

Abstract
Recent studies suggest that circulating tumor cells and cell-free DNA may represent powerful non-invasive tools for monitoring disease in patients with solid and hematologic malignancies. Here, we conducted a pilot study in 27 myeloma patients to explore the clonotypic V(D)J rearrangement for monitoring circulating myeloma cells and cell-free myeloma DNA. Next-generation sequencing was used to define the myeloma V(D)J rearrangement and for subsequent peripheral blood tracking after treatment initiation. Positivity for circulating myeloma cells/cell-free myeloma was associated with conventional remission status (P<0.001) and 91% of non-responders/progressors versus 41% of responders had evidence of persistent circulating myeloma cells/cell-free myeloma DNA (P<0.001). About half of the partial responders showed complete clearance of circulating myeloma cells/cell-free myeloma DNA despite persistent M-protein, suggesting that these markers are less inert than the M-protein, rely more on cell turnover and, therefore, decline more rapidly after initiation of effective treatment. Positivity for circulating myeloma cells and for cell-free myeloma DNA were associated with each other (P=0.042), but discordant in 30% of cases. This indicates that cell-free myeloma DNA may not be generated entirely by circulating myeloma cells and may reflect overall tumor burden. Prospective studies need to define the predictive potential of high-sensitivity determination of circulating myeloma cells and DNA in the monitoring of multiple myeloma.
Introduction
The introduction of novel proteasome inhibitors,1 immunomodulatory drugs2 and monoclonal antibodies3,4 has led to deeper and longer-lasting responses in patients with multiple myeloma.5–7 The detection of minimal residual disease has, therefore, become increasingly important for the management of this disease.8 Bone marrow minimal residual disease studies using multicolor flow cytometry9 or next-generation sequencing (NGS) of the clonotypic V(D)J immunoglobulin (Ig) rearrangement10,11 suggest that minimal residual disease predicts progression-free and overall survival. The latter technology is currently the most sensitive, with a detection rate of one per 10−6 bone marrow cells. It does, however, requires repetitive bone marrow sampling, a procedure that is painful for the majority of patients.
