

Abstract
The discovery of mechanisms that alter genetic information via RNA editing or introducing covalent RNA modifications points towards a complexity in gene expression that challenges long-standing concepts. Understanding the biology of RNA modifications represents one of the next frontiers in molecular biology. To this date, over 130 different RNA modifications have been identified, and improved mass spectrometry approaches are still adding to this list. However, only recently has it been possible to map selected RNA modifications at single-nucleotide resolution, which has created a number of exciting hypotheses about the biological function of RNA modifications, culminating in the proposition of the ‘epitranscriptome’. Here, we review some of the technological advances in this rapidly developing field, identify the conceptual challenges and discuss approaches that are needed to rigorously test the biological function of specific RNA modifications.
Keywords: RNA, transcriptome, chemical modification, epigenetics
1. Introduction
Gene expression is a multi-layered process that starts with controlling access to particular sequence information encoded in DNA, followed by copying this information to RNA molecules, which then branch off into transferring their sequence information into polypeptides (as coding RNAs, cRNAs), or which function as non-coding RNAs (ncRNAs). Importantly, the functionality of RNA does not only rely on sequence information. While splicing, and in particular alternative splicing, can diversify RNAs, each and every RNA nucleoside can be chemically modified or even interchanged (RNA-edited). Presently, over 130 post-synthetic RNA nucleoside modifications can be distinguished in different kingdoms [1]. Although the functions of most of these modifications are largely unknown, their presence in many species point towards evolutionarily conserved molecular toolboxes that may modulate the flow of genetic information or allow reacting to environmental challenges. Deciphering how exactly such modulation is achieved is an exciting new frontier in biology. Most of the known RNA modifications map to abundant RNAs (transfer RNA, tRNA; ribosomal RNA, rRNA). Therefore, our understanding of the molecular function of RNA modifications has mostly been shaped by work on tRNAs and rRNAs. In addition, modifications of eukaryotic cRNA ends (i.e. 5′-capping, 3′-tailing) have long been considered to be the only relevant post-transcriptional changes to mRNA. However, already the discovery of intricate and cell-type-specific RNA splicing patterns and the widespread occurrence of RNA editing events indicated that RNAs do not necessarily function as primary transcripts [2,3]. Moreover, internal cRNA modifications such as methyl-6-adenosine (m6A), methyl-5-cytosine (m5C), ribose-methylation (2'-O-Me) and pseudo-uridine (Ψ), although known for over 50 years [4–8], have not been accessible to molecular investigation until very recently. Thanks to improved methodology, including next-generation sequencing (NGS) as well as mass spectrometry, an ‘explosion’ of activity in RNA modification research has started a feverish race aiming to comprehensively map specific RNA editing and modification patterns transcriptome-wide and in various tissues and cell types. These are exciting times, not only for RNA biologists, but also for structural and systems biologists. However, it is important to point out that many of the conclusions in this rapidly developing field are still based on manipulating a limited number of experimental model systems with the focus on a few tractable RNA modifications, while the function of the majority of RNA modification has not been addressed (figure 1). And even though thousands to millions of specific RNA modification events (covalent and edited, respectively) have now been mapped to particular sequences, very little or rigorous follow-up experimentation has been conducted. Consequently, while quantitatively more is known about the position of particular RNA modification sites, obtaining functional insight is lagging behind. Despite these shortcomings, already the mapping of inosine, m6A, Ψ, m5C or m1A, especially to low-abundance RNAs (i.e. cRNAs and long ncRNAs), has given rise to testable hypotheses and has opened new avenues for exploration. Many recent reviews described the technical details of current RNA modification mapping approaches and also discussed the seminal studies in the field. Instead of reiterating the content of other reviews, we will only briefly introduce the technological advances for mapping RNA modifications, but will instead focus on conceptual and technical challenges that arise when studying complex biological systems involving only a few catalytic entities affecting a multitude of substrates. Because of the recent introduction of terms such as ‘RNA epigenetics’ and ‘epitranscriptomics’ into the field, we also aim to critically discuss their definition, especially in the light of an invoked similarity to the complexities observed for epigenetic gene regulation systems.
Figure 1.

Modifications under the surface of detection. Improved sequencing methods have led to the discovery of millions of modification sites in all classes of RNAs. However, efficient detection of modifications is mostly possible at sites that undergo deamination. Given that more than 130 types of modified ribonucleotides are known to date, it can be expected that novel technologies will lead to a huge increase in detectable RNA modifications that are currently hidden underneath the (detection) surface.
2. RNA modification research on the move: recent advances
Understanding the development of organisms has been the focus of most experimental biology during the last 100 years. Ever since covalent RNA modifications were linked to amphibian oocyte development [9], conceptualizing their molecular function centred mostly on cellular proliferation and differentiation. Especially, mutations in tRNA modification enzymes caused developmental aberrations, affected organismal fitness or were incompatible with life altogether (reviewed in [10–13]), which is not surprising, given their prominent roles in protein synthesis. By contrast, no single rRNA modification was found to be essential for ribosome function under standard conditions [14,15]. In support of this notion, ribosomal assembly can be achieved using in vitro-transcribed 23S rRNA [16], which even allows for peptidyl transfer [17], but also in vitro-transcribed tRNAs lacking all modifications are functional in reconstituted protein translation assays [18]. In addition, some tRNA modification mutant phenotypes can be rescued by overexpressing respective tRNAs [19,20], indicating that post-transcriptional RNA modifications are not essential to the basic function of RNAs (at least in protein translation), pointing towards context-dependent functions. Indeed, the phenomenon of codon bias has placed tRNAs and their modifications into a context-dependent light [21–24]. Four major advances have contributed to conceptualizing biologically relevant functions of RNA modifications (figure 2).
Figure 2.

Advances in conceptualizing the dynamic ‘epitranscriptome’. While sequence context-dependent mapping has become a technical reality (a), it is currently not clear how RNA modifications influence each other. Some (but not all) modifications have been shown to be reversible (b), while other modifications have been shown to be inducible (c). The mechanisms of induction and removal of modifications, and in particular the regulatory mechanisms underlying the dynamic landscape of RNA modifications, are poorly understood. As some RNA modifications are specific for certain phyla, their presence or absence can be interpreted as pathogen-associated molecular patterns (PAMPs) and therefore help to distinguish self- from non-self RNAs (d).
2.1. Improved methods for sequence context detection
While physico-chemical detection methods can only report on the presence or absence of a modified nucleoside, adapted NGS technology provided evidence for the notion that virtually every RNA species, including lowly expressed RNAs, is decorated with specific RNA modifications. NGS data mining also pointed to commonalities in the local clustering of particular RNA modifications at functional sequence features, especially in cRNAs such as in untranslated regions (UTRs), transcriptional start sites, exons and introns. For instance, m6A was mostly mapped to the last exon in the majority of cRNAs indicating regulation of 3′ UTR function [25]. By contrast, m1A was enriched in the 5′ UTR and around start codons of human and mouse mRNAs [26,27], indicating roles that are different from m6A. Furthermore, while Ψ appeared to cluster in mRNA coding sequences [28], m5C was mapped at 5′ and 3′ UTRs in mRNAs of a highly unstable cancer cell line [29], at translation start sites in mouse embryonic stem cells (mESCs) and whole brain tissues [30], but also to coding sequences in different mouse tissues [31] and in Arabidopsis [32]. NGS data also revealed that the majority of A-to-I RNA editing events occur in mobile element-derived sequences [33], and not in cRNAs to which editing events had been assigned earlier [34].
2.2. The potential for reversibility
The discovery of enzymatic activities that remove RNA modifications, although presently only documented as integral parts of adenosine methylation systems (reversing m6A, m6Am and m1A) [27,35–37], pointed towards the responsiveness of these modification systems to signals eliciting RNA repair [38] or removal of modified entities when required. In addition, modifications, if not reversed to the unmodified state, can be further modified such as, for instance, Ψ by N1 methylation [39], m5C to various oxidation products by the activity of ten-eleven translocation family enzymes [40] or 3-methylcytosine (m3C) to 3-methyluridine (m3U) [41].
