

Abstract
Aldehyde dehydrogenase 1A1 (ALDH1A1) activity is high in hematopoietic stem cells and functions in part to protect stem cells from reactive aldehydes and other toxic compounds. In contrast, we found that approximately 25% of all acute myeloid leukemias expressed low or undetectable levels of ALDH1A1 and that this ALDH1A1− subset of leukemias correlates with good prognosis cytogenetics. ALDH1A1− cell lines as well as primary leukemia cells were found to be sensitive to treatment with compounds that directly and indirectly generate toxic ALDH substrates including 4-hydroxynonenal and the clinically relevant compounds arsenic trioxide and 4-hydroperoxycyclophosphamide. In contrast, normal hematopoietic stem cells were relatively resistant to these compounds. Using a murine xenotransplant model to emulate a clinical treatment strategy, established ALDH1A1− leukemias were also sensitive to in vivo treatment with cyclophosphamide combined with arsenic trioxide. These results demonstrate that targeting ALDH1A1− leukemic cells with toxic ALDH1A1 substrates such as arsenic and cyclophosphamide may be a novel targeted therapeutic strategy for this subset of acute myeloid leukemias.
Introduction
The Aldehyde Dehydrogenase (ALDHs) gene family contains at least 19 isoforms that metabolize retinoids, reactive oxygen species (ROS) and endogenous and exogenous aldehydes.1,2 High levels of the ALDH1A1 isoform are expressed in hematopoietic stem cells (HSCs) as measured by mRNA analysis and staining with the fluorescent ALDH1A1 substrate Aldefluor.3 Despite this, ALDH1A1 is dispensable in murine HSCs, as it is compensated for by increased expression of the ALDH3A1 isoform and possibly others.4,5 Murine HSCs deficient in both ALDH1A1 and ALDH3A1 (Aldh1a1/3a1 DKOs) have a blocked B-cell development and reduced numbers of HSCs.4 The early B cells in these mice have increased levels of ROS and reactive aldehydes along with abnormalities in cell cycling, intracellular signaling and gene expression. Notably, HSCs and progenitors in these mice have elevated p38MAPK activity, increased sensitivity to DNA damage, and aberrant cell cycle distribution.4 These findings suggest an important role for ALDHs in metabolizing ROS and reactive aldehydes in primitive and differentiated hematopoietic cells, and show that the ALDHs and their substrates impact a variety of cellular processes in hematopoiesis.
The role that ALDHs play in normal HSCs suggests they may have important roles in leukemia as well.6,7 Several prior reports support this contention. In Fanconi anemia, loss of activity of the ALDH2 isoform in concert with FANC-D deficiency predisposes to the development of acute myeloid leukemia (AML) and bone marrow failure.8,9 ALDH activity in human leukemia also mediates resistance to a number of drugs10 and high levels of ALDH activity, as measured by Aldefluor staining, predict for a poor outcome to treatment.11–14 Leukemic stem cells (LSCs) that drive leukemia growth and disease relapse may also be identified with Aldefluor staining with either high or intermediate levels of staining.15,16
Given these prior observations, in the present study, we sought to further clarify and define the role and significance of ALDH activity in acute leukemia with a particular focus on determining whether ALDH biology could be exploited for new treatment approaches.
Methods
Analysis of public databases
Analysis of ALDH family genes for mRNA expression, DNA methylation, and survival outcomes was performed with data available from The Cancer Genome Atlas (TCGA) (cancergenome.nih.gov/cancersselected/acutemyeloidleukemia).17 Analysis of the expression of ALDH genes in leukemic versus normal hematopoietic cells was performed using the GSE9476 data set.18
Cell lines and primary specimens
Acute myeloid leukemia specimens were obtained from apheresis products of patients who gave Institutional Review Board approved informed consent for sample procurement at the University of Rochester and the University of Colorado in Denver. Normal 34+ HSCs were enriched from umbilical cord blood (UCB) specimens collected and distributed by the University of Colorado Cord Blood Bank (UCCBB), which is accredited by the American Association of Blood Banks and licensed by the US Food & Drug Administration. Cell lines were commercially supplied by ATCC (Manassas, VA, USA).
Flow cytometry analysis
Human samples were stained with CD34, CD38, CD123 and Aldefluor as previously described.3,19,20 AML cells were routinely gated through the blast window and it was confirmed that pheresis derived samples were not contaminated with significant numbers of normal HSCs based on staining with anti-CD123, a marker that distinguishes AML from normal HSCs together with other AML directed markers including HLA-DR, CD71 and CD7 (Figure 1C and Online Supplementary Figure S4A).21–23 Anti-mouse antibodies were as described previously.4,24 All antibodies and staining reagents were from BD Biosciences (San Jose, CA, USA). Intracellular 4HNE staining and visualization of gamma (γ) H2AX by flow cytometry were performed as previously described.4 All cell staining and analytical procedures were performed either manually or using high content semi-automated flow cytometry, as previously described.25,26
Figure 1.
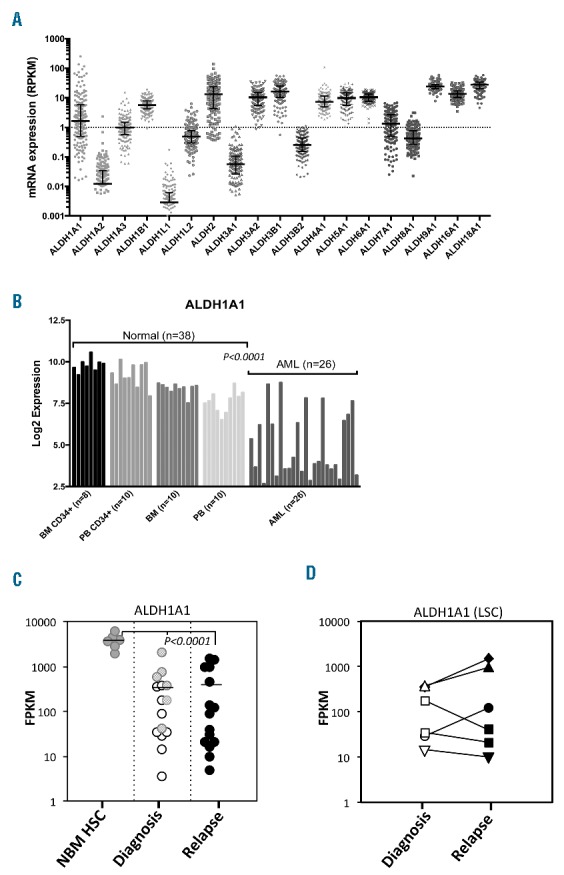
(A) Variable expression of ALDH1A1 and other aldehyde dehydrogenase (ALDH) isoforms in human acute myeloid leukemia (AML). The AML dataset from The Cancer Genome Atlas (TCGA) was analyzed for differential expression of ALDH isoforms in human AML blasts. (B) The GSE9476 dataset was analyzed for differential expression of ALDH1A1 between normal human hematopoietic cells (HSCs) and AML samples. (C) ALDH1A1 expression based on RNA-Seq analysis in LSC (white) and non-LSC (light gray) samples at diagnosis and LSC samples at relapse (black). The control is similarly analyzed normal bone marrow CD34+ cells (NBM HSC) (dark gray) (****P<0.0001). (D) ALDH1A1 expression using RNA-Seq analysis between paired diagnostic and relapsed LSC samples. (each symbol represents a different patient).