In solid tumors but also some hematologic cancers, circulating nucleic acids (cell-free DNA) and circulating tumor cells are becoming a promising minimally-invasive tool for monitoring tumor burden and response to treatment.12–14 To date, diffuse-large B-cell lymphoma is a prototype hematologic disease in which the detection of the disease-specific V(D)J rearrangement in peripheral blood cell-free DNA by NGS has been shown to predict relapses before established radiological staging methods demonstrate evidence of disease recurrence.15,16 The general applicability of this monitoring concept to multiple myeloma does, however, remain to be determined, since disease distribution, vascularity, spreading and cell turnover differ from those in lymphoma, potentially affecting the circulating malignant cell and cell-free DNA compartments.
Here, we investigated the clinical utility of circulating myeloma cells and DNA in the monitoring of multiple myeloma using the clonotypic V(D)J rearrangement as a highly patient-specific detection marker.
Methods
Patients’ characteristics and ethics statement
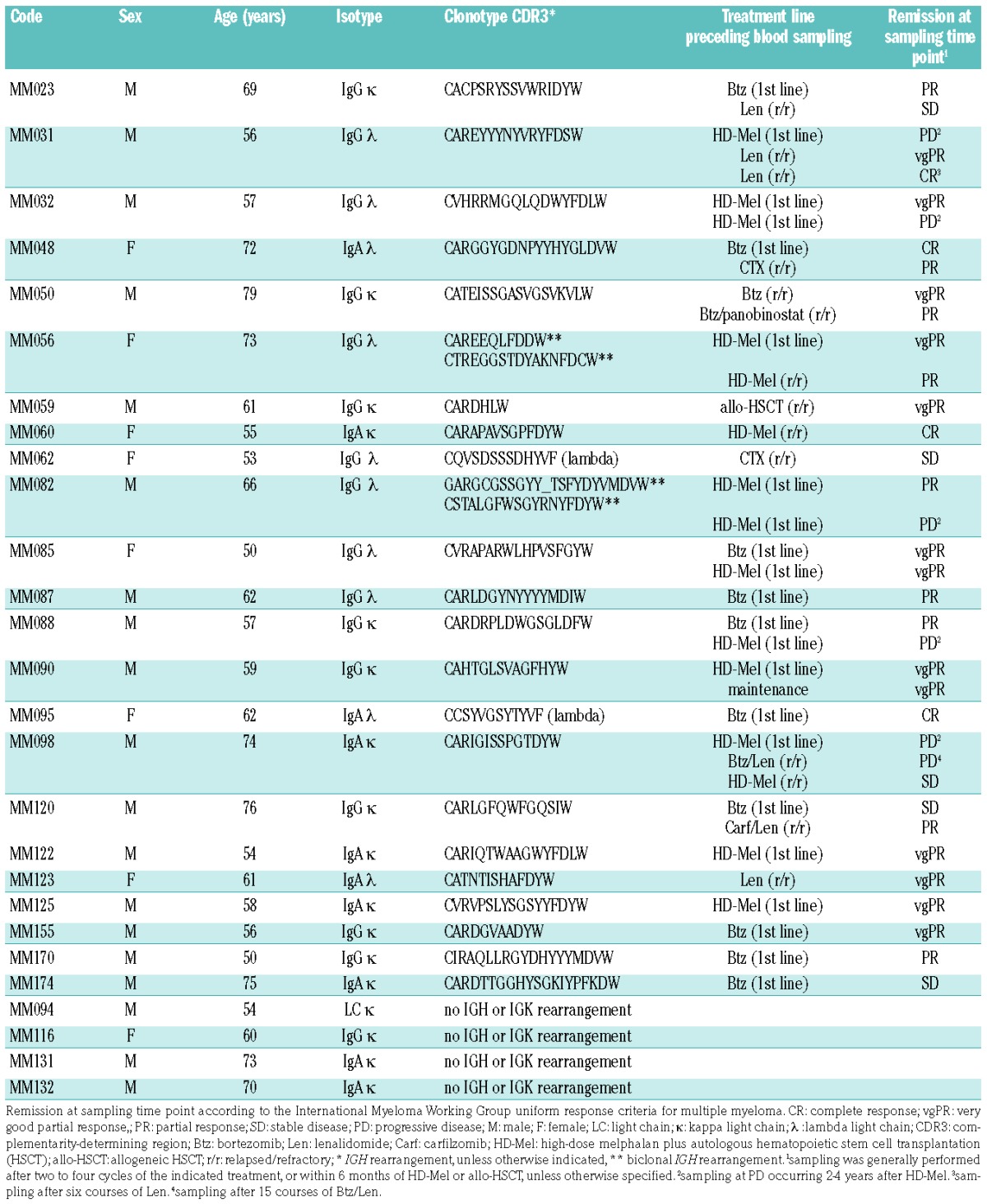
A cohort of 27 patients with multiple myeloma treated at the University Medical Center Hamburg-Eppendorf was investigated as approved by the ethics committee (Ethikkommission der Landesärztekammer Hamburg) and after informed consent from the patients (Table 1). All patients underwent bone marrow sampling, performed at a time point with active disease, to determine the myeloma clonotype sequence (IGH, IGL or IGK) as well as peripheral blood sampling for analysis of V(D)J rearrangements before and during the course of treatment. Baseline clinical characteristics and remission status, according to the International Myeloma Working Group (IMWG), were assessed for all patients.
Table 1.
Patients’ characteristics.
DNA preparation from peripheral blood and bone marrow leukocytes and plasma
Whole blood or bone marrow aspirate was collected into tubes containing heparin and processed within 2 h. Plasma was separated by centrifugation and stored at −80°C. Leukocytes were isolated with erythrocyte lysis using a standard lysis buffer (ammonium chloride 8.29 g/L, EDTA 0.372 g/L, potassium hydrogen carbonate 1 g/L) and frozen in freezing medium (90% fetal bovine serum, Biochrom, VWR, Darmstadt, Germany; 10% dimethyl sulfoxide, Sigma-Aldrich, Taufkirchen, Germany). Genomic DNA was extracted from frozen peripheral blood or bone marrow leukocytes using a Gen Elute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich, Taufkirchen, Germany). Cell-free DNA was extracted from plasma using a QIAamp Circulating Nucleic Acid Kit (QIAGEN, Hilden, Germany). DNA was quantified by absorbance (Nano Drop ND-1000, Peqlab) for genomic DNA or fluorometric methods (Qubit 3.0, Thermo Fischer Scientific) for cell-free DNA.