2.3. Dynamic regulation
Exposure of cells or organisms to non-laboratory conditions revealed dynamic responses of RNA modification systems to various stresses. For instance, mass spectrometric analyses detected tRNA modification changes upon exposure to mechanistically different toxins [42], which affected codon usage indicating that stress-specific reprogramming of nucleoside modification contributes to translational control [23,43]. Of note, modifications in rRNA and tRNA are especially abundant in thermophilic organisms, suggesting functional roles at elevated temperatures [44]. For instance, RrmJ (FtsJ), a well-conserved heat-shock protein, is highly induced upon heat stress when it catalyses 2′-O-Me at exactly one U in 23S rRNA affecting the (A)1-site of bacterial ribosomes [45]. Interestingly, Cfr, an enzyme generating C8-methyladenosine, targets bacterial 23S rRNA upon environmental insult, causing resistance to several ribosome-targeted antibiotics [46]. Furthermore, heat shock increased m6A (or m6Am) in 5′ UTRs of mammalian cRNAs, thereby promoting cap-independent translation [47,48]. Also, stress conditions resulting in growth arrest increased m5C at specific positions in yeast tRNA [49], and the activity of Pmt1, a (cytosine-5) RNA methyltransferase homologue, was strongly stimulated by the microbiome-dependent tRNA modification queuosine [50]. Similarly, nutrient deprivation in yeast and serum starvation of human cells induced RNA pseudo-uridylation [51,52]. These findings and the identification of RNA editing and modification events in post-mitotic and adult tissues [53–56] bear witness to the notion that RNA modifications are dynamically placed, can be further modified, repaired or even removed in response to events that are not developmentally programmed, but allow organisms to react to changing environments.
2.4. Molecular pattern recognition determination
RNA modifications contribute to immune system function by acting as discriminators between RNAs originating from different phyla. For instance, modified nucleosides such as m5C, m6A, m5U, s2U or Ψ suppressed signalling of innate RNA sensors such as human toll-like receptors TLR3, TLR7 and TLR8 [57]. Furthermore, a link between MDA5-mediated viral mRNA sensing and 2'-O-Me suggested that RNA modifications act as molecular signatures for the discrimination between RNAs [58]. Supporting this notion, a single 2′-O-Me on Gm18 in tRNA was sufficient to suppress immune stimulation through human TLR7, indicating that, beyond its primary structural role, 2'-O-Me acts as TLR7 signalling antagonist [59,60]. Of note, one isoform of mammalian ADAR1 (p150) contains an interferon-inducible promoter, p150 shuttles between the nucleus and cytoplasm, and activated p150 in virus-infected cells caused an increase in detectable inosines in cellular RNAs [61]. Furthermore, single-stranded and inosine-containing RNAs, after uptake by scavenger class-A receptors and signalling through TLR3 and dsRNA-activated protein kinase, can stimulate the innate immune system [62]. These findings established that RNA modifications facilitate distinguishing host RNAs (self) from foreign RNAs (non-self) [63,64]. Importantly, the biological effects of specific RNA modifications, when introduced into synthetic RNAs, have contributed to the ‘second coming’ of RNA therapeutics [65]. For instance, replacement of every fourth uridine and cytidine with 2-thiouridine and m5C, respectively, decreased binding of synthetic mRNAs to pattern recognition receptors (i.e. TLR3, TLR7, TLR8, RIG-I) in human blood cells [66]. Developing small-interfering RNAs, RNA-based vaccines or mRNA therapeutics regularly includes modifying component RNA strands, which decreases nuclease sensitivity and reduces the activation of the innate immune response [67–70]. Furthering our understanding of the discriminatory potential of specific RNA modifications is holding immense promise for increasing tissue delivery and cellular uptake properties of RNAs, which are still important hurdles to the informed design of RNA-based therapeutics [71]. In summary, recent insights into the placing, dynamics, potential reversibility as well as the immunogenicity of specific RNA modifications are major advances in the field, which are crucial for defining common functional denominators that are needed when conceptualizing the context-dependent roles of RNA modifications.
3. RNA modification research comes of age: conceptual challenges
Understanding the biological impact of modified RNA nucleosides that are often non-abundant and respond dynamically to environmental conditions poses various conceptual challenges (figure 3).
Figure 3.
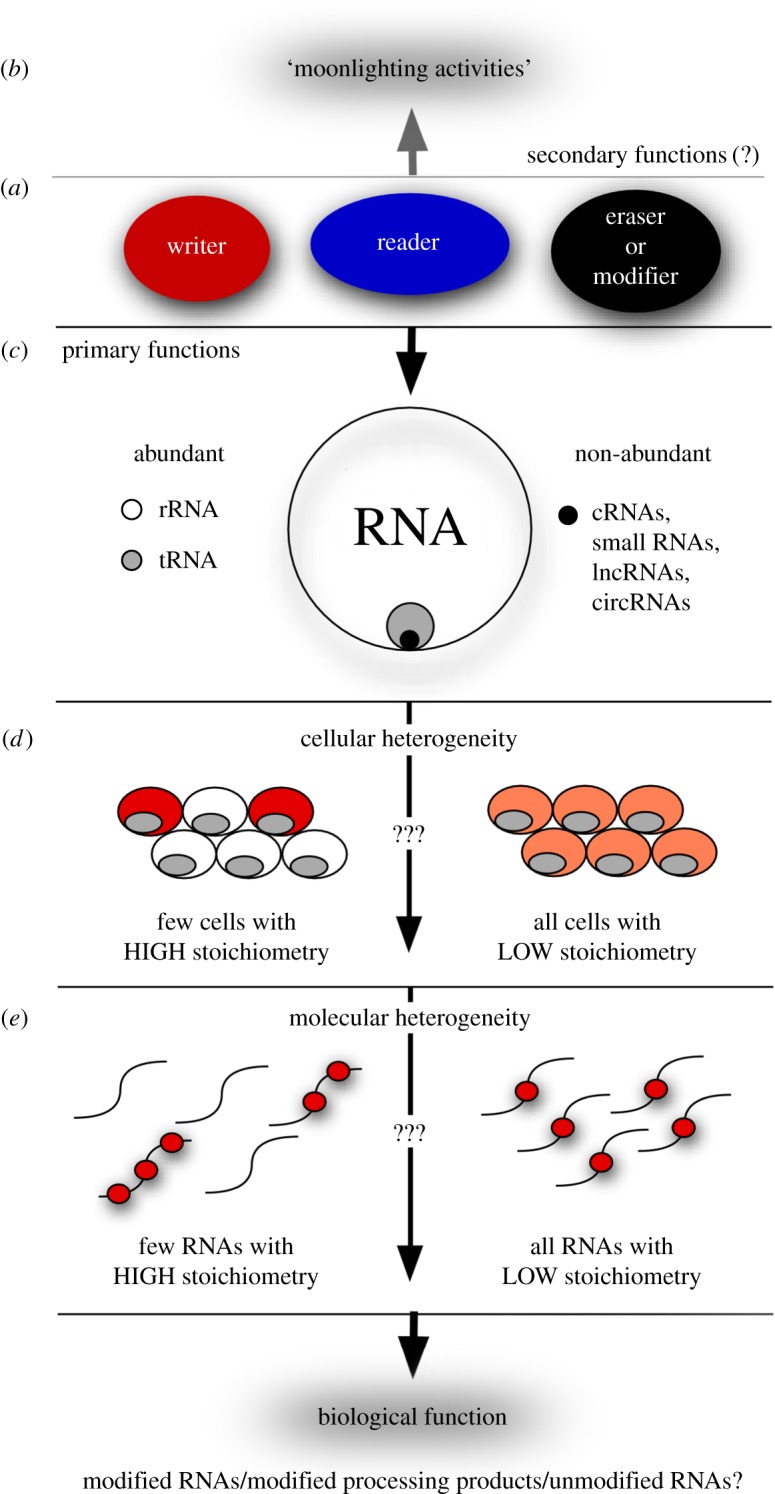
The challenges of the multi-layered world of RNA modifications. RNA modifications are introduced by ‘writer’ enzymes that can then be interpreted by ‘reader’ proteins before being physically removed by ‘erasers’ or further modified by ‘modifiers’ of modifications (a). While few modifications have been described that employ all three classes of proteins, it is conceivable that readers and erasers exist for many more modification types than is currently appreciated. Moreover, all classes of proteins may exhibit moonlighting functions (b), in addition to their reported modification-related activity. The majority of RNA modifications occur in abundant RNA species, such as rRNAs (greater than 90% of all RNAs) and tRNAs (greater than 5% of all RNAs), but also non-abundant RNAs (less than 5% of all RNAs including cRNA, small RNAs, lncRNAs, circRNAs) carry modifications (c). Presently, it is impossible to distinguish whether a given RNA modification in a population of cells is very abundant in some but completely absent from other cells, or alternatively, whether all cells in a population contain a rather low abundance of this particular modification (d). Lastly, even within individual cells, it is unclear whether individual RNAs carry multiple marks of the same modification, while other RNAs remain unmodified, or alternatively, whether modifications are distributed rather homogeneously among all transcripts (e). Addressing these questions will be the next challenge in RNA modification research.
