

Abstract
Defective autophagy has been linked with many pathologies, including cancer, diabetes, infectious disease, myopathies, heart, liver, lung, and neurodegenerative disease. Autophagy has therefore become an important target in drug discovery. Recent advances have identified new ways to monitor autophagy in vitro and in vivo. Many assays rely on visualizing autophagy-related intracellular markers such as microtubule-associated protein light chain 3 (LC3) II, which have posed issues with in vivo and clinical translation of the in vitro assays. Here, we present an overview of current in vitro and in vivo methodologies to measure autophagy with a special focus on the tumor microenvironment.
Keywords: Autophagy, Tumor microenvironment, Autophagy modulators, Autophagic flux, Automated quantitative analyses (AQUA), Comparative secretome analysis, High-autophagy cells, Low-autophagy cells
9.1 Introduction
Autophagy is a dynamic and tightly regulated homeostatic process. Autophagy is characterized by sequestration of cellular organelles and proteins into autophagic vesicles and fusion of these vesicles with lysosomes. Under normal conditions, basal autophagy plays an important role in the housekeeping of the cell via disposal of waste, which includes removal of damaged subcellular organelles and protein aggregates. Under stress conditions, including starvation, intracellular pathogens, and reactive oxygen species, autophagy supplies the cell with an internal source of nutrients (amino acids, sugars, and nucleic acids). This recycling capacity helps cells adapt and survive in the face of unfavorable conditions. The autophagy pathway occurs in many distinct stages, including autophagic vesicle initiation, nucleation, elongation, fusion, and degradation (Fig. 9.1). These stages are tightly regulated by several autophagy-related (Atg) proteins. Activation or suppression of autophagic machinery is regulated through a number of signaling networks including the mammalian target of rapamycin (mTOR) or AMPK signaling, the ER stress response, and the DNA damage response. For example a signaling kinase called unc-like kinase 1 (Ulk1)/Atg 1 complex is responsible for initiation autophagy. Phosphorylation of Ulk1 at Ser 317 and Ser 777 by the energy-sensing AMP-activated protein kinase (AMPK) activates autophagy, whereas phosphorylation of Ulk1 at ser 757 by mammalian target of rapamycin (mTOR) dissociates Ulk1 from AMPK and suppresses autophagy [1]. In addition, many posttranslational modifications including acetylation, ubiquitination, and oxidation of sulfhydryls regulate the autophagic process. After initiation and activation, the Ulk1/Atg 1 complex guides the localization of class III phosphoinositide 3-kinase (PI3K) complex for the regulation of nucleation and assembly of the phagophore membrane. In addition, a ubiquitin-like conjugation system involving ATG7 and ATG3 guides the formation of the Atg5-Atg12-Atg16L complex. This complex in turn is responsible for conjugating LC3 to phosphatidylethanolamines (PE), thereby promoting the elongation and closing of the autophagosomal membrane [1]. The sequestering of cargo substrates (protein aggregates, mitochondria, and other organelles) into the autophagosomes is mainly guided by cargo receptor proteins such as P62/SQSTM1. Finally, autophagosomes fuse with lysosomes to form autolysosomes, where the degradation of cargo is mainly achieved by acid hydrolases (for more details of molecular machinery of autophagy see [2]).
Fig. 9.1.

Formation of autophagic vesicles and the site of autophagy measurement
Given the well-known role of autophagy in normal physiology, recent evidence suggests that autophagy in cancer cells plays a dual role, to either suppress early carcinogenesis or support the survival and growth of advanced tumors [3]. The role of autophagy within the tumor microenvironment (TME) is being increasingly recognized as a key factor in cancer progression. Evidence indicates that autophagy promotes tumor progression [4], resistance to therapy [5, 6], a blunting of immune effector cell function [7–9], and metastasis [10]. A study shows that in a mouse model of ductal adenocarcinoma of the pancreas (PDAC), high levels of basal autophagy support malignant growth. Inhibition of autophagy by chloroquine (CQ) or silencing of Atg5 induced tumor regression [11]. In addition, recent data suggest that autophagy is involved in the cross talk between the cancer cell and tumor stroma, where Atg5 expressed in endothelial cells mediates starvation and hypoxia-induced angiogenesis by high mobility group box1 (HMGB1), a pro-autophagic factor [12]. In pancreatic cancer cells a protein called receptor for advanced glycation end products (RAGE) overexpression and interaction with HMGB1 causes resistance to apoptosis and induces autophagy as pro-survival mechanism [13]. Taken together these capacities of autophagy contribute to its role as a resistance mechanism to multiple cancer therapeutics. Despite a large volume of data that supports targeting autophagy in cancer, development of autophagy inhibitors has been slow. One reason for this is that the methods used to monitor autophagy have significant limitations. Despite these limitations some of these assays have been applied to human samples effectively to fuel translational research. Here we will provide detailed information for each assays and report on their strengths and weaknesses.
9.2 Autophagy Modulators
Even though numerous reports have validated the role of autophagy as a potential therapeutic target for diseases [14], the role of autophagy in anticancer treatment is complex and may be dependent heavily on the tumor microenvironment [15]. On the one hand unfettered autophagy induced by specific anticancer agents can promote autophagic cell death in vitro. On the other hand, there is mounting evidence that in the context of an advanced cancer, intratumoral autophagy promotes growth and resistance of cancers [16]. Although a growing number of reports on the discovery of small-molecule autophagy modulators have been published, concern is raised that some of the hits in these screens modulate autophagy through off-target or indirect effects. Many new autophagy modulators appear to be effective only in cell culture systems but little is known about their safety or efficacy in preclinical and clinical uses. Given the complexity of interpreting the autophagy process, one should consider using multiple methods to discover more selective agents that can efficiently modulate autophagy either alone or in combination. At present, many autophagy modulating agents serve as a potential anticancer therapeutics and here is the partial list of widely used autophagic modulators (Table 9.1) and site of action in the autophagy pathway.
