

Abstract
Homeodomain-only protein homeobox (HOPX) is the smallest homeodomain protein. It was regarded as a stem cell marker in several non-hematopoietic systems. While the prototypic homeobox genes such as the HOX family have been well characterized in acute myeloid leukemia (AML), the clinical and biological implications of HOPX in the disease remain unknown. Thus we analyzed HOPX and global gene expression patterns in 347 newly diagnosed de novo AML patients in our institute. We found that higher HOPX expression was closely associated with older age, higher platelet counts, lower white blood cell counts, lower lactate dehydrogenase levels, and mutations in RUNX1, IDH2, ASXL1, and DNMT3A, but negatively associated with acute promyelocytic leukemia, favorable karyotypes, CEBPA double mutations and NPM1 mutation. Patients with higher HOPX expression had a lower complete remission rate and shorter survival. The finding was validated in two independent cohorts. Multivariate analysis revealed that higher HOPX expression was an independent unfavorable prognostic factor irrespective of other known prognostic parameters and gene signatures derived from multiple cohorts. Gene set enrichment analysis showed higher HOPX expression was associated with both hematopoietic and leukemia stem cell signatures. While HOPX and HOX family genes showed concordant expression patterns in normal hematopoietic stem/progenitor cells, their expression patterns and associated clinical and biological features were distinctive in AML settings, demonstrating HOPX to be a unique homeobox gene. Therefore, HOPX is a distinctive homeobox gene with characteristic clinical and biological implications and its expression is a powerful predictor of prognosis in AML patients.
Introduction
HOPX was first identified in the expression sequence tag database for transcripts encoding proteins related to the development of the heart in mice and zebrafish.1,2 Human HOPX, located in chromosome 4q12, has five isoforms. The predominant one encodes a short protein of 73 amino acids with a molecular weight of 12 kd. This is by far the smallest homeodomain protein, with conservation of the homeodomain of 60 amino acids. But the difference between this and other homeodomain proteins is that the HOPX protein does not bind DNA directly. Rather, it exerts its transcriptional inhibition through sequestration of serum responsive factor by physical interaction and by recruitment of histone deacetylase.3
Recently, the HOPX gene has been regarded as a stem cell marker in intestine, hair follicles, and pulmonary alveolar cells.4–7 Some studies suggested a role for HOPX in tumorigenesis with clinical implications. HOPX has been suggested to be a tumor suppressor gene in lung, colon, esophagus, pancreas, uterine and stomach cancers.8–13 Most of the studies showed silencing of HOPX through hypermethylation of the promoter as a mechanism of its downregulation in cancer cells.9–13 However, the mechanisms for the tumor suppression remain largely unknown.
The HOX family and HOPX belong to homeobox genes. However, while HOX family genes have been well studied in acute myeloid leukemia (AML),14 the clinical and biological significance of HOPX in human hematopoiesis remains undefined. We are interested in exploring the roles of HOPX in AML patients, as well as comparing HOX and HOPX in the pathophysiology of malignant hematopoiesis. In this study, we compared clinical and biological characteristics between de novo AML patients with higher and lower HOPX expression and found that higher HOPX expression was strictly correlated with unfavorable prognosis of AML patients in ours and the other two independent cohorts. Multivariate analysis revealed higher HOPX expression as an independent unfavorable prognostic factor, and independent of several published gene signatures derived from multiple cohorts. Using bioinformatics approaches, we found that HOPX expression was closely associated with known hematopoietic stem cell (HSC) signatures. While HOPX and HOX family genes were highly expressed in normal hematopoietic stem/progenitor cells (HSPC), the expression patterns and associated clinical and biological features between these two classes of homeobox genes differed dramatically in AML settings. Taken together, our study suggests that HOPX has a significant impact on various clinical and biological aspects of AML, and that there is a distinction between HOX family genes and HOPX in the AML setting.
Methods
Patients
A total of 347 adult patients diagnosed with de novo AML according to the 2008 World Health Organization classification in the National Taiwan University Hospital (NTUH) who had cryopreserved bone marrow (BM) cells and complete clinical and laboratory data available for analysis were retrospectively enrolled. Among them, 227 patients received standard induction chemotherapy. Non-M3 (acute promyelocytic leukemia, APL) patients received idarubicin 12 mg/m2 per day for 2–3 days and cytarabine 100 mg/m2 per day for 5–7 days, as described previously.15 APL patients received concurrent all-trans retinoic acid and idarubicin. The remaining 120 patients received palliative therapy with supportive care or low-dose chemotherapy due to underlying comorbidity or in accordance with patient decision. We also prospectively enrolled another cohort of 56 newly diagnosed adult de novo AML patients with adequate BM samples for more detailed studies of the HOPX gene, including expression pattern of HOPX isoforms in AML. The study was approved by the Research Ethics Committee of the NTUH.
Cytogenetic and mutation analysis
Chromosomal abnormalities16 and mutation analyses were performed as previously described.15–20
Gene expression microarray datasets and data analysis
We profiled global gene expression of BM mononuclear cells from the 347 patients (NTUH dataset) using Illumina HumanHT-12 v.4 Expression BeadChip (Illumina, San Diego, CA, USA) (GSE68469 and GSE71014).21–23 Two large microarray datasets of AML with overall survival (OS) data, including The Cancer Genome Atlas (TCGA) dataset (n=186)24 and GSE12417 [all with cytogenetically normal (CN) AML; n=162],25 were utilized to validate the prognostic significance of HOPX. We used TCGA-normalized level-2 intensity and GSE12417 GPL96 data (profiled with Affymetrix Human Genome U133A Array), normalized as described by Metzeler et al.25 Gene expression profiles GSE12662 (n=91),26 GSE24006 (n=54),27 and GSE24759 (n=211)28 were also included to investigate the gene expression patterns in normal hematopoiesis.