3.1. The impact of modifications in high versus low abundance RNAs
The majority of documented RNA modifications map to abundant ncRNAs (rRNA, and tRNA) [72]. For instance, up to 25% of all nucleotides in tRNAs can be modified. Measured RNA modification stoichiometry in ncRNAs indicates biological relevance [73]. About 1% cellular RNA has coding potential and only a fraction of these cRNAs are modified. In addition, other low-abundance RNAs such as small RNAs and long ncRNAs (lncRNAs) carry modifications. Even circular RNAs (circRNAs) are m6A-modified [74], and high expression of circular RNAs in the nervous system, robust A-to-I editing in brain tissues and ADAR1 knockdown-induced circRNA expression point towards a role for RNA editing in circRNA function [75]. Of note, only a fraction of these low-abundance RNA species appears to be modified by the same enzymatic machineries that also target abundant ncRNAs. This poses the challenge of how to separate the biological effects of an RNA modification at a modifiable position in abundant versus low-abundance RNA.
3.2. Biological function of modified RNA turnover products
Secondary products originating from mature RNAs that were considered to be metabolically stable might also be functionally relevant. For instance, stress-induced processing of various small RNAs (i.e. tRNAs, snoRNAs, vaultRNAs) into smaller RNAs has been observed [76–78]. Whether or not the modification status of these secondary RNAs is different from that of their parental molecules and contributes to their potential functions remains to be tested [79]. The challenge will be separating the functional impact of RNA modifications on molecules of origin and their processing products.
3.3. Appreciating the heterogeneity of RNA modifications
The high stoichiometry of some tractable RNA modifications, especially in ncRNAs, gave rise to the notion that RNA modifications are either present or absent from an RNA species. However, this binary view was challenged by findings showing that tRNA isoacceptors can be incompletely modified at specific positions [80–82]. Recent NGS-based mapping data also indicated the substoichiometric occurrence of RNA modifications in cRNAs. Is the seemingly low occurrence of RNA modifications at sequence-identical RNAs a result of analysing heterogeneous cell populations, failure to exclude technical and analytical artefacts, or does it reflect reality? These questions underscore that accurate quantification of modified nucleosides at high sensitivity remains a pressing technical problem. However, if such substoichiometry reflects cellular reality, how can one conceptualize the biological impact of an RNA modification that only occurs on a few of many sequence-identical RNAs?
3.4. Approaching the single cell ‘epitranscriptome’
Presently, the detection and functional analysis of RNA modifications uses and addresses entire cell populations. Given the low stoichiometry of cRNA modifications, key to assigning biological function will be quantifying actual copy numbers of individual RNAs that are modified, preferably in single cells. Single molecule detection using advanced hybridization and imaging techniques revealed that, on average, mammalian cells only contain 50–70 copies of an individual and well-expressed mRNA, while some mRNAs (i.e. coding for transcription factors) are only present in single-digit copy numbers [83,84]. Furthermore, single-cell RNA sequencing approaches revealed not only quantitative expression differences between cell types [85] but also expression fluctuations in cell populations of the same genetic origin and subtype [86,87]. Therefore, a combination of quantifying RNA copy number and RNA modification status is needed to arrive at new concepts that help distinguishing biologically meaningful signals from background noise.
3.5. Understanding loss-of-function phenotypes
Deducing biological function often relies on interpreting the effects of gene knockouts or targeted mutations reducing catalytic activity of enzymes. In the case of RNA-modifying enzymes with multiple substrates, it is unclear how to assign mutant phenotypes to loss of modification in specific RNA species. For instance, the NOP/Sun2RNA methyltransferase family member 2 (NSUN2) targets the majority of nuclear-encoded tRNAs, but also other cRNAs and ncRNAs [78,88]. A genetic knock-out in mice caused a variety of phenotypes ranging from impaired cellular differentiation to sex-specific infertility [89,90], while human individuals with congenital mutations in a splice-acceptor site of the NSUN2 gene display pleiotropic phenotypes defined by intellectual disability and skin differentiation defects [91,92]. Which methylation site(s) in which RNA substrate(s) cause(s) these phenotypes? While NSUN2 is a robust tRNA methyltransferase on (many) tRNAs, it is unclear which non-modified tRNA species or if any other RNA substrate contributes to the observed pleiotropy of mutant phenotypes. Also, loss of inosines from the transcriptome results in pleiotropic effects. In mammals, adenosines are deaminated to inosines by ADAR1 or ADAR2 [93]. Inosines are interpreted as guanosines during splicing or translation. Most interestingly, ADAR1 and ADAR2 have partially overlapping and distinct substrate specificities [94–96], while loss-of-function phenotypes of both enzymes differ considerably. ADAR2 deletion causes early post-natal lethality in mice, which can be rescued by genomic replacement of a single A-to-G in the Gria2 gene mimicking an abundant editing event at the corresponding site in the mRNA [97]. By contrast, ADAR1-deficient mice die during embryogenesis but can be rescued by deletion of viral RNA sensors, indicating that the presence of inosines in cellular RNAs is required to suppress the activation of innate immunity sensors [98–100]. Interestingly, inosines introduced by ADAR2 are either numerically insufficient or are wrongly placed to effectively suppress innate immune signalling. Furthermore, rescue of ADAR2 deficiency by a ‘pre-edited’ Gria2 allele raises the question as to the functional significance of the many other ADAR2 sites in coding regions, the elucidation of which may require further experimentation beyond the observation of loss-of-function phenotypes under standard laboratory conditions. Similar considerations apply to any multi-substrate RNA modification system, which presents an important conceptual challenge when interpreting RNA modification system mutant phenotypes.
3.6. Distinguishing primary and secondary protein functions
Some RNA modification enzyme null mutant phenotypes are only partially recapitulated in mutants harbouring catalytically dead versions, indicating limits to simplified genotype-to-phenotype inference [101,102]. For instance, a full deletion of the yeast m6A methyltransferase IME4 displayed more severe phenotypes than those caused by a catalytic mutant, suggesting that IME4 may have RNA methylation-independent functions [103,104]. Similarly, METTL3 promotes translation in human cancer cells but independently of m6A catalysis [105]. Null mutations in the human FTO gene, an AlkB subfamily member that reverses m6A methylation through oxidation, link to an autosomal-recessive lethal syndrome in humans [106], and individuals with intron mutations in FTO displayed increased risk of obesity and type 2 diabetes [107,108], indicating that m6A RNA methylation plays a role in the aetiology of metabolic diseases. However, a recent study showed that single-nucleotide polymorphisms (SNPs) in this FTO intron affect the promoter of IRX3, a transcription factor linked to the regulation of body mass in mice, and not FTO expression [109]. Even ADAR1 may have editing-independent functions because a catalytically dead ADAR1 mutation can be fully rescued by a mutation in the viral RNA sensor MDA5, while the same sensor mutation is unable to fully rescue a complete deletion of ADAR1 [100,110]. Supporting the notion of editing-independent functions for ADAR1, overexpression of a catalytic mutant ADAR1 protein in ADAR1 null human ESCs rescued neuronal differentiation defects by affecting miRNA processing [111]. This suggests that ADAR1 may be required for other functions beyond adenosine deamination. Indeed, enzymes can gain activities that are unrelated to their canonical functions. Such ‘moonlighting’ activities have been reported for glycolytic enzymes that shuttle into the nucleus (reviewed in [112]) and metabolic enzymes that display context-dependent RNA binding activities (reviewed in [113]), which might even affect the regulation of RNA-modifying enzymes as observed for mitochondrial tRNA methylation [114]. Therefore, a critical challenge is to link RNA modification system mutant phenotypes to a particular nucleotide modification and to separate them from secondary functions of the associated proteins.
3.7. Semantics
The term ‘RNA epigenetics’ has been coined by invoking similarities between the reversibility of epigenetic DNA or protein modifications and the dynamic nature of one RNA modification, m6A [115]. The potential for modification reversibility is a major part in the epigenetic concept, which poses that functionally relevant changes to the genome brought about by epigenetic mechanisms do not involve nucleotide sequence changes and can be inherited (reviewed in [116]). Accordingly, ‘RNA epigenetics’ would encompass inheritable and functionally relevant RNA modifications, which do not involve nucleotide sequence changes and are reversible. However, such a stringent definition would exclude a number of RNA modifications, especially those that have not been observed to be directly reversible (i.e. Ψ to U, m5C to C, and various editing events by, for instance, reamination) unless one considers further modification of chemical groups as a kind of reversal which could account for neutralizing the impact of the original modification-intrinsic feature. In addition, the heritability of modified RNAs (through cell divisions or generations) has only recently been experimentally addressed. While RNA modifications have been detected in sperm-derived RNA [117], a maternally provided m6A reader protein was shown to mediate accelerated degradation of maternal RNAs in zebra fish, thereby contributing to maternal–zygotic transition (MZT) [118]. However, experimental proof for reversibility and inheritance-related biological functions of other RNA modification systems needs to be provided before labelling all RNA modifications as functional in an epigenetic sense. A more general term, ‘epitranscriptomics’, was introduced after m6A mapping studies showed widespread occurrence in every RNA sequence context [119–121]. Meanwhile, research into the molecular function of adenosine modification systems is fast progressing, implicating m6A, m1A and m6Am in regulating mRNA splicing, export, stability and translation. In fact, the term ‘epitranscriptomics’ is now almost exclusively used for RNA modification systems that target cRNAs [122–125]. This raises the question of whether RNA modifications on abundant ncRNAs should be excluded from the concept of the ‘epitranscriptome’. The recent reporting on the function of tRNA m1A demethylation in the control of protein translation is a reminder that this would be a mistake [126]. Furthermore, agreeing on an all-encompassing ‘epitranscriptome’ definition will be a more daunting task than defining the ‘epigenome’, which, by utmost simplification, is present only on one or two copies of nuclear DNA. By contrast, RNA modification systems act on more than two copies of sequence-identical RNAs, and often on different RNA species. Dissecting such complexities on individual RNA modification systems first might reveal common denominators allowing the synthesis of a more widely applicable definition for the ‘epitranscriptome’ in the future.
