

Abstract
Mast cell leukemia is a rare variant of advanced systemic mastocytosis characterized by at least 20% of mast cells in a bone marrow smear. We evaluated clinical and molecular characteristics of 28 patients with (n=20, 71%) or without an associated hematologic neoplasm. De novo mast cell leukemia was diagnosed in 16 of 28 (57%) patients and secondary mast cell leukemia evolving from other advanced systemic mastocytosis subtypes in 12 of 28 (43%) patients, of which 7 patients progressed while on cytoreductive treatment. Median bone marrow mast cell infiltration was 65% and median serum tryptase was 520 μg/L. C-findings were identified in 26 of 28 (93%) patients. Mutations in KIT (D816V, n=19; D816H/Y, n=5; F522C, n=1) were detected in 25 of 28 (89%) patients and prognostically relevant additional mutations in SRSF2, ASXL1 or RUNX1 (S/A/Rpos) in 13 of 25 (52%) patients. Overall response rate in 18 treatment-naïve patients was 5 of 12 (42%) on midostaurin and 1 of 6 (17%) on cladribine, and after switch 1 of 4 (25%) on midostaurin and 0 of 3 on cladribine, respectively. S/A/Rpos adversely affected response to treatment and progression to secondary mast cell leukemia (n=6) or acute myeloid leukemia (n=3) while on treatment (P<0.05). The median overall survival from mast cell leukemia diagnosis was 17 months as compared to 44 months in a control group of 124 patients with advanced systemic mastocytosis but without mast cell leukemia (P=0.03). In multivariate analyses, S/A/Rpos remained the only independent poor prognostic variable predicting overall survival (P=0.007). In conclusion, the molecular signature should be determined in all patients with mast cell leukemia because of its significant clinical and prognostic relevance.
Introduction
According to the World Health Organization (WHO) classification, advanced systemic mastocytosis (SM) comprises patients with aggressive SM (ASM), SM with an associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL).1–3 MCL is characterized by at least 20% of mast cells (MC) in a bone marrow (BM) smear and a particularly poor prognosis with median survival time of less than six months. In the majority of patients with MCL, the aleukemic variant (MCs represent less than 10% of all blood leukocytes) is diagnosed.2,4–6
Systemic mastocytosis is characterized by somatically acquired, activating mutations in the gene encoding the receptor tyrosine kinase KIT, most commonly KIT D816V which is identified in over 80% of all patients with SM.7 Recent data, however, have highlighted that the molecular pathogenesis of advanced SM is complex with one or more additional mutations, eg. in ASXL1, TET2, JAK2 V617F, RUNX1, SRSF2, present in over 60% of advanced SM patients.8–10 These additional mutations are usually acquired by neoplastic (stem) cells prior to KIT D816V, thereby indicating a multi-mutated stem cell disease and a step-wise process of oncogenesis.11 Molecular aberrations, eg. in SRSF2, ASXL1, or RUNX1 (S/A/R gene panel) have a strong adverse impact on disease phenotype and prognosis.9,12 A new risk score was therefore proposed for patients with SM. This score includes clinical variables, eg. splenomegaly and elevated alkaline phosphatase (AP), as well as molecular markers, eg. mutations in the S/A/R gene panel (S/A/Rpos).13
Our current knowledge of MCL, including clinical and molecular characteristics, treatment options, survival, and prognostic factors is limited to case reports, case-series and/or literature reviews.14–16 We here report on a relatively large group of patients with recently diagnosed MCL. The aim of the present study was: a) to characterize the clinical and molecular features in patients with MCL; b) to evaluate response and resistance to various treatments; and c) to define prognostic factors.
Methods
Diagnosis of mast cell leukemia
The diagnosis of SM was established according to the WHO classification.2,5,6,17 BM biopsies and BM smears were evaluated by reference pathologists of the European Competence Network on Mastocytosis, ECNM (H-PH and KS). Diagnosis of MCL was based on the presence of at least 20% MCs in BM smears (Figure 1). Twenty-eight patients were diagnosed between 2008 and 2016. The study design adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Review Board of the Medical Faculty of Mannheim, University of Heidelberg, as part of the ‘German Registry on Disorders of Eosinophils and Mast Cells’. All patients gave written informed consent.
Figure 1.
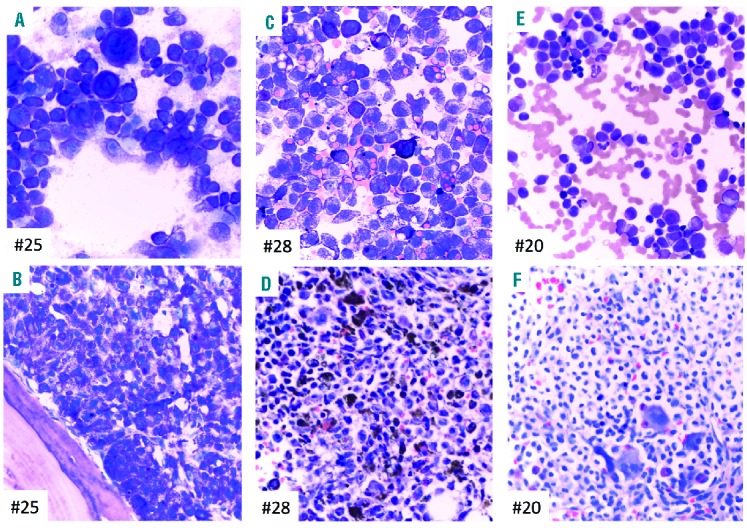
Bone marrow morphology and phenotype of 3 patients with mast cell leukemia (MCL). Patients #25: MCL, #28: MCL, #20: MCL with an associated hematologic neoplasm (AHN). (A, C and E) Bone marrow (BM) smears show an abundance of pleomorphic mast cells (MCs) exhibiting metachromatic granules. Quantitative criteria for diagnosis of MCL are completely fulfilled since MC make up more than 90% of all nucleated cells. Note the bizarre giant metachromatic MCs in (A), the prominent hemophagocytic activity of MC in (C), and the marked cytological atypia of MCs with pronounced hypogranulation in (E). Case (E) also exhibits immature atypical eosinophils enabling the diagnosis of an AHN, probably myelodysplastic/myeloproliferative neoplasm with eosinophilia. (B, D and F) BM sections show extreme hypercellularity and packed MC infiltrates. Fat cells and normal blood cell precursors are subtotally depleted. The cytomorphological aspects are fully reflected by the histomorphological findings. Note the extreme siderosis in (D) and the clear-cell aspect of atypical MC in (F) with the possibility of a misdiagnosis (hairy cell leukemia, histiocytosis or even metastatic infiltrates of a renal cell carcinoma) unless appropriate immunohistochemistry is performed. (A–F) Wright-Giemsa staining.