Table 9.1.
List of compounds with known mechanism of autophagy modulation
| Autophagy inducers | ||
|---|---|---|
| AMPK activators and mTORC1 inhibitors |
|
|
| Inhibitors of class I PI3K signaling |
|
|
| Modulators of intracellular Ca2+ |
|
|
| Proteasome inhibitors |
|
|
| Histone deacetylase (HDACs) inhibitors |
|
|
| Synthetic and plant-based chemotherapeutic agents |
|
|
| Autophagy inhibitors | ||
| Class III PI3K inhibitors blocks autophagy |
|
|
| Microtubule modulators |
|
|
| Lysosomal inhibitors |
|
|
| Vacuolar H+-ATPase inhibitors |
|
|
| Inhibitors of lysosomal hydrolases and proteases |
|
|
| c-Jun N-terminal kinase (JNK) inhibitors |
|
|
9.3 Measuring Autophagy Markers by Immunoblotting
In autophagy research, Western blot data are widely used as a semiquantitative method to analyze autophagic marker proteins in cells, animals, and in human tissues. When appropriate precautions are taken before and after sample preparation, Western blot provides useful information for monitoring autophagy in cancer research. Here we provide basic information of some well-known autophagic marker proteins and key points that should be considered when performing Western blots for the detection and analysis of autophagy markers.
-
Microtubule-associated protein 1A/1B-light chain 3 (LC3)
Detecting LC3 by immunoblotting has become a reliable method for monitoring autophagy and autophagy-related processes. Readers are advised to refer Chap. 10 for details on LC3 immunoblotting. To assess autophagic flux using LC3, autophagy modulators are used to perturb autophagy and rationally conclude the effects on autophagy flux in the given system (see below).
-
Other autophagy marker proteins
Depending on the cell types and the stimuli, the autophagic marker proteins are differentially regulated during autophagy induction. During autophagy, the ratio of LC3-II/LC3-I, or the amount of LC3-II, is generally increased, but other autophagy-related proteins, such as Beclin 1, Atg5–Atg12, Atg4, and Atg7, may or may not be increased.
9.3.1 p62 (SQSTM1/Sequestosome 1)
p62 is a ubiquitin- and LC3-binding protein. The analysis of p62 (also called as SQSTM1/sequestosome 1) protein levels along with LC3 can provide insights about autophagic flux in a tissue-dependent manner. Total cellular levels of p62 inversely correlate with autophagic activity in most conditions, because p62 directly binds to LC3 and gets selectively incorporated into autophagosomes and degraded by autophagy [17]. Thus, in a given condition, low autophagic flux can be determined by high basal levels of p62 and low LC3-II levels. Conversely, high flux can be inferred by high LC3-II and decreased p62 levels after autophagy stimulation. However, it should be noted that changes in p62 levels are also used to indicate a defect in the turnover of poly-ubiquitinated protein aggregates. Thus, to monitor autophagic flux, analysis of p62 protein levels should be used only in combination with other methods such as LC3-II turnover [18]. P62 is also regulated by oxidative stress and the NF-kB pathway. Therefore there are often discordant behaviors between autophagic flux and p62 levels. The interpretation of changes in p62 should therefore be made cautiously.
9.3.2 Beclin, ATG5, ATG7, and Other Core Autophagy Proteins
Beclin (ATG8) is monoallelically deleted in cancers which demonstrate a loss of the neighboring tumor suppressor gene BRCA1 [19]. Beclin levels have been used by some as a measure of autophagy, but this should be avoided. In all cases of Beclin allelic loss, the retained allele always harbors wild-type Beclin, and beclin levels can be transcriptionally increased to wild-type levels even in beclin heterozygous cells. ATG5 levels have been shown to be regulated by promoter methylation in melanoma cells [20]. The measurement of ATG proteins in general is not a reliable reflection of autophagic flux, unless the protein levels of these enzymes are close to zero, as would be the case in genetically engineered cell lines, or as mentioned epigenetically silenced cell lines. Even in these cases, confirmation of LC3 levels is necessary. In our experience >70 % knockdown of ATG5 in melanoma cells blunted but did not completely abrogate LC3 lipidation [21].
9.3.3 Phosphorylation Status of ULK
There are multiple ULK isoforms which exist in mammalian cells, including ULK1, ULK2, ULK3, ULK4, and ULK5. ULK1 may be the predominant isoform responsible for the initiation of autophagy. Phosphorylation of ULK1 on Ser 317 and Ser777 in certain contexts can serve as marker for autophagy activity [22]. One problem with the detection of phospho-ULK1 by western blot is the low abundance of endogenous ULK1 in many systems.
9.4 Monitoring the Number of Autophagosomes and Autophagy Flux by Western Blot
9.4.1 Measuring the Number of Autophagosomes by LC3-I to LC3-II Conversion
The number of autophagosomes observed at any specific time point is a function of the balance between the rate of their generation and the rate of their conversion into autolysosomes. Thus, a number of autophagosomes can be monitored simply by detecting the conversion of LC3-I to LC3-II by immunoblotting. Due to the association of LC3-II with the autophagosome membrane, the levels of LC3-II correlates with the number of autophagosome formation (the amount of autophagic membrane labeled with LC3-II). The conversion of LC3-I to LC3-II provides an indicator of autophagic process but not the activity [23], because LC3-II increases during early autophagy (autophagosome formation), but at later steps it decreases (due to self-degradation). Therefore, the amount of LC3-II does not reflect the activity of autophagy as shown in Fig. 9.2. Increased LC3-II levels in a cell line or tissue could be associated with any of the following reasons: (1) enhanced autophagosome synthesis, (2) reduced autophagosome turnover, (3) delayed lysosome trafficking, (4) reduction of fusion between compartments (lysosomes with autophagosomes), (5) impaired proteolytic activity in lysosomal compartment. The factors which affect the interpretation of the LC3 Western blot data are as follows: (1) differential affinities of antibodies for LC3-I compared to LC3-II, (2) different expression levels of LC3-I and LC3-II depending on cell line and tissue, (3) LC3-II itself is subject to autophagic degradation at the lysosome [24].