4. Detecting RNA modifications: a prerequisite for addressing function
Determining the biological function of RNA modifications requires methodology that allows distinguishing unmodified from modified nucleosides, mapping their distribution and quantifying modified positions. For many years, RNA modifications could mostly be detected by means of their physico-chemical properties using chromatographic methods and mass spectrometry [127]. While these techniques reported mostly on abundant modifications in equally abundant RNA species (i.e. tRNAs, rRNAs), rare modification events, especially in low-abundance RNAs, went largely unnoticed. The single major technological advance that ‘restarted’ and crucially expanded the scope of the RNA modification field was the application of massive parallel sequencing technologies to the detection of low-abundance RNA modification events. Most importantly, NGS-based technologies either preserve or allow deducing RNA sequence context, a necessary prerequisite for experimentally testing the function of a particular RNA modification in a particular RNA sequence. Because the last 5 years of RNA modification research can be described as one large mapping expedition, we will briefly summarize the three basic approaches to using NGS technology for RNA modification mapping.
4.1. Direct RNA modification mapping
Currently, only RNA editing events, which all involve deamination reactions, can be mapped directly because the identity of a deaminated nucleotide can be inferred from its cognate pairing during the reverse transcription reaction, which, when compared with the genomic reference sequence, allows for modification calling. The very first attempt to map RNA modifications on a transcriptome-wide scale made use of sequence information of expressed sequence tags and predicted thousands of A-to-I editing events in humans [128]. Only the introduction of NGS technology allowed more cost-effective experimentation, resulting in crucial insights into the extent of A-to-I editing [129] and revealing that editing is a tissue-specific and developmentally regulated phenomenon [130,131].
4.2. Indirect modification mapping without prior enrichment
All indirect RNA modification-mapping methods without prior RNA enrichment rely on the differential reactivity of a canonical nucleoside when compared with its modified nucleoside. Many such methods exploit the fidelity and/or processivity of reverse transcriptases (RT) allowing ‘RNA modification calling’ at sites that interfered with RT [132]. For instance, all detection methods for pseudouridine (Ψ) employ treatment of RNA with N-cyclohexyl-N′-(2-morpholinoethyl)-carbodiimidemetho-p-toluenesulfonate (CMCT), which introduces Ψ-CMC adducts that terminate cDNA synthesis, thereby revealing potential Ψ positions. This selective labelling chemistry has been combined with NGS to develop Ψ-Seq, Pseudo-seq and PSI-seq, which allow transcriptome-wide Ψ mapping [28,52,133]. A variation for A-to-I editing detection, termed Inosine Chemical Erasing-Seq (ICE-Seq), uses cyanoethylation of exiting inosines, which introduces bulky groups allowing to map inosines indirectly at terminated cDNA reads [55]. In addition, RT-mediated errors at specific sites were shown to be caused by RNA modifications rather than the infidelity rate of RTs [134]. Indeed, positional information on m1A can be inferred indirectly both from aborted cDNA synthesis and from increased nucleotide misincorporation [135]. In addition, chemical deamination using sodium bisulfite is the basis for indirect m5C mapping in RNA using NGS. Methylated cytosines are more refractory to deamination than non-methylated cytosines, which allows determining m5C in its sequence context by a method called RNA bisulfite sequencing [82]. Furthermore, differential RNA stability has been exploited to indirectly map specific RNA modifications. For instance, chemical resistance to nucleophilic cleavage under alkaline conditions of phosphodiester bonds located at 3′ of 2′-O-Me residues is the basis for RiboMethSeq, which allows indirect mapping of 2′-O-Me in RNA using NGS [136].
4.3. Indirect modification mapping after prior enrichment
Various approaches aim at enrichment of modified RNAs before NGS. The most widely used employ antibodies against nucleic acids and their modifications. Because the majority of covalent RNA modifications involve methyl groups [72,137], various antibodies against methylated nucleosides are used for enrichment of RNA fragments containing methylated nucleotides. In addition, UV-induced cross-linking strategies have been incorporated into antibody-mediated enrichment strategies. Photo-cross-linking-assisted m6A sequencing (PA-m6A-seq [138]) combines photoactivatable and ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP). Alternatively, m6A-CLIP and m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (m6A-iCLIP) use cross-linking-induced mutation and RT truncation profiles to reveal the precise position of m6A [25,139]. Furthermore, various mechanism-based enrichment approaches exploit the catalytic steps performed by RNA modification enzymes. For instance, pyrimidine-modifying enzymes can be trapped in denaturant-resistant complexes on substrate molecules containing nucleotide analogues, which has been exploited to enrich for RNA substrates of human Dnmt2/Trdmt1 and NSun2 enzymes using a mechanism-based enrichment method termed Aza-immunoprecipitation, Aza-IP [140]. In addition, mutating particular cysteines in (cytosine-5) RNA methyltransferases allows covalent trapping of substrate RNAs by a method termed methylation-induced cross-linking and immunoprecipitation (miCLiP) [78]. Alternative enrichment strategies employ chemical modification of already modified nucleosides. For instance, selective labelling of Ψ using a chemically synthesized CMC derivative, azido-CMC (N3-CMC), allows conjugating biotin followed by enrichment of biotinylated Ψ-containing RNAs and sequencing by a method called CeU-seq [141]. Also, repurposing the chemical reactivity of known naturally modified nucleosides to create hyper-modifications amenable to selective physical enrichment has been discussed by Phelps et al. [142]. The interested reader is referred to recent excellent reviews that detail the involved methodologies [143,144].
5. Where might RNA modifications be?
Although major advances have been made in developing NGS-based modification mapping technologies, extensive bioinformatics analysis is needed for extracting experimental noise from potential RNA modification signatures. These algorithms employ user-defined stringencies/thresholds and use diverse statistical methods for ‘modification calling’. Bioinformatic output is often taken for granted and not further addressed using alternative methodologies.
5.1. Trust the mapping data?
For instance, RT-mediated ‘modification calling’ relies on significant numbers of reads with specific mis-incorporations or terminations at particular positions. Still, the error rate of Illumina-based sequencing [145] might call artefacts or preclude the accurate detection of less abundant modifications or of modifications that only occur in a small subset of cells or transcripts. Chemical pre-treatment of RNAs might introduce artefacts, as has been observed for RNA bisulfite sequencing [146]. In addition, genomic SNPs might be interpreted as actual RNA modification-induced RT mismatches, and mapping reads across splice junctions or at polyadenylation sites may lead to inaccurately calling false positives, which is a major pitfall for RNA editing mapping, especially in non-model organisms. Considerations of arising problems and suitable mapping tools are reviewed by Conesa et al. [147].
5.2. Trust the antibodies?
Importantly, RNA enrichment approaches are prone to the creation and inclusion of experimental artefacts. For example, antibody-based enrichment of modified RNA fragments critically depends on sufficient antibody specificity. Antibody specificity, although often taken for granted, is indeed a great concern in medical sciences [148]. Serious problems with insufficient antibody specificity have also been encountered in epigenetics research, which crucially relies on mapping histone modifications. For instance, one of the first and widely used antibodies against methylated H3K9 was later shown to significantly cross-react with H3K27 methylation [149], and the quantitative assessment of antibody specificities used for modification detection showed substantial cross-reactivity between antibody preparations [150,151]. Most natural antibodies against nucleic acids are directed either against double-stranded or ribonucleoprotein structures [152]. However, antibody titres against modifications can be induced by immunization with nucleic acid conjugates containing modifications [153,154]. Importantly, only a small portion of a nucleoside (such as a chemical modification) frequently represents the major antigen epitope surface [155], indicating that antibodies to such restricted epitopes will probably bind to other (structurally related) haptens. For example, anti-m7G antibodies cross-reacted with guanosine [155], and electron microscopy revealed that a single anti-m7G antibody bound to the m7G-cap, while an average of three anti-m7G antibodies bound randomly to the remaining RNA molecule [156]. As many covalent RNA modifications involve methyl groups, antibodies against methylated nucleosides could cross-react with similar methyl group-containing epitopes. In addition, some nucleotides contain more than one chemical modification and antibody preparations against one epitope might enrich nucleotides with additional modifications. As an example, anti-m6A antibody preparations were unable to distinguish m6Am (ribose-methylated m6A) from m6A in RNA [37].