Assessment of KIT D816V
Qualitative and quantitative assessment of the KIT D816V expressed allele burden was performed using allele-specific quantitative real-time reverse transcriptase polymerase chain reaction analyses (qRT-PCR) as previously described.12 Molecular analyses were performed in all patients at baseline. In KIT D816V negative patients, exon 17 was sequenced by conventional Sanger sequencing. In selected patients, targeted next-generation deep amplicon sequencing (NGS) was used for identification of alternative mutations in all KIT exons.
Targeted next-generation sequencing analysis
Next-generation sequencing analysis was either performed by 454 FLX amplicon chemistry (Roche, Penzberg, Germany) or library preparation based on the TruSeq Custom Amplicon Low Input protocol (Illumina, San Diego, CA, USA) and sequencing on the MiSeq instrument (Illumina, San Diego, CA, USA) to investigate the KIT gene and the following 18 genes: ASXL1, CBL, ETV6, EZH2, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, SETBP1, SF3B1, SRSF2, TET2, TP53, U2AF1, ZRSR2.8 Gene mutations were annotated compared to the reference sequence based on the Ensembl Transcript ID (Ensembl release 85: July 2016).
Cytogenetic analysis and fluorescence in situ hybridization analysis
Cytogenetic analyses of at least 20 Giemsa-banded bone marrow (BM) metaphases (24 hour and/or 48 hour culture) were analyzed and interpreted according to the International System for Human Cytogenetic Nomenclature.18 If necessary, chromosome banding analysis was combined with fluorescence in situ hybridization (FISH) analysis according to the manufacturer’s instructions (Metasystems, Altlussheim, Germany).19
Statistical analyses
Statistical analyses considered clinical, laboratory or molecular parameters obtained at time of MCL diagnosis. Overall survival (OS) analysis was determined as time from date of diagnosis to date of death or last visit. For analysis of survival differences between the various subtypes of advanced SM, we used the current MCL patient cohort and a patient cohort with advanced SM but without MCL (n=124), who were enrolled within the ‘German Registry on Disorders of Eosinophils and Mast Cells’. Pearson correlation analysis was performed for the correlation between two parameters. Differences in the distribution of continuous variables between categories were analyzed by Mann-Whitney test (for comparison of two groups). For categorical variables, Fishers’s exact test was used. OS probabilities were estimated with the Kaplan-Meier method and compared by the log-rank test in univariate analysis. For the estimation of hazard ratios (HRs) and multivariate analysis, the Cox proportional hazard regression model was used. P<0.05 (two-sided) was considered significant. There was no adjustment for multiple testing as all analyses were explorative. SPSS version 22.0.0 (IBM Corporation, Armonk, NY, USA) was used for statistical analysis.