Fig. 9.2.
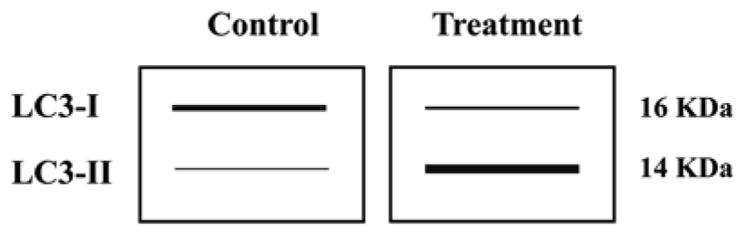
Schematic representation of LC3-I to LC3-II conversion during a typical treatment condition
9.4.2 Monitoring Autophagic Flux Using the Bafilomycin Clamp Assay
Given the fact that measuring the conversion of LC3-I to LC3-II and/or LC3-II normalization with loading control (actin, tubulin, or GAPDH) is not always a good indicator of cellular autophagic activity, it is mandatory to discriminate whether or not LC3-II accumulation is due to induction of autophagy or due to the blockage in the later stages of autophagy by conducting “autophagic flux” assays in their experimental condition [25]. Here, we discuss how LC3 Western blot experiment can be designed and used to measure autophagic flux with some examples. LC3 turnover can be examined in a cell line or tissues by inducing starvation (amino acids depletion/serum) alone or in combination with inhibitors that inhibit degradation of autolysosome content. Table 9.2 gives a partial list of conditions and inhibitors commonly used to study autophagic flux.
Table 9.2.
Partial list of conditions and inhibitors commonly used to study autophagic flux
| Category | Compounds |
|---|---|
| Starvation (induce autophagy) |
|
| Lysosomotropic agents |
|
| Lysosomal proteases |
|
To better understand and interpret the changes in levels of LC3-II processing (autophagic flux) in a given condition, we have provided an example (Fig. 9.3) where cells with low and high autophagic levels were cultured in regular nutrient- rich and starved media with or without inhibitor such as bafilomycin A1 (BafA1). As shown in Fig. 9.3, the differences in the level of LC3-II can be used to distinguish the autophagic rate between low (Cell Line A Fig. 9.3) and high (Cell Line B Fig. 9.3) autophagy cells under starvation/nonstarvation, with and without BafA1. Levels of LC3-II are elevated by treatment with BafA1 even under nutrient-rich condition in both cells lines [low (lane 2 vs. 1) and high (lane 6 vs. 5)]. Starvation induces LC3B processing in both cell lines when compared with nutrition-rich media [low (lane 3 vs. 1) and high (lane 7 vs. 5)] and treatment with BafA1 increased the amount of processed LC3B-II in both cell lines, but it is even higher in highly autophagic cells with lower autophagic capacity [low (lane 2) and high (lane 6)]. This finding would indicate that Cell Line B has a high basal rate of autophagy, which is further increased by starvation [low (lane 4) and high (lane 8)]. From the Western blot, we can interpret that accumulation of LC3-II positive autophagosomes in the presence of BafA1 would be evidence of efficient autophagic flux. Whereas if there was any failure in the increase of LC3-II protein during the presence of BafA1, this would indicate that there is a defect or delay in the early autophagic process (prior to degradation of LC3-II at autolysosome).
Fig. 9.3.
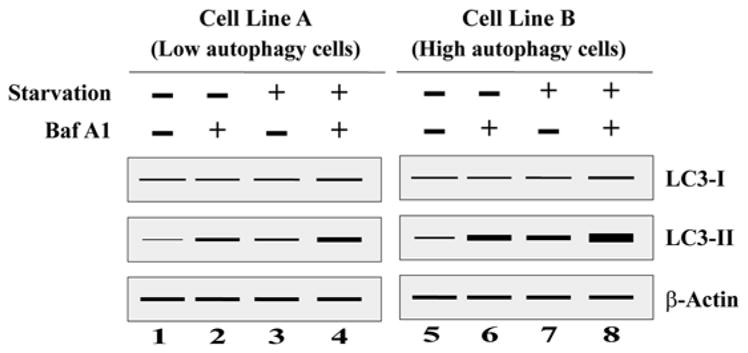
A schematic representation of a Western blot after LC3 turnover assay to distinguish autophagy level in low and high autophagic cells. Cells were cultured in regular nutrition-rich media without Baf A1 (lane 1 and 5), regular nutrition-rich media with Baf A1 (lane 2 and 6), starvation media without Baf A1 (lane 3 and 7), and starvation media with Baf A1 (lane 4 and 8). Baf A1 bafilomycin A1
9.4.3 Discovery of New Autophagy Modulators via the Bafilomycin Clamp Assay
Even though several pharmacological compounds are known to inhibit autophagy in vitro, only CQ or its derivative hydroxychloroquine (HCQ) have been used in clinical trials. So, there is the clear need for more work on the development of selective inhibitors that target autophagy proteins directly. At the same time, careful consideration is needed when targeting these proteins; because some of the autophagic proteins are also involved in autophagy-independent functions (e.g., UVRAG inhibits translocation of Bax from the cytosol to mitochondria, thereby preventing apoptosis). Also, better methods or multiple methods should be combined in the discovery and screening of new specific small-molecule autophagy modulators, both inducers and inhibitors. For example we have previously reported that BRAF inhibitors induce autophagy in BRAF mutant melanoma. To ensure that BRAF inhibition was in fact inducing autophagy and not blocking distal clearance of AVs, a bafilomycin clamp experiment was conducted to determine how PLX4720 impacted autophagic flux (Fig. 9.4). A375P cells were treated with DMSO, PLX4720 1 μM, rapamycin 500 nM, or HCQ 10 μM, in the absence or presence of BafA1. At 24 h, PLX4720 or rapamycin (a proximal autophagy inducer) treatment resulted in a threefold increase in the LC3II/LC3 ratio in Baf A1-treated cells compared to control cells whereas HCQ (distal autophagy inhibitor) did not demonstrate an increase in LC3II/LC3I ratio in Baf A1-treated cells compared to control, providing further evidence that BRAF inhibition induces autophagy.