Next-generation sequencing of V(D)J repertoires
Ig V(D)J segments were amplified with BIOMED2-FR1/-FR3 (IGH), -Ig kappa (IGK) or -Ig lambda (IGL) primer pools17 containing Illumina-compatible adapters and barcodes as described previously with an input of 500 ng (75000 genomes) genomic DNA or 250 ng cell-free DNA.18 Figure 1 shows a scheme of the amplification strategy. The polymerase chain reaction (PCR) product was cleaned-up using SPRIselect reagent (Beckmann Coulter, Brea, CA, USA) and 2 μL eluted DNA were used for a second PCR, during which Illumina adapter sequences were extended and a sample-specific barcode was added. The final PCR product was size-separated with 1.5% agarose gel electrophoresis and amplicons were purified using the NucleoSpin® Gel and PCR Clean-up Kit (Macherey-Nagel, Düren, Germany). The concentration of the final PCR products was determined on Qubit 3.0 (Thermo Fischer Scientific) and amplicon purity was controlled on an Agilent 2100 Bioanalyzer (Agilent Technologies, Böblingen, Germany). NGS was performed on an Illumina MiSeq sequencer with 500 or 600 cycle single-indexed, paired-end runs.
Figure 1.

Scheme of V(D)J DNA amplification and next-generation sequencing from peripheral blood cellular and cell-free DNA. Illumina adapters are shown in green and blue, barcode sequences are shown in red. cfDNA: cell-free DNA; IGH: immunoglobulin heavy chain; IGK/L: immunoglobulin kappa/lambda chain; FW: forward; RV: reverse; R1/R2: read 1/2; NGS: next-generation sequencing.
Data analysis and statistics
Demultiplexing and Fastq formatted data output was generated by the MiSeq reporter. Raw sequences were processed to Ig V(D)J clonotypes based on the MiXCR analysis tool,19 and the different sequencing samples were compared using the tcR R package for Ig analysis.20 Data were plotted using R statistical software tools as well as GraphPad Prism 5. A sample was considered positive when the clonotypic rearrangement was detected at least twice. Differences between two groups were analyzed using the two-sided Student t test and categorical data were compared by the Fisher exact test. Linear regression analyses were performed to evaluate an association between response to treatment and positivity for circulating myeloma cell V(D)J [cmc-V(D)J] and/or cell-free myeloma V(D)J [cfm-V(D)J]. Analyses were carried out using IBM SPSS version 22. A P value of <0.05 was considered statistically significant.
Results and Discussion
Study design
A total of 27 myeloma patients requiring myeloma-directed treatment were included in this investigation. Clonotypic V(D)J rearrangements of the malignant plasma cell were determined from the bone marrow and subsequently used for clonal tracking in peripheral blood leukocyte DNA [cmc-V(D)J] and cell-free DNA [cfm-V(D)J] before and after treatment initiation at routine clinical remission assessments. Blood sampling was performed after two to four courses of the indicated treatment or within 6 months after high-dose melphalan/allogeneic stem cell transplantation, unless specified otherwise. The patients’ characteristics, treatments and sampling time points are summarized in Table 1 and Online Supplementary Table S1. A diagram of the study workflow is shown in Figure 2.
Figure 2.
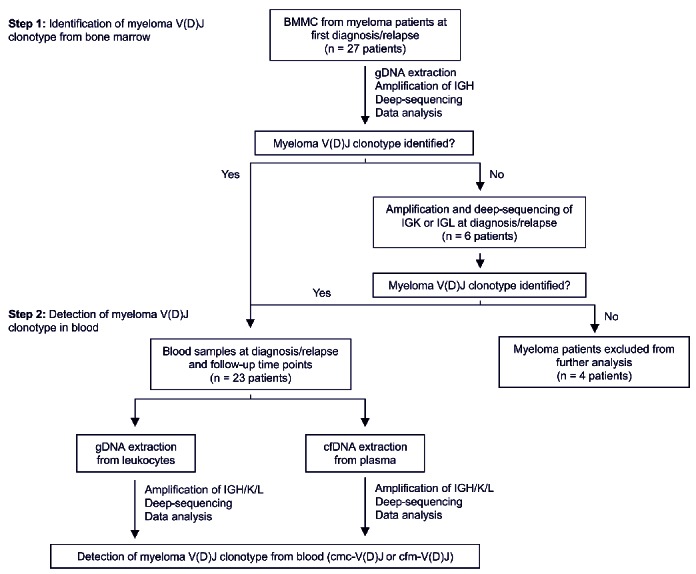
Schematic illustration of study workflow. BMMC: bone marrow mononuclear cells; IGH: immunoglobulin heavy chain; IGK/L: immunoglobulin kappa/lambda chain; gDNA: genomic DNA; cfDNA: cell-free DNA.