Results
Clinical and morphological characteristics
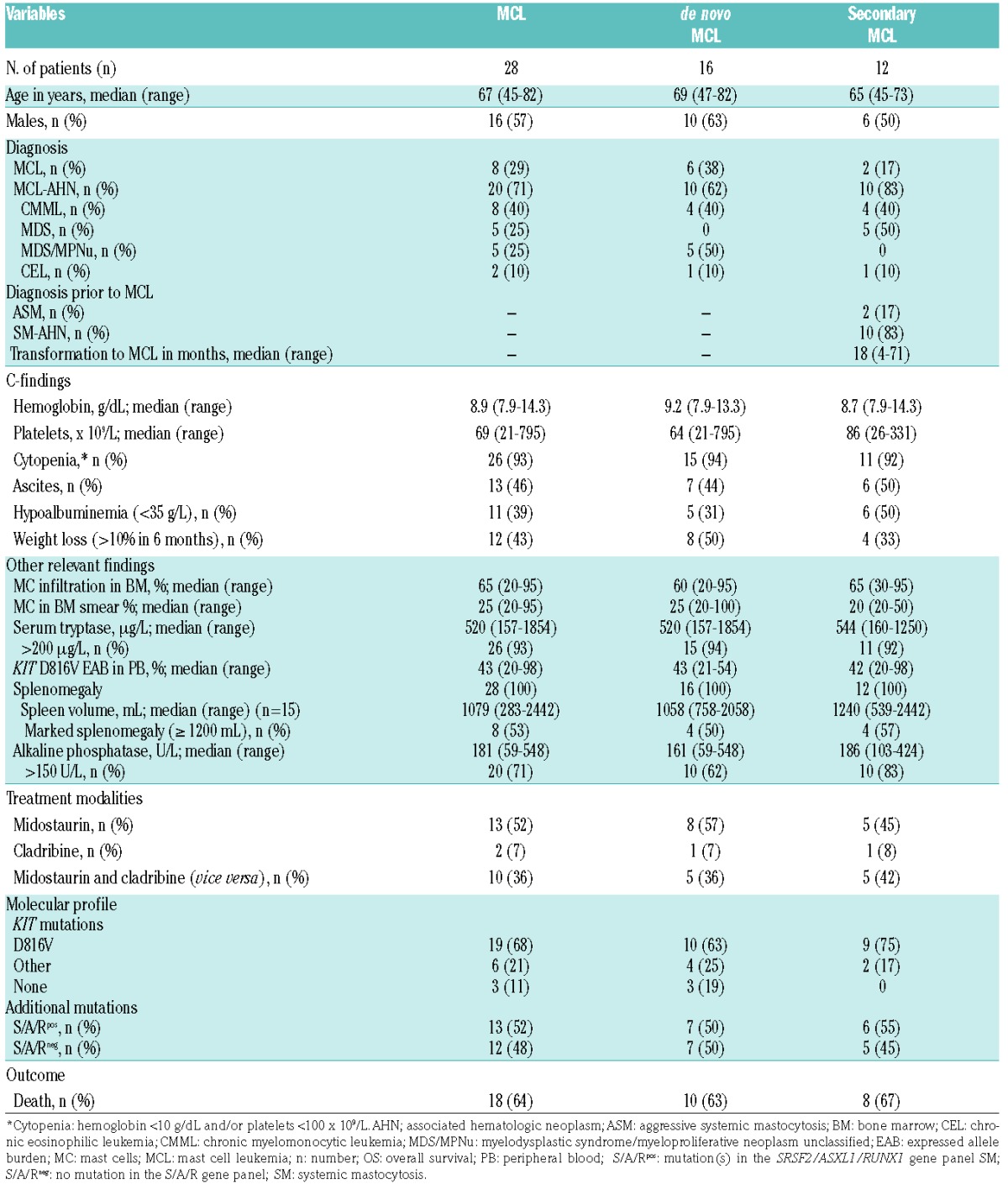
The median age of the 28 patients was 67 years (range 45–82; male 57%). The median percentage of MCs in BM smears and trephine biopsies was 25% (range 20–95) and 65% (range 20–95; 82% of patients >50%), respectively. MCs in peripheral blood (PB) 10% or more (leukemic MCL) were only seen in 2 of 28 (7%) patients. MCL-AHN was diagnosed in 20 of 28 (71%) patients: chronic myelomonocytic leukemia (CMML, n=8), myelodysplastic myeloproliferative neoplasm unclassifiable (MDS/MPNu, n=5), MDS (n=5) or chronic eosinophilic leukemia (CEL, n=2). Blood parameters analyzed in this study included: leukocytes (median 6.1×109/L, range 1.3–47.6), hemoglobin (median 8.9 g/dL, range 7.9–14.3; <10 in 71% of patients), platelets (median 69×109/L, range 21–795; <100 in 68% of patients), eosinophils (0.1×109/L, range 0–12.9; >1.0 in 14%), and monocytes (0.6×109/L, range 0.1–2.9; >1.0 in 25%). Hematologic C-findings such as hemoglobin less than 10 g/dL and/or platelets less than 100×109/L were identified in 26 of 28 (93%) patients. Median serum tryptase level (normal value <11.4 μg/L) was 520 μg/L (range 157–1854; 93% ≥200) and median KIT D816V expressed allele burden was 43% (range 20–98) in PB. Signs of non-hematologic organ dysfunction included elevated AP (20 of 28, 71%, median 181; range 59–548) and splenomegaly in 28 of 28 (100%) patients. Spleen volumetry results obtained by magnetic resonance imaging were available in 16 patients and showed marked splenomegaly (≥1200 mL) in 8 of 16 cases (50%) (Table 1).
Table 1.
Clinical, laboratory, molecular and treatment characteristics and outcome of 28 patients with de novo mast cell leukemia (MCL, n=16) or secondary MCL (n=12).
De novo mast cell leukemia and secondary mast cell leukemia
De novo MCL was diagnosed in 16 of 28 (57%) patients. Secondary MCL (sMCL) evolving from other advanced SM subtypes (SM-AHN, n=10; ASM, n=2) was observed in 12 of 28 (43%) patients (Table 1) with a median time to progression of 18 months (range 4–71). Of note, 3 of 28 patients (11%) progressed into secondary acute myeloid leukemia (MCL-sAML) 18, 28 and 34 months, respectively, after the diagnosis of MCL-CMML had been established. Overall, no statistically significant differences were observed between de novo MCL and sMCL regarding clinical, morphological or molecular characteristics (Table 1).
KIT mutations
Overall, 25 of 28 (89%) patients had mutations in KIT (Figure 2). KIT D816V was identified in 19 of 28 patients (68%), and alternative KIT mutations (D816H, n=3; D816Y, n=2; F522C, n=1) in 6 of 28 (21%) patients by Sanger sequencing or targeted NGS (KIT F522C), respectively.
Figure 2.
