

Abstract
Purpose
Genetic variability in KRAS and EGFR predicts response to cetuximab in irinotecan refractory colorectal cancer. Whether these markers or others remain predictive in combination biologic therapies including bevacizumab is unknown. We identified predictive biomarkers from patients with irinotecan refractory metastatic colorectal cancer treated with cetuximab plus bevacizumab.
Methods
Patients who received cetuximab plus bevacizumab for irinotecan refractory colorectal cancer in either of two Phase II trials conducted were identified. Tumor tissue was available for 33 patients. Genomic DNA was extracted and used for mutational analysis of KRAS, BRAF, and p53 genes. Fluorescence in situ hybridization was performed to assess EGFR copy number. The status of single genes and various combinations were tested for association with response.
Results
Seven of 33 patients responded to treatment. KRAS mutations were found in 14/33 cases, and 0 responded to treatment (p=0.01). EGFR gene amplification was seen in 3/33 of tumors and in every case was associated with response to treatment (p<0.001). TP53 and BRAF mutations were found in 18/33 and 0/33 tumors, respectively, and there were no associations with response to either gene.
Conclusions
EGFR gene amplification and KRAS mutations are predictive markers for patients receiving combination biologic therapy of cetuximab plus bevacizumab for metastatic colorectal cancer. One marker or the other is present in the tumor of half of all patients allowing treatment response to be predicted with a high degree of certainty. The role for molecular markers in combination biologic therapy seems promising.
Introduction
Colorectal cancer (CRC) is the third most common malignancy diagnosed in males and females and remains the third leading cause of cancer deaths in the United States, accounting for 49,700 deaths in 2015 (1). Approximately 20% of patients have distant metastases at time of diagnosis, and an equal number will develop metastatic disease after treatment for clinically localized disease (2). The management of patients with metastatic CRC has evolved considerably due to the introduction of new cytotoxic drug combinations and of novel targeted agents (3).
Cetuximab is a monoclonal antibody that blocks dimerization of epidermal growth factor receptor (EGFR), thereby inhibiting downstream signal transduction. Bevacizumab is a monoclonal antibody that binds to and sequesters vascular endothelial growth factor (VEGF). The VEGF pathway, which indirectly regulates the EGFR pathway, plays a central role in promoting tumor angiogenesis (34). Treatment with bevacizumab can enhance clinical response rate to cytotoxic drug combinations and deter tumorigenesis (4). On the other hand, cetuximab has efficacy as a single agent in a minority of patients (10%) and, when combined with irinotecan, provides significant improvements in progression-free survival (PFS) and overall survival (5–8). Previous reports by Saltz et al. showed that the combination of cetuximab, bevacizumab and irinotecan yield 20% response rates in patients with metastatic CRC who have failed prior chemotherapy (6). Furthermore, since the success of this so-called Bond-2 study, there is limited data on molecular studies looking at the role of biomarkers in combination biologic therapy in metastatic CRC. The identification of molecular genetic variables that could accurately predict response could dramatically improve patient outcomes by assuring effective drug treatment for those who respond and by avoiding the toxicity and economic cost of ineffective therapy.
Several publications have examined the usefulness of tumor-derived molecular markers for prediction of response and resistance to cetuxamib (10–14). EGFR protein is over-expressed in 60–80% of CRCs detected by immunohistochemistry (IHC) but does not predict response to cetuximab (5, 15, 16, 17). More recent studies have shown that EGFR gene copy number and KRAS mutational status predict response to cetuximab in advanced CRC (10, 11). Specifically, for patients with irinotecan refractory CRC, cancers harboring KRAS mutations do not respond to cetuximab. Furthermore, several reports show that tumors that have upregulated the EGFR signaling pathway by receptor amplification or ligand overexpression are drug sensitive. These data are important and exciting, but at present not all responses to cetuximab can be explained by molecular profiling. In addition, it is unclear how reliable these tumor markers will be in the setting of drug combinations that include bevacizumab.