5.3. One method to rule them all?
A much-awaited alternative for single-molecule and single-nucleotide resolution mapping is direct RNA sequencing, which does not require RT. This so-called ‘nanopore’ technology was able to directly detect RNA modifications [157–159]. Direct RNA sequencing would also allow addressing the spatial relationship between different RNA modifications on the same transcript, information that no other available technology can provide, but which will be crucial for proving the suggested ‘epitranscriptomic’ interplay of different RNA modifications. However, even if direct RNA sequencing becomes available, the need and urgency for orthogonal validation of detected RNA modifications, preferentially using physico-chemical methods, will remain the same.
6. How many modifications, exactly? Determining stoichiometry will be key
Methods allowing absolute quantification and determining correct stoichiometry of individual RNA modifications are still very much needed because, presently, statements about the actual number of individual RNA modifications are made only in approximation, taking into account transcriptome-wide NGS-derived data and mass spectrometry analysis of species-enriched RNA. Especially, NGS data appear to differ when it comes to reproducibility in modification calling, which is exemplified by the low overlap of Ψ signatures obtained from different carbodiimide-based analyses [160], by discrepancies in m6A mapping results using MeRIP-seq and m6A-seq [121], and by publications that reach contradictory conclusions about the existence of m5C in cRNA of mESCs when using RNA bisulfite sequencing [30,161]. Recently, two methods have been developed that aim to address the stoichiometry of m6A. One low-throughput method, named site-specific cleavage and radioactive-labelling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET), quantifies m6A at candidate loci [162]. Another method, called m6A-level and isoform-characterization sequencing (m6A-LAIC-seq), allows measuring m6A/A stoichiometry in a transcriptome-wide fashion [163]. Present estimates put the ratio of Ψ/U or m6A/A at 0.2–0.6% in cRNA. Estimates for m1A/A range from 0.015 to 0.054% in cell lines to 0.16% in tissues [26], for m5C/C from 0.025 to 0.1% [164], and to about 0.003% for m6Am/A [163]. Such values, which, for instance, translate into an average of three modified positions per 1000 nucleotides per mRNA molecule for m6A, raise questions as to whether only a few sequence-identical RNAs are modified at the same position or if they are modified only in a few analysed cells (figure 3). Even the absolute quantification of RNA editing sites is still problematic because editing is probably a cell-specific phenomenon creating different mapping readouts in different experimental systems. Ultimately, high-sensitivity mass spectrometry methods will be the most accurate technology to correctly quantify RNA modifications. However, enrichment of specific RNAs at sufficient purity will still be required. Purification of mitochondrial tRNAs using a sequential set-up of NHS-coupled DNA oligonucleotides has already been achieved by ‘chaplet column chromatography’ followed by mass spectrometry analysis of tRNA modifications [165]. Hybridization-based enrichment has been applied to more RNA species using ‘reciprocal circulating chromatography’, which allows automated and parallel isolation of multiple RNAs from a complex RNA mixture [166]. However, determining and quantifying RNA modification patterns in such purified RNAs using mass spectrometry still suffers from low sensitivity. Even advanced mass-spectrometry approaches that also conserve RNA sequence information (LC/MS on RNA fragments obtained from RNase T1 digest) are lagging behind in sensitivity (by several orders of magnitude) when compared with methods that quantify single nucleosides. Still, the development of mass-spectrometry technology is ongoing and the interested reader is referred to excellent reviews that summarize the current state of the art of these approaches [73,167].
7. Start to think post-mapping: time for functional experiments
Despite these advances in the RNA modification field, the toolbox for functional studies is still underdeveloped. Thus, molecular insights into the function of individual RNA modifications are still lagging behind. In addition, identifying the molecular interaction networks of RNA modification components, including their expression regulation and subcellular localization, has not been thoroughly addressed. However, deducing these interactions for every modification in every transcript in different cellular and environmental contexts will be an industrial-scale project involving coordinated research efforts akin to the ‘Encode’ and ‘ModEncode’ projects.
7.1. Pick an RNA modification site and … disturb it
With thousands of already mapped RNA modification sites, it is time to address their biological functions, which, surprisingly, only a few studies have attempted. A handful of cRNAs have been studied for the consequences of editing at specific sites [168–172]. Similarly, while it is known that misplacing particular modifications in rRNA affects ribosome function [173], no single site-specific mutagenesis for any covalent cRNA modification that would provide evidence for its function has been reported. In fact, only one report exists, in which two m6A sites were changed in Rous sarcoma virus-derived RNA. However, the mutant virus was as infectious as controls, indicating no impact of these m6A residues on virus infectivity [174]. Importantly, observations that m6A methyltransferases might tolerate degeneracy in the extended consensus sequence indicating compensatory methylation at adjacent sites [175] could not be confirmed when point-mutating various m6A sites in yeast in vivo [176]. Therefore, rather than perturbing the entire editing/modification machinery using genetic mutations, approaches need to be developed that allow specifically depositing, removing or preventing RNA modifications in specific RNAs. Genome editing by, for instance, using modified CRISPR/Cas9 systems [177] will become a pivotal tool. CRISPR/Cas9 technology has already been used to target epigenetic modulators to DNA [178] or to visualize specific RNAs [179], and could be adapted to modulate RNA modification systems accordingly. Importantly, as virtually nothing is known about the interactions of different RNA modifications that have been mapped to the same RNA sequence (but not necessarily to the same RNA molecule), manipulating RNA modifications in close proximity to each other using genome or transcriptome-editing techniques should make in vivo testing of their cis-interactions feasible.
7.2. Choose your experimental playground
The choice of experimental design and model system will influence not only the detection of RNA modifications, but also how to address their dynamics and biological functions. This is highly relevant in the light of the fact that all known RNA modifications contribute to the fine-tuning rather than to the absolute molecular function/fate/destiny of an RNA. For instance, the use of fast-growing, immortalized cell culture systems, while useful for biochemical approaches, might be ill-suited for the robust detection and manipulation of context-dependent RNA modifications. For instance, A-to-I editing studies in most tissue culture cells require ectopic expression of ADARs [180]. By contrast, cancer cell lines often accumulate advantageous genetic aberrations such as duplications of chromosomal regions that also contain RNA modification enzymes such as, for example, the (cytosine-5) RNA methyltransferase NSun2 [181]. In addition, the origin of cells, the donor genotype and their passage number might all influence cellular behaviour, as evidenced by different potentials of induced pluripotent stem cell lines for self-renewal and in vitro differentiation [182]. For instance, it remains unclear how m6A impacts embryonic stem cell (ESC) pluripotency. It was reported that m6A methyltransferase mutants promote ESC self-renewal by maintaining their ground state [183], but also displayed impaired exit from self-renewal and the naive state [184,185], results that indicated different experimental conditions or cell types. Furthermore, context-dependent functions often remain hidden because of limitations as to how to model life in the laboratory. Thus, finding the correct challenges (i.e. stressors) that create conditions amenable for robust experimentation will be an important experimental design goal in RNA modification research. Also, the use of non-model organisms may provide different insights when addressing the biological function of RNA modification systems [186]. In addition, the observed low stoichiometry of many RNA modifications at specific sites necessitates the development of ‘single cell’ approaches. Observed patterns in the localization of individual RNAs (reviewed in [187]) indicate compartmentalized RNA metabolism and the possibility of localized translation [188], including the possibility of localized RNA modification events. Finally, measuring the actual effects of individual RNA modifications in specific RNA molecules on protein translation is presently the biggest knowledge gap, which cannot be bridged by simply correlating RNA expression data with mapped RNA modification patterns but will require the use of ribosome profiling and stable isotope labelling techniques or the development of additional, preferably single-cell technologies, allowing reproducible quantification of protein translation.