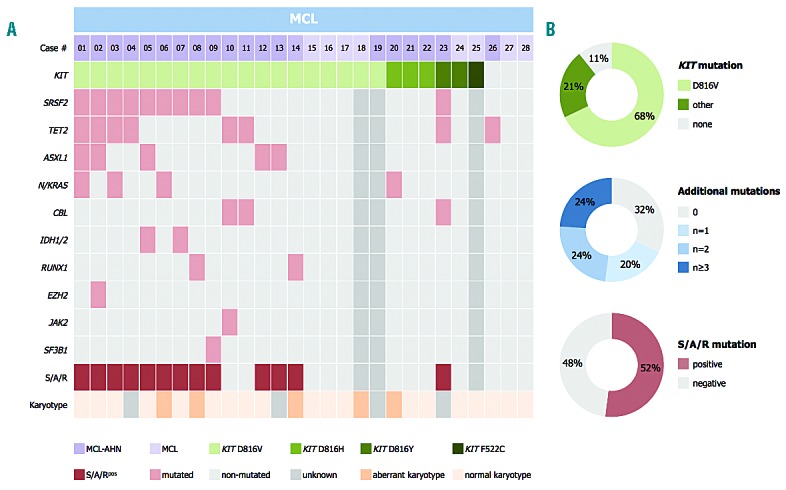
Mutational profile of 28 patients with mast cell leukemia (MCL). (A) Alignment of gene mutations in 28 patients with MCL. Each column represents an individual patient. Results of genetic analyses are depicted in different colors and (B) relative frequency distribution of KIT mutations, additional mutations and mutations in the SRSF2/ASXL1/RUNX1 (S/A/R) gene panel.
Additional mutations
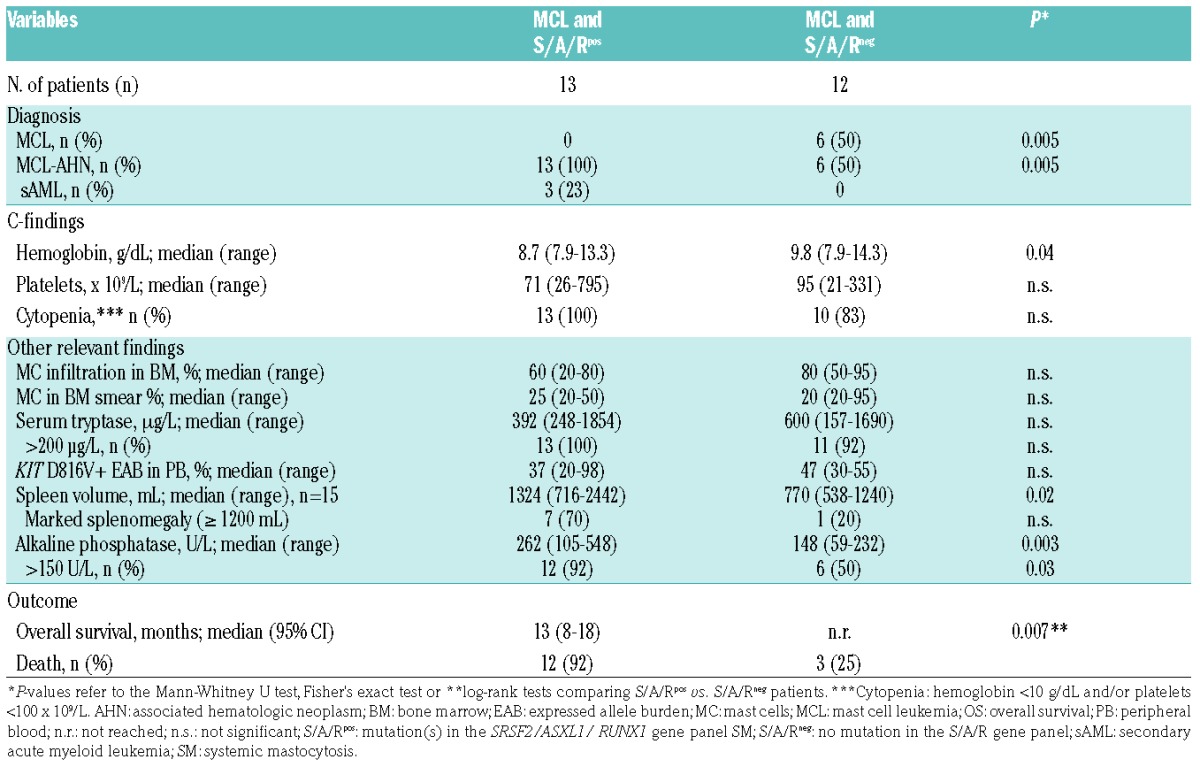
Data on additional mutations were available in 25 of 28 (89%) patients. Seventeen of 25 (68%) patients with MCL had at least one additional mutation (median 2, range 1–4) in 10 of 18 analyzed genes, and 8 of 25 (32%) patients were negative for additional mutations (Figure 2). The most frequently affected genes were SRSF2 (n=10, 40% of patients), TET2 (n=8, 32%), ASXL1 (n=5, 20%), N/KRAS (n=4, 16%), CBL (n=3, 12%) IDH1/2 (n=2, 8%), and RUNX1 (n=2, 8%). EZH2, JAK2 and SF3B1 were less frequently affected (<5%). Only the concurrent presence of CBL and TET2 achieved significance (P<0.05). At least one mutation in the S/A/R gene panel (S/A/Rpos) was identified in 13 of 25 (52%) patients. Clinical and morphological features in S/A/Rpos and S/A/Rneg patients with MCL were significantly different. In particular, S/A/Rpos patients had lower median hemoglobin levels, a higher median spleen volume, and higher AP levels (Table 2).
Table 2.
Clinical, laboratory characteristics and outcome of 25 patients with mast cell leukemia (MCL) depending on the mutational status in the SRSF2/ASXL1/RUNX1 (S/A/R) gene panel.
Cytogenetic analysis
A karyotype was available in 24 of 28 (86%) patients with MCL which was normal in 19 of 24 (79%) and aberrant in 5 of 24 (21%) patients; 3 patients had a complex karyotype (≥ 3 aberrations) and 2 patients a del(5q) or del(12p), respectively. According to these findings, there is no recurrent cytogenetic abnormality in patients with MCL.