Western blot analysis
Lysates were prepared from Kasumi-1 and UCB CD34+ enriched populations. Primary mouse monoclonal antibodies to 4HNE adducts (Ab48506, Abcam; Cambridge, MA, USA) were used in a 1:500 dilution. Western blots were performed as previously described.27
Overexpression studies
For restoring ALDH1A1 gene function in Kasumi-1 cells, the HA-ALDH1A1 gene was excised from Addgene plasmid # 11610 using HindIII/Xba1 and sub-cloned into the pLVX-EF1a-IRES- mCherry Vector from Clontech (Cat# 631987), using EcoR1/Xba1 cloning sites. The sequence was verified using EF1a-Fwd and IRES-Rev sequencing primers. High titer lentiviral production was performed in 293TN cells and AML cells were transduced as previously described.28
Xenotransplant studies
To test the drug sensitivity of MOLM-13 and primary AML cells in vivo, NSGS mice29 were treated intraperitoneally (IP) with busulfan (30 mg/kg) then injected intravenously (IV) with 5–7×106 AML cells. After marrow engraftment was confirmed, control mice were treated IP with saline while the rest of the mice were treated IP with 5 μg/g ATO daily for four days together with 150 μg/g Cy at day 1 and day 4. The mice were sacrificed by CO2 asphyxiation and the bone marrow harvested for flow cytometry analysis. Further details are available in the Online Supplementary Methods.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7.0. Typically, unpaired two-tail Student t-tests were performed and P<0.05 was considered significant. Aggregate data were presented as means and standard deviations (SD).
Results
ALDH isoform expression is variable between AMLs and correlates with cytogenetic subtype
Variable levels of Aldefluor staining in AML have been reported and may correlate with outcomes to treatment.11,13 However, given the large number of ALDH isoforms and the findings that Aldefluor is a substrate for several of them, we sought to more precisely determine if variability in expression of specific individual ALDH isoforms occurs in AML and if so, whether expression of specific isoforms correlated with prognosis.4,30 First, we analyzed 165 leukemia specimens from The Cancer Genome Atlas (TCGA) dataset for levels of expression of all 19 human ALDH family genes.31 A 4–5 log difference in expression of the ALDH1A1 isoform was observed among this cohort of AML specimens, with 39.4% (65 of 165) expressing ALDH1A1 at less than 1 RPKM (Figure 1A). None of the AMLs expressed significant levels of ALDH3A1; however, a number of other isoforms including ALDH1B1, ALDH2, ALDH3A2, ALDH3B1, ALDH4A1, ALDH5A1, ALDH6A1, ALDH9A1, ALDH16A1 and ALDH18A1 were expressed at abundant levels in a majority of AML samples (Figure 1A). To confirm these findings, qPCR for ALDH isoforms was performed on 7 primary AML samples and 3 of 7 had no detectable ALDH1A1 expression, 5 of 7 had no ALDH3A1 expression and, again, a number of other ALDH isoforms were variably expressed similar to the TCGA data (Online Supplementary Figure S1A). When ALDH1A1 expression in AML was compared to that in normal HSCs and progenitors via analysis of another publicly available dataset,18 ALDH1A1 was consistently found at high levels in normal CD34+ cells from all the bone marrow (BM) and peripheral blood (PB) samples while ALDH1A1 levels were again quite variable in AMLs with a minority having low levels of expression (Figure 1B). ALDH1A3, ALDH2, ALDH3B1 and ALDH8A1 expression was also significantly different between normal hematopoietic cells and AMLs (Online Supplementary Figure S1B). Lastly, several ALDH isoforms including ALDH3B1 and ALDH1A3 had significant negative and positive associations, respectively, with ALDH1A1 (Online Supplementary Figure S1B).
Given the utility of using ALDH functional activity as a marker of HSCs and other cancer stem cells, ALDH may be utilized as a marker of LSCs as well.11,32,33 To assess the level of ALDH1A1 expression in LSCs both at diagnosis and at relapse, AML samples from 6 patients were analyzed with RNA-Seq and the level of ALDH1A1 mRNA transcripts measured in both LSCs and non-LSCs as previously described.34 In these AMLs, ALDH1A1 was expressed at significantly lower levels than HSCs controls in both LSCs and non-LSCs and no significant differences in ALDH1A1 expression between LSCs and non-LSCs were noted (Figure 1C). In order to further test whether ALDH1A1 expression increased following treatment with standard chemotherapy as a mechanism of drug resistance, LSCs from relapse samples from the same 6 patients were similarly examined for ALDH1A1 expression. Again, ALDH1A1 expression was significantly lower in relapse LSCs than the HSC controls, and analysis of paired diagnosis and relapse LSC samples did not show consistent or significant increases in ALDH1A1 expression (Figures 1D). In addition, no significant differences were noted in any other ALDH isoforms between diagnosis and relapse in either LSCs or non-LSCs (Online Supplementary Figure S1D).
Together these data demonstrate that at least in a subset of AMLs, ALDH1A1 expression is not elevated in LSCs and does not increase following standard chemotherapy treatment.
Given the significant ALDH isoform variability at the mRNA level between different AMLs observed in these studies, we next sought to investigate whether there was similar variability in ALDH enzymatic activity between AML samples. Eighteen primary AMLs were stained with Aldefluor dye and analyzed by flow cytometry. Consistent with the gene expression data, substantial differences in the overall content of Aldefluor staining cells between the different AML samples were observed (Figure 2A and B). As with the gene expression studies, a subset of AMLs had very low or absent numbers of cells with detectable Aldefluor staining [Aldefluor (0–0.1%)] (Figure 2A) demonstrating that they lacked ALDH1A1 and any other ALDH isoforms that may use Aldefluor as a substrate. Together, these findings demonstrate considerable variability in expression of different ALDH isoforms between different AMLs as well as relative to the HSCs and progenitors from which they were presumably derived. The most striking finding was the low to undetectable levels of expression and activity of ALDH1A1 in any cells in a substantial subset of AMLs (25%–40%) despite its universal high-level expression in normal HSCs and progenitors (henceforth termed ALDH1A1− AML).
Figure 2.