7.3. Mechanistic details will be revealed in vitro
However, it remains to be seen if in vivo manipulation of single RNA modification sites (within a transcriptomic ‘ocean’) will make understanding mechanistic principles easier. Alternatively, further development of simplified in vitro reconstitution assays using specifically modified but homogeneous RNA species for functional tests will be useful for identifying mechanistic details such as site selection, protein-binding capabilities as well as structural consequences of particular RNA modifications. Importantly, parameters such as cap-dependency of translation, decapping efficiency, codon usage or splicing in the context of specific mRNA modifications have already been addressed using in vitro assays [37,70,189,190], and could be further modified, including real-time kinetic read-outs.
8. Conclusion
The explosion in RNA modification-related research during the last decade has mostly been fuelled by the success in identifying RNA modifications in their sequence context. First common conceptual denominators as to why RNA modification systems are so abundant and diverse have been delineated. These include that RNA modification systems facilitate discriminating RNAs from one another and that their activities become necessary when adjusting the functionality of specific RNAs to cellular processes (i.e. translation) that need fine-tuning, especially after environmental perturbation. However, neither transcriptome-wide maps of RNA modifications nor simply disrupting RNA modification systems by mutating writers/readers/erasers, while providing positional information and a diversity of phenotypes, will necessarily lead to a better understanding of the probably complex interplay of RNA modifications, their molecular consequences or their context-dependent regulation. Developing functional assays that incorporate positional information from existing high-throughput data will therefore be the challenge for future generations of RNA biologists.
Acknowledgements
We thank the vibrant RNA community in Vienna but also around the world for exciting discussions and for contributing to the immense knowledge gain that has spurred RNA research.
Authors' contributions
M.S. and U.K. designed and drafted the structure of the manuscript. U.K. helped drafting the figures. M.S. and M.F.J. wrote the manuscript. All authors gave final approval for publication.
Competing interests
We declare we have no competing interests.
Funding
Funding was provided by Austrian Science Fund (grant nos. P26845 and P29094).
References
- 1.Machnicka MA, et al. 2013. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 41, D262–D267. (doi:10.1093/nar/gks1007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fersht AR. 1981. Enzymic editing mechanisms and the genetic code. Proc. R. Soc. Lond. B 212, 351–379. (doi:10.1098/rspb.1981.0044) [DOI] [PubMed] [Google Scholar]
- 3.Benne R, Van den Burg J, Brakenhoff JP, Sloof P, Van Boom JH, Tromp MC. 1986. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 46, 819–826. (doi:10.1016/0092-8674(86)90063-2) [DOI] [PubMed] [Google Scholar]
- 4.Desrosiers R, Fridrich K, Rottmann F. 1974. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl Acad. Sci. USA 71, 3971 (doi:10.1073/pnas.71.10.3971) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry RP, Kelley DE, LaTorre J. 1974. Synthesis and turnover of nuclear and cytoplasmic polyadenylic acid in mouse L cells. J. Mol. Biol. 82, 315–331. (doi:10.1016/0022-2836(74)90593-2) [DOI] [PubMed] [Google Scholar]
- 6.Wei CM, Gershowitz A, Moss B. 1975. Methylated nucleotides block 5' terminus of HeLa cell messenger RNA. Cell 4, 379–386. (doi:10.1016/0092-8674(75)90158-0) [DOI] [PubMed] [Google Scholar]
- 7.Cohn WE. 1960. Pseudouridine, a carbon–carbon linked ribonucleoside in ribonucleic acids: isolation, structure, and chemical characteristics. J. Biol. Chem. 235, 1488–1498. [PubMed] [Google Scholar]
- 8.Amos H, Korn M. 1958. 5-methyl cytosine in the RNA of Escherichia coli. Biochim. Biophys. Acta 29, 444–445. (doi:10.1016/0006-3002(58)90214-2) [DOI] [PubMed] [Google Scholar]
- 9.Ozban N, Tandler J, Sirlin JL. 1964. Methylation of nucleolar RNA during development of the amphibian ooecyte. J. Embryol. Exp. Morphol. 12, 373–380. [PubMed] [Google Scholar]
- 10.Freed EF, Bleichert F, Dutca LM, Baserga SJ. 2010. When ribosomes go bad: diseases of ribosome biogenesis. Mol. BioSyst. 6, 481–493. (doi:10.1039/b919670f) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarin LP, Leidel SA. 2013. Modify or die?—RNA modification defects in metazoans. RNA Biol. 11, 1555–1567. (doi:10.4161/15476286.2014.992279) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirchner S, Ignatova Z. 2014. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 16, 98–112. (doi:10.1038/nrg3861) [DOI] [PubMed] [Google Scholar]
- 13.Tuorto F, Lyko F. 2016. Genome recoding by tRNA modifications. Open Biol. 6, 160287 (doi:10.1098/rsob.160287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Decatur WA, Fournier MJ. 2002. rRNA modifications and ribosome function. Trends Biochem. Sci. 27, 344–351. (doi:10.1016/S0968-0004(02)02109-6) [DOI] [PubMed] [Google Scholar]
- 15.Sharma S, Lafontaine DLJ. 2015. ‘View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem. Sci. 40, 560–575. (doi:10.1016/j.tibs.2015.07.008) [DOI] [PubMed] [Google Scholar]
- 16.Green R, Noller HF. 1999. Reconstitution of functional 50S ribosomes from in vitro transcripts of Bacillus stearothermophilus 23S rRNA. Biochemistry 38, 1772–1779. (doi:10.1021/bi982246a) [DOI] [PubMed] [Google Scholar]
- 17.Semrad K, Green R. 2002. Osmolytes stimulate the reconstitution of functional 50S ribosomes from in vitro transcripts of Escherichia coli 23S rRNA. RNA 8, 401–411. (doi:10.1017/S1355838202029722) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cload ST, Liu DR, Froland WA, Schultz PG. 1996. Development of improved tRNAs for in vitro biosynthesis of proteins containing unnatural amino acids. Chem. Biol. 3, 1033–1038. (doi:10.1016/S1074-5521(96)90169-6) [DOI] [PubMed] [Google Scholar]
- 19.Esberg A, Huang B, Johansson MJO, Byström AS. 2006. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell 24, 139–148. (doi:10.1016/j.molcel.2006.07.031) [DOI] [PubMed] [Google Scholar]
- 20.Leidel S, et al. 2009. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 458, 228–232. (doi:10.1038/nature07643) [DOI] [PubMed] [Google Scholar]
- 21.Hopper AK, Phizicky EM. 2003. tRNA transfers to the limelight. Genes Dev. 17, 162–180. (doi:10.1101/gad.1049103) [DOI] [PubMed] [Google Scholar]
- 22.Duechler M, Leszczyńska G, Sochacka E, Nawrot B. 2016. Nucleoside modifications in the regulation of gene expression: focus on tRNA. Cell. Mol. Life Sci. 73, 3075–3095. (doi:10.1007/s00018-016-2217-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan CTY, Pang YLJ, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC. 2012. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3, 937 (doi:10.1038/ncomms1938) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu C, Begley TJ, Dedon PC. 2014. tRNA modifications regulate translation during cellular stress. FEBS Lett. 588, 4287–4296. (doi:10.1016/j.febslet.2014.09.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ke S, et al. 2015. A majority of m6A residues are in the last exons, allowing the potential for 3' UTR regulation. Genes Dev. 29, 2037–2053. (doi:10.1101/gad.269415.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dominissini D, et al. 2016. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 530, 441–446. (doi:10.1038/nature16998) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, Yi C. 2016. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 12, 311–316. (doi:10.1038/nchembio.2040) [DOI] [PubMed] [Google Scholar]
- 28.Schwartz S, et al. 2014. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162. (doi:10.1016/j.cell.2014.08.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Squires JEJ, Patel HRH, Nousch MM, Sibbritt TT, Humphreys DTD, Parker BJB, Suter CMC, Preiss TT. 2012. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40, 5023–5033. (doi:10.1093/nar/gks144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amort T, Rieder D, Wille A, Khokhlova-Cubberley D, Riml C, Trixl L, Jia X-Y, Micura R, Lusser A. 2017. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 18, 1 (doi:10.1186/s13059-016-1139-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X, et al. 2017. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 27, 606–625. (doi:10.1038/cr.2017.55) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.David R, Burgess A, Parker B, Li J, Pulsford K, Sibbritt T, Preiss T, Searle IR. 2017. Transcriptome-wide mapping of RNA 5-methylcytosine in Arabidopsis mRNAs and non-coding RNAs. Plant Cell 29, 445–460. (doi:10.1105/tpc.16.00751) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bazak L, et al. 2014. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24, 365–376. (doi:10.1101/gr.164749.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Connell MA. 1997. RNA editing: rewriting receptors. Curr. Biol. 7, R437–R439. (doi:10.1016/S0960-9822(06)00212-0) [DOI] [PubMed] [Google Scholar]
- 35.Zheng G, et al. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29. (doi:10.1016/j.molcel.2012.10.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia G, et al. 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887. (doi:10.1038/nchembio.687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mauer J, et al. 2017. Reversible methylation of m(6)Am in the 5' cap controls mRNA stability. Nature 541, 371–375. (doi:10.1038/nature21022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falnes PØ. 2005. RNA repair—the latest addition to the toolbox for macromolecular maintenance. RNA Biol. 2, 14–16. (doi:10.4161/rna.2.1.1602) [DOI] [PubMed] [Google Scholar]
