2703–2713. doi: 10.1091/mbc.E12-12-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alvarez G, Lu B, Yap KA, Wong LC, Thevathasan JV, Lim L, Ji F, Tan KW, Mancuso JJ, Tang W, et al. STIM2 regulates PKA-dependent phosphorylation and trafficking of AMPARs. Mol Biol Cell. 2015a;26:1141–1159. doi: 10.1091/mbc.E14-07-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alvarez G, Shetty MS, Lu B, Yap KA, Oh-Hora M, Sajikumar S, Bichler Z, Fivaz M. Impaired spatial memory and enhanced long-term potentiation in mice with forebrain-specific ablation of the Stim genes. Front Behav Neurosci. 2015b;9:180. doi: 10.3389/fnbeh.2015.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Karl RM, Alexander RP, Adelsberger H, Brill MS, Ruhlmann C, Ansel A, Sakimura K, Baba Y, Kurosaki T, et al. STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron. 2014;82:635–644. doi: 10.1016/j.neuron.2014.03.027. [DOI] [PubMed] [Google Scholar]
- Holbro N, Grunditz A, Oertner TG. Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc Natl Acad Sci U S A. 2009;106:15055–15060. doi: 10.1073/pnas.0905110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland TM, Saggau P. Facilitation of L-type Ca2+ channels in dendritic spines by activation of β2 adrenergic receptors. J Neurosci. 2004;24:8416–8427. doi: 10.1523/JNEUROSCI.1677-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulot JS, Fauconnier J, Ramanujam D, Chaanine A, Aubart F, Sassi Y, Merkle S, Cazorla O, Ouille A, Dupuis M, et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation. 2011;124:796–805. doi: 10.1161/CIRCULATIONAHA.111.031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klejman ME, Gruszczynska-Biegala J, Skibinska-Kijek A, Wisniewska MB, Misztal K, Blazejczyk M, Bojarski L, Kuznicki J. Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca2+ store in neurons. Neurochem Int. 2009;54:49–55. doi: 10.1016/j.neuint.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Frotscher M, Segal M. Synaptopodin regulates spine plasticity: mediation by calcium stores. J Neurosci. 2014;34:11641–11651. doi: 10.1523/JNEUROSCI.0381-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers GJ, Goedhart J, van den Heuvel DJ, Gerritsen HC, Gadella TW., Jr Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry (Mosc) 2007;46:3775–3783. doi: 10.1021/bi0622874. [DOI] [PubMed] [Google Scholar]
- Lalonde J, Saia G, Gill G. Store-operated calcium entry promotes the degradation of the transcription factor Sp4 in resting neurons. Sci Signal. 2014;7:ra51. doi: 10.1126/scisignal.2005242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, Zakharenko SS. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc Natl Acad Sci U S A. 2004;101:16665–16670. doi: 10.1073/pnas.0407581101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey A, Daniel JA, Hadzic G, Chau N, Clayton EL, Mariana A, Whiting A, Gorgani NN, Lloyd J, Quan A, et al. Building a better dynasore: the dyngo compounds potently inhibit dynamin and endocytosis. Traffic. 2013;14:1272–1289. doi: 10.1111/tra.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Bito H, Deisseroth K, Tsien RW. Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci. 2000;20:266–273. doi: 10.1523/JNEUROSCI.20-01-00266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, et al. Role of hippocampal CaV1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell’Acqua ML. AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep. 2014;7:1577–1588. doi: 10.1016/j.celrep.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- Ng AN, Doherty AJ, Lombroso PJ, Emptage NJ, Collingridge GL. Rapid regulation of endoplasmic reticulum dynamics in dendritic spines by NMDA receptor activation. Mol Brain. 2014;7:60. doi: 10.1186/s13041-014-0060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell’Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dittmer PJ, Youn DH, Dell’Acqua ML, Sather WA. Localized calcineurin confers Ca2+-dependent inactivation on neuronal L-type Ca2+ channels. J Neurosci. 2012;32:15328–15337. doi: 10.1523/JNEUROSCI.2302-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padamsey Z, Emptage N. Two sides to long-term potentiation: a view towards reconciliation. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130154. doi: 10.1098/rstb.2013.0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci U S A. 2004;101:17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science. 2010;330:101–105. doi: 10.1126/science.1191027. [DOI] [PubMed] [Google Scholar]
- Pigott BM, Garthwaite J. Nitric oxide is required for L-type Ca2+ channel-dependent long-term potentiation in the hippocampus. Front Synaptic Neurosci. 2016;8:17. doi: 10.3389/fnsyn.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Store-operated calcium channels. Physiol Rev. 2015;95:1383–1436. doi: 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Konnerth A. Stores not just for storage: intracellular calcium release and synaptic plasticity. Neuron. 2001;31:519–522. doi: 10.1016/s0896-6273(01)00402-0. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13:549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram A, Shum AK, McBride HJ, Kessler JA, Feske S, Miller RJ, Prakriya M. Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J Neurosci. 2014;34:9107–9123. doi: 10.1523/JNEUROSCI.0263-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiber J, Hawkins A, Zhang ZS, Wang S, Burch J, Graham V, Ward CC, Seth M, Finch E, Malouf N, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10:688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL, 3rd, Bezprozvanny I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 2014;82:79–93. doi: 10.1016/j.neuron.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toresson H, Grant SG. Dynamic distribution of endoplasmic reticulum in hippocampal neuron dendritic spines. Eur J Neurosci. 2005;22:1793–1798. doi: 10.1111/j.1460-9568.2005.04342.x. [DOI] [PubMed] [Google Scholar]
- von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science. 2010;330:105–109. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. J Neurosci. 2008;28:5740–5751. doi: 10.1523/JNEUROSCI.3998-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE. An expanded palette of genetically encoded Ca2+ indicators. Science. 2011;333:1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.