Fig. 9.4.
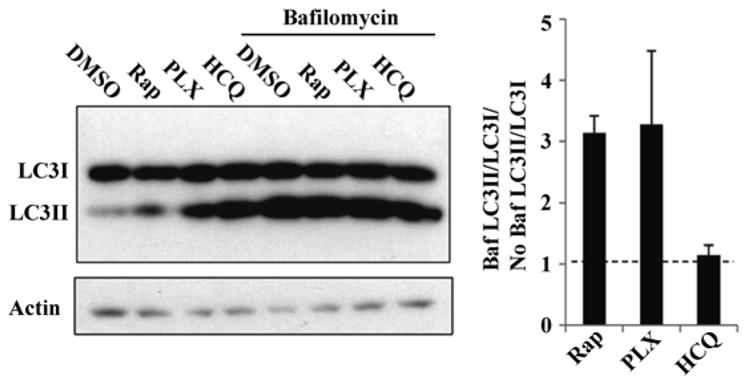
A bafilomycin clamp demonstrates BRAF inhibitor-induced autophagy. Immunoblotting against LC3 was performed using A375 (BRAF mutant melanoma) cell lysate 24 h after treatment as indicated. Representative image of one out three experiments is shown. Quantification of the bands demonstrates that in the presence of Baf A1, LC3II levels increased with PLX treatment compared no Baf A1
Figure 9.5 shows another example of how to interpret the results of LC3-I to LC3-II conversion, the bafilomycin clamp assay, and integrate this information with the level of p62 when screening the efficacy of autophagy modulators (inhibitor and activator). To elucidate the mode of action of any modulators, one should do the bafilomycin clamp assay to compare the new modulators with known autophagy modulators such as BafA1 (inhibitor) or Rapamycin (inducer). As depicted in Fig. 9.5a, treatment of cells with BafA1 increased the level of LC3-II (lane 2) by blocking the late stage of autophagy. Both compound A (inducer) and compound B (inhibitor) increased LC3-II level (Fig. 9.5, lanes 3 and 5). However, co-incubation of these compounds with BafA1 showed drastic differences. Treatment of compound A along with BafA1 induced further accumulation of LC3-II (lane 4) when compared with LC3-II levels after treatment with BafA1 alone or compound A alone (lanes 2 and 3 respectively). This result suggests the increased level of LC3-II after treatment with compound A was further enhanced when autophagic flux was further blocked by BafA1, which confirms that compound A is an autophagy activator. On the other hand, no synergistic increase of LC3-II level was observed after co-incubation of compound B with BafA1 (lane 6) when compared with BafA1 alone treatment (lane 2) or compound B alone (lane 5). This confirms that compound B is neither an inducer nor an early stage inhibitor, because the LC3-II level induced by compound B was unaffected by the additional blockage of late-stage autophagic flux by BafA1. Here an additional experiment is needed to prove the mode of action of compound B (inhibitor). As shown in Fig. 9.5b, treatment of cells with rapamycin (Rap), an activator of autophagy (through mTOR inhibition), induced autophagy and accumulated the LC3-II level (lane 2). When compound B was co-incubated with Rap, it induced a greater increase in LC3-II (lane 6) than the LC3-II level of either Rap alone (lane 2) or compound B alone (lane 5), which denotes the compound B blocked autophagic flux. Interestingly, the compound A (activator) co-incubation with Rap induced a greater level of LC3-II (lane 4) when compared with either compound A alone (lane 3) or Rap alone (lane 2), even though both compounds activate autophagy.
Fig. 9.5.

A schematic representation of a Western blot to show how the results of LC3-I to LC3-II conversion in an bafilomycin clamp assay in the presence of (a) bafilomycin (Baf A1) or (b) rapamycin (Rap) and p62 degradation could be used to identify the mode of action of new autophagic modulators. Compound A (inducer) and compound B (Inhibitor)
When cells were treated with compound A as shown in Fig. 9.5a, it increased the level of LC3II and assisted the degradation of p62 whereas treatment with compound B also increased the level of LC3II, but inhibited the degradation of p62 level in the cells Fig. 9.5b. From the quantification of LC3II and p62 levels, we can tell that compound A acts as a autophagy inducer and compound B as inhibitor. This synergistic effect by compound A proves its effectiveness in activation of autophagy pathway. Therefore, in any experimental condition, one should be able to distinguish whether autophagosomes accumulation is due to autophagy induction or a block of the late stages by performing “autophagy flux” assays.