- 39.Meyer B, et al. 2011. The Bowen-Conradi syndrome protein Nep1 (Emg1) has a dual role in eukaryotic ribosome biogenesis, as an essential assembly factor and in the methylation of Ψ1191 in yeast 18S rRNA. Nucleic Acids Res. 39, 1526–1537. (doi:10.1093/nar/gkq931) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu L, et al. 2014. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 136, 11 582–11 585. (doi:10.1021/ja505305z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubio MAT, Gaston KW, McKenney KM, Fleming IMC, Paris Z, Limbach PA, Alfonzo JD. 2017. Editing and methylation at a single site by functionally interdependent activities. Nature 542, 494–497. (doi:10.1038/nature21396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan CTY, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. 2010. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 6, e1001247 (doi:10.1371/journal.pgen.1001247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patil A, Dyavaiah M, Joseph F, Rooney JP, Chan CTY, Dedon PC, Begley TJ. 2012. Increased tRNA modification and gene-specific codon usage regulate cell cycle progression during the DNA damage response. Cell Cycle 11, 3656–3665. (doi:10.4161/cc.21919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noon KR, Bruenger E, McCloskey JA. 1998. Posttranscriptional modifications in 16S and 23S rRNAs of the archaeal hyperthermophile Sulfolobus solfataricus. J. Bacteriol. 180, 2883–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bügl H, Fauman EB, Staker BL, Zheng F, Kushner SR, Saper MA, Bardwell JC, Jakob U. 2000. RNA methylation under heat shock control. Mol. Cell 6, 349–360. (doi:10.1016/S1097-2765(00)00035-6) [DOI] [PubMed] [Google Scholar]
- 46.Giessing AMB, Jensen SS, Rasmussen A, Hansen LH, Gondela A, Long K, Vester B, Kirpekar F. 2009. Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA 15, 327–336. (doi:10.1261/rna.1371409) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian S.-B, Jaffrey SR. 2015. 5' UTR m(6)A promotes cap-independent translation. Cell 163, 999–1010. (doi:10.1016/j.cell.2015.10.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao G, et al. 2015. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 526, 591–594. (doi:10.1038/nature15377) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Preston MAM, D'Silva SS, Kon YY, Phizicky EME. 2013. tRNAHis 5-methylcytidine levels increase in response to several growth arrest conditions in Saccharomyces cerevisiae. RNA 19, 243–256. (doi:10.1261/rna.035808.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Müller M, et al. 2015. Dynamic modulation of Dnmt2-dependent tRNA methylation by the micronutrient queuine. Nucleic Acids Res. 43, 10 952–10 962. (doi:10.1093/nar/gkv980) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu G, Xiao M, Yang C, Yu Y-T. 2011. U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. EMBO J. 30, 79–89. (doi:10.1038/emboj.2010.316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. 2014. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146. (doi:10.1038/nature13802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hwang T, et al. 2016. Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 19, 1093–1099. (doi:10.1038/nn.4337) [DOI] [PubMed] [Google Scholar]
- 54.Dillman AA, Hauser DN, Gibbs JR, Nalls MA, McCoy MK, Rudenko IN, Galter D, Cookson MR. 2013. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat. Neurosci. 16, 499–506. (doi:10.1038/nn.3332) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sakurai M, et al. 2014. A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res. 24, 522–534. (doi:10.1101/gr.162537.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Widagdo J, et al. 2016. Experience-dependent accumulation of N6-methyladenosine in the prefrontal cortex is associated with memory processes in mice. J. Neurosci. 36, 6771–6777. (doi:10.1523/JNEUROSCI.4053-15.2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karikó K., Buckstein M, Ni H, Weissman D. 2005. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175. (doi:10.1016/j.immuni.2005.06.008) [DOI] [PubMed] [Google Scholar]
- 58.Zust R, et al. 2011. Ribose 2'-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12, 137–143. (doi:10.1038/ni.1979) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jöckel S, et al. 2012. The 2'-O-methylation status of a single guanosine controls transfer RNA-mediated Toll-like receptor 7 activation or inhibition. J. Exp. Med. 209, 235–241. (doi:10.1084/jem.20111075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gehrig S, et al. 2012. Identification of modifications in microbial, native tRNA that suppress immunostimulatory activity. J. Exp. Med. 209, 225–233. (doi:10.1084/jem.20111044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hood JL, Morabito MV, Martinez CR, Gilbert JA, Ferrick EA, Ayers GD, Chappell JD, Dermody TS, Emeson RB. 2014. Reovirus-mediated induction of ADAR1 (p150) minimally alters RNA editing patterns in discrete brain regions. Mol. Cell. Neurosci. 61, 97–109. (doi:10.1016/j.mcn.2014.06.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao J-Y, Thakur SA, Zalinger ZB, Gerrish KE, Imani F. 2011. Inosine-containing RNA is a novel innate immune recognition element and reduces RSV infection. PLoS ONE 6, e26463 (doi:10.1371/journal.pone.0026463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dalpke A, Helm M. 2012. RNA mediated Toll-like receptor stimulation in health and disease. RNA Biol. 9, 828–842. (doi:10.4161/rna.20206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Connell MA, Mannion NM, Keegan LP. 2015. The epitranscriptome and innate immunity. PLoS Genet. 11, e1005687 (doi:10.1371/journal.pgen.1005687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conde J, Artzi N. 2015. Are RNAi and miRNA therapeutics truly dead? Trends Biotechnol. 33, 141–144. (doi:10.1016/j.tibtech.2014.12.005) [DOI] [PubMed] [Google Scholar]
- 66.Kormann MSD, et al. 2011. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 29, 154–157. (doi:10.1038/nbt.1733) [DOI] [PubMed] [Google Scholar]
- 67.Tiemann K, Rossi JJ. 2009. RNAi-based therapeutics-current status, challenges and prospects. EMBO Mol. Med. 1, 142–151. (doi:10.1002/emmm.200900023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shukla S, Sumaria CS, Pradeepkumar PI. 2010. Exploring chemical modifications for siRNA therapeutics: a structural and functional outlook. ChemMedChem 5, 328–349. (doi:10.1002/cmdc.200900444) [DOI] [PubMed] [Google Scholar]
- 69.Singh MS, Peer D. 2016. RNA nanomedicines: the next generation drugs? Curr. Opin. Biotechnol. 39, 28–34. (doi:10.1016/j.copbio.2015.12.011) [DOI] [PubMed] [Google Scholar]
- 70.Karikó K., Muramatsu H, Ludwig J, Weissman D. 2011. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 39, e142 (doi:10.1093/nar/gkr695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reautschnig P, Vogel P, Stafforst T. In press. The notorious R.N.A. in the spotlight—drug or target for the treatment of disease. RNA Biol. (doi:10.1080/15476286.2016.1208323) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Motorin Y, Helm M. 2011. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2, 611–631. (doi:10.1002/wrna.79) [DOI] [PubMed] [Google Scholar]
- 73.Kellner S, Ochel A, Thüring K, Spenkuch F, Neumann J, Sharma S, Entian K-D, Schneider D, Helm M. 2014. Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res. 42, e142 (doi:10.1093/nar/gku733) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Y, et al. 2017. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 27, 626–641. (doi:10.1038/cr.2017.31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rybak-Wolf A, et al. 2015. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 58, 870–885. (doi:10.1016/j.molcel.2015.03.027) [DOI] [PubMed] [Google Scholar]
- 76.Thompson DM, Lu C, Green PJ, Parker R. 2008. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 14, 2095–2103. (doi:10.1261/rna.1232808) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taft RJ, Glazov EA, Lassmann T, Hayashizaki Y, Carninci P, Mattick JS. 2009. Small RNAs derived from snoRNAs. RNA 15, 1233–1240. (doi:10.1261/rna.1528909) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hussain S, et al. 2013. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 4, 255–261. (doi:10.1016/j.celrep.2013.06.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Durdevic ZZ, Schaefer MM. 2013. tRNA modifications: necessary for correct tRNA-derived fragments during the recovery from stress? Bioessays 35, 323–327. (doi:10.1002/bies.201200158) [DOI] [PubMed] [Google Scholar]