Response and resistance to treatment
Twenty-five of 28 (89%) patients were treated with various cytoreductive drugs (Table 3). Three patients were not treated because of death at diagnosis (n=1) or comorbidity (n=2). Overall, 23 of 25 (92%) patients were treated with midostaurin [international phase II study on the efficacy and safety of midostaurin in advanced SM (clinicaltrial.gov identifier: 00782067), n=9; compassionate use program by Novartis Pharmaceuticals, n=14]20 at any time prior to or after diagnosis of MCL. Twelve of 25 (48%) patients received cladribine.
Table 3.
Treatment modalities and responses in 25 patients with mast cell leukemia (MCL).
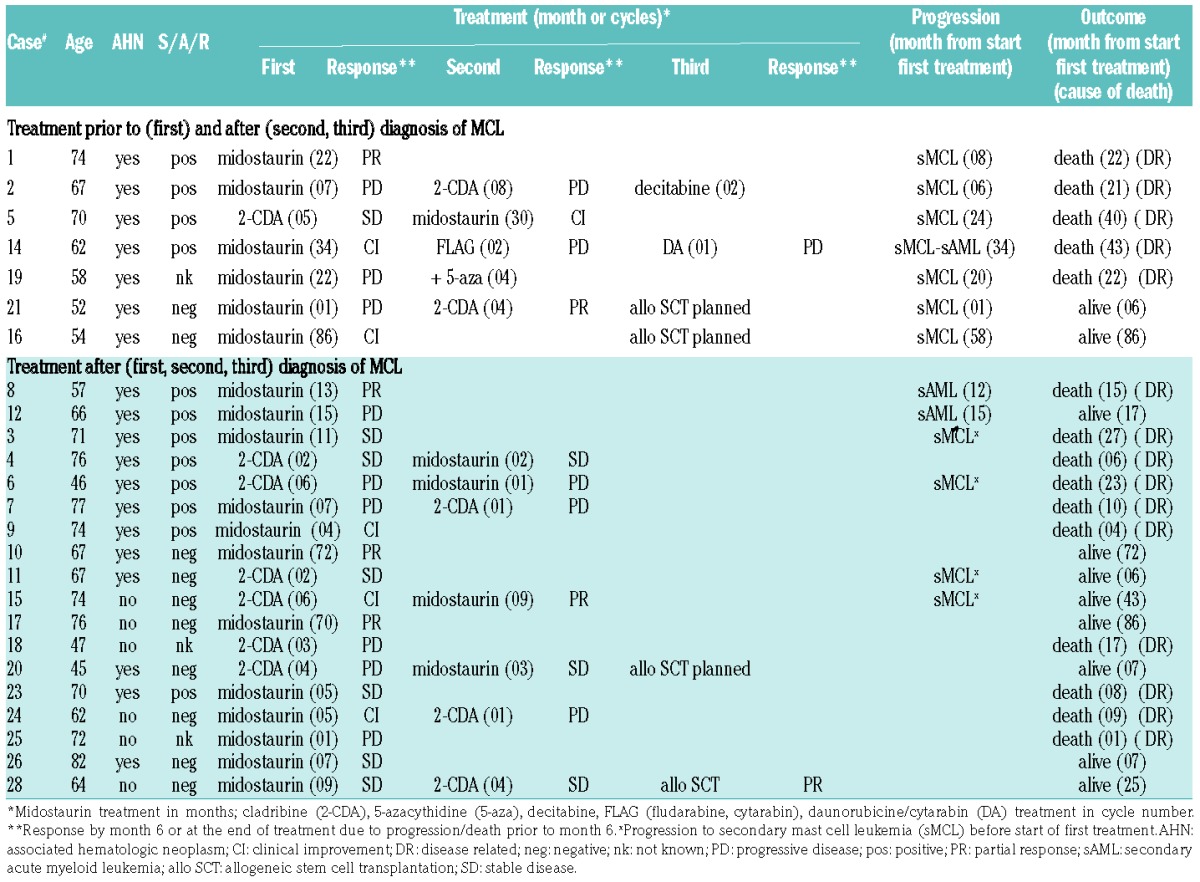
In 18 treatment-naïve MCL patients, the overall response rate (ORR) according to IWG-MRT and ECNM criteria was 5 of 12 (42%) on midostaurin and 1 of 6 (17%) on cladribine, respectively, and after switching, 1 of 4 (25%) on midostaurin and 0 of 3 on cladribine, respectively. Independently of midostaurin (n=16) or cladribine (n=13), there was no significant difference in ORR during those 29 individual treatment periods between MCL-AHN and MCL patients [6 of 20 (30%) vs. 4 of 9 (44%); P=0.2]. In contrast, there was a significant difference in ORR in 27 individual treatment periods (the S/A/R status was unknown in 2 patients) between S/A/Rpos and S/A/Rneg patients [2 of 13 (15%) vs. 8 of 14 (57%); P<0.05] (Table 3). Overall, progression while on treatment (n=9) occurred predominantly in patients with AHN (8 of 9 patients) after a median of 15 months (range 1–58) from start of treatment and was significantly associated with S/A/Rpos (6 of 8 patients; P<0.05).
Prognostic factors and overall survival
The median OS of all 28 MCL patients from diagnosis was 17 months [95% confidence interval (CI): 10–24] with a 2-year OS probability of 24%. The median OS of 124 patients with advanced SM (ASM and SM-AHN) but without MCL was 44 months [95%CI: 31.3–56.7; HR 1.8 (1.1–3.1); P=0.03] (Figure 3). These data confirm the particularly poor prognosis of patients with MCL.