Analysis of gene expression in next-generation sequencing datasets
To investigate the absolute levels of gene expression in AML, we analyzed expression data of 179 AML samples profiled with Illumina Genome Analyzer RNA Sequencing in TCGA dataset.24 Reads per kilobase per million mapped reads (RPKM) levels of gene expression were extracted from TCGA database.24
Gene signature analysis
The association of HOPX gene with stem cell characteristics was analyzed by the Gene Set Enrichment Analysis (GSEA; a Java application that can be down-loaded at http://www.broadinstitute.org/gsea/index.jsp)29 and as detailed in the Online Supplementary Appendix. In order to examine whether genes are involved in HSC quiescence, we employed another gene set enrichment scoring method that averages z-values of all involved genes.30
Methylation microarray datasets and analysis
DNA methylation data from Illumina Infinium HumanMethylation450 BeadChips of AML (n=194) were downloaded from the TCGA database.24 We transformed methylation beta-values to normally distributed M-values for further analysis.31
Expression of HOPX isoforms
Human HOPX has five isoforms including HOPXa (NM_032495), HOPXb (three variants including NM_139212, NM_139211 and NM_001145459; abbreviated hereafter as b1, b2, and b3, respectively), and HOPXc (NM_001145460) (UCSC genomic database; www.genome.ucsc.edu) (Online Supplementary Figure S1). Analysis of HOPX isoform expression was performed by quantitative real time-polymerase chain reaction as detailed in the Online Supplementary Appendix, Online Supplementary Table S1 and Online Supplementary Figure S1.
Bisulfite treatment and methylation analysis of HOPX
We interrogated the methylation status of the CpG islands of HOPX-b2 isoform NM_139211 from −15 to +109 bp region around the transcription start site (TSS).10 Methods are described in the Online Supplementary Appendix.
Statistical analysis
Statistical analysis was carried out as described previously;21–23 a brief description is available in the Online Supplementary Appendix.
Results
Correlation of HOPX expression with clinical features
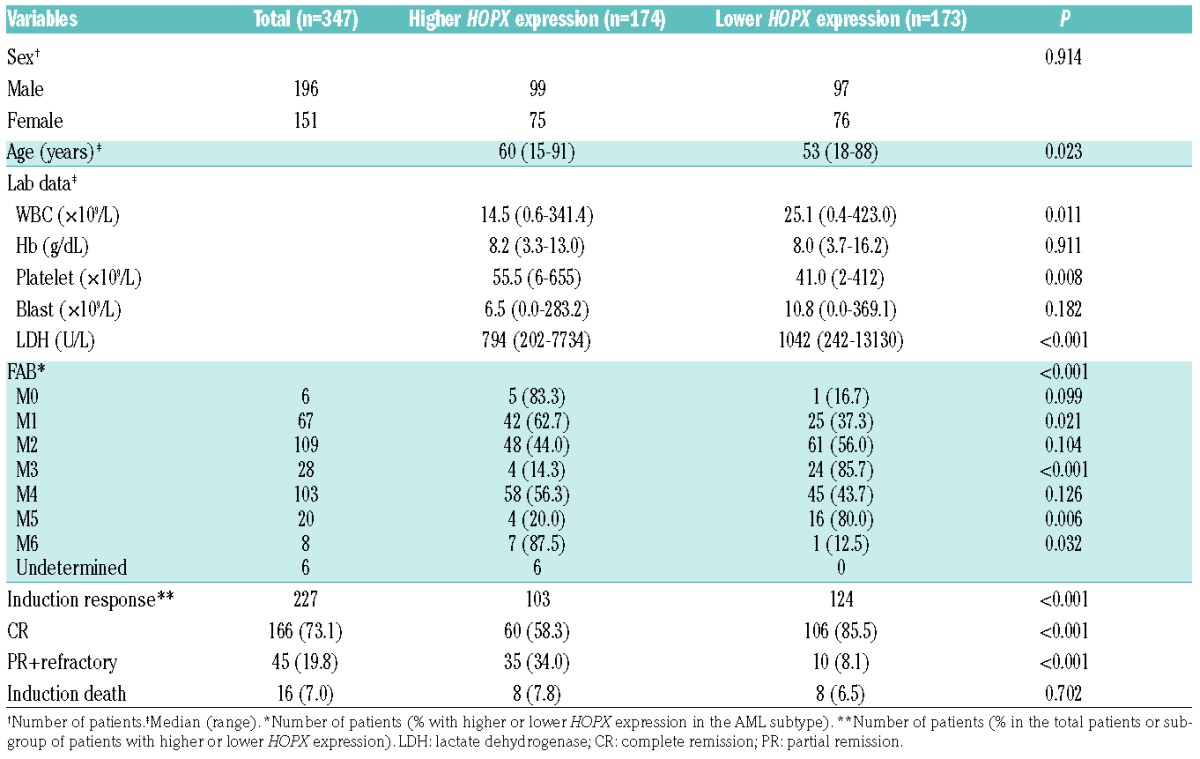
The 347 AML patients were divided into two groups based on the HOPX expression levels above (higher expression group) or below (lower expression group) the median level of HOPX expression on the arrays. Higher HOPX expression was associated with older age (P=0.023), higher platelet counts (P=0.008), lower white blood cell (WBC) counts (P=0.011), and lower lactate dehydrogenase (LDH) levels (P<0.001) at diagnosis (Table 1). Patients with M1 and M6 according to the French-American-British (FAB) classification more frequently had higher HOPX expression (P=0.021 and P=0.032, respectively), while those with M3 and M5 had significantly lower levels of HOPX expression (P<0.001 and P<0.006, respectively). The comparison of clinical features between higher and lower HOPX expression groups in those receiving standard chemotherapy (n=227) is shown in the Online Supplementary Table S2. The association of higher HOPX expression with higher platelet counts, lower LDH levels, and FAB subtypes remained the same in this group of patients as that of the total cohort.
Table 1.
Comparison of clinical manifestations between acute myeloid leukemia patients with higher and lower HOPX expression.