In this study we provide data obtained from 33 patients with irinotecan refractory colorectal cancer who were treated with cetuximab plus bevacizumab to see if predictive markers for response to cetuximab remain valid. We test for the presence of four genetic alterations: EGFR gene amplification, KRAS mutations (codons 12, 13), BRAF mutations (V600E) and p53 mutations (exons 5–9) for their potential use as predictive markers for cetuximab and bevacizumab in metastatic CRC.
Methods
Patient Selection
Patient selection and tumor block accrual were approved by the Institutional Review Board and Human Tissue Utilization Committee of Memorial Sloan-Kettering Cancer Center (MSKCC), respectively. Cases were obtained from multi-institutional studies (with MSKCC as lead site) for which tumor specimens were available (6). Clinical trial eligibility included clinical documentation of failure after receiving at least one chemotherapy regimen for metastatic disease that contained either irinotecan or irinotecan and bevacizumab. Failure on therapy for metastatic disease was defined as progression on treatment or within 6 weeks after receiving the last dose of a given therapy. Progression was defined as any enlargement of a measurable or evaluable lesion, or any new lesion, which was felt by the treating physician to represent a clinical failure.
Gene mutation analysis
Genomic DNA was extracted from paraffin tumor blocks using the DNA isolation kits (QIAGEN) and used for KRAS, BRAF and TP53 mutation analysis. KRAS and BRAF mutations were detected using PCR/LDR approaches as previously described (18, 19, 20, 41).
Fluorescence in situ hybridization
EGFR amplification was detected using fluorescence in situ hybridization (FISH) as previously described (42). All 33 samples were detected EGFR gene and CEP 7 signals by two-color FISH. At least 30 cells of each sample were counted, and the ratio of 3.0 or more was taken as evidence of gene amplification.
Statistical analysis
Patients were scored as responders if they showed evidence of partial response (PR) or complete response (CR) to treatment on computed tomography (CT) scan as defined by the response evaluation criteria in solid tumors (RECIST) criteria. Non-responders had evidence of either stable disease (SD) or progression of disease (POD) as defined by this criterion. Skin toxicity was scored according to the common terminology criteria for adverse events (CTCAE) as defined by the National Cancer Institute.
Fisher’s exact test (2-tailed) was used to examine associations of response to treatment. All statistical calculations were done using either SPSS version 12.0 for windows (SPSS, Inc., Chicago, IL) or GraphPad Prism version 3.03 for Windows (GraphPad Software, Inc. San Diego, CA). P values of <0.05 were considered significant.
Results
Patient Characteristics (Supplemental table)
The cohort consisted of 20 males and 13 females with a median age of 58 years (range 32 to 74 years). All patients had metastatic CRC that was irinotecan-resistant based on a poor response to prior chemotherapeutic regimens. Eleven individuals received a combination of cetuximab and bevacizumab (CB) while 22 received irinotecan in addition to cetuximab and bevacizumab (CBI). As defined by the RECIST criteria, 7 of 33 (21%) patients responded to treatment of CB or CBI. The non-responders included 20 with stable disease (SD) and 6 with progressive disease (POD). The median duration of response to cetuximab and bevacizumab was 223 days (range, 121–529 days). In the six patients with stable disease, the median duration of stabilization was 125 days (range, 41–387 days). Ten patients had treatment associated with grade 1 skin toxicity and 11 had grade 2 or 3 skin toxicity.
Genetic Analysis and Association with Response to Therapy (Table 1)
TABLE 1.
Genetic characteristics of KRAS, BRAF, p53, and EGFR in relationship to chemotherapeutic response
| KRAS | BRAF | TP53 | EGFR | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Wild type | Mutant | Wild type | Mutant | Wild type | Mutant | Amplification | No amplification | |
| Response | 7 | 0 | 7 | 0 | 4 | 3 | 3 | 4 |
| No Response | 12 | 14 | 26 | 0 | 11 | 15 | 0 | 26 |
| p=0.01 | NS | NS | p<0.001 | |||||
KRAS mutations were found in the tumors of 14 patients (42%). All mutations were found in the no response group while no mutations were found in the response group (p=0.01). There were no BRAF mutations found in the entire cohort. The tumors of 18 patients (55%) were found to have p53 mutations within exons 5–9. Three of the 7 responders (43%) were found to have p53 mutations while 15 of 26 (58%) non-responders possessed this mutation. Three of 33 (9%) patient tumors possessed EGFR amplification and all amplifications were in the response group (p<0.001).