Figure 3.
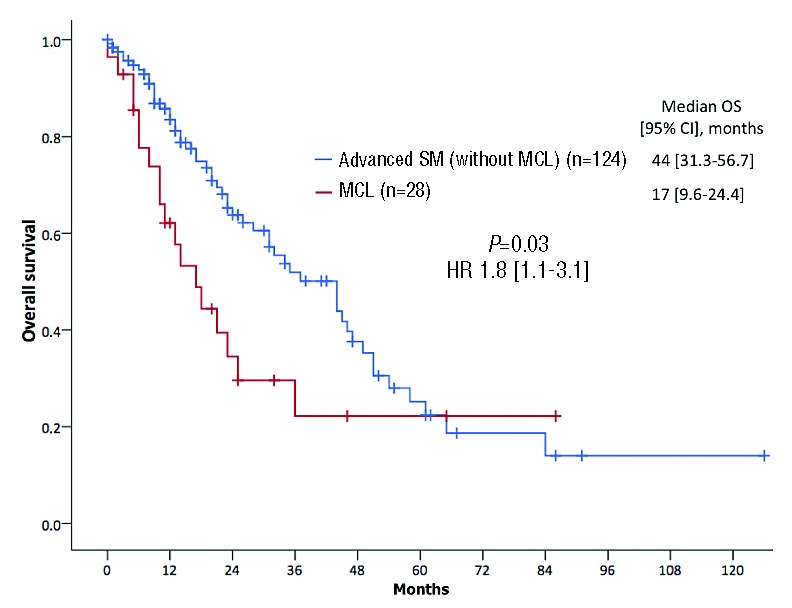
Overall survival (OS). Kaplan-Meier estimates of OS depending on comparisons between patients with mast cell leukemia (MCL, n=28) and a control group of patients with advanced systemic mastocytosis (without MCL) (n=124) enrolled within the ‘German Registry on Disorders of Eosinophils and Mast Cells’. CI: confidence interval; HR: hazard ratio.
In univariate analyses of multiple clinical, morphological and molecular variables only bicytopenia [hemoglobin <10 g/dL and platelets <100×109/L, n=13 vs. hemoglobin ≥10 g/dL or platelets ≥100×109/L, n=13; P=0.02, HR 3.2 (1.2–8.9)], elevated AP [P=0.009, HR 3.3 (1.3–8.3)] and S/A/Rpos [P=0.007, HR 5.0 (1.8–18.1)] were identified as poor prognostic variables regarding OS. In multivariate analyses, S/A/Rpos remained the only independent poor risk factor concerning OS (median 13 months, 95%CI: 8–18 vs. median not reached) (Figure 4).
Figure 4.
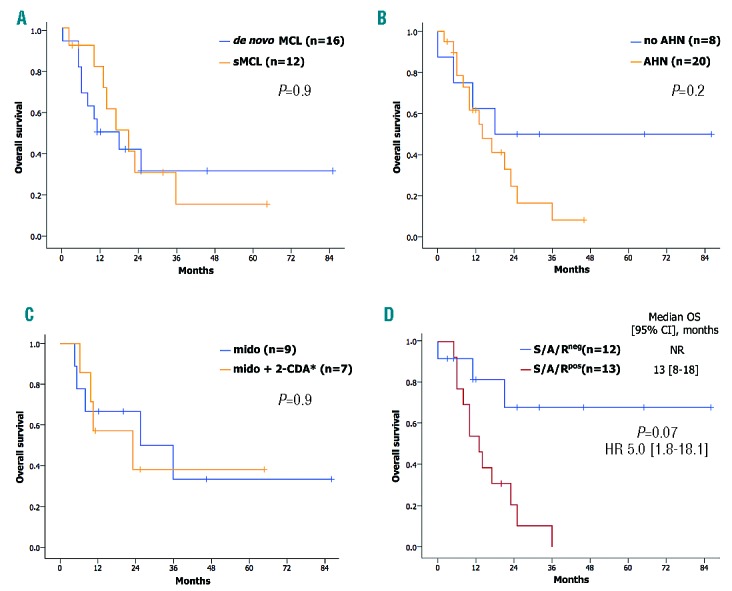
Kaplan-Meier estimates of overall survival (OS) depending on comparisons between multiple variables. (A) De novo mast cell leukemia (MCL) and secondary MCL (sMCL), (B) MCL with or without an associated hematologic neoplasm (AHN), (C) midostaurin treatment or cladribine (2-CDA) and/or midostaurin vice versa and (D) with mutations in the SRSF2/ASXL1/RUNX1 (S/A/Rpos) or without mutation in S/A/R (S/A/Rneg) gene panel. CI: confidence interval; HR: hazard ratio; NR: not reached.
There was no difference in OS between de novo MCL vs. sMCL (P=0.9) or MCL-AHN vs. MCL (P=0.2). In treatment-naïve MCL patients, no difference was observed between midostaurin monotherapy (n=9) and sequential midostaurin/cladribine treatment or vice versa (n=7) (P=0.9) (Figure 4).