Correlation of HOPX expression with cytogenetics and molecular alterations
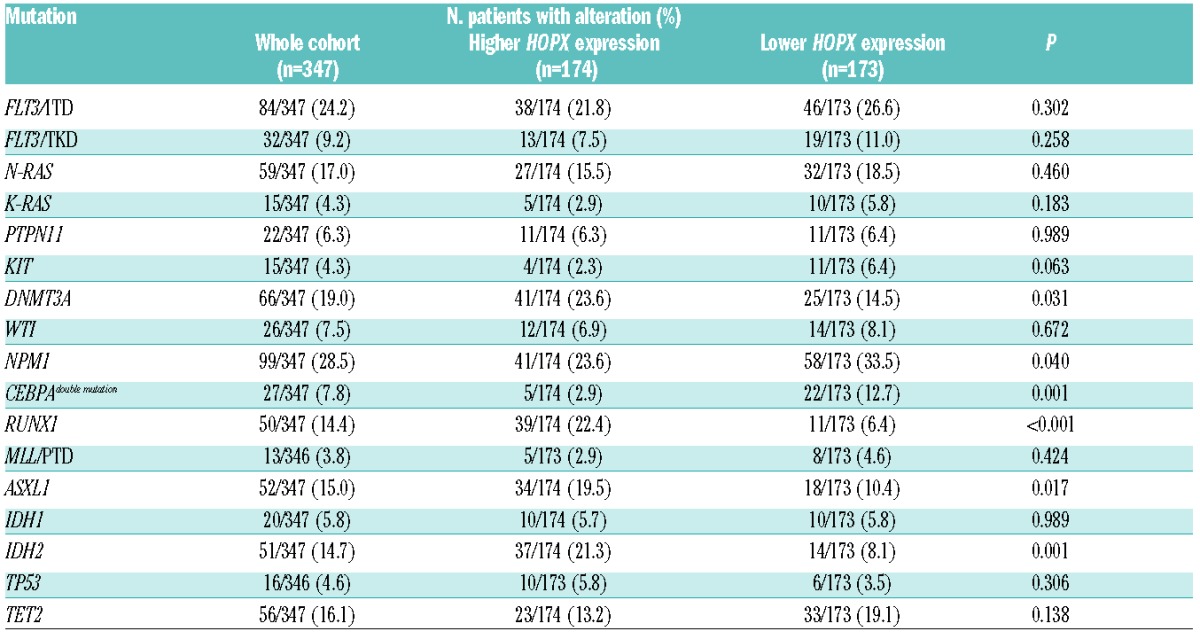
Chromosome data were available in 325 patients at diagnosis (Online Supplementary Table S3). Higher HOPX expression was negatively associated with favorable karyotypes, including t(8;21) and t(15;17) (both P<0.001). We also analyzed the mutation status of 16 genes and found that the patients with higher HOPX expression had significantly higher incidences of mutations in RUNX1 (P<0.001), IDH2 (P=0.001), ASXL1 (P=0.017), and DNMT3A (P=0.031), but less frequently had CEBPA double mutations (P=0.001) and NPM1 mutation (P=0.040) (Table 2).
Table 2.
Association of HOPX expression levels with other genetic alterations.
Higher HOPX expression predicts poor clinical outcome in de novo AML patients
Among the 227 patients who received standard chemotherapy, those with higher HOPX expression had a lower complete remission (CR) rate (58.3% vs. 85.5%; P<0.001) (Table 1), shorter OS (median 23.7 months vs. 116.8 months; log-rank P<0.001) and disease-free survival (DFS) (median 5.9 months vs. not reached; log-rank P<0.001) than those with lower HOPX expression after a median follow up of 57.0 months (Figure 1A and B). Univariate Cox proportional hazards analysis confirmed the prognostic value of HOPX expression as a continuous variable in predicting patients’ OS [Hazard Ratio (HR): 1.44; 95%CI: 1.21–1.71; P<0.001] and DFS (HR: 1.55; 95%CI: 1.31–1.82; P<0.001). The prognostic significance of HOPX expression could be validated in another two independent cohorts: TCGA24 and GSE1241725 (Figure 1C and D). The unfavorable prognostic effects of higher HOPX expression were also seen in the subgroup of patients with AML other than APL (median OS 23.7 vs. 108.1 months; P<0.001) and those with a normal karyotype (median OS 39.1 vs. 116.8 months; P=0.004) (Figure 1E and F). The results could also be validated by the TCGA cohort (Online Supplementary Figure S2A and B).
Figure 1.
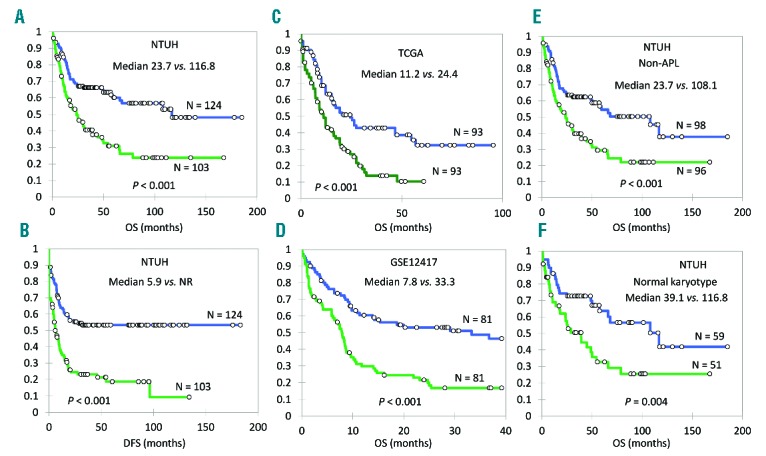
HOPX expression levels and acute myleoid leukemia (AML) patients’ survival. (A and B) In NTUH cohort, the overall survival (OS) and disease-free survival (DFS) of AML patients with higher HOPX expression are significantly shorter than those with lower expression: median OS 23.7 versus 116.8 months, P<0.001; median DFS 5.9 months versus not reached (NR); P<0.001. (C and D) The observation is validated by TCGA (median OS 11.2 months vs. 24.4 months; P<0.001) and GSE12417 (median OS 7.8 months vs. 33.3 months; P<0.001) cohorts. (E and F) When we restrict the analysis in non-acute promyelocytic leukemia (APL) patients or patients with a normal karyotype in NTUH cohort, HOPX levels still significantly correlate with OS: median OS 23.7 versus 108.1 months (P=0.0003) and median OS 39.1 versus 116.8 months (P=0.004), respectively. Green line: higher HOPX expression group; blue line: lower HOPX expression group.