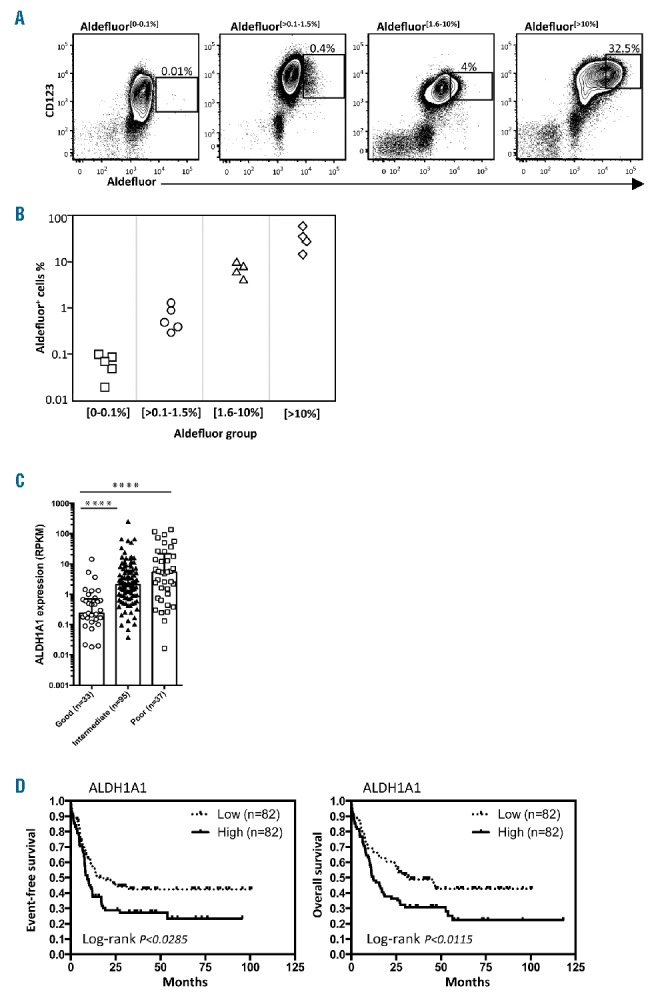
Levels of ALDH1A1 are variable between acute myeloid leukemias (AMLs) and have prognostic significance. (A) Aldefluor staining was performed to analyze single cell ALDH activity in representative samples of human primary AMLs that co-expressed CD123, a useful discriminator of AML and normal hematopoietic stem cells (HSCs). (B) Summary of Aldefluor staining in 18 different AML samples. (C) Association of ALDH1A1 expression level and poor, intermediate and good risk cytogenetic categories (TCGA dataset; ****P<0.0001). (D) Event-free survival (EFS) and overall survival (OS) Kaplan-Meier curve plots showing prognostic value of ALDH1A1 expression in all AML samples analyzed in the Cancer Genome Atlas dataset.
We next analyzed whether variability in the expression of the different ALDH isoforms, particularly ALDH1A1, had prognostic significance in predicting outcomes to treatment. When TCGA AML samples were grouped into Good, Intermediate and Poor Risk cytogenetic categories and analyzed for levels of ALDH1A1 expression, a significant association between the Good Risk cytogenetic category and lower levels of ALDH1A1 expression was observed (Figure 2C). In addition, low-level expression of the ALDH1A1 isoform was correlated with improved event-free survival (EFS) and overall survival (OS) in the entire cohort of AML patients (Figure 2D). To evaluate whether the prognostic value of ALDH1A1 was independent from cytogenetics, intermediate risk samples were fractionated based on ALDH1A1 expression and analyzed for EFS and OS. A trend to an association between better EFS or OS and lower expression of ALDH1A1 was found within intermediate risk cytogenetic subgroups, although the sample sizes were not big enough to formally confirm this association (Online Supplementary Figure S1E).
Absence of ALDH1A1 in human leukemia cell lines renders them sensitive to toxic ALDH substrates
The finding that there is a subset of AMLs that do not express ALDH1A1 (ALDH1A1− AMLs) while normal HSCs and progenitors express high levels of ALDH1A1 suggested that exploiting low level ALDH1A1 expression may offer a new avenue to selectively target this subgroup of AMLs.35 Previously we had shown that primary murine marrow cells lacking Aldh1a1 and Aldh3a1 were sensitive to the reactive aldehyde 4-hydroxynonenal (4HNE), likely because these ALDHs are the primary metabolizers of this toxic compound.4 In addition, an in silico screen of compounds active in human leukemic stem cells revealed that 4HNE has potent human anti-leukemic activity.36 Together, these findings suggested that human ALDH1A1− AMLs may be particularly sensitive to 4HNE treatment given their predicted limited ability to metabolize this toxic compound. To test this, first, a series of human AML cell lines were screened by Aldefluor staining along with qPCR analysis and both Kasumi-1 and MOLM-13 were identified as representative ALDH1A1− AML cell lines suitable for further analysis (Online Supplementary Figure S2A and B). At baseline, Kasumi-1 had high levels of endogenous reactive aldehyde adducts relative to normal UCB CD34+ cells (Figure 3A), which further increased after 4HNE treatment. DNA damage and ROS were also increased in Kasumi-1 cells treated with 4HNE while normal CD34+ cells were relatively resistant to these effects (Figure 3B and C). In addition, when Kasumi-1 cells were exposed to increasing concentrations of 4HNE, an LD50 of 4 μM was found, while in contrast, CD34+ normal cord blood cells were almost completely resistant to comparable doses of 4HNE treatment (Figure 3D).
Figure 3.
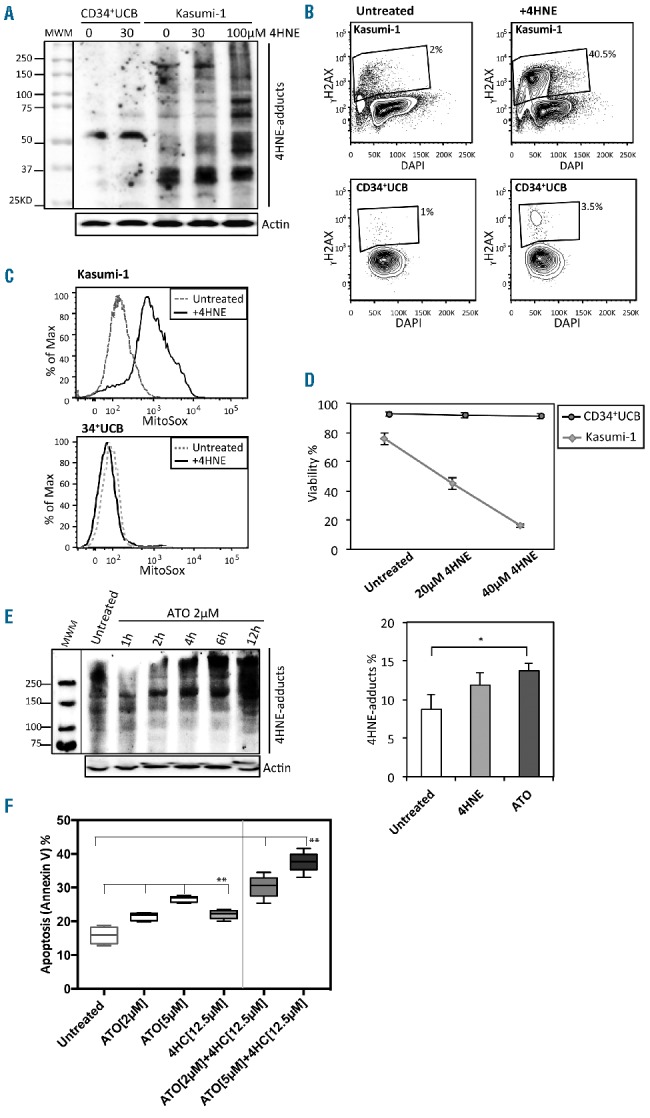
ALDH1A1− acute myeloid leukemia (AML) cell lines are sensitive to toxic substrates of aldehyde dehydrogenase (ALDH). (A) Western Blot measurement of 4HNE protein adducts in Kasumi-1 and normal CD34+ umbilical cord blood (UCB) cells treated with 30 μM 4HNE or vehicle. Kasumi-1 cells treated with 100 μM 4HNE were included as a positive control. (B) Flow cytometric assessment of DNA damage (γH2AX) in Kasumi-1 with and without 4HNE treatment compared to normal CD34+ UCB cells. (C) Flow cytometric assessment of ROS in Kasumi-1 with and without 30–40 μM 4HNE treatment compared to normal CD34+ UCB cells. (D) 4HNE dose response curves in Kasumi-1 and normal CD34+ cells treated with 20 and 40 μM 4HNE [10 μM 4HNE was added to cell media every hour, data is mean and standard deviation (SD) of triplicate samples]. (E) Western blot analysis of 4HNE protein adducts generated by treating Kasumi-1 cells with 2 μM ATO over 12 hours and flow cytometric analysis of 4HNE adducts in Kasumi-1 induced by 2.5 μM 4HNE and 2 μM ATO treatment at 12 hours (n=3; *P<0.05). (F) Apoptosis of Kasumi-1 cells following overnight treatment with ATO (2 μM or 5 μM) or 4HC (12.5 μM) alone and in combination (n=6 replicates; **P<0.0001). (D–F) Bars represent mean±SD. (D and E) Results of technical triplicates are shown.
In order to begin to translate this concept into a clinically relevant treatment strategy for ALDH1A1− AML, we sought to identify known drugs that would induce intracellular 4HNE in ALDH1A1− AML cells, as 4HNE itself is not clinically useful due to non-specific toxicity and a very short half-life.37 Arsenic trioxide (ATO) is a well-characterized compound used in the treatment of acute promyelocytic leukemia that has significant pro-oxidant activity, suggesting that it could elevate intracellular reactive aldehyde levels, as these are frequent byproducts of ROS.38 Kasumi-1 cells treated with ATO had elevated intracellular 4HNE adduct levels (Figure 3E)39 and ATO induced a concentration-dependent increase of apoptosis in AML cells (Figure 3F). Next, we sought to determine whether we could further amplify the anti-leukemic effects of ATO in ALDH1A1− AML by combining it with 4-hydroperoxycyclophosphamide (4HC), an active metabolite of cyclophosphamide (Cy) and known toxic substrate of ALDH1A1. The combination of ATO and even sublethal levels of 4HC led to higher levels of cell killing than control or either drug alone (Figure 3F).40
To determine whether these effects were dependent on ALDH1A1 expression, Kasumi-1 cells were transduced with a lentiviral vector that over-expressed ALDH1A1 (Online Supplementary Figure S2C) and tested over a wide range of 4HC and ATO concentrations, alone and together. Restoration of ALDH1A1 expression at least partially blocked the effects of 4HNE mediated generation of ROS and reactive aldehyde adducts in Kasumi-1 cells (Figure 4A and B, respectively). Expression of ALDH1A1 also protected Kasumi-1 cells to varying degrees from 4HC and ATO, depending on their concentrations (Figure 4C). Similar effects were noted in MOLM-13 cells transduced to express ALDH1A1 (Online Supplementary Figure S2D and E). Furthermore, Kasumi-3 cells, which express high levels of ALDH1A1, were rendered more sensitive to 4HC and ATO following treatment with an ALDH inhibitor (DEAB) (Figure 4D and Online Supplementary Figure S2F). In addition, differences in DNA damage were also noted between ALDH1A1− and ALDH1A1+ Kasumi-1 cells treated in vitro with 4HC and ATO (Figure 4E). Lastly, we tested the in vivo anti-leukemia activity of Cy plus ATO on MOLM-13 AML cells. MOLM-13 were used for these studies as opposed to Kasumi-1 as they consistently engraft NSGS mice, while Kasumi-1 do not engraft well in the conditions tested. Briefly, MOLM-13 cells were transplanted into NSGS mice and then treated daily with ATO for four days followed by Cy at day 1 and day 4. When the marrow content of MOLM-13 was analyzed by flow cytometry, two weeks later, the overall content of MOLM-13 cells was dramatically reduced in Cy plus ATO treated mice relative to placebo-treated controls (Figure 4F). Consequently, these results demonstrate that loss of ALDH1A1 renders AML cell lines more sensitive to 4HC and ATO.
Figure 4.
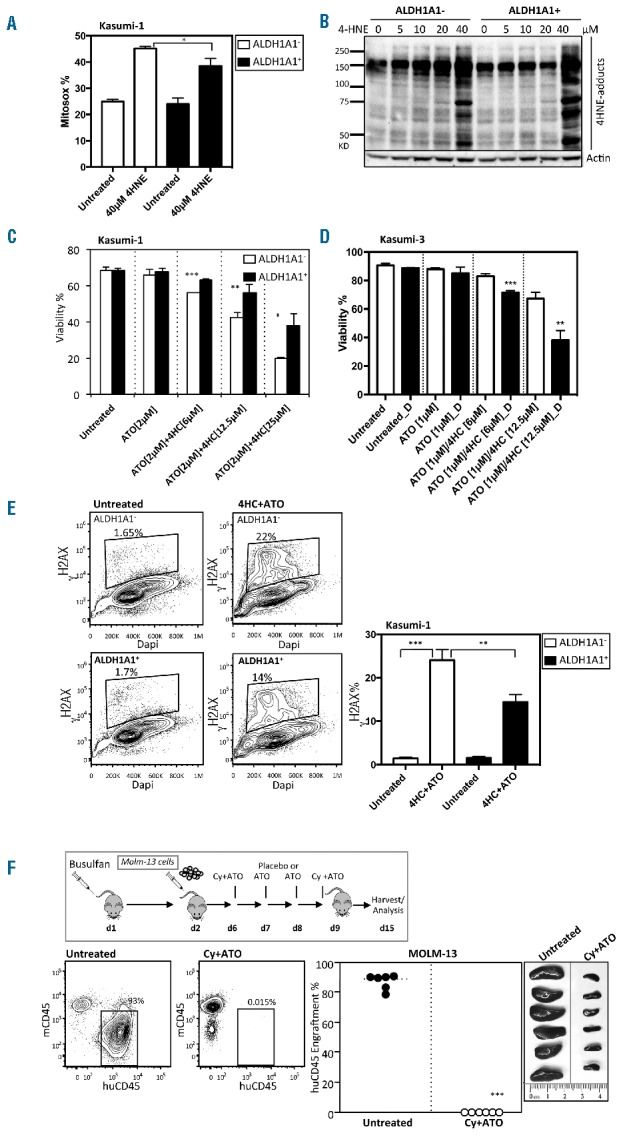
Overexpression of ALDH1A1+ partially rescues ALDH1A1− acute myeloid leukemia (AML) cell sensitivity to toxic substrates of ALDH. (A) ROS (Mitosox) detection in ALDH1A1+ and ALDH1A1- Kasumi-1 cells after six hours of treatment with 40 μM 4-HNE (n=3;*P<0.05). (B) Western blot analysis of 4-HNE protein adducts generated by treating ALDH1A1+ and ALDH1A1− Kasumi-1 cells with different concentrations of 4-HNE for 1 hour. (C) Viability of Kasumi-1 cells engineered to express ALDH1A1 through lentiviral gene transfer and treated with various combinations and doses of 4HC and ATO (n=3 replicates; ***P<0.0005, **P<0.005, *P<0.05). (D) Viability of Kasumi-3 cells treated with various combinations and doses of 4HC and ATO with (black) or without (white) the ALDH inhibitor DEAB (D) (n=3; **P<0.005, ***P<0.0005). (E) Flow cytometric assessment and summary graph of DNA damage (γH2AX) in ALDH1A1+ and ALDH1A1− Kasumi-1 cells treated with 4HC+ATO (n=3; **P<0.005, ***P<0.0005). (A–C) Bars represent mean±SD. Technical triplicates were performed. (F) In vivo treatment with Cy and ATO of NSGS mice with established ALDH1A1− MOLM-13 leukemia (n=6; ***P<0.0005).
Absence of ALDH1A1 in primary AMLs renders them sensitive to toxic ALDH substrates
To determine if the results obtained from the AML cell line studies were relevant to authentic primary AMLs, first, in vitro sensitivity studies were performed. Peripheral blood samples from apheresis collections of 4 ALDH1A1− AML patients were treated overnight with 4HC and ATO, alone or in combination. The 4HC plus ATO combination generated more DNA damage and cell killing than either agent alone or controls (Figure 5A). In addition, when 3 samples known to have both ALDH1A1+ and ALDH1A1− subsets were sorted into Aldefluor+ and Aldefluor− fractions (Online Supplementary Figure S3A–C) and treated in vitro with 4HC plus ATO, the Aldefluor− cells were sensitive to treatment while the Aldefluor+ cells were relatively resistant as measured by cell killing and DNA damage (Figure 5B and C). To further determine whether ALDH1A1 expression modulates the effects of 4HC and ATO in primary AML cells, Aldefluor+ AML cells were treated with DEAB, an inhibitor of ALDH1A1, and then the 4HC/ATO drug combination was added.41 Again, the Aldefluor+ controls were relatively resistant to treatment with 4HC/ATO, while in contrast the DEAB-treated cells had a greater sensitivity to 4HC/ATO than control (Figure 5D). Together these data further demonstrate that ALDH1A1, at least in part, mediates resistance to these agents.
Figure 5.
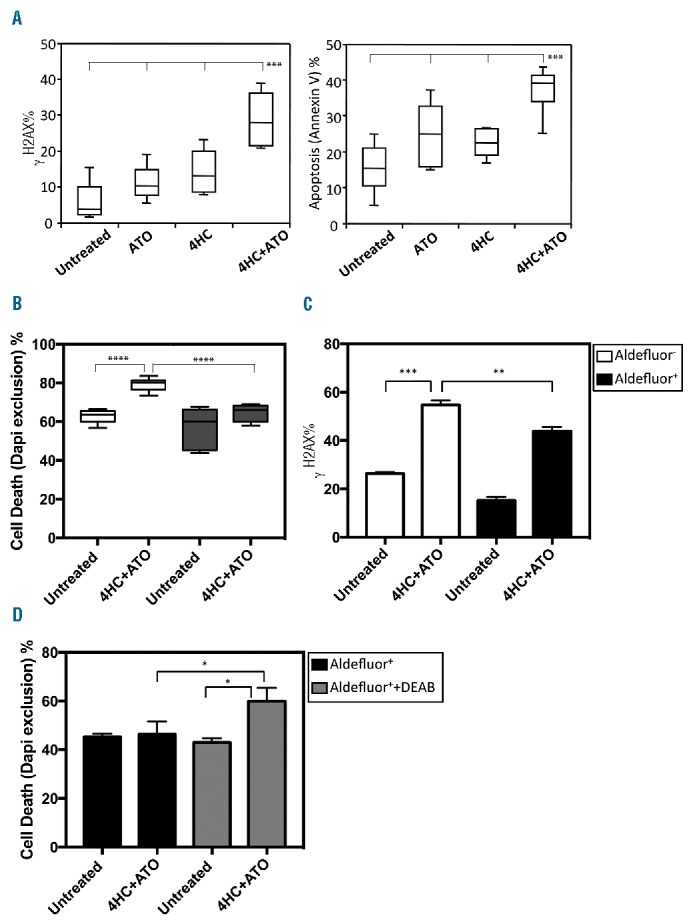
Primary human ALDH1A1− acute myeloid leukemias (AMLs) are sensitive to 4HC+ATO while ALDH1A1+ AMLs are relatively resistant. (A) In vitro sensitivity of primary human AMLs to 4HC and ATO. Primary human AMLs were treated overnight with 4HC+ATO (12.5 μM+2 μM) and DNA damage and cell viability were measured (4 AMLs in triplicate; ***P<0.0005). (B) Sensitivity of Aldefluor+ and Aldefluor− fractions from 2 representative AMLs purified by FACS and then treated overnight with 4HC+ATO (12.5 μM+2 μM) (n=6; **P<0.005, ***P<0.0005). (C) DNA damage (γH2AX) of Aldefluor+ and Aldefluor− fractions from an ALDH1A1+ AML following overnight treatment with 4HC+ATO (n=3; **P<0.005, ***P<0.0005). (D) Relative sensitivity of Aldefluor+ fraction from an ALDH1A1+ AML to 4HC+ATO with and without 5 μM DEAB at 18 hours (n=3; *P<0.05). (A–D) Bars represent mean±SD. (C–D) Results of technical triplicates are shown.