Discussion
In addition to the confirmation of some known clinical and molecular characteristics of MCL, we identified several new and important aspects in this heterogeneous disorder, which may improve understanding of the clinical phenotype, prediction of response to treatment and prognosis. Confirmatory results included the prognostic relevance of the defining criterion of at least 20% of MCs in a BM smear and the presence of the KIT D816V mutation in approximately 60–70% of patients, which is significantly lower than other SM subtypes. However, alternative mutations at position 816, eg. D816H or D816Y, were identified in a significant proportion of KIT D816V negative patients leading to an overall incidence of KIT D816 mutations in approximately 90% of patients with MCL. We therefore recommend that KIT exon 17 and, if negative, other exons of KIT gene should be sequenced in KIT D816V negative patients with advanced SM and particularly MCL.14,21,22
In our series, the proposed subtypes of MCL without C-findings (chronic MCL) and leukemic MCL were only observed in 2 patients each, in line with previously published series,14,15 the latter clearly demonstrating that presence or absence of MCs in PB is only of minor importance for the diagnosis of MCL. We have, however, observed a much higher relative frequency of MCL-AHN than that reported in the literature (71% vs. 10%),14 which is most likely due to the fact that BM aspirates (cytology) and BM sections (histology and immunohistochemistry) of all our cases were centrally reviewed. Because of the high MC burden (eg. MC infiltration in BM, serum tryptase level, KIT D816 allele burden) in MCL and the multilineage involvement of the KIT D816V mutation,11 MCL and AHN almost always derive from the same myeloid progenitors, and the question as to whether C-findings are attributable to MCL or to AHN is not relevant.
Moreover, there are distinct differences between de novo MCL and sMCL evolving from other advanced SM subtypes.14 In our series, approximately one-third of patients had sMCL, in all cases progressing from other advanced SM subtypes, with 40% of those patients progressing while on cytoreductive treatment, predominantly midostaurin. We did not observe direct evolution from indolent SM (ISM) to MCL, confirming previous reports that progression usually occurs from SM-AHN or ASM.4 Importantly, there was no difference in OS between de novo MCL and sMCL.
Consistent with previous reports, cladribine has only shown a little activity in our patients with MCL,14,21,23–25 with only 2 cases showing durable partial response or clinical improvement. On the other hand, the median OS of 17 months is certainly better than the survival reported for historical controls.4,14,26 Because the vast majority of patients (>90%) have also been treated with midostaurin, either prior to or following cladribine, the relatively high ORR of midostaurin compared with cladribine is likely to be associated with the improved survival. Most of the previously reported MCL cases were frequently treated with imatinib, dasatinib or masitinib, which are definitely without any substantial efficacy in KIT D816V advanced SM.14
At the moment, there are no distinct parameters to predict the response to treatment and the risk for progression to sMCL or MCL-sAML. Overall, the most significant prognostic factor identified for MCL patients was the presence or absence of mutations in S/A/R. In particular, S/A/Rpos patients with MCL had a more aggressive phenotpye, a lower response rate, more (intrinsic) resistance to disparate treatment modalities, and a poor survival compared to S/A/Rneg MCL. The patients with progression to sMCL or to MCL-AML while on treatment were predominantly S/A/Rpos. Moreover, durable remissions and long-term OS were exclusively observed in S/A/Rneg patients. This also explains why no significant differences were observed between MCL and MCL-AHN, because MCL-AHN was also associated with other, potentially more favorable mutations than in S/A/R (eg. TET2, CBL, JAK2). It is, therefore, not the presence of AHN per se, but the molecular background of AHN that impacts on prognosis.
Because of resistant and rapidly progressive disease followed by poor OS, particularly in S/A/Rpos patients with MCL, intensive chemotherapy and allogeneic stem cell transplantation (SCT) is recommended as the only curative treatment option for young and fit patients.23 Allogeneic SCT should be performed early because responses on midostaurin, cladribine and/or myeloid-type induction chemotherapy may only be short-lived.26,27 However, only a minority of patients are eligible for allogeneic SCT because of advanced age, SM-related organ damage (eg. liver cirrhosis), comorbidity, and frequently poor response or progression on various treatment options, including intensive chemotherapy. In addition, Ustun et al. have reported a 5-year survival rate of only 14% for patients with MCL, which may have been influenced by several negative factors, including the S/A/R mutation status and a poor remission status prior to transplant. Clinicial trials are warranted to define the optimal intensity, duration, and efficacy of combined strategies with midostaurin and chemotherapy for treatment of MCL in general, and also for debulking and bridging prior to allogeneic SCT. In this respect, new drugs such as brentuximab-vedotin in CD30 positive advanced SM,28 gemtuzumab ozogamicin29 and possibly also hypomethylating agents30 should be investigated.
We conclude that: a) MCL has the worst prognosis of the various advanced SM subtypes; b) leukemic MCL and MCL without C-findings are rare; c) sMCL is frequent and evolves from other advanced SM subtypes but not directly from ISM; d) KIT D816 mutations are more frequent than previously reported and KIT D816V negative patients should be tested for other KIT mutations; e) mutations in the S/A/R gene panel are present in approximately 50% of patients with MCL and are adversely associated with phenotype, response to treatment, progression, and OS; and f) the median OS of approximately 1.5 years, achieved predominantly in midostaurin-treated patients, is superior compared to historical controls.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/6/1035
Funding
This work was supported by the ‘Deutsche José Carreras Leukämie-Stiftung e.V.’ (grant n. DJCLS R 13/05) and by the SEED program of the Medical Faculty Mannheim, Heidelberg University, and by the Austrian Science Fund (FWF) SFB project F4704-B20.
References
- 1.Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435–453. [DOI] [PubMed] [Google Scholar]
- 2.Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25(7):603–625. [DOI] [PubMed] [Google Scholar]
- 3.Theoharides TC, Valent P, Akin C. Mast Cells, Mastocytosis, and Related Disorders. N Engl J Med. 2015;373(2):163–172. [DOI] [PubMed] [Google Scholar]
- 4.Lim KH, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009;113(23):5727–5736. [DOI] [PubMed] [Google Scholar]
- 5.Horny HP, A C, Metcalfe DD, Swerdlow SH, Campo E, Harris NL, et al. World Health Organization (WHO) Classification of Tumours. Mastocytosis (Mast cell disease). Pathology & Genetics. Tumours of Haematopoietic and Lymphoid Tissues, vol. 2: Lyon, France: IARC Press, 2008, p. 54–63. [Google Scholar]
- 6.Pardanani A. Systemic mastocytosis in adults: 2017 update on diagnosis, risk stratification and management. Am J Hematol. 2016;91(11):1146–1159. [DOI] [PubMed] [Google Scholar]
- 7.Gleixner KV, Mayerhofer M, Cerny-Reiterer S, et al. KIT-D816V-independent oncogenic signaling in neoplastic cells in systemic mastocytosis: role of Lyn and Btk activation and disruption by dasatinib and bosutinib. Blood. 2011;118(7):1885–1898. [DOI] [PubMed] [Google Scholar]
- 8.Schwaab J, Schnittger S, Sotlar K, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122(14):2460–2466. [DOI] [PubMed] [Google Scholar]
- 9.Jawhar M, Schwaab J, Schnittger S, et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia. 2016;30(1):136–143. [DOI] [PubMed] [Google Scholar]
- 10.Pardanani AD, Lasho TL, Finke C, et al. ASXL1 and CBL mutations are independently predictive of inferior survival in advanced systemic mastocytosis. Br J Haematol. 2016;175(3):534–536. [DOI] [PubMed] [Google Scholar]
- 11.Jawhar M, Schwaab J, Schnittger S, et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia. 2015;29(5):1115–1122. [DOI] [PubMed] [Google Scholar]
- 12.Erben P, Schwaab J, Metzgeroth G, et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis. Ann Hematol. 2014;93(1):81–88. [DOI] [PubMed] [Google Scholar]
- 13.Jawhar M, Schwaab J, Hausmann D, et al. Splenomegaly, elevated alkaline phosphatase and mutations in the SRSF2/ASXL1/RUNX1 gene panel are strong adverse prognostic markers in patients with systemic mastocytosis. Leukemia. 2016;30(12):2342–2350. [DOI] [PubMed] [Google Scholar]
- 14.Georgin-Lavialle S, Lhermitte L, Dubreuil P, et al. Mast cell leukemia. Blood. 2013; 121(8):1285–1295. [DOI] [PubMed] [Google Scholar]
- 15.Valent P, Berger J, Cerny-Reiterer S, et al. Chronic mast cell leukemia (MCL) with KIT S476I: a rare entity defined by leukemic expansion of mature mast cells and absence of organ damage. Ann Hematol. 2015;94(2):223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valent P, Blatt K, Eisenwort G, et al. FLAG-induced remission in a patient with acute mast cell leukemia (MCL) exhibiting t(7;10)(q22;q26) and KIT D816H. Leuk Res Rep. 2014;3(1):8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 18.Simons A, Shaffer LG, Hastings RJ. Cytogenetic Nomenclature: Changes in the ISCN 2013 Compared to the 2009 Edition. Cytogenet Genome Res. 2013;141(1):1–6. [DOI] [PubMed] [Google Scholar]
- 19.Schoch C, Schnittger S, Bursch S, et al. Comparison of chromosome banding analysis, interphase- and hypermetaphase-FISH, qualitative and quantitative PCR for diagnosis and for follow-up in chronic myeloid leukemia: a study on 350 cases. Leukemia. 2002;16(1):53–59. [DOI] [PubMed] [Google Scholar]
- 20.Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N Engl J Med. 2016;374(26):2530–2541. [DOI] [PubMed] [Google Scholar]
- 21.Mital A, Piskorz A, Lewandowski K, et al. A case of mast cell leukaemia with exon 9 KIT mutation and good response to imatinib. Eur J Haematol. 2011;86(6):531–535. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez-Twose I, Matito A, Morgado JM, et al. Imatinib in systemic mastocytosis: a phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget. 2016. July 19 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kluin-Nelemans HC, Oldhoff JM, Van Doormaal JJ, et al. Cladribine therapy for systemic mastocytosis. Blood. 2003; 102(13):4270–4276. [DOI] [PubMed] [Google Scholar]
- 24.Pardanani A, Hoffbrand AV, Butterfield JH, Tefferi A. Treatment of systemic mast cell disease with 2-chlorodeoxyadenosine. Leuk Res. 2004;28(2):127–131. [DOI] [PubMed] [Google Scholar]
- 25.Valentini CG, Rondoni M, Pogliani EM, et al. Mast cell leukemia: a report of ten cases. Ann Hematol. 2008;87(6):505–508. [DOI] [PubMed] [Google Scholar]
- 26.Ustun C, Reiter A, Scott BL, et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol. 2014;32(29):3264–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ustun C, Gotlib J, Popat U, et al. Consensus Opinion on Allogeneic Hematopoietic Cell Transplantation in Advanced Systemic Mastocytosis. Biol Blood Marrow Transplant. 2016;22(8):1348–1356. [DOI] [PubMed] [Google Scholar]
- 28.Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis. Blood. 2015;126(26):2832–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez-Twose I, Martinez-Barranco P, Gotlib J, et al. Complete response to gemtuzumab ozogamicin in a patient with refractory mast cell leukemia. Leukemia. 2016;30(8):1753–1756. [DOI] [PubMed] [Google Scholar]
- 30.Ghanim V, Herrmann H, Heller G, et al. 5-azacytidine and decitabine exert proapoptotic effects on neoplastic mast cells: role of FAS-demethylation and FAS re-expression, and synergism with FAS-ligand. Blood. 2012;119(18):4242–4252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.