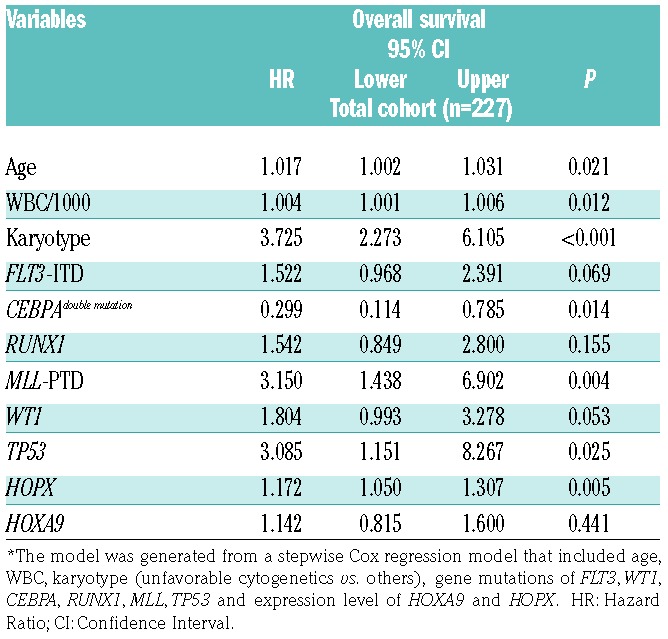
By univariate analysis, HOPX expression levels and several parameters exhibited a significant impact on OS (Online Supplementary Table S4). When we combined all these prognostic factors together in a multivariate analysis, higher expression of HOPX remained a poor prognostic factor for OS (P=0.005) (Table 3), independent of age, WBC counts, karyotypes, mutation statuses of FLT3, CEBPA, MLL, TP53, WT1, and RUNX1 and expression levels of HOXA9.
Table 3.
Multivariate analysis (Cox regression) on overall survival.*
Further analysis showed much higher HOPX expression in those patients who failed to achieve CR than those who obtained a CR (by array signal intensity; P<0.001) (Figure 2A) suggesting a tight association of higher HOPX expression and drug resistance. Furthermore, HOPX expression was lower in APL, which consisted mainly of leukemic cells that are blocked at the differentiation stage of promyelocytes, indicating a possible relationship between HOPX expression and maturation stages of AML cells (Figure 2B).
Figure 2.
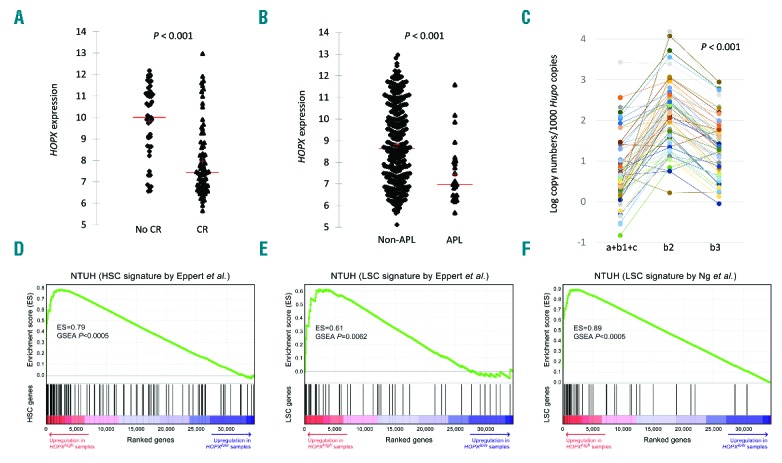
HOPX expression levels, its correlation with treatment response, and its role as a stem cell marker by gene set enrichment analysis. (A) There was a significant difference in HOPX levels between patients with (n=166) and without (n=61) complete remission (CR) after induction chemotherapy. (B) HOPX expression is also much higher in non-APL patients. (C) Real-time PCR of HOPX in the cohort of 56 acute myeloid leukemia (AML) patients prospectively recruited showing predominant expression of isoform HOPXb2 (NM_139211) (left, isoforms a+b1+c; middle, b2; right, b3). (D, E, and F) GSEA plots of curated HSC and LSC signatures in the NTUH dataset.35,36 Red-to-indigo bars denote the genome-wide gene list ranked based on their P-values (t-test) between samples with high (top quartile) and low (bottom quartile) expression of HOPX. Significant positive GSEA enrichment scores indicate that HOPX expression is positively associated with HSC and LSC signatures.
Comparisons between HOPX expression and published prognostic gene signatures in predicting prognosis
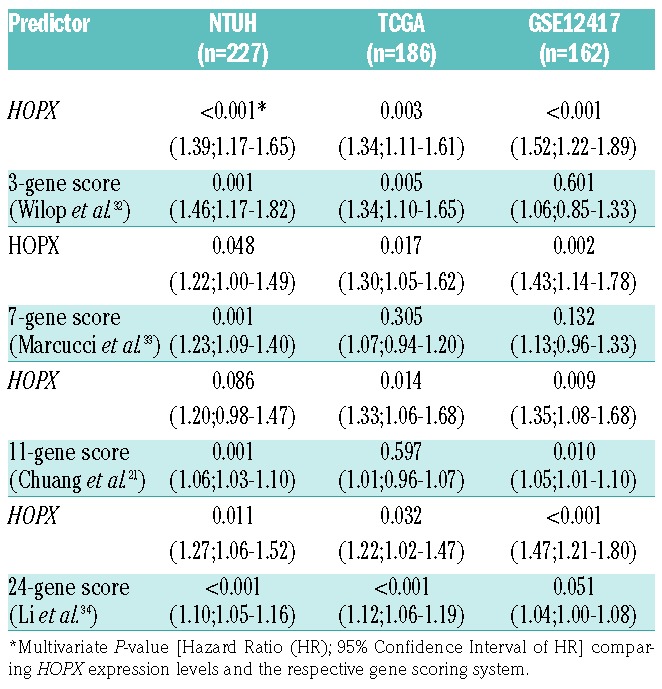
Several gene expression-based prognostic predictors have been developed from various study designs in AML. To compare the performance of prognostic prediction of HOPX expression with those published predictors, we performed pairwise multivariate Cox analysis between HOPX expression and each of the 3-gene, 7-gene, 11-gene, and 24-gene predictors in three datasets.21,32–34 Remarkably, the HOPX expression remained independent (with Cox multivariate analysis P<0.05) in most of the comparison settings (11 of 12 comparisons) (Table 4). Our data suggest HOPX to be a simple and powerful alternative for prognostication in AML.
Table 4.
Comparisons of HOPX to published prognostic gene signatures.