9.5 In Vivo Labeling by Acidotropic Dyes
9.5.1 Monodansylcadaverine (MDC) or Dansylcadaverine
MDC is a dye which emits a blue fluorescence and is thought to preferentially accumulate in autophagic vesicles but not in the early and late endosome. MDC accumulation in autophagic vesicles is exclusively due to a combination of ion trapping and specific interactions with vesicle membrane lipids. The mechanism of MDC accumulation is mainly dependent on the type of cells, condition, and whether it is fixed or non-fixed and so on. MDC’s autofluorescent property (excitation wavelength at 365 nm) is due to the presence of a dansyl group conjugated to (diamine-pentane) cadaverine. MDC was initially characterized as a selective marker of autolysosomes [26] and later as a marker of earlier autophagic compartments [27, 28]. Both in cultured cells and in animal tissues, accumulation of MDC-positive vesicles can be used as a marker to characterize autophagy by various different autophagy modulators. The size and number of MDC-fluorescent vesicles markedly increases or decreases in response to several autophagy modulators. For example, in cells subjected to a physiological or pharmacological stimulus of autophagy, such as amino acid deprivation or rapamycin treatment, the MDC-labeled compartments increases [28]. On the other hand, incorporation of MDC is almost completely inhibited when cells are treated with well-known inhibitors of autophagy, such as 3-methyladenine (3-MA) and wortmannin (WM). In addition to the morphological analysis of the autophagy pathway by fluorescence microscopy, autophagy modulation can be quantitatively monitored by flow cytometry. Therefore, MDC can be used as a good marker of autophagic vesicles; however in our experience MDC likely also accumulates in late endosomes and potentially other vesicular compartments. MDC fluorescence is variable and often not uniform across fields. Therefore MDC staining is not accepted as a stand-alone measurement of autophagy [29].
9.5.2 Acridine Orange
Acridine orange is a lysosomotropic green fluorescent dye that accumulates in acidic organelles in a pH-dependent manner. Within acidic vesicles (autolysosomes), acridine orange becomes protonated and trapped within the organelle and forms aggregates that emit bright red fluorescence, while in the nucleus acridine orange fluoresces bright green. At neutral pHs, Acridine Orange is a hydrophobic green fluorescent molecule. Thus, it is possible to track the formation of acidic vesicular organelles with acridine orange staining in any cultured cells [30]. But, it should be noted that acridine orange can also accumulate in other acidic nonlysosomal compartments such as endocytic vesicles and the trans-Golgi apparatus. Therefore, acridine orange staining should be used with other autophagic markers. Acridine orange labeled cells can be measured by flow cytometry or fluorescence microscopy. The advantage of labeling cells with acridine orange in this assay is not dependent on the availability of specific antibodies or probes. The precaution in using acridine orange is that one should not over-digest the cells with trypsin-ethylenediaminetetraacetic acid (EDTA) as EDTA will change the permeability of the cells and cause the cells to lose acridine orange staining.
9.5.3 Bovine Serum Albumin Derivatives (DQ-BSAs)
DQ Green BSA and DQ Red BSA dyes by Molecular Probes are self-quenching derivatives of bovine serum albumin (BSA) that are labeled with BODIPY ® dyes, BODIPY FL and BODIPY TR-X. These fluorophores can be used to visualize a vesicular compartment known as amphisome, a combination of autophagic and endocytic pathways [31]. Subsequently, this compartment fuses with a lysosome, in which the trapped materials are degraded by lysosomal enzymes. After proteolytic digestion, DQ Green BSA and DQ Red BSA probes release fragments with excitation and emission maxima of ~505 nm and ~515 nm and ~590 nm and ~620 nm respectively [32]. Thus, these dyes can be used to analyze the convergence between autophagic compartments with a functional lysosome, determination of the intracellular proteolytic activity and the completion of the autophagic pathway in real time.
9.5.4 Staining with LysoTracker and LysoSensor Probes
Invitrogen’s LysoTracker and LysoSensor probes are weakly basic amine based fluorescent dye; they selectively accumulate in the acidic subcellular compartments with low internal pH (below 5) of the cell by endocytosis. They are mainly used for labeling and tracing acidic organelles in live cells. LysoSensor reagents display a pH-dependent increase in fluorescence intensity upon acidification which is not seen in LysoTracker probes. LysoTracker staining can be used to detect abnormalities in vesicular pH [33] and efficiency of autophagosome/lysosome fusion in live cells [34]. The combination of fluorescently tagged LC3 and Lysotracker staining has been used to obtain insights about the fusion between autophagosomes and acidic endo/lysosomal compartments [35]. LysoSensor probes are mainly used to investigate the acidification of lysosomes and alterations of lysosomal function or trafficking that occur in cells. For example, tumor cells with lower pH [36] in their lysosomes can be compared with one having higher pH [37].
9.6 Autophagy Detection by Electron Microscopy
9.6.1 Conventional Transmission Electron Microscopy (TEM)
Analysis of autophagy by conventional transmission electron microscopy (TEM) is considered as a sensitive and reliable semiquantitative method, to show the increase in the number of autophagic vesicles (AV) in a given cell or tissue area. Also, it can be used for both qualitative and quantitative analysis of changes in various autophagic structures. At high magnification (20,000–25,000×) EM can potentially identify and distinguish autophagosomes from autolysosomes [38]. Double-membrane autophagosomes indicate immature AVs and degradative single-membrane autolysosomes are late AVs. The single-membrane autolysosomes mostly contain cytoplasmic material and/or organelles at various stages of degradation. Sometimes secretory vesicles (SV) are confused with AVs, because both morphology and size (500–1 mm) are similar in both. So the very important distinguishing feature that separates AV from SV is that the AV (autophagosomes) should have a clear double-membrane structure separated by an electron-lucent cleft, with encapsulated organelles (mitochondria) and undigested cytoplasmic contents (glycogen granules).
9.6.1.1 Key Steps in TEM Protocol for Cultured Cells
The ideal number of cells for EM is enough cells to form a pellet in an Eppendorf tube when spun down.
For tumor and nontumor tissue: a sesame seed size piece of tissue is ideal for fixation. Larger tissues may lead to fixation artifact.
Fix the cells with a solution containing 3 % glutaraldehyde plus 2 % paraformaldehyde in 0.1 M cacodylate buffer.
Most electron microscopy core facilities will process samples, embed, cut, and stain grids for EM imaging. Methods detailing this process can be found in [39].
Examine the images in a transmission electron microscope at an accelerating voltage of 80 kV.
9.6.1.2 Quantification of Autophagic Vesicles in Clinical Trial Specimens
Quantitative EM to measure autophagic vesicles was incorporated into the first five clinical trials involving hydroxychloroquine for cancer patients [40–44]. In these studies peripheral blood mononuclear cells were used as a surrogate for tumor tissue. In one study of temsirolimus and hydroxychloroquine [41] tumor tissue was analyzed for AVs via EM. Successful translation of this assay into the clinic was demonstrated in these clinical trials where, for the first time, a formal pharmacokinetic-pharmacodynamic relationship between HCQ exposure and AV accumulation was made. The following methods were adapted from these papers, and demonstrate the feasibility of using an EM-based assay to obtain useful information even in a multi-institution clinical trial setting.
9.6.1.3 Quantification of AV in PBMC
Collect venous blood in cell preparation tubes with a ficoll gradient from patients before treatment and at specific time points on treatment. PBMCs isolated should be separated into 2–4 cell pellets. PBMC pellet one should be immediately fixed with 2 % glutaraldehyde and stored at 4 °C until embedding. Embedding can be performed as described above.
Image capture: High-powered micrographs (10,000–12,000×) of 20–25 mono-nuclear cells from multiple distinct low-powered fields in each sample should be obtained.
Scoring of Vesicles: Autophagy vesicles should be scored by two independent experienced investigators who are blinded to treatments and time points. Morphological criteria for AV include (1) circularity, (2) contrast with structures that were white or lighter than the cytoplasm, (3) vesicles with contents, (4) vesicles >200 nm in size and, (5) vesicles >200 nm interior to the plasma membrane. Vesicular structures with cristae characteristic of mitochondria in cross section should be excluded. The average of two investigators counts along with the standard deviation should be reported for vesicle counts.
PBMC pellets 2–4 can then be used for immunoblotting against LC3 as described in Chap. 10.
For tumor tissue a similar process can be used. Quantification of a number of AV per cell is much more challenging in tumor tissue if the sample is not well fixed because cell boundaries can be vague.
9.6.1.4 Disadvantage of TEM
Identifying and classifying structures need expertise and experience.
When quantifying AVs on a per cell basis, the magnification (8000–12,000×) usually precludes determination of whether or not candidate vesicles do in fact have a double membrane. Therefore there is an error rate with every count for labeling structures AV that upon much higher magnification (20,000×) would in fact be reclassified as another subcellular structure.
Ex vivo tissue should be fixed immediately and systematically across samples to avoid changes in autophagy that may occur simply due to elapsed time ex vivo.
EM pictures should also be taken over different times to ensure detection of autophagosomes. Artifacts may be easily generated during fixation and staining processes. Thus, repeating the processing of samples especially cutting and staining the grids ultimately used for visualization is often needed [45].
The cost and labor intensive nature of EM analysis is prohibitive.
9.6.2 Immuno-electron Microscopy (IEM)
The combination of EM and immunogold labeling can facilitate the identification of vesicles such as endosomes and golgi vesicles/tubules that are difficult to distinguish from AV in conventional EM. In IEM, an antibody against precisely defined subcellular proteins can be used to monitor AVs in both basal and stress conditions [46]. Table 9.3 shows a partial list of proteins whose antibodies which have been successfully used for IEM in autophagic research. Using the combination of classical TEM and IEM, different vesicular compartments (double membrane, multivesicular, multilamellar) and subpopulations of autophagic bodies (autophagosomes, autophagolysosomes, multilamellar bodies and dense residual bodies) can be identified by various morphologic and biochemical criteria as described elsewhere [47].
Table 9.3.
Partial list of some key proteins which are commonly targeted for immunogold labeling
| Protein | Conditions and interpretation |
|---|---|
| Beclin-1 |
|
| Calnexin |
|
| Protein disulfide isomerase (PDI) |
|
| Cathepsin D |
|
9.7 Monitoring Autophagy by Fluorescence Microscopy
Readers are kindly advised to refer Chap. 10 for details on imaging of LC3-GFP and Tandem RFP-LC3-GFP constructs (tfLC3) by fluorescence microscopy.
9.8 Quantitative Assay for Monitoring Autophagy in Live Cells Using Enzo’s Cyto-ID ® Autophagy Detection Kit
The Cyto-ID® Autophagy Detection Kit from Enzo Life Sciences can be used to visualize autophagic vacuoles and to monitor the autophagic flux in living cells using fluorescence microscopy and flow cytometry [48]. The probe is a cationic amphiphilic tracer (CAT) dye, selectively and rapidly labels autophagic vacuoles. When the dye enters into autophagy-associated vacuoles [pre-autophagosomes, autophagosomes, and autolysosomes (autophagolysosomes)], they show bright fluorescence, while it stains minimally within lysosomes. The Cyto-ID ® dye specifically labels autophagic vacuoles independent of LC3 protein and eliminates the need for transfection. It is excluded from lysosomes as seen with other lysosomotropic dye-based assays such as MDC.
9.9 Immunohistochemical Analysis of Autophagy-Related Proteins
Recently, the role of autophagy in cancer development and progression has been investigated by many researchers using immunohistochemistry. Many antibodies are commercially available and are routinely used for immunohistochemistry against proteins involved in autophagy pathways such as LC3. Our group developed a novel staining method for analysis of LC3B in formalin-fixed, paraffin-embedded (FFPE) tissues, which uses light microscopy and quantitative immunofluorescence [automated quantitative analyses (AQUA)] [49]. AQUA analysis allowed for objective computer-assisted quantification of LC3 staining intensity and puncta. When applied to a tissue microarray of breast cancer and melanoma specimens, a linear correlation was made between LC3 staining intensity and LC3 puncta. Other groups have found that LC3 staining intensity does not correlate with LC3 puncta in other tumor types.
9.9.1 Assessment of LC3 Staining by AQUA
For analysis of LC3B in a multitumor tissue microarray (TMA), rabbit anti-LC3 B (1:5000, overnight at 4 °C) was used and rabbit horseradish peroxidase-conjugated secondary antibody and Cy5 tyramide signal amplification were used. The immuno-fluorescence intensity of LC3B was averaged for each pixel on the basis of the surrounding 15 pixels (~2 μm2). Pixels with an LC3B expression level above the local mean were designated as “punctate” and the remaining pixels as “diffuse.” Oversaturated pixels were excluded from the analysis. The expression of LC3B in each designation was then calculated as the percentage of pixels assigned to the punctate category. In a given tissue the AQUA program computes fluorescence-conjugated anti-LC3B staining patterns by designating pixels yellow for punctate and blue for diffuse LC3B staining. Other groups also have reported the LC3 punctate staining in FFPE of colorectal tumors, lymph node, and liver metastasis [50]. Also strong LC3 expression was observed in esophageal, gastric, colorectal cancers and correlated with high Ki-67 expression [51 ]. Taken together, it seems that the quantification of autophagic marker LC3 punctate is correlated with tumor cell proliferation, metastasis, high nuclear grade, and worse patient outcome [49 ] and the staining method described coupled with the AQUA system could be incorporated into clinical trials of drugs that are known to modulate autophagy. If AQUA is not available, quantification by traditional histological scoring has been successful as well [52].
9.10 Secreted Cytokines as Biomarkers of Autophagy Levels in Melanoma
So far in autophagic research, there are no quantitative preclinical assays that have been developed, which can be easily translated into clinics for testing the efficacy of autophagy modulators. In addition, it is not realistic to use traditional two-dimensional (2D) culture system for developing effective autophagy markers, because it does not accurately recapitulate the tumor microenvironment. In this regard, recently, we developed a three-dimensional (3D) cell culture system and a molecular profiling method to identify and quantitate autophagy-related secretion of proteins [53]. This 3D cell culture system appears to more faithfully reproduce autophagy dynamics found in the tumor microenvironment compared to traditional 2D culture.
9.10.1 Sample Acquisition for Secretome Analysis
Here we summarize the workflow of 3D culture and comparative secretome analysis of melanoma cells (Fig. 9.6). Genetically identical melanoma cell lines, WM793 (a primary melanoma) and 1205Lu (derived from WM793 metastases in vivo), were used for the development of this method. Electron micrographs, fluorescent images of melanoma cells expressing tandem fluorescent tagged LC3 (mCherry-eGFP-LC3), and Western blot analysis on WM793 and 1205Lu show that 1205Lu (subclone of WM793) has higher autophagic flux than WM793 [53].
Fig. 9.6.

Schematic workflow of 3D cell culture and sample (overlaying media and interstitial fluid) collection for comparative secretome analysis of melanoma cells
9.10.1.1 Uses of 3D Culture and Comparative Secretome Analysis
By mass spectrometry (MS) analysis, there are 599 differentially secreted proteins identified across high- and low-autophagy cell lines, 571 were present at similar levels in both cell lines, 26 proteins were elevated in 1205Lu, and 2 proteins were elevated in WM793.
After MS analysis, the proteins with minimum 6-fold increase in abundance were further characterized by ELISA. The proteins such as IL1B, CXCL8, LIF, FAM3C, and DKK3 were elevated in the media of high-autophagy cell lines.
Consistent with the cell culture system, using commercially available ELISA kits, the above five candidate biomarkers were found to be significantly elevated in the serum of melanoma patients who had high-autophagy tumors compared to patients who had low-autophagy tumors as measured by EM [53].
This demonstrates that our comparative secretome analysis provides a unique window into how this method can be utilized to identify novel prognostic, predictive, or pharmacodynamic biomarkers of autophagy (Fig. 9.7). Development of such circulating markers of autophagy could enhance laboratory research in autophagy and eventually could aid cancer therapy decisions in melanoma or in any type of cancer. However, it needs to be validated in a larger number of patients and in other cancer types.
Fig. 9.7.

Scheme of autophagic dynamics and the release of autophagy-related secreted proteins and vesicles in the circulation
9.11 Concluding Remarks and Future Perspectives
Due to the intense interest in the role of autophagy in human diseases, numerous methods have been developed in the past decade to monitor autophagy in mammalian cells. Even though we have discussed most of the currently available methods and techniques in autophagy detection, there are new methods being developed constantly. So readers are advised to keep up with these advances and must always consider the variables into account when designing the experiment and data interpretation. In addition, we recommend the use of more than one assay to measure autophagosomes number or to measure autophagic flux, whenever possible. In future, we look forward to the development of more quantitative noninvasive assays to measure autophagy within the tumor microenvironment. For instance with increased knowledge about the autophagic pathway, it may be possible to develop radiological technologies such as positron emission tomography that can detect autophagy for use in clinical medicine.
References
- 1.Chen Y, Klionsky DJ. The regulation of autophagy—unanswered questions. J Cell Sci. 2011;124:161–70. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys. 2015;44:101–22. doi: 10.1146/annurev-biophys-060414-034248. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–80. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen N, Debnath J. Autophagy and tumorigenesis. FEBS Lett. 2010;584:1427–35. doi: 10.1016/j.febslet.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, et al. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy. 2009;5:400–3. doi: 10.4161/auto.5.3.7784. [DOI] [PubMed] [Google Scholar]
- 6.Yang ZJ, Chee CE, Huang S, et al. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akalay I, Janji B, Hasmim M, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013;73:2418–27. doi: 10.1158/0008-5472.CAN-12-2432. [DOI] [PubMed] [Google Scholar]
- 8.Akalay I, Janji B, Hasmim M, et al. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy. 2013;9:1104–6. doi: 10.4161/auto.24728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baginska J, Viry E, Berchem G, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013;110:17450–5. doi: 10.1073/pnas.1304790110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol. 2010;22:241–5. doi: 10.1016/j.ceb.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang R, Livesey KM, Zeh HJ, et al. HMGB1: a novel Beclin 1-binding protein active in autophagy. Autophagy. 2010;6:1209–11. doi: 10.4161/auto.6.8.13651. [DOI] [PubMed] [Google Scholar]
- 13.Kang R, Tang D, Schapiro NE, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17:666–76. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–30. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–16. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2015 doi: 10.1038/onc.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 18.Bjorkoy G, Lamark T, Pankiv S, et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–97. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 19.Laddha SV, Ganesan S, Chan CS, et al. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res. 2014;12:485–90. doi: 10.1158/1541-7786.MCR-13-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, He Z, von Rutte T, et al. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med. 2013;5:202ra123. doi: 10.1126/scitranslmed.3005864. [DOI] [PubMed] [Google Scholar]
- 21.Ma XH, Piao S, Wang D, et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res. 2011;17:3478–89. doi: 10.1158/1078-0432.CCR-10-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanida I, Minematsu-Ikeguchi N, Ueno T, et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 24.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 25.Ni HM, Bockus A, Wozniak AL, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14. [PubMed] [Google Scholar]
- 27.Iwai-Kanai E, Yuan H, Huang C, et al. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4:322–9. doi: 10.4161/auto.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–29. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 29.Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet. 2007;16:1495–503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- 30.Han J, Burgess K. Fluorescent indicators for intracellular pH. Chem Rev. 2010;110:2709–28. doi: 10.1021/cr900249z. [DOI] [PubMed] [Google Scholar]
- 31.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Voss EW, Jr, Workman CJ, Mummert ME. Detection of protease activity using a fluorescence-enhancement globular substrate. Biotechniques. 1996;20:286–91. doi: 10.2144/96202rr06. [DOI] [PubMed] [Google Scholar]
- 33.Eskelinen EL, Schmidt CK, Neu S, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell. 2004;15:3132–45. doi: 10.1091/mbc.E04-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez-Polo RA, Boya P, Pauleau AL, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 35.Pierzynska-Mach A, Janowski PA, Dobrucki JW. Evaluation of acridine orange, LysoTracker Red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytometry A. 2014;85:729–37. doi: 10.1002/cyto.a.22495. [DOI] [PubMed] [Google Scholar]
- 36.Chauhan SS, Liang XJ, Su AW, et al. Reduced endocytosis and altered lysosome function in cisplatin-resistant cell lines. Br J Cancer. 2003;88:1327–34. doi: 10.1038/sj.bjc.6600861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang LW, Maher VM, McCormick JJ, et al. Alkalinization of the lysosomes is correlated with ras transformation of murine and human fibroblasts. J Biol Chem. 1990;265:4775–7. [PubMed] [Google Scholar]
- 38.Eskelinen EL, Reggiori F, Baba M, et al. Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy. 2011;7:935–56. doi: 10.4161/auto.7.9.15760. [DOI] [PubMed] [Google Scholar]
- 39.Yla-Anttila P, Vihinen H, Jokitalo E, et al. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009;452:143–64. doi: 10.1016/S0076-6879(08)03610-0. [DOI] [PubMed] [Google Scholar]
- 40.Mahalingam D, Mita M, Sarantopoulos J, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10:1403–14. doi: 10.4161/auto.29231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391–402. doi: 10.4161/auto.29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rangwala R, Leone R, Chang YC, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1369–79. doi: 10.4161/auto.29118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenfeld MR, Ye X, Supko JG, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10:1359–68. doi: 10.4161/auto.28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogl DT, Stadtmauer EA, Tan KS, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10:1380–90. doi: 10.4161/auto.29264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eskelinen EL. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy. 2008;4:257–60. doi: 10.4161/auto.5179. [DOI] [PubMed] [Google Scholar]
- 46.Dunn WA., Jr Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–45. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunn WA., Jr Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol. 1990;110:1923–33. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan LL, Shen D, Wilkinson AR, et al. A novel image-based cytometry method for autophagy detection in living cells. Autophagy. 2012;8:1371–82. doi: 10.4161/auto.21028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lazova R, Camp RL, Klump V, et al. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18:370–9. doi: 10.1158/1078-0432.CCR-11-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato K, Tsuchihara K, Fujii S, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007;67:9677–84. doi: 10.1158/0008-5472.CAN-07-1462. [DOI] [PubMed] [Google Scholar]
- 51.Yoshioka A, Miyata H, Doki Y, et al. LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int J Oncol. 2008;33:461–8. [PubMed] [Google Scholar]
- 52.Ma XH, Piao SF, Dey S, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124:1406–17. doi: 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kraya AA, Piao S, Xu X, et al. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy. 2015;11:60–74. doi: 10.4161/15548627.2014.984273. [DOI] [PMC free article] [PubMed] [Google Scholar]