Determination of clonotypic myeloma V(D)J rearrangements
In 19 of 27 patients, an unambiguous IGH VDJ rearrangement could be identified, two cases were biclonal and another two patients only showed an IGL VJ rearrangement (Table 1). The four cases without discernable rearrangements were excluded from further analysis.
Next-generation screening for circulating myeloma cell-V(D)J and cell-free myeloma-V(D)J
Based on previous studies,21–23 we established optimal PCR and NGS conditions for comprehensive immune repertoire analysis and high-sensitivity detection of V(D)J rearrangements from leukocyte DNA. A sequencing depth of 80,000 reads per sample was sufficient to comprehensively analyze the B-lineage repertoire from 500 ng genomic or 250 ng cell-free DNA, which can typically be extracted from 1–5 mL of blood (Online Supplementary Figure S1A). To measure the sensitivity of our approach, we spiked monoclonal B-cell DNA, derived from the Burkitt lymphoma cell line DG75, into polyclonal leukocyte DNA and determined detection rates of the clonotypic DG75 V(D)J rearrangement by NGS. These experiments showed high fidelity detection even if only low amounts of clonotypic genomes were spiked into the polyclonal background (Online Supplementary Table S2).
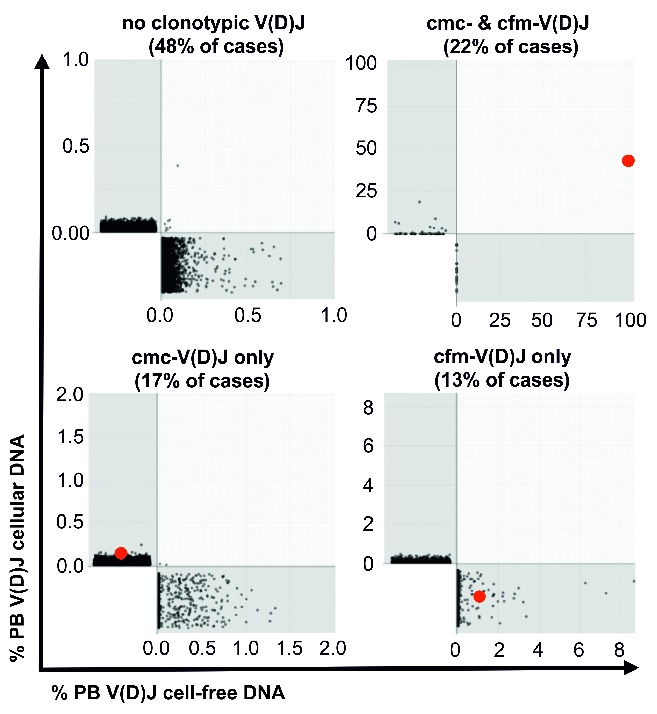
Using these high-sensitivity detection conditions, the 23 cases with a definable myeloma V(D)J rearrangement underwent further screening of blood samples before and after initiation of treatment when routine remission evaluation was performed (Table 1). Figure 3 shows representative baseline bone marrow and peripheral blood V(D)J plots of patient MM123 with evidence of cmc-V(D)J and cfm-V(D)J. Overall, cmc-V(D)J was detectable in 71% and cfm-V(D)J in 100% of cases at the baseline screening (Figure 4A). At the follow-up time points after treatment initiation, cmc-V(D)J was detectable in 40% and cfm-V(D)J in 34% of samples (Figure 4A). For further analyses, V(D)J sampling was considered positive if cmc-V(D)J or cfm-V(D)J or both resulted positive, which was the case in 47% of follow-up samples (Figure 4B). Clear associations were observed between poor remission status (assessed by M-protein-based IMWG criteria) and positive cmc-V(D)J sampling (regression coefficient 1.60; 95% CI: 0.68–2.50; P=0.002) (Figure 4A), evidence of cfm-V(D)J (regression coefficient 1.49; 95% CI: 0.70–2.27; P=0.001) (Figure 4A) as well as detection of V(D)J in at least one compartment (regression coefficient 1.67; 95% CI: 0.82–2.53; P<0.001) (Figure 4B), and 91% of non-responders (patients with stable or progressive disease) remained positive for cmc-/cfm-V(D)J, compared to 41% of responders (patients with partial remission or better) (P<0.001) (Figure 4B). The percentage of clonotypic to polyclonal cellular and cell-free V(D)J DNA did not differ significantly between responders and non-responders/progressors (P=0.170) (Figure 4C).
Figure 3.

Representative V(D)J bone marrow and peripheral blood repertoires of patient MM123 at diagnosis. Every dot represents a clonotypic V(D)J rearrangement within the immunoglobulin repertoire. The size of each dot represents the size of the clone. The malignant plasma cell clone is highlighted in the bone marrow as well as in the cellular and cell-free peripheral blood compartments. The plot was generated using R statistical software tools. BM: bone marrow; PB: peripheral blood.
Figure 4.

Monitoring of circulating myeloma cells [(cmc-V(D)J)] and cell-free myeloma DNA [(cfm-V(D)J)] after myeloma treatment by next-generation sequencing. (A) Positivity of patients’ samples for cmc-V(D)J and cfm-V(D)J at diagnosis/relapse and after treatment, respectively. Remission status is indicated according to the IMWG criteria. (B) Positivity of patients’ samples for V(D)J at diagnosis/relapse and after treatment. Time points were considered V(D)J-positive if the malignant clone was detectable in at least one compartment (cellular or cell-free). (C) Quantification of cmc-/cfm-V(D)J per global number of V(D)J rearrangements per compartment. Patients with PD and SD were summarized as non-responders/progressors and patients with PR, vgPR and CR were summarized as responders. PD: progressive disease; SD: stable disease; PR: partial response; vgPR: very good partial response; CR: complete response.
Detection of cmc-V(D)J was significantly associated with cfm-V(D)J-positivity (P=0.042). Nevertheless, polyclonal V(D)J repertoires were virtually non-overlapping and a discordance of about 30% was noted between cmc-V(D)J and cfm-V(D)J positivity (example repertoire overlaps are shown in Figure 5). This suggests that circulating cellular and cell-free compartments contain complementary information and that cfm-V(D)J may not be generated entirely within the vascular space (from circulating myeloma cells), but that it reflects myeloma burden in extravascular sites such as the bone marrow or extramedullary manifestations. Concurrent investigation of both compartments does, therefore, appear reasonable.
Figure 5.
Overlap of V(D)J repertoires from cellular and cell-free peripheral blood compartments. Shared V(D)J clonotypes from different compartments were calculated using the tcR tool20 and overlap repertoires were plotted using the R statistical software tool. The V(D)J rearrangement of the malignant plasma cell clone [(cmc- and/or cfm-V(D)J)] is shown in red.
One unexpected aspect of our findings was the rather low rate of positivity for cmc-V(D)J (45%) or cfm-V(D)J (39%) or both (68%) in patients with no or incomplete M-protein responses (very good partial response or less). Since our technical approach showed high repertoire coverage as well as high sensitivity of detection, we concluded that this reflects true absence of the clonotypic DNA in the blood-derived genomes used for sequencing library preparation and that only higher genomic input could eventually enhance the sensitivity of detection. To study whether higher genomic PCR input resulted in higher detection rates of the clonotypic V(D)J DNA, we selected two cfm-V(D)J negative cases with progressive disease and scaled up the PCR input from 250 ng (37,500 genomes) to 1250 ng (187,500 genomes), typically extractable from >10 mL of blood. As expected, repertoire diversity increased, but the clonotypic myeloma V(D)J rearrangement could still not be detected in these samples (Online Supplementary Figures S1B–D). This confirmed our previous hypothesis that a fraction of patients with no or incomplete M-protein responses may not release any myeloma DNA into the plasma and that this biomarker potentially has different biological implications than those of M-protein.
Taken together, our pilot study gives valuable biological insights into the circulating cellular and cell-free compartments that can be explored by “liquid biopsy” in multiple myeloma. It indicates that cmc-V(D)J and cfm-V(D)J may decline more promptly in response to effective treatments than the relatively inert M-protein and may, therefore, be more informative regarding cell turnover and potentially suitable for immediate estimation of treatment efficacy or even early prediction of minimal residual disease negativity, not only in patients with low- or asecretory myeloma. Due to the limitations of this study (small cohort size, heterogeneous treatments), the actual predictive significance of rapid clearance of cmc-V(D)J or cfm-V(D)J, but also its persistence in M-protein responders cannot be reliably assessed. Future prospective studies will need to address whether this noninvasive diagnostic tool is of predictive importance and therefore of additional value to the established protein-based monitoring approach in multiple myeloma.
Supplementary Material
Acknowledgments
This study was supported by the Eppendorfer Krebs- und Leukämiehilfe e.V. and the Deutsche Krebshilfe (grant 110906 to MB).
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/6/1105
References
- 1.Dou QP, Zonder JA. Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr Cancer Drug Targets. 2014;14(6):517–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Yang F, Shen Y, et al. Maintenance therapy with immunomodulatory drugs in multiple myeloma: a meta-analysis and systematic review. J Natl Cancer Inst. 2016;108(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621–631. [DOI] [PubMed] [Google Scholar]
- 4.Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373(13):1207–1219. [DOI] [PubMed] [Google Scholar]
- 5.Kristinsson SY, Anderson WF, Landgren O. Improved long-term survival in multiple myeloma up to the age of 80 years. Leukemia. 2014;28(6):1346–1348. [DOI] [PubMed] [Google Scholar]
- 6.Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2013. In: NCI ed. Bethesda, MD; 2016. [Google Scholar]
- 8.Fulciniti M, Munshi NC, Martinez-Lopez J. Deep response in multiple myeloma: a critical review. Biomed Res Int. 2015; 2015:832049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rawstron AC, Gregory WM, de Tute RM, et al. Minimal residual disease in myeloma by flow cytometry: independent prediction of survival benefit per log reduction. Blood. 2015;125(12):1932–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Lopez J, Lahuerta JJ, Pepin F, et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. 2014;123(20):3073–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paiva B, van Dongen JJ, Orfao A. New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood. 2015;125(20):3059–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368(13):1199–1209. [DOI] [PubMed] [Google Scholar]
- 13.Armand P, Oki Y, Neuberg DS, et al. Detection of circulating tumour DNA in patients with aggressive B-cell non-Hodgkin lymphoma. Br J Haematol. 2013;163(1):123–126. [DOI] [PubMed] [Google Scholar]
- 14.Ramirez JM, Fehm T, Orsini M, et al. Prognostic relevance of viable circulating tumor cells detected by EPISPOT in metastatic breast cancer patients. Clin Chem. 2014;60(1):214–221. [DOI] [PubMed] [Google Scholar]
- 15.Roschewski M, Dunleavy K, Pittaluga S, et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: a correlative biomarker study. Lancet Oncol. 2015;16(5): 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurtz DM, Green MR, Bratman SV, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood. 2015;125(24):3679–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. [DOI] [PubMed] [Google Scholar]
- 18.Schliffke S, Akyuz N, Ford CT, et al. Clinical response to ibrutinib is accompanied by normalization of the T-cell environment in CLL-related autoimmune cytopenia. Leukemia. 2016;30(11):2232–2234. [DOI] [PubMed] [Google Scholar]
- 19.Bolotin DA, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12(5):380–381. [DOI] [PubMed] [Google Scholar]
- 20.Nazarov VI, Pogorelyy MV, Komech EA, et al. tcR: an R package for T cell receptor repertoire advanced data analysis. BMC Bioinformatics. 2015;16:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braig F, Marz M, Schieferdecker A, et al. Epidermal growth factor receptor mutation mediates cross-resistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget. 2015;6(14):12035–12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braig F, Voigtlaender M, Schieferdecker A, et al. Liquid biopsy monitoring uncovers acquired RAS-mediated resistance to cetuximab in a substantial proportion of patients with head and neck squamous cell carcinoma. Oncotarget. 2016;7(28):42988–42995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiele B, Kloster M, Alawi M, et al. Next-generation sequencing of peripheral B-lineage cells pinpoints the circulating clonotypic cell pool in multiple myeloma. Blood. 2014;123(23):3618–3621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.