The expression pattern and promoter methylation of HOPX isoforms in AML patients
The pattern of expression of the 5 isoforms of HOPX and CpG methylation status in primary AML are still unknown. Because of the low levels of expression and the impossibility of separating isoforms a, b1, and c, we merged these three together for quantification. We quantified the expression levels of these isoforms in prospectively recruited AML patients’ marrow by real time-PCR and found that HOPXb2 (NM_139211) was the predominant isoform in human AML cells; the other four variants were markedly under-represented (Figure 2C). In addition, quantification of the CpG methylation in HOPXb2 promoter regions revealed low levels of methylation in most AML patients (Online Supplementary Figure S3). This finding seemed to differ from that in some studies of solid cancers in which hypermethyation of HOPX in this region caused gene silencing and was associated with poor prognosis.9–13 Further studies are needed to confirm this hypothesis.
Correlation of HOPX expression with stem cell signatures
We curated xenotransplantation-derived HSC and leukemia stem cell (LSC) gene signatures and a recently up-dated LSC signature from previous reports35,36 and employed the gene set enrichment analysis (GSEA) method29 to analyze their associations with HOPX expression. GSEA tests the enrichment of each gene signature in the list of global genes ranked by HOPX-associated differential expression. Higher HOPX expression was associated with upregulation of HSC and LSC genes in the NTUH dataset (enrichment scores, 0.79, 0.61, and 0.89; P<0.0005, P=0.006, and P<0.0005, respectively) (Figure 2D–F). Concordant significant enrichments were identified in TCGA and GSE12417 AML datasets (all P-values ≤0.015) (Online Supplementary Figure S4). Seventeen and five genes appeared as leading-edge genes (ie. a subset of core-enrichment genes) of the HSC and LSC signatures (Figure 2D and E), respectively, in all the three cohorts (Online Supplementary Table S5). Interestingly, an ATP-binding-cassette (ABC) transporter gene, ABCB1, was a common leading-edge gene of HSC signature (Online Supplementary Table S5). ABC transporter genes were reported to be associated with chemoresistance in AML, with higher ABCB1, ABCG1, ABCG2 expression levels being independently poor prognostic factors.37 Expression of these three ABC genes was significantly higher in samples with higher HOPX expression (all P-values <0.001) (Online Supplementary Table S6), but the mechanistic link between ABC and HOPX expression still has to be explored in further studies.
Expression patterns of HOPX and HOX genes in normal hematopoietic cells
HOPX and HOX family genes all encode homeodomain proteins and HOX genes are well-known HSC markers.38–40 To further delineate the similarities and the distinctions between HOPX and the HOX gene family, we first analyzed their expression patterns in normal hematopoietic cells using arrays derived from public data. We curated three public gene expression datasets derived from normal hematopoietic cells26–28 and we chose 12 HOX genes with at least moderate expression levels (RPKM> 5 according to TCGA RNA seq data) for further analysis.24 In GSE2400627 and GSE12662,26 HOPX and HOX genes were generally expressed in a concordant manner (mean correlation coefficient 0.37 and 0.35, respectively) (left panels, Online Supplementary Figure S5A and B). We further analyzed a dataset of 9 distinct normal hematopoietic cell populations (GSE24759) (Online Supplementary Figure S6).28 HOPX and HOX family genes were all highly expressed in normal CD34+ hematopoietic cells (average z-values = 0.82 and 0.75; 1-sample t-test both P<0.001) (Online Supplementary Figure S6). The concordant expression patterns between HOPX and HOX family suggest their shared roles in normal hematopoiesis.
Expression patterns of HOPX and HOX family genes in AML
We investigated the absolute gene expression levels of HOPX and the HOX family in AML from the TCGA RNA sequencing dataset. Among them, HOPX was the second highest expressed gene (average RPKM = 25.6 in TCGA RNA sequencing dataset; n=179), after the most abundant gene HOXA9 (RPKM = 43.3). We then compared the expression patterns between HOPX and HOX family genes in AML cells. The concordance of expression patterns shown in normal hematopoietic cells were no longer present in AML cells (correlation in GSE24006 and GSE12662 −0.31 and 0.07, respectively) (Online Supplementary Figure S5A and B). We sought to investigate the similarities/distinctions between HOPX and HOX family genes by clustering of AML patients in our dataset (NTUH) according to their expression levels. Because of the unequal numbers of genes between HOPX and HOX family (1 vs. 12), we performed a 2-step hierarchical clustering to balance the potential bias in unsupervised clustering. Briefly, patients were first clustered only by the 12 HOX family genes. Subsequently, each cluster was subject to the second round of clustering with inclusion of HOPX. As a result, we were able to identify and focus on 4 distinct groups of patients for further analysis (HOXhigh/HOXlow by HOPXhigh/HOPXlow) (Figure 3A) in whom the high/low expressions of HOX and HOPX were confirmed significant in each cluster (comparisons of average z-scores against zero; 1-sample t-test P<0.0001) (Figure 3B). The 4 groups also showed a significantly different prognosis: HOXlow/HOPXlow patients had the longest OS, while HOXhigh/HOPXhigh patients had the poorest outcome (P<0.0001) (Figure 3C).
Figure 3.
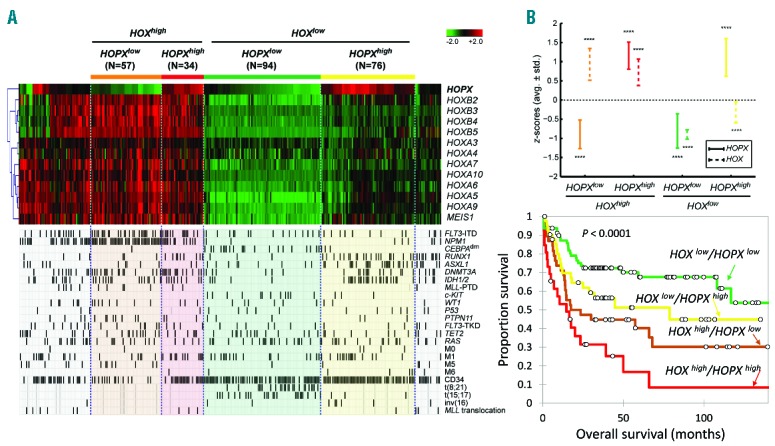
Comparison of expression patterns, clinical and biological features among subgroups of NTUH AML patients based on hierarchical clustering of HOPX and HOX family genes. (A) Heatmap of HOPX and HOX family genes in NTUH data. We identify 4 groups of patients based on a 2-step hierarchical clustering: HOXhigh/HOPXlow by HOPXhigh/HOPXlow. Molecular and clinical variables including gene mutations, cytogenetic abnormalities, and leukemia classifications are compared among the clusters. (B) Average z-scores of HOPX and HOX genes in each group. Whiskers denote average±standard deviation of the z-scores. Statistical significance of the z-scores against zero is assessed by the 1-sample t-test (****P<0.0001). (C) Overall survival of the 4 groups of patients. HOXlow/HOPXlow patients have the best overall survival (OS), followed by HOXlow/HOPXhigh, HOXhigh/HOPXlow, and HOXhigh/HOPXhigh (P<0.001).