FISH Analysis and Association with Response to Therapy (Figure 1)
Figure 1.
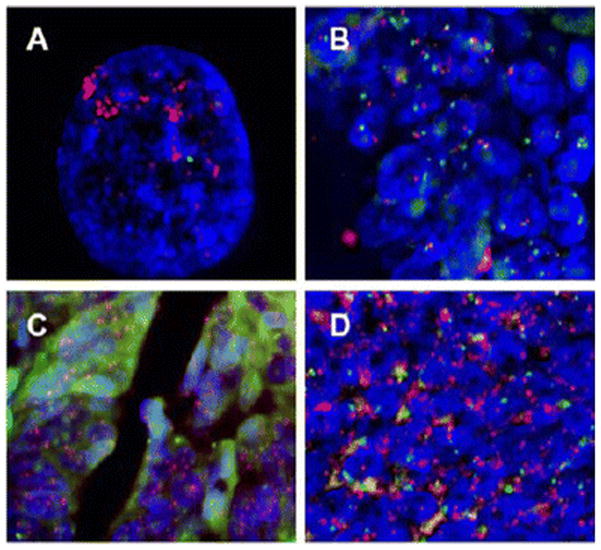
Representative FISH analysis performed on formalin-fixed, paraffin-embedded human colon cancer samples. Dual-color FISH probes contain EGFR (red signals) and centromere of chromosome 7 (green signals). DAPI (blue) was used as counterstain. A shows EGFR gene amplification in human epidermoid carcinoma cell line A-431 with well-documented EGFR gene amplification. B shows colon cancer without EGFR amplification. C and D show colon cancer with EGFR amplification.
EGFR amplification was seen in 3 of 33 (9%) cases. Figure 1a represents a positive control for EGFR amplification via FISH in human epidermoid carcinoma cell line A-431. Figure 1b is an example of 1 of 30 non-amplifications. Figures 1c and 1d portray 2 patients for which EGFR amplification was seen in representative cases. EGFR amplification was significantly associated with favorable response to target therapy (p<0.001).
Progression-Free Survival
Median time-to-tumor-progression was increased in responders compared to non-responders (291.7 vs. 124.4 days, P=0.0012). Possession of EGFR amplification was not associated with a longer progression-free survival compared to patients without EGFR amplification (log-rank, P = 0.91) (Figure 2A). Patients with tumors that had WT KRAS did not have a significantly longer progression-free survival than patients with mutant KRAS (log-rank, P = 0.23) (Figure 2B).
Figure 2.
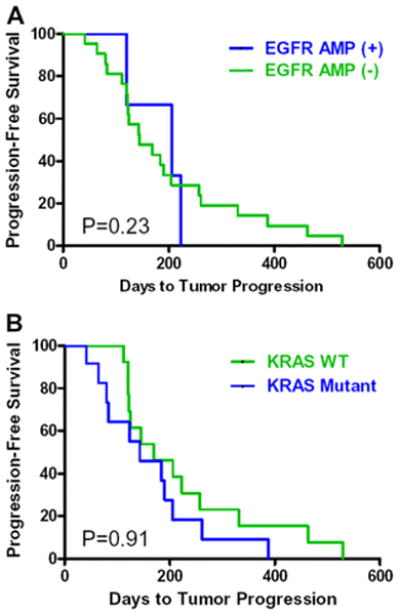
Progression-free survival in patients with cetuximab and bevacizumab. The progression-free survival time was calculated by the Kaplan-Meier method. Panel A shows progression-free survival with EGFR amplification status (P=0.91). Panel B shows progression-free survival with KRAS mutation status (P=0.23).
Discussion
Multiple studies over the last decade using biomarker analysis support the feasibility of refining risk stratification in CRC by incorporating tumor pathology stage with molecular characteristics (35–38). Though the benefit of targeted drugs has often been realized in metastasis, there remain conflicting reports hampering commonplace use of personalized therapy by genomic profiling (39). Thus further studies are required to refine the role and candidacy of biomarkers to individualize patient therapy. Our primary aim was to determine if EGFR gene amplification and KRAS mutation are predictive of tumor response to cetuximab in combination with bevacizumab. Our secondary aim was to see if other genetic markers - mutations in the BRAF oncogene and mutations in the P53 tumor suppressor gene - might also be correlated with tumor response. Our study shows treatment response to cetuximab and bevacizumab in irinotecan refractory, metastatic CRC correlates with KRAS mutation status and EGFR gene amplification.
Patients carrying tumors with KRAS mutations have been reported to have a poorer prognosis and a diminished response to adjuvant chemotherapy (11–13, 27, 35). Initial studies on patients with metastatic CRC showed no relation between KRAS mutation and response to cetuximab treatment, whereas subsequent studies showed that KRAS mutations were associated with lack of response (10, 11–13, 27, 35). Further, EGFR is overexpressed in 60–80% of CRC detected by immunohistochemistry (IHC). However, clinical studies demonstrated that many patients whose tumors express EGFR on IHC fail to respond to EGFR targeted therapy, and conversely, patients who respond may have tumors without EGFR expression (5, 15–17). These discrepancy are probably explained by the different methods of KRAS and EGFR analysis in these studies, since it is more difficult to obtain high-quality DNA from paraffin-embedded tissue samples.
In our study, a KRAS mutation was identified in 41% of CRC tumors. Strikingly, none of the 14 patients with a KRAS mutation responded to cetuximab, while 7 (35%) of the 20 non-mutated patients had a response (p = 0.04). These results demonstrate that KRAS mutational status is highly predictive of tumor resistance to cetuximab. Partial responses meeting RECIST criteria were observed in 21% (7/33) of all patients. The three patients whose tumors had EGFR amplification were wild-type for KRAS, and notably all showed major drug responses. Four patients showed drug responses in the absence of EGFR amplification and also possessed a non-mutated KRAS gene. P53 mutations were detected in 55% of tumors, but these mutations had no predictive value for drug response. Similar findings in regards to EGFR amplification and KRAS status were reported by Lievre and colleagues showing that EGFR amplification and a wild-type KRAS genotype are predictors of response to cetuximab (11). Similar trends were also seen by Moroni et al and Sartore-Bianchii et al (10, 45). Our data indicate that EGFR gene amplification and KRAS mutation remain powerful and independent markers for predicting tumor response when bevacizumab is added to cetuximab.
Our data are concordant with results published for irinotecan refractory patients treated with cetuximab alone, panitumumab alone, or cetuximab plus irinotecan (5, 7, 8, 24, 35). Though earlier studies supported a favorable response of bevacizumab to cetuximab, recent data has been mixed (36). Investigators have recently shown clinical utility in the use of combination biologic therapy with these two agents (43, 44). Thus, suggestions of a potential role for combination biologic therapy in the future treatment of metastatic CRC remain and warrant further testing. Though we are not the first group to identify predictive markers to cetuximab treatment, we are the first group to show that the predictive roles of KRAS and EGFR in cetuximab and bevacizumab treatment hold true – tumors with KRAS mutations remain drug resistant while those with wild-type KRAS and EGFR amplification remain drug sensitivity. If the sparing use of combination biologic therapy becomes standardized treatment in the future treatment of metastatic CRC, we believe biomarkers such as these should play a role in directing patient management.
The use of tumor markers to select patients for treatment with combination targeted therapy is promising, and clinical trials utilizing predictive markers to stratify drug treatment and optimize benefit are warranted. This study underlines the important role that molecular markers can play in predicting response to biologic agents in the treatment of CRC. In addition to guiding patient selection, these and other markers may prove useful in assessing the benefit of novel drug combinations designed to overcome resistance to biological agents.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Program Project Grant PO1-CA65930 and T32 surgical oncology training grant CA 09501 of the National Cancer Institute.
We thank Dr. Margaret Leversha (Cytogenetic Core Facility Lab, Memorial Sloan-Kettering Cancer Center) for valuable technical assistance for FISH analysis.
Footnotes
Compliance with Ethical Standards
Conflict of Interest: Sajid Khan, Zhaoshi Zeng, Jinru Shia, and Philip Paty declare no conflicts of interest.
Ethical approval: This retrospective study does not contain any studies with human participants by any of the authors. Tissue was obtained from patients enrolled in clinical trials. These trials were in accordance with the ethical standards of the institutional review board and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Tumor block accrual were approved by the institutional review board.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Macdonald JS. Adjuvant therapy of colon cancer. CA Cancer J Clin. 1999;49:202–19. doi: 10.3322/canjclin.49.4.202. [DOI] [PubMed] [Google Scholar]
- 3.Meyerhardt JA, Mayer RJ. Systemic Therapy for Colorectal Cancer. N Engl J Med. 2005;352:476–87. doi: 10.1056/NEJMra040958. [DOI] [PubMed] [Google Scholar]
- 4.Hicklin DJ, Ellis LM. Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis. J Clin Oncol. 2005;23:1011–27. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham D, Humblet Y, Siena S, et al. Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 6.Saltz LB, Lenz HJ, Kindler HL, et al. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: the BOND-2 study. J Clin Oncol. 2007;25(29):4557–61. doi: 10.1200/JCO.2007.12.0949. [DOI] [PubMed] [Google Scholar]
- 7.Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the Treatment of Colorectal Cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 8.Tabernero J, Van Cutsem E, Diaz-Rubio E, et al. Phase II Trial of Cetuximab in Combination With Fluorouracil, Leucovorin, and Oxaliplatin in the First-Line Treatment of Metastatic Colorectal Cancer. J Clin Oncol. 2007;25:5225–32. doi: 10.1200/JCO.2007.13.2183. [DOI] [PubMed] [Google Scholar]
- 9.Schrag D. The Price Tag on Progress -- Chemotherapy for Colorectal Cancer. N Engl J Med. 2004;351:317–9. doi: 10.1056/NEJMp048143. [DOI] [PubMed] [Google Scholar]
- 10.Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. The Lancet Oncology. 2005;6:279–86. doi: 10.1016/S1470-2045(05)70102-9. [DOI] [PubMed] [Google Scholar]
- 11.Lievre A, Bachet J-B, Le Corre D, et al. KRAS Mutation Status Is Predictive of Response to Cetuximab Therapy in Colorectal Cancer. Cancer Res. 2006;66:3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 12.Di Fiore F, Blanchard F, Charbonnier F, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer. 2007;96:1166–9. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of Epiregulin and Amphiregulin and K-ras Mutation Status Predict Disease Control in Metastatic Colorectal Cancer Patients Treated With Cetuximab. J Clin Oncol. 2007;25:3230–7. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 14.Sartore-Bianchi A, Moroni M, Veronese S, et al. Epidermal Growth Factor Receptor Gene Copy Number and Clinical Outcome of Metastatic Colorectal Cancer Treated With Panitumumab. J Clin Oncol. 2007;25:3238–45. doi: 10.1200/JCO.2007.11.5956. [DOI] [PubMed] [Google Scholar]
- 15.Chung KY, Shia J, Kemeny NE, et al. Cetuximab Shows Activity in Colorectal Cancer Patients With Tumors That Do Not Express the Epidermal Growth Factor Receptor by Immunohistochemistry. J Clin Oncol. 2005;23:1803–10. doi: 10.1200/JCO.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 16.Shia J, Klimstra DS, Li AR, et al. Epidermal growth factor receptor expression and gene amplification in colorectal carcinoma: an immunohistochemical and chromogenic in situ hybridization study. Mod Pathol. 2005;18:1350–6. doi: 10.1038/modpathol.3800417. [DOI] [PubMed] [Google Scholar]
- 17.Cerea G, Ricotta R, Schiavetto I, et al. Cetuximab for treatment of metastatic colorectal cancer. Ann Oncol. 2006;17:vii66–7. doi: 10.1093/annonc/mdl954. [DOI] [PubMed] [Google Scholar]
- 18.Khanna M, Park P, Zirvi M, et al. Multiplex PCR/LDR for detection of K-ras mutations in primary colon tumors. Oncogene. 1999;18:27–38. doi: 10.1038/sj.onc.1202291. [DOI] [PubMed] [Google Scholar]
- 19.Sorlie T, Johnsen H, Vu P, Lind GE, Lothe R, Borresen-Dale AL. Mutation screening of the TP53 gene by temporal temperature gradient gel electrophoresis. Methods Mol Biol. 2005;291:207–16. doi: 10.1385/1-59259-840-4:207. [DOI] [PubMed] [Google Scholar]
- 20.Guldberg P, Nedergaard T, Nielsen HJ, Olsen AC, Ahrenkiel V, Zeuthen J. Single-step DGGE-based mutation scanning of the p53 gene: application to genetic diagnosis of colorectal cancer. Hum Mutat. 1997;9:348–55. doi: 10.1002/(SICI)1098-1004(1997)9:4<348::AID-HUMU8>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 21.Pao W, Miller VA. Epidermal Growth Factor Receptor Mutations, Small-Molecule Kinase Inhibitors, and Non-Small-Cell Lung Cancer: Current Knowledge and Future Directions. J Clin Oncol. 2005;23:2556–68. doi: 10.1200/JCO.2005.07.799. [DOI] [PubMed] [Google Scholar]
- 22.Barber TD, Vogelstein B, Kinzler KW, Velculescu VE. Somatic Mutations of EGFR in Colorectal Cancers and Glioblastomas. N Engl J Med. 2004;351:2883. doi: 10.1056/NEJM200412303512724. [DOI] [PubMed] [Google Scholar]
- 23.Tsuchihashi Z, Khambata-Ford S, Hanna N, Janne PA. Responsiveness to Cetuximab without Mutations in EGFR. N Engl J Med. 2005;353:208–9. doi: 10.1056/NEJM200507143530218. [DOI] [PubMed] [Google Scholar]
- 24.Lenz H-J, Van Cutsem E, Khambata-Ford S, et al. Multicenter Phase II and Translational Study of Cetuximab in Metastatic Colorectal Carcinoma Refractory to Irinotecan, Oxaliplatin, and Fluoropyrimidines. J Clin Oncol. 2006;24:4914–21. doi: 10.1200/JCO.2006.06.7595. [DOI] [PubMed] [Google Scholar]
- 25.De Pas T, Pelosi G, de Braud F, et al. Modulation of Epidermal Growth Factor Receptor Status by Chemotherapy in Patients With Locally Advanced Non-Small-Cell Lung Cancer Is Rare. J Clin Oncol. 2004;22:4966–70. doi: 10.1200/JCO.2004.01.195. [DOI] [PubMed] [Google Scholar]
- 26.Ono M, Kuwano M. Molecular Mechanisms of Epidermal Growth Factor Receptor (EGFR) Activation and Response to Gefitinib and Other EGFR-Targeting Drugs. Clin Cancer Res. 2006;12:7242–51. doi: 10.1158/1078-0432.CCR-06-0646. [DOI] [PubMed] [Google Scholar]
- 27.Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic Activation of the RAS/RAF Signaling Pathway Impairs the Response of Metastatic Colorectal Cancers to Anti-Epidermal Growth Factor Receptor Antibody Therapies. Cancer Res. 2007;67:2643–8. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 29.Moasser MM, Basso A, Averbuch SD, Rosen N. The Tyrosine Kinase Inhibitor ZD1839 (“Iressa”) Inhibits HER2-driven Signaling and Suppresses the Growth of HER2-overexpressing Tumor Cells. Cancer Res. 2001;61:7184–8. [PubMed] [Google Scholar]
- 30.Cappuzzo F, Varella-Garcia M, Shigematsu H, et al. Increased HER2 Gene Copy Number Is Associated With Response to Gefitinib Therapy in Epidermal Growth Factor Receptor-Positive Non-Small-Cell Lung Cancer Patients. J Clin Oncol. 2005;23:5007–18. doi: 10.1200/JCO.2005.09.111. [DOI] [PubMed] [Google Scholar]
- 31.Nathanson DR, Culliford AT, Shia J, et al. HER 2/neu expression and gene amplification in colon cancer. International Journal of Cancer. 2003;105:796–802. doi: 10.1002/ijc.11137. [DOI] [PubMed] [Google Scholar]
- 32.Bunz F, Hwang PM, Torrance C, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–9. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS. Effect of p53 status on tumor response to antiangiogenic therapy. Science. 2002;295:1526–8. doi: 10.1126/science.1068327. [DOI] [PubMed] [Google Scholar]
- 34.Ruzzo A, Graziano F, Canestrari E, Magnani E. Molecular predictors of efficacy to anti-EGFR agents in colorectal cancer patients. Curr Ca Drug Targets. 2010;10:68–79. doi: 10.2174/156800910790980205. [DOI] [PubMed] [Google Scholar]
- 35.Nash GM, Gimbel M, Shia J, et al. KRAS mutation correlates with accelerated metastatic progression in patients with colorectal liver metastases. Ann Surg Oncol. 2010 Feb;17(2):572–8. doi: 10.1245/s10434-009-0605-3. [DOI] [PubMed] [Google Scholar]
- 36.Sinicrope FA, Mahoney MR, Yoon HH, et al. Analysis of molecular markers by anatomic tumor site in stage III colon carcinomas from adjuvant chemotherapy trial NCCTG N0147 (Alliance) Clin Cancer Res. 2015 Dec 1;21(23):5294–304. doi: 10.1158/1078-0432.CCR-15-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato J, Futamura M, Kanematsu M, et al. Combination therapy with zoledronic acid and cetuximab effectively suppresses growth of colorectal cancer cells regardless or KRAS status. Int J Cancer. 2016 Mar 15;138(6):1516–27. doi: 10.1002/ijc.29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dienstmann R, Salazer R, Tabernero J. Personalizing colon cancer adjuvant therapy: selecting optimal treatments for individual patients. J Clin Oncol. 2015 Jun 1;33(16):1787–96. doi: 10.1200/JCO.2014.60.0213. [DOI] [PubMed] [Google Scholar]
- 39.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumoral heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012 Mar 8;366(10):883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabbinavar FF, Schulz J, McCleod M, et al. Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results of a randomized phase II trial. J Clin Oncol. 2005 Jun 1;23(16):3697–705. doi: 10.1200/JCO.2005.05.112. [DOI] [PubMed] [Google Scholar]
- 41.Khan SA, Idrees K, Forslund A, et al. Genetic variants in germline TP53 and MDM2 SNP309 are not associated with early onset colorectal cancer. J Surg Oncol. 2008 Jun 1;97(7):621–5. doi: 10.1002/jso.20996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spindler KL, Lindebjerg J, Nielsen JN, et al. Epidermal growth factor receptor analyses in colorectal cancer: a comparison of methods. Int J Oncol. 2006 Nov;29(5):1159–65. [PubMed] [Google Scholar]
- 43.Chapman SJ, McKavanagh D, Burge ME, et al. Effectiveness of bevacizumab and cetuximab in metastatic colorectal cancer across selected public hospitals in Queensland. Asia Pac J Clin Oncol. 2016 Jul 20; doi: 10.1111/ajco.12518. [DOI] [PubMed] [Google Scholar]
- 44.Larsen FO, Pfeiffer P, Nielsen D, et al. Bevacizumab in combination with cetuximab and irinotecan after failure of cetuximab and irinotecan in patients with metastatic colorectal cancer. Acta Oncol. 2011 May;50(4):574–7. doi: 10.3109/0284186X.2010.546369. [DOI] [PubMed] [Google Scholar]
- 45.Sartore-Bianchi A, Moroni M, Veronese V, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007 Aug 1;25(22):3238–45. doi: 10.1200/JCO.2007.11.5956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.