Next, to test whether 4HC plus ATO was also effective against human primary AML cells capable of engrafting immunodeficient mice, 3 primary Aldefluor[0–0.1%] AMLs were treated in vitro overnight with either vehicle control or 4HC plus ATO and transplanted into busulfan-treated NSGS mice. All control-treated AMLs engrafted at levels of 20%–100% while 4HC plus ATO treated AMLs failed to engraft any of 25 mice (Figure 6A). In contrast, when normal UCB derived CD34+ cells were treated with 4HC plus ATO in a similar fashion, although engraftment was reduced in NSGS mice, it could be readily detected in 75% of recipients (Figure 6B). Lastly, to replicate the clinical scenario as closely as possible, we tested in vivo the effects of Cy and ATO on an Aldefluor[0–0.1%] AML that had been previously engrafted in NSGS mice compared to an Aldefluor[>10%] AML positive control known to express high levels of ALDH1A1. Two weeks following in vivo treatment of Aldefluor[0–0.1%] AML engrafted mice with Cy plus ATO, the overall marrow content of human AML was significantly reduced in mice relative to placebo-treated controls (Figure 7A). In contrast, the Aldefluor[>10%] AML engrafted mice showed no differences in engraftment between treated and untreated groups (Figure 7B).
Figure 6.
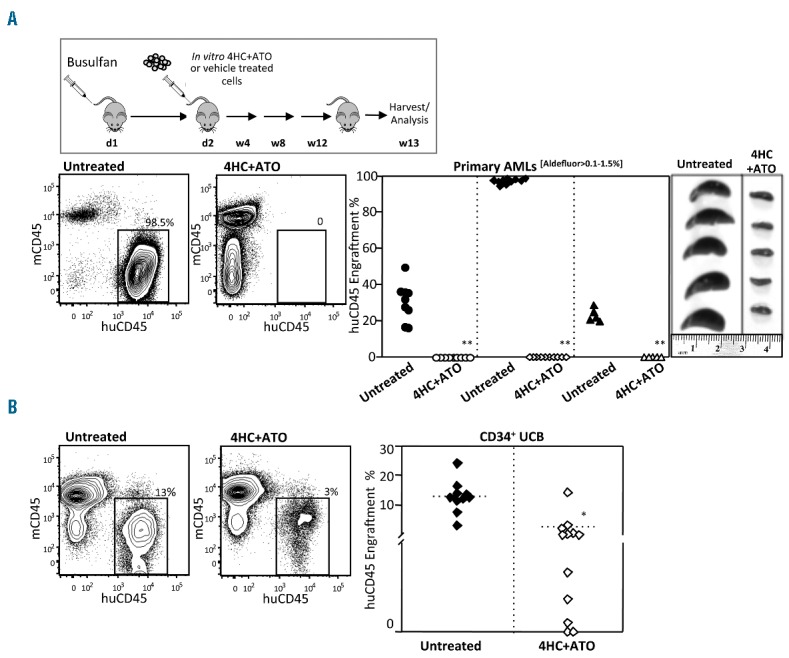
ALDH1A1− acute myeloid leukemias (AMLs) are sensitive to 4HC+ATO while normal HSCs are relatively resistant. (A) 4HC+ATO in vitro treatment eliminated engraftment of primary AMLs in NSGS mice. Representative flow cytometric analysis of harvested bone marrow of Aldefluor[0–0.1%] AMLs that underwent overnight ex vivo treatment with 4HC+ATO (30 μM+ 5 μM) or vehicle (left panels) and summary of xenotransplant data from the 3 AMLs treated in vitro and transplanted into NSGS mice (right panels). The data are a summary of 25 of 30 control and 25 of 30 treated mice analyzed after 12 weeks (**P<0.0005). (B) In contrast, 4HC+ATO in vitro treatment did not completely eliminate engraftment of normal CD34+UCB cells in NSGS mice. NSGS mice transplanted with 2 pooled UCBs following 4HC+ATO or vehicle treatment as above (treated: n=12; untreated: n=10). While level of engraftment was reduced, in contrast to the AML experiments above, 7 of 12 mice treated with normal CD34+UCB cells displayed more than 1% engraftment (*P<0.005). Analysis of engraftment in bone marrow was performed at 13 weeks using flow cytometry.
Figure 7.
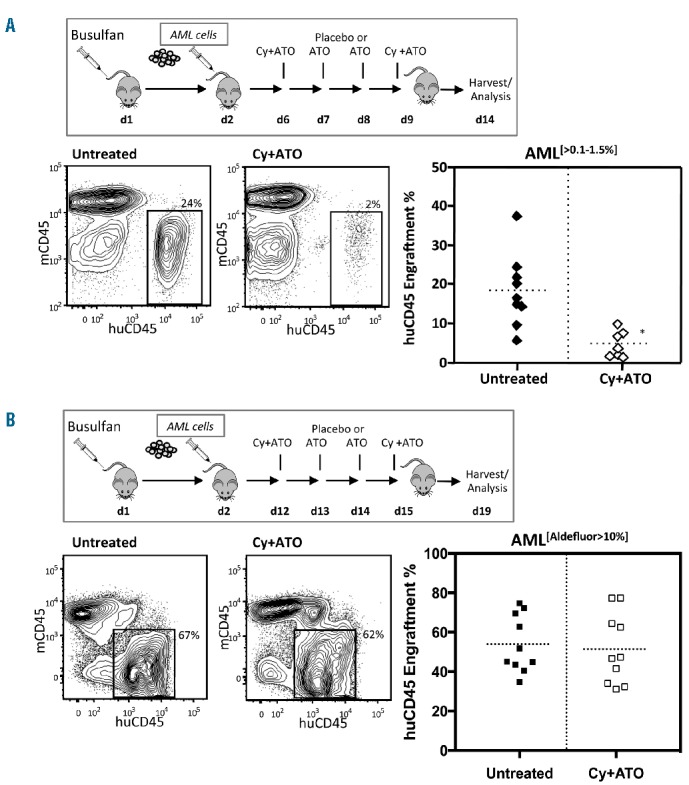
ALDH1A1− acute myeloid leukemias (AMLs) are sensitive to Cy+ATO while ALDH1A1+ AMLs are relatively resistant. (A) In vivo Cy+ATO treatment reduced leukemic burden in NSGS mice engrafted with primary ALDH1A1− AML cells (treated: n=7; untreated: n=9; *P<0.005). (B) In vivo Cy+ATO treatment shows no differences in leukemic burden in NSGS mice engrafted with primary ALDH1A1+ AML cells (n=10; not significant).
Discussion
In this report, we demonstrate that there is expression of a number of ALDH isoforms in AML and several have substantial variability in expression. The most prominent of these is ALDH1A1, which has low levels of expression and activity in a subset of AMLs, despite its nearly universal expression in normal HSCs and progenitors. In fact, in 25%–40% of AMLs we could find few or no cells expressing any ALDH1A1 as determined by Aldefluor staining and flow cytometry as well as qPCR analysis (termed ALDH1A1−AML). This includes finding low to absent levels of ALDH1A1 in LSCs as well. The factors governing the variability of ALDH1A1 expression in AMLs are unknown; however, the majority of ALDH1A1− AMLs in public databases do not have obvious ALDH1A1 gene deletions (data not shown) suggesting that loss of ALDH1A1 expression may occur as a consequence of gene regulation either through epigenetic or gene expression/repression mechanisms.42 Further studying the underlying mechanisms of ALDH1A1 gene regulation in HSCs, as well as AMLs, may provide useful insights into the biology of each. We also found that loss of ALDH1A1 may have prognostic implications as ALDH1A1− AMLs correlate with good prognosis cytogenetics and there was a trend to improved outcomes in intermediate risk cytogenetic AML patients based on level of ALDH1A1 expression, although we could not formally prove an association. It would be interesting to seek to extend and confirm these observations in larger prospective studies. If this trend is confirmed, then ALDH1A1 status may become a useful tool to stratify patients to different treatment approaches. These observations also raise additional questions regarding why ALDH1A1 expression would be associated with cytogenetic status. In addition, if outcomes are indeed impacted by ALDH1A1 status, it would be important to determine whether this was because loss of ALDH1A1 renders ALDH1A1− AMLs more sensitive to chemotherapies that induce ROS and/or reactive aldehydes or whether this acts through a different mechanism.
Since a substantial subgroup of AMLs is deficient in ALDH1A1 expression at different stages of disease (diagnosis and relapse), we speculated that these ALDH1A1− AMLs may specifically be more sensitive to compounds that directly or indirectly generate toxic ALDH1A1 substrates. First, 4HNE, a prototypical toxic reactive aldehyde substrate of ALDH1A1 was quite effective at killing ALDH1A1− cell lines while CD34+ UCB, which expresses high levels of ALDH1A1, was quite resistant to this compound. Of more clinical relevance, we found that ATO, a widely used drug in the treatment of APL, could induce the intracellular production of 4HNE, leading to selective killing of ALDH1A1− AMLs. This effect was at least partially blocked by restoration of ALDH1A1 activity through gene transfer, further supporting a role for ALDH1A1 in this effect. The finding that ALDH1A1 gene transfer did not fully block the effect of ATO is likely because ATO potentially works through a variety of mechanisms, of which generating 4HNE is only one.43 To amplify the effect of ATO in ALDH1A1− AML, it was combined with 4HC for in vitro studies and Cy for in vivo studies as these generate a toxic intermediate substrate termed aldophosphamide that is primarily metabolized by ALDH1A1. ALDH1A1− AML cell lines as well as primary ALDH1A1− AMLs were particularly sensitive to the ATO and 4HC/Cy combinations both in vitro and in vivo, while, again, normal HSCs were relatively resistant. The in vivo studies ultimately may be extended to determine mechanisms of resistance to ALDH-directed therapy.
The finding that there is an apparent therapeutic window between normal HSCs and ALDH1A1− AMLs treated with toxic ALDH1A1 substrates, suggests a novel targeted therapy strategy for this newly defined AML subset (modeled in Figure 8). In this strategy, we propose that AMLs lacking ALDH1A1 are more sensitive to compounds that directly or indirectly generate toxic ALDH substrates including ROS, reactive aldehydes and others, while normal HSCs with high levels of ALDH1A1 are relatively resistant. Consequently, rational drug combinations focusing on these agents may be particularly effective in ALDH1A1− AML patients. As a first step towards exploring this treatment strategy, the findings in this report provide a rationale for performing a phase I/II clinical trial of Cy plus ATO in ALDH1A1− AML patients to determine efficacy and toxicity. In addition, these observations suggest the utility of further pre-clinical studies to identify even more effective and less toxic combinations of compounds that exploit the differences in ALDH1A1 expression between normal HSCs and AMLs. Ultimately, it will be interesting to determine whether therapeutic targeting of the differences in ALDH isoform expression between malignant cells and normal tissue stem cells can be extended to other hematologic cancers such as myeloma and lymphoma, and solid tumors as well.
Figure 8.
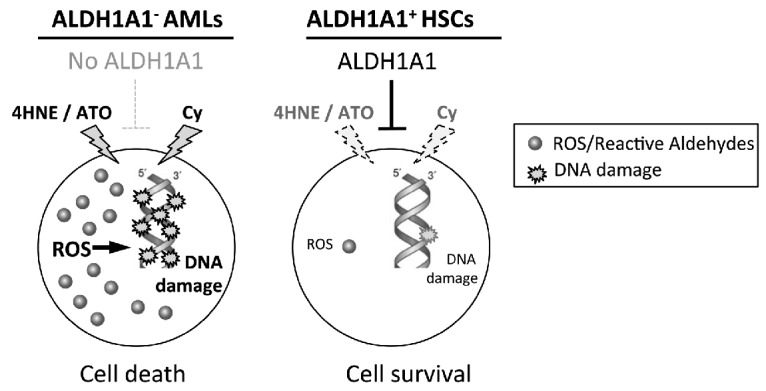
Model for selectively targeting ALDH1A1− acute myeloid leukemias (AMLs). In this treatment strategy, AMLs will be profiled for aldehyde dehydrogenase (ALDH) isoform expression and those lacking ALDH1A1 expression will be treated with agents that directly and indirectly generate lethal levels of ALDH substrates including ROS, reactive aldehydes and others. In contrast, normal hematopoietic stem cells express high levels of ALDH1A1 that could metabolize these compounds resulting in relative sparing.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge technical assistance by the Flow Cytometry Cores (CFAR and UCCC) and the Animal Housing & Care Vivarium of the University of Colorado Denver (Anschutz Medical Campus).
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/6/1054
References
- 1.Vasiliou V, Thompson DC, Smith C, Fujita M, Chen Y. Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells. Chem Biol Interact. 2013;202(1–3):2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh S, Brocker C, Koppaka V, et al. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic Biol Med. 2013;56:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Storms RW, Trujillo AP, Springer JB, et al. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci USA. 1999;96(16):9118–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gasparetto M, Sekulovic S, Brocker C, et al. Aldehyde dehydrogenases are regulators of hematopoietic stem cell numbers and B-cell development. Exp Hematol. 2012; 40(4):318–329.e312. [DOI] [PubMed] [Google Scholar]
- 5.Levi BP, Yilmaz OH, Duester G, Morrison SJ. Aldehyde dehydrogenase 1a1 is dispensable for stem cell function in the mouse hematopoietic and nervous systems. Blood. 2009;113(8):1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fritz KS, Petersen DR. Exploring the biology of lipid peroxidation-derived protein carbonylation. Chem Res Toxicol. 2011;24(9):1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias¿ Blood. 2011;117(22):5816–5826. [DOI] [PubMed] [Google Scholar]
- 8.Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, Patel KJ. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489(7417):571–575. [DOI] [PubMed] [Google Scholar]
- 9.Hira A, Yabe H, Yoshida K, et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood. 2013; 122(18):3206–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koelling TM, Yeager AM, Hilton J, Haynie DT, Wiley JM. Development and characterization of a cyclophosphamide-resistant subline of acute myeloid leukemia in the Lewis × Brown Norway hybrid rat. Blood. 1990;76(6):1209–1213. [PubMed] [Google Scholar]
- 11.Cheung AM, Wan TS, Leung JC, et al. Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia. 2007;21(7):1423–1430. [DOI] [PubMed] [Google Scholar]
- 12.Hoang VT, Hoffmann I, Borowski K, et al. Identification and separation of normal hematopoietic stem cells and leukemia stem cells from patients with acute myeloid leukemia. Methods Mol Biol. 2013; 1035:217–230. [DOI] [PubMed] [Google Scholar]
- 13.Ran D, Schubert M, Pietsch L, et al. Aldehyde dehydrogenase activity among primary leukemia cells is associated with stem cell features and correlates with adverse clinical outcomes. Exp Hematol. 2009;37(12):1423–1434. [DOI] [PubMed] [Google Scholar]
- 14.Schuurhuis GJ, Meel MH, Wouters F, et al. Normal hematopoietic stem cells within the AML bone marrow have a distinct and higher ALDH activity level than co-existing leukemic stem cells. PLoS One. 2013;8(11):e78897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleischman AG. ALDH marks leukemia stem cell. Blood. 2012;119(15):3376–3377. [DOI] [PubMed] [Google Scholar]
- 16.Gerber JM, Smith BD, Ngwang B, et al. A clinically relevant population of leukemic CD34(+)CD38(−) cells in acute myeloid leukemia. Blood. 2012;119(15):3571–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stirewalt DL, Meshinchi S, Kopecky KJ, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer. 2008;47(1):8–20. [DOI] [PubMed] [Google Scholar]
- 19.Fallon P, Gentry T, Balber AE, et al. Mobilized peripheral blood SSCloALDHbr cells have the phenotypic and functional properties of primitive haematopoietic cells and their number correlates with engraftment following autologous transplantation. Br J Haematol. 2003;122(1):99–108. [DOI] [PubMed] [Google Scholar]
- 20.Storms RW, Green PD, Safford KM, et al. Distinct hematopoietic progenitor compartments are delineated by the expression of aldehyde dehydrogenase and CD34. Blood. 2005;106(1):95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000; 14(10):1777–1784. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q, Wang M, Hu Y, et al. Significance of CD71 expression by flow cytometry in diagnosis of acute leukemia. Leuk Lymphoma. 2014;55(4):892–898. [DOI] [PubMed] [Google Scholar]
- 23.van Rhenen A, Moshaver B, Kelder A, et al. Aberrant marker expression patterns on the CD34+CD38- stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia. 2007;21(8):1700–1707. [DOI] [PubMed] [Google Scholar]
- 24.Gasparetto M, Sekulovic S, Zakaryan A, et al. Varying levels of aldehyde dehydrogenase activity in adult murine marrow hematopoietic stem cells are associated with engraftment and cell cycle status. Exp Hematol. 2012;40(10):857–866 e855. [DOI] [PubMed] [Google Scholar]
- 25.Brinkman RR, Gasparetto M, Lee SJ, et al. High-content flow cytometry and temporal data analysis for defining a cellular signature of graft-versus-host disease. Biol Blood Marrow Transplant. 2007;13(6):691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gasparetto M, Gentry T, Sebti S, et al. Identification of compounds that enhance the anti-lymphoma activity of rituximab using flow cytometric high-content screening. J Immunol Methods. 2004;292(1–2):59–71. [DOI] [PubMed] [Google Scholar]
- 27.Pei S, Minhajuddin M, Callahan KP, et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem. 2013;288(47): 33542–33558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashton JM, Balys M, Neering SJ, et al. Gene sets identified with oncogene cooperativity analysis regulate in vivo growth and survival of leukemia stem cells. Cell Stem Cell. 2012;11(3):359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wunderlich M, Chou FS, Link KA, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24(10):1785–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreb JS. Aldehyde dehydrogenase as a marker for stem cells. Curr Stem Cell Res Ther. 2008;3(4):237–246. [DOI] [PubMed] [Google Scholar]
- 31.Miller CA, Wilson RK, Ley TJ. Genomic landscapes and clonality of de novo AML. N Engl J Med. 2013;369(15):1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearce DJ, Taussig D, Simpson C, et al. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells. 2005;23(6):752–760. [DOI] [PubMed] [Google Scholar]
- 33.Hoang VT, Buss EC, Wang W, et al. The rarity of ALDH(+) cells is the key to separation of normal versus leukemia stem cells by ALDH activity in AML patients. Int J Cancer. 2015;137(3):525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho TC, LaMere M, Stevens BM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood. 2016;128(13):1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith C, Gasparetto M, Humphries K, Pollyea DA, Vasiliou V, Jordan CT. Aldehyde dehydrogenases in acute myeloid leukemia. Ann NY Acad Sci. 2014;1310(1):58–68. [DOI] [PubMed] [Google Scholar]
- 36.Hassane DC, Guzman ML, Corbett C, et al. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood. 2008;111(12): 5654–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fritz KS, Petersen DR. An overview of the chemistry and biology of reactive aldehydes. Free Radic Biol Med. 2013;59:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeanne M, Lallemand-Breitenbach V, Ferhi O, et al. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18(1):88–98. [DOI] [PubMed] [Google Scholar]
- 39.Chou WC, Jie C, Kenedy AA, Jones RJ, Trush MA, Dang CV. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc Natl Acad Sci USA. 2004;101(13):4578–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andersson BS, Mroue M, Britten RA, Farquhar D, Murray D. Mechanisms of cyclophosphamide resistance in a human myeloid leukemia cell line. Acta Oncol. 1995;34(2):247–251. [DOI] [PubMed] [Google Scholar]
- 41.Morgan CA, Parajuli B, Buchman CD, Dria K, Hurley TD. N, N-diethylaminobenzaldehyde (DEAB) as a substrate and mechanism-based inhibitor for human ALDH isoenzymes. Chem Biol Interact. 2015;234 (18–28). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao D, Mo Y, Li MT, et al. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J Clin Invest. 2014;124(12):5453–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar S, Yedjou CG, Tchounwou PB. Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL-60) cells. J Exp Clin Cancer Res. 2014;33:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.