To further compare the clinical and biological characteristics among AML patients with different expression levels of HOPX/HOX family genes, we analyzed patients’ gene mutations, cytogenetic abnormalities, and other clinical and lab parameters. WBC counts and LDH levels varied significantly among groups (ANOVA P=0.026 and 0.0009, respectively) (Figure 4A and B). HOXlow/HOPXhigh patients had the lowest WBC counts and LDH levels (median 5765/μL vs. 24,720/μL and 643 U/L vs. 1027 U/L, respectively; both P<0.001). Each subgroup also had distinct biological characteristics, including CD34 expression, gene mutations of FLT3, NPM1, CEBPA, RUNX1, DNMT3A, IDH1/2 (all P<0.0001) and ASXL1 (P=0.0002) (Figure 3A and 4C). FLT3-ITD and mutations in NPM1 and DNMT3A are more common in HOXhigh patients regardless of HOPX expression levels; RUNX1 mutation is more frequent in HOPXhigh regardless of HOX expression levels; CEBPA double mutation is predominantly seen in HOXlow/HOPXlow patients; ASXL1 mutation is mainly present in HOXlow/HOPXhigh subgroup; IDH1/2 mutations are particularly rare in HOXlow/HOPXlow patients; CD34+ blasts are low in HOXhigh/HOPXlow patients. Compared with other patients, HOXhigh/HOPXhigh patients had higher incidences of FAB M0 (4 of 76 vs. 1 of 185; P=0.011), CD34 expression on leukemic cells (64 of 71 vs. 104 of 175; P<0.001), and mutations in ASXL1 (23 of 76 vs. 16 of 185; P<0.001), RUNX1 (21 of 76 vs. 15 of 185; P<0.001), and IDH1/2 (25 of 76 vs. 30 of 185; P=0.003), while HOXhigh/HOPXlow patients, when compared with others, had more FAB M5 (8 of 57 vs. 2 of 204; P<0.001) and mutations in NPM1 (47 of 57 vs. 19 of 204; P<0.001), FLT3 (FLT3-ITD) (26 of 57 vs. 38 of 204; P<0.001), MLL (MLL-PTD) (5 of 57 vs. 2 of 203; P=0.001), PTPN11 (8 of 57 vs. 8 of 203; P=0.005), and WT1 (8 of 57 vs. 11 of 204; P=0.026) (Figure 3A and data not shown). These results demonstrated marked distinctions between HOPX and HOX family genes in their association with genetic alterations and clinical features in AML.
Figure 4.
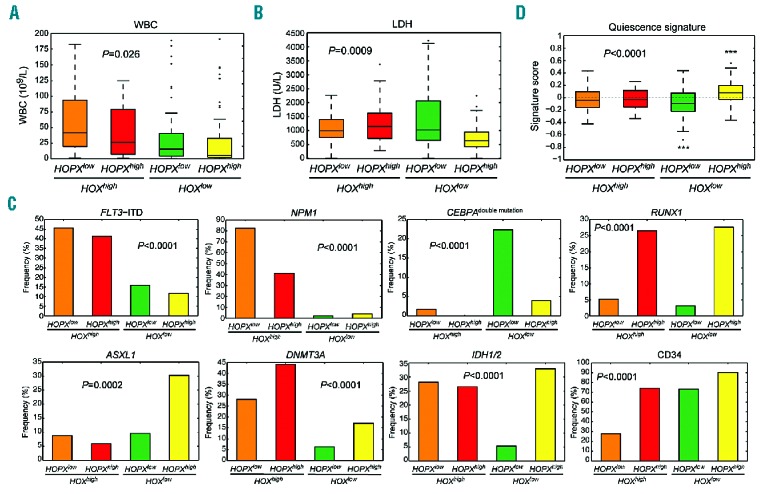
The distinct clinical and genetic features among acute myeloid leukemia (AML) patients stratified by expression of HOPX and HOX family genes. (A and B) Box plots of white blood cell (WBC) counts and serum lactate dehydrogenase (LDH) levels. There is a significant difference in both variables among the 4 clusters. The highest median of WBC count appears in HOXhigh/HOPXlow group, while that of HOXlow/HOPXhigh falls to the bottom of all clusters. Similar trends are seen in LDH levels. Statistical significance were tested by ANOVA tests. (C) The 4 clusters are associated with distinctive gene mutations. FLT3-ITD and mutations in NPM1 and DNMT3A are more common in HOXhigh patients regardless of HOPX expression levels; the RUNX1 mutation is more frequent in HOPXhigh regardless of HOX expression levels; the CEBPA double mutation is predominantly seen in HOXlow/HOPXlow patients; the ASXL1 mutation is mainly present in HOXlow/HOPXhigh subgroup; IDH1/2 mutations are particularly rare in HOXlow/HOPXlow patients. CD34+ blasts are low in HOXhigh/HOPXlow patients. (D) Association of quiescence HSC signature with expression of HOPX and HOX family genes. We employ a gene set enrichment scoring to quantify the overall activity of the gene signature in each sample; a positive/negative score represents a tendency toward quiescent/proliferative HSC state. Inter-group changes are tested by an ANOVA test. Asterisks denote significant differences from zero assessed by one-sample t-tests (***P<0.001).
Distinct associated HSC gene signatures between HOPX and HOX family genes
Although all HOPX and HOX family genes are stem cell markers, our data showed that the two were associated with distinct features in AML. Stem cell signatures could be divided into two groups with either quiescence or proliferation characteristics.41 The low LDH levels and WBC counts in the AML patients with high HOPX and low HOX family gene expression (Figure 4A and B) raises the possibility that expression of HOPX may favor quiescence of stem cells. To test this hypothesis, we examined the expression profiles of each subgroup of our AML patients by a gene set scoring30 based on a known quiescence signature in HSC.41 A positive signature score denotes a tendency toward a quiescent HSC state. The significance level of a score against zero (representing no tendency) was tested by 1-sample t-test. Patients with high expression of HOX family genes did not exhibit a significant tendency toward the quiescence state regardless of the abundance of HOPX expression (P=0.22 and 0.45 for HOXhigh/HOPXhigh and HOXhigh/HOPXlow, respectively) (Figure 4D). However, when HOX expression is low, HOPXhigh and HOPXlow were significantly associated with quiescence and non-quiescence, respectively (P=0.0007 and 0.0003, respectively) (Figure 4D). Overall, our data suggested a fundamental difference between HOPX and HOX family genes in their stem cell properties in the AML setting.
Comparison of methylation patterns between HOPX and HOX family genes in AML
Besides the different expression patterns and associated clinical and biological features in HOPX compared with HOX family genes, we also sought to find out whether there was any difference in their methylation patterns by analyzing the TCGA epigenome-wide microarray dataset (n=194). Generally, these genes formed 3 clusters according to the methylation patterns (Online Supplementary Figure S7A). HOPX was largely unmethylated in most AML patients (mean methylation M-value −1.63; 1-sample t-test against zero P<0.001; area under curve with negative M 69.05%) (Online Supplementary Figure S7B). Methylation levels of HOXA3, HOXA4, HOXA5, and HOXB3 were generally high, while other HOX genes, including HOXA7, HOXA9, and HOXB4, were uniformly hypomethylated (Online Supplementary Figure S7A and B). Taken together, our data highlighted the different molecular and clinical features that distinguish between HOPX and HOX family genes in AML.
Discussion
To our knowledge, this is the first report regarding the prognostic significance of HOPX expression in de novo AML patients and the direct comparison between HOPX and the HOX family in normal and malignant hematopoiesis. The prognostic significance of HOPX expression is independent of common known clinical and genetic factors as well as several published gene signatures. We also showed that the promoter region was barely methylated in leukemic cells from AML patients, in contrast to heavy methylation in solid cancers,8–10,12,13,42 indicating that CpG methylation is not one of the main mechanisms of regulation of HOPX gene expression in primary human AML cells. Finally, HOPX appeared to be a distinct homeobox gene in AML cells when compared with HOX family genes.
Studies have shown that HOPX is a stem cell marker of hair follicle, intestine, and lung alveolar cells.5–7 Through functional annotation, our current study showed that HOPX expression was associated with HSC and LSC signatures in AML cells from our cohort and also two other validation cohorts, indicating that HOPX was an LSC marker in AML. Stemness is an established property pertaining to drug resistance and poor prognosis in cancer patients.43 LSC signature is associated with unfavorable prognosis in AML patients. The underlying mechanisms by which stem cell signatures in AML cells predict poor treatment outcome have been postulated to be related to their association with chemotherapy resistance,27 probably due to the quiescent nature of these cells. The tight association between HOPX expression and stem cell properties is likely a major reason for the unfavorable prognosis in AML patients with higher HOPX expression shown in this study. In addition, higher HOPX expression was significantly associated with the expression of some ABC transporters, a family of proteins that bind ATP as energy source to transport the endogenous or exogenous molecules through the cell membranes.44,45 They are abundant in stem cells, including HSCs and LSCs, and are responsible for multidrug resistance in cancer treatment.46 Therefore, leukemia patients with higher HOPX are less likely to obtain CR after induction chemotherapy. Further functional studies are needed to throw light on its significance in leukemia stemness and drug-resistance.
We showed that HOPX had distinct expression pattern and associated clinical and biological features when compared with other homeobox genes such as the HOX family in the AML setting. While they were both enriched in normal CD34+ HSPCs, their expression in AML was asynchronous. The findings that higher HOPX expression, accompanied with lower HOX expression, was closely associated with FAB M0 subtype, CD34 expression on leukemic cells, lower WBC counts, LDH levels, and quiescence stem cell signature indicates its relationship with more immature and quiescent stem cell characters.
Our study was mainly based on a retrospective cohort although we validated our results from other public array cohorts and 56 prospectively enrolled patients. Further studies in large prospective cohorts are warranted to confirm our observations. Moreover, in vivo studies are necessary to delineate the pathophysiological effects of HOPX in hematopoiesis and leukemogenesis.
Supplementary Material
Acknowledgments
The authors would like to thank the FACS Core of National Taiwan University Hospital and Kai-Ting Yang, the sorting technician, for performing cell sorting and FACS analysis.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/6/1044
Funding
The study was supported by a National Taiwan University Hospital–National Taiwan University joint research grant (UN103-051), Ministry of Science and Technology of Taiwan (MOST102-2325-B-002-028, 103-2314-B-002-130-MY3, 103-2314-B-002-131MY3 and 104-2923-B-002-001), Far Eastern Hospital and NTUH joint grant 105-FTN24, NTUH and NTUMC joint grant UN106-024, and Ministry of Health and Welfare of Taiwan (MOHW102-TD-C-111-001 and MOHW103-TD-B-111-04).
References
- 1.Shin CH, Liu ZP, Passier R, et al. Modulation of cardiac growth and development by HOP, an unusual homeodomain protein. Cell 2002;110(6):725–735. [DOI] [PubMed] [Google Scholar]
- 2.Chen F, Kook H, Milewski R, et al. Hop is an unusual homeobox gene that modulates cardiac development. Cell. 2002; 110(6):713–723. [DOI] [PubMed] [Google Scholar]
- 3.Kook H, Yung WW, Simpson RJ, et al. Analysis of the structure and function of the transcriptional coregulator HOP. Biochemistry. 2006;45(35):10584–10590. [DOI] [PubMed] [Google Scholar]
- 4.Takeda N, Jain R, Leboeuf MR, et al. Hopx expression defines a subset of multipotent hair follicle stem cells and a progenitor population primed to give rise to K6+ niche cells. Development. 2013;140(8):1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334(6061):1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munoz J, Stange DE, Schepers AG, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012; 31(14):3079–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain R, Barkauskas CE, Takeda N, et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat Commun. 2015;6:6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Pacyna-Gengelbach M, Deutschmann N, Niesporek S, Petersen I. Homeobox gene HOP has a potential tumor suppressive activity in human lung cancer. Int J Cancer. 2007;121(5):1021–1027. [DOI] [PubMed] [Google Scholar]
- 9.Harada Y, Kijima K, Shinmura K, et al. Methylation of the homeobox gene, HOPX, is frequently detected in poorly differentiated colorectal cancer. Anticancer Res. 2011;31(9):2889–2892. [PubMed] [Google Scholar]
- 10.Yamashita K, Kim MS, Park HL, et al. HOP/OB1/NECC1 promoter DNA is frequently hypermethylated and involved in tumorigenic ability in esophageal squamous cell carcinoma. Mol Cancer Res. 2008;6(1):31–41. [DOI] [PubMed] [Google Scholar]
- 11.Katoh H, Yamashita K, Waraya M, et al. Epigenetic silencing of HOPX promotes cancer progression in colorectal cancer. Neoplasia. 2012;14(7):559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaguchi S, Asanoma K, Takao T, Kato K, Wake N. Homeobox gene HOPX is epigenetically silenced in human uterine endometrial cancer and suppresses estrogen-stimulated proliferation of cancer cells by inhibiting serum response factor. Int J Cancer. 2009;124(11):2577–2588. [DOI] [PubMed] [Google Scholar]
- 13.Ooki A, Yamashita K, Kikuchi S, et al. Potential utility of HOP homeobox gene promoter methylation as a marker of tumor aggressiveness in gastric cancer. Oncogene. 2010;29(22):3263–3275. [DOI] [PubMed] [Google Scholar]
- 14.Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27(5):1000–1008. [DOI] [PubMed] [Google Scholar]
- 15.Tang JL, Hou HA, Chen CY, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114(26):5352–5361. [DOI] [PubMed] [Google Scholar]
- 16.Chou WC, Chou SC, Liu CY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118(14):3803–3810. [DOI] [PubMed] [Google Scholar]
- 17.Chou WC, Tang JL, Lin LI, et al. Nucleophosmin mutations in de novo acute myeloid leukemia: the age-dependent incidences and the stability during disease evolution. Cancer Res. 2006; 66(6):3310–3316. [DOI] [PubMed] [Google Scholar]
- 18.Hou HA, Lin CC, Chou WC, et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia. 2014; 28(1):50–58. [DOI] [PubMed] [Google Scholar]
- 19.Chou WC, Huang HH, Hou HA, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116(20):4086–4094. [DOI] [PubMed] [Google Scholar]
- 20.Chou WC, Hou HA, Chen CY, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010;115(14):2749–2754. [DOI] [PubMed] [Google Scholar]
- 21.Chuang MK, Chiu YC, Chou WC, et al. An mRNA expression signature for prognostication in de novo acute myeloid leukemia patients with normal karyotype. Oncotarget. 2015;6(36):39098–39110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chuang MK, Chiu YC, Chou WC, Hou HA, Chuang EY, Tien HF. A 3-microRNA scoring system for prognostication in de novo acute myeloid leukemia patients. Leukemia. 2015;29(5):1051–1059. [DOI] [PubMed] [Google Scholar]
- 23.Chiu YC, Tsai MH, Chou WC, et al. Prognostic significance of NPM1 mutation-modulated microRNA-mRNA regulation in acute myeloid leukemia. Leukemia. 2016;30(2):274–284. [DOI] [PubMed] [Google Scholar]
- 24.Network CGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Eng J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metzeler KH, Hummel M, Bloomfield CD, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112(10):4193–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Payton JE, Grieselhuber NR, Chang LW, et al. High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J Clin Invest. 2009;119(6):1714–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304(24):2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Novershtern N, Subramanian A, Lawton LN, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144(2):296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsiao TH, Chiu YC, Hsu PY, et al. Differential network analysis reveals the genome-wide landscape of estrogen receptor modulation in hormonal cancers. Sci Rep. 2016;6:23035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilop S, Chou WC, Jost E, et al. A three-gene expression-based risk score can refine the European LeukemiaNet AML classification. J Hematol Oncol. 2016;9(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcucci G, Yan P, Maharry K, et al. Epigenetics meets genetics in acute myeloid leukemia: clinical impact of a novel seven-gene score. J Clin Oncol. 2014; 32(6):548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z, Herold T, He C, et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol. 2013;31(9):1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–1093. [DOI] [PubMed] [Google Scholar]
- 36.Ng SW, Mitchell A, Kennedy JA, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433–437. [DOI] [PubMed] [Google Scholar]
- 37.Marzac C, Garrido E, Tang R, et al. ATP Binding Cassette transporters associated with chemoresistance: transcriptional profiling in extreme cohorts and their prognostic impact in a cohort of 281 acute myeloid leukemia patients. Haematologica. 2011; 96(9):1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26(47):6766–6776. [DOI] [PubMed] [Google Scholar]
- 39.Abramovich C, Pineault N, Ohta H, Humphries RK. Hox genes: from leukemia to hematopoietic stem cell expansion. Ann N Y Acad Sci. 2005;1044:109–116. [DOI] [PubMed] [Google Scholar]
- 40.Owens BM, Hawley RG. HOX and non-HOX homeobox genes in leukemic hematopoiesis. Stem Cells. 2002;20(5):364–379. [DOI] [PubMed] [Google Scholar]
- 41.Venezia TA, Merchant AA, Ramos CA, et al. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004;2(10):e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waraya M, Yamashita K, Katoh H, et al. Cancer specific promoter CpG Islands hypermethylation of HOP homeobox (HOPX) gene and its potential tumor suppressive role in pancreatic carcinogenesis. BMC Cancer. 2012;12:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005; 5(4):275–284. [DOI] [PubMed] [Google Scholar]
- 44.Costello RT, Mallet F, Gaugler B, et al. Human acute myeloid leukemia CD34+/CD38- progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000; 60(16):4403–4411. [PubMed] [Google Scholar]
- 45.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001; 11(7):1156–1166. [DOI] [PubMed] [Google Scholar]
- 46.Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotech. 2007;25(11):1315–1321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.