- 80.Chan JC, Yang JA, Dunn MJ, Agris PF, Wong TW. 1982. The nucleotide sequence of a glutamine tRNA from rat liver. Nucleic Acids Res. 10, 3755–3758. (doi:10.1093/nar/10.12.3755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Helm M, Florentz C, Chomyn A, Attardi G. 1999. Search for differences in post-transcriptional modification patterns of mitochondrial DNA-encoded wild-type and mutant human tRNALys and tRNALeu(UUR). Nucleic Acids Res. 27, 756–763. (doi:10.1093/nar/27.3.756) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schaefer M, Pollex T, Hanna K, Lyko F. 2009. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 37, e12 (doi:10.1093/nar/gkn954) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, Wold BJ. 2014. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 24, 496–510. (doi:10.1101/gr.161034.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zenklusen D, Larson DR, Singer RH. 2008. Single-RNA counting reveals alternative modes of gene expression in yeast. Nat. Struct. Mol. Biol. 15, 1263–1271. (doi:10.1038/nsmb.1514) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Islam S, Kjällquist U, Moliner A, Zajac P, Fan J-B, Lönnerberg P, Linnarsson S. 2011. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res 21, 1160–1167. (doi:10.1101/gr.110882.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saliba A-E, Westermann AJ, Gorski SA, Vogel J. 2014. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 42, 8845–8860. (doi:10.1093/nar/gku555) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vera M, Biswas J, Senecal A, Singer RH, Park HY. 2016. Single-cell and single-molecule analysis of gene expression regulation. Annu. Rev. Genet. 50, 267–291. (doi:10.1146/annurev-genet-120215-034854) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Khoddami V, Cairns BR. 2013. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 31, 458–464. (doi:10.1038/nbt.2566) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blanco S, Kurowski A, Nichols J, Watt FM, Benitah SA, Frye M. 2011. The RNA-methyltransferase Misu (NSun2) poises epidermal stem cells to differentiate. PLoS Genet. 7, e1002403 (doi:10.1371/journal.pgen.1002403) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hussain S, et al. 2013. The mouse cytosine-5 RNA methyltransferase NSun2 is a component of the chromatoid body and required for testis differentiation. Mol. Cell. Biol. 33, 1561–1570. (doi:10.1128/MCB.01523-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khan MA, et al. 2012. Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. Am. J. Hum. Genet. 90, 856–863. (doi:10.1016/j.ajhg.2012.03.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, Frye M, Al-Gazali L, Gleeson JG. 2012. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 49, 380–385. (doi:10.1136/jmedgenet-2011-100686) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nishikura K. 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 79, 321–349. (doi:10.1146/annurev-biochem-060208-105251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 279, 4894–4902. (doi:10.1074/jbc.M311347200) [DOI] [PubMed] [Google Scholar]
- 95.Riedmann EM, Schopoff S, Hartner JC, Jantsch MF. 2008. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 14, 1110–1118. (doi:10.1261/rna.923308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stefl R, Allain FHT. 2005. A novel RNA pentaloop fold involved in targeting ADAR2. RNA 11, 592–597. (doi:10.1261/rna.7276805) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406, 78–81. (doi:10.1038/35017558) [DOI] [PubMed] [Google Scholar]
- 98.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944. (doi:10.1016/j.immuni.2015.11.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. (doi:10.1126/science.aac7049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mannion NM, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9, 1482–1494. (doi:10.1016/j.celrep.2014.10.041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tollervey D, Lehtonen H, Jansen R, Kern H, Hurt EC. 1993. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell 72, 443–457. (doi:10.1016/0092-8674(93)90120-F) [DOI] [PubMed] [Google Scholar]
- 102.Zebarjadian Y, King T, Fournier MJ, Clarke L, Carbon J. 1999. Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol. Cell. Biol. 19, 7461–7472. (doi:10.1128/MCB.19.11.7461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA. 2002. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 30, 4509–4518. (doi:10.1093/nar/gkf573) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Agarwala SD, Blitzblau HG, Hochwagen A, Fink GR. 2012. RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLoS Genet. 8, e1002732 (doi:10.1371/journal.pgen.1002732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin S, Choe J, Du P, Triboulet R, Gregory RI. 2016. The m(6)A Methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345. (doi:10.1016/j.molcel.2016.03.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Boissel S, et al. 2009. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am. J. Hum. Genet. 85, 106–111. (doi:10.1016/j.ajhg.2009.06.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dina C, et al. 2007. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 39, 724–726. (doi:10.1038/ng2048) [DOI] [PubMed] [Google Scholar]
- 108.Frayling TM, et al. 2007. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894. (doi:10.1126/science.1141634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smemo S, et al. 2014. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375. (doi:10.1038/nature13138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liddicoat BJ, Chalk AM, Walkley CR. 2016. ADAR1, inosine and the immune sensing system: distinguishing self from non-self. Wiley Interdiscip. Rev. RNA 7, 157–172. (doi:10.1002/wrna.1322) [DOI] [PubMed] [Google Scholar]
- 111.Chen T, et al. 2015. ADAR1 is required for differentiation and neural induction by regulating microRNA processing in a catalytically independent manner. Cell Res. 25, 459–476. (doi:10.1038/cr.2015.24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boukouris AE, Zervopoulos SD, Michelakis ED. 2016. Metabolic enzymes moonlighting in the nucleus: metabolic regulation of gene transcription. Trends Biochem. Sci. 41, 712–730. (doi:10.1016/j.tibs.2016.05.013) [DOI] [PubMed] [Google Scholar]
- 113.Castello A, Hentze MW, Preiss T. 2015. Metabolic enzymes enjoying new partnerships as RNA-binding proteins. Trends Endocrinol. Metab. 26, 746–757. (doi:10.1016/j.tem.2015.09.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J, Rossmanith W. 2012. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase—extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 40, 11 583–11 593. (doi:10.1093/nar/gks910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.He C. 2010. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 6, 863–865. (doi:10.1038/nchembio.482) [DOI] [PubMed] [Google Scholar]
- 116.Bird A. 2007. Perceptions of epigenetics. Nature 447, 396–398. (doi:10.1038/nature05913) [DOI] [PubMed] [Google Scholar]
- 117.Chen Q, et al. 2016. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 351, 397–400. (doi:10.1126/science.aad7977) [DOI] [PubMed] [Google Scholar]
- 118.Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C. 2017. m(6)A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 52, 475–478. (doi:10.1038/nature21355) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Meyer KDK, et al. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149, 1635–1646. (doi:10.1016/j.cell.2012.05.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dominissini D, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. (doi:10.1038/nature11112) [DOI] [PubMed] [Google Scholar]
- 121.Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE. 2012. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 13, 175 (doi:10.1186/gb-2012-13-10-175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li S, Mason CE. 2013. The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genomics Hum. Genet. 15, 127–150. (doi:10.1146/annurev-genom-090413-025405) [DOI] [PubMed] [Google Scholar]
- 123.Meyer KD, Jaffrey SR. 2014. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 15, 313–326. (doi:10.1038/nrm3785) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schwartz S. 2016. Cracking the epitranscriptome. RNA 22, 169–174. (doi:10.1261/rna.054502.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roundtree IA, He C. 2016. RNA epigenetics—chemical messages for posttranscriptional gene regulation. Curr. Opin. Chem. Biol. 30, 46–51. (doi:10.1016/j.cbpa.2015.10.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu F, et al. 2016. ALKBH1-mediated tRNA demethylation regulates translation. Cell 167, 816–828. (doi:10.1016/j.cell.2016.09.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kellner S, Burhenne J, Helm M. 2010. Detection of RNA modifications. RNA Biol. 7, 237–247. (doi:10.4161/rna.7.2.11468) [DOI] [PubMed] [Google Scholar]
- 128.Levanon EY, et al. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22, 1001–1005. (doi:10.1038/nbt996) [DOI] [PubMed] [Google Scholar]
- 129.Li JB, Levanon EY, Yoon J-K, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. 2009. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324, 1210–1213. (doi:10.1126/science.1170995) [DOI] [PubMed] [Google Scholar]
- 130.Huntley MA, Lou M, Goldstein LD, Lawrence M, Dijkgraaf GJP, Kaminker JS, Gentleman R. 2016. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genomics 17, 61 (doi:10.1186/s12864-015-2291-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Picardi E, Manzari C, Mastropasqua F, Aiello I, D'Erchia AM, Pesole G. 2015. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci. Rep. 5, 14941 (doi:10.1038/srep14941) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Behm-Ansmant I, Helm M, Motorin Y. 2011. Use of specific chemical reagents for detection of modified nucleotides in RNA. J. Nucleic Acids 2011, 408053 (doi:10.4061/2011/408053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lovejoy AF, Riordan DP, Brown PO. 2014. Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE 9, e110799 (doi:10.1371/journal.pone.0110799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ebhardt HA, Tsang HH, Dai DC, Liu Y, Bostan B, Fahlman RP. