

Chronic neutrophilic leukemia (CNL) is an uncommon BCR-ABL1-negative myeloproliferative neoplasm (MPN). Its true incidence remains unknown with only ~200 cases described in literature, and it is most likely under-diagnosed. The disease course is often heterogeneous, ranging from a relatively indolent disorder in some individuals, to an aggressive disease with rapid blast transformation in others.1 Accordingly, treatment strategies are often heterogeneous, ranging from observation alone, cytoreductive therapy and, in younger and fitter individuals, allogeneic stem cell transplant.2 Recent studies have shown that between 60%–80% of CNL patients harbor somatic activating mutations in the granulocyte-colony stimulating factor receptor (CSF3R) gene.3 Two types of mutations exist: point mutations in the CSF3R extracellular domain (‘membrane proximal mutations’), and nonsense or frame-shift mutations leading to shortening of the receptor tail (‘truncation mutations’).
Previous work has suggested differential sensitivity to kinase inhibitor.3–4 Primary cells from a CNL patient with a truncating mutation CSF3R S783fs displayed marked in vitro sensitivity to dasatinib (Bristol-Myers Squibb, New York City, USA) yet was insensitive to a range of JAK inhibitors.3 In contrast, drug sensitivity profiling of cells derived from CNL individuals with the CSF3R T618I mutation, the most common membrane proximal mutation, displayed resistance to dasatinib but were sensitive to JAK inhibition. Moreover, in vitro assays demonstrated that membrane proximal mutations (T615A and T618I) had augmented ability to transform Ba/F3 cells to interleukin-3 independent growth when compared with truncating mutations (Q741X and S783fs), and were associated with higher levels of phosphorylated JAK2 and STAT3.3 Currently, it is speculated that membrane proximal mutations have the ability to signal in the absence of ligand, whereas truncation mutations lead to receptor overexpression and hypersensitivity to ligands. Analyses revealed that the most common proximal membrane mutation T618I attenuated O-glycosylation of the G-CSFR, leading to greater receptor dimerization and subsequent aberrant signaling. Mutations of SETBP1 have also been described in CNL,4 and both SETBP1 and CSF3R mutations were noted in up to 24% of CNL cases. The prognosis in such cases appears worse, but numbers remain too small for meaningful interpretation. We hereby report, for the first time, a patient with CNL displaying dual proximal membrane and truncating mutations who demonstrated an initial hematological response coupled with a reduction in both mutation allelic burdens following treatment with the JAK1/JAK2 inhibitor ruxolitinib (Novartis Pharmaceuticals, Basel, Switzerland) until molecular relapse occurred.
A 74-year old Caucasian female was referred for investigation of a two-year history of progressive neutrophilia. She described weight loss and progressive fatigue for more than one year. Examination revealed no lymphadenopathy or organomegaly. Full blood count demonstrated hemoglobin (Hb) 118 g/l, white blood cell count (WBC) 54.6 × 109/l, neutrophil count 51.9 × 109/l, and platelet count 181 × 109/l. Peripheral blood film examination confirmed a neutrophil leukocytosis (<10% myelocytes), with no excess of eosinophils or basophils, and no dysplastic features. Bone marrow trephine histology showed a florid increase in granulopoiesis with normal maturation. No dysplastic features were evident. There was no excess of blast cells, or of eosinophils or mast cells. Reticulin fibers were not increased. Conventional karyotypic analysis was normal and there was no evidence of either the JAK2 V617F mutation or BCR-ABL rearrangement.
Amplification and sequencing of CSF3R from exons 14 and 17 from peripheral blood leukocyte DNA using methodology described previously3 identified two mutations - a proximal membrane mutation T618I occurring within exon 14 and G739stop, a truncating mutation, occurring within exon 17. RT-PCR of exons 14–17, followed by cloning and sequencing, indicated that both mutations were on the same allele. No mutations were identified in SETBP1 exon 4. Clinical and laboratory features were therefore in keeping with a diagnosis of CNL.
Given her symptom burden and baseline anemia, ruxolitinib 5 mg twice daily was commenced in February 2014, inducing a rapid hematological response, yet no attenuation of symptom burden. Dose augmentation to 10mg mane and 5mg nocte was attempted for two months to try and improve the symptom response, but this was poorly tolerated, predominantly due to (presumed drug-related) worsening of pre-existing urinary symptoms. Ruxolitinib was temporarily withheld, but this led to a rapid rebound leukocytosis (day 119); the drug was recommenced and the patient subsequently reachieved hematological control 10 days later. Despite her excellent hematological response, it is important to emphasize that the patient did not obtain relief from her fatigue and persistent urinary tract symptoms. She then proceeded to have a urological intervention (removal of right renal chromophobe tumour) and suffered a perioperative infective complication requiring prolonged antibiotic use. At this time, she had haematological relapse from her CNL as shown in Figure 1a at day 784.
Figure 1.
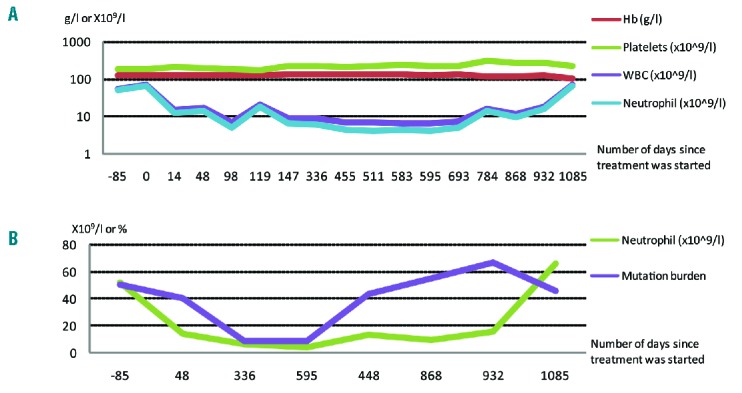
Hematological parameter and mutation burden changes over time. (A) Hematological perimeters of patient before and after treatment with ruxolitinib with the x-axis denoting the day from ruxolitinib commencement; * indicates commencement of ruxolitinib, **indicates temporary withdrawal of ruxolitinib, *** indicates re-initiation of ruxolitinib, **** indicates urological intervention. Y-axis is drawn on logarithmic scale. (B) Neutrophil count and mutation burden expressed as normalized percentage of T618I of patient before and after treatment with ruxolitinib, with the x-axis denoting the day from ruxolitinib commencement.
Remarkably, sequential monitoring of both the CSF3R T618I and CSF3R G739 stop mutation by sequence analysis and digital PCR (T618I only) revealed initial progressive reductions in the allelic burden for both mutations followed by molecular relapse around the perioperative period (day 784), as shown in Figure 1b. The 50% mutation burden prior to treatment is consistent with heterozygous mutations in virtually all cells. Eleven months after starting ruxolitinib, the mutation burden had fallen to 8%, suggesting 16% of cells were mutated, as shown in Figure 2. This level remained essentially constant for the following 9 months. At the time of clinical relapse, the mutation allelic burdens returned to baseline.
Figure 2.
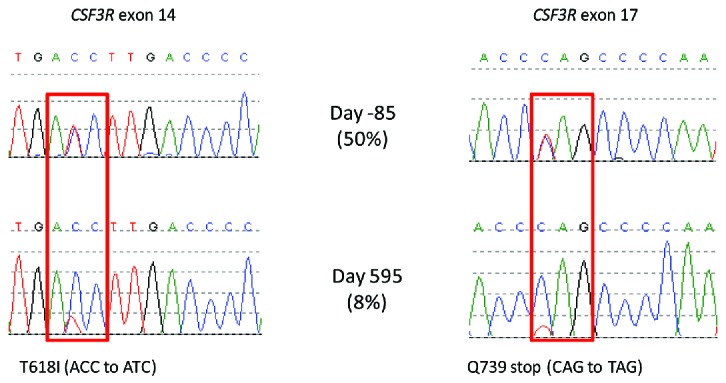
Different CSF3R mutations and their initial responses to treatment. CSF3R exons 14 and 17 were amplified from total peripheral blood leukocyte DNA and Sanger sequenced.5 To quantify the level of the T618I mutation, we designed a droplet digital PCR (ddPCR) assay. The ddPCR reaction mixture consisted of 10μl ddPCR supermix (Bio-Rad, Hemel Hempstead, UK), 250nm wild type probe ([HEX]ACCC[T]GA[T]GA[C]CT[T]GACC[BHQ1]), and mutation specific probe ([6FAM]ACCC[T]GA[T]GA[T]CT[T]GACC[BHQ1]), 900nm forward (CCACCAACAGTACAGTCC) and reverse (ACCAGGGGATTCAAAGTC) primers, and 7.2μl of fragmented DNA at 16.25ng/μl (bases in square brackets are locked nucleic acids). The entire reaction was loaded into a cartridge (Bio-Rad) together with 70μl droplet generation oil (Bio-Rad) and placed into the droplet generator (Bio-Rad). After processing, the droplets were transferred to a 96 well plate which was sealed with a heat sealer. PCR amplification was performed in a Thermal Cycler (Applied Biosystems 2720) using the following cycling conditions: 95°C for 10 mins, 40 cycles of 94°C for 30s and 60.8C for 1 min, 1 cycle of 98°C for 10 mins and ending at 4°C. After amplification, the plate was loaded onto the droplet reader (Bio-Rad) and analysed immediately with Quantasoft analysis software (Bio-Rad).
Experience to date with the clinical and molecular responses induced by ruxolitinib in both CNL and atypical CML (aCML) are limited to single case reports. Maxson et al. were the first to report on a CNL patient with a CSF3R T618I mutation who demonstrated a sustained and significant reduction in neutrophil counts on dose-titrated ruxolitinib.5 In contrast, Lasho et al. reported on a CNL patient with concurrent CSF3R and SETBP1 mutations where single agent ruxolitinib was ineffective in gaining a hematological response.6 Dao et al. reported on an aCML patient with aCSF3R T618I mutation who gained both symptomatic, splenic and hematological improvement following therapy with ruxolitinib yet no significant change in mutation allelic burden when measured at 4-months.7
Our case is unique in that we have demonstrated an excellent, but temporary, hematological response to ruxolitinib coupled with a 9-month reduction in dual mutation allelic burdens. Clinical response was very closely linked with molecular response in our patient. The reason for loss of response to ruxolitinib is unknown at present, but we speculate that it may be akin to heterodimeric JAK-STAT activation as a mechanism of persistence as described by Koppikar et al.8 Of relevance, in the setting of myelofibrosis, longer-term follow up of the phase III COMFORT studies, investigating the efficacy of ruxolitinib, revealed differential reductions in the JAK2 V617F mutation allelic burden in a proportion of JAK inhibitor treated patients.9 However, in CNL there remain many unanswered questions as regards the effects of JAK inhibitor therapy. Though we think achieving molecular response is important, it remains unknown if JAK-inhibitor mediated reductions in mutation allelic burden, as demonstrated temporarily in our case, will alter the natural history of the disease. Does the presence of dual mutations in CSF3R augment aberrant signaling, affecting the disease biology and risk of transformation? Can these be altered by JAK-inhibitor therapy? Moreover, whether the cytokine milieu in CNL is deregulated or not requires evaluation and correlation with patient symptom burden. A prospective phase II trial investigating the safety and efficacy of ruxolitinib in CNL is currently open (clinicaltrials.gov Identifier:02092324) and will enhance our understanding of how JAK inhibitors may affect both disease phenotype and natural history of this intriguing disorder.
Supplementary Material
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Elliott MA, Hanson CA, Dewald GW, Smoley SA, Lasho TL, Tefferi A. WHO-defined chronic neutrophilic leukemia: a long-term analysis of 12 cases and a critical review of the literature. Leukemia. 2005; 19(2):313–317. [DOI] [PubMed] [Google Scholar]
- 2.Bohm J, Schaefer HE. Chronic neutrophilic leukaemia: 14 new cases of an uncommon myeloproliferative disease. J Clin Pathol. 2002; 55(11):862–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gotlib J, Maxson JE, George TI, Tyner JW. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013;122(10):1707–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piazza R, Valletta S, Winkelmann N, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013; 45(1):18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lasho TL, Mims A, Elliott MA, Finke C, Pardanani A, Tefferi A. Chronic neutrophilic leukemia with concurrent CSF3R and SETBP1 mutations: single colony clonality studies, in vitro sensitivity to JAK inhibitors and lack of treatment response to ruxolitinib. Leukemia. 2014;28(6):1363–1365. [DOI] [PubMed] [Google Scholar]
- 7.Dao KH, Solti MB, Maxson JE, et al. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk Res Rep. 2014;3(2):67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koppikar P, Bhagwat N, Kilpivaara O, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012;489(7414):155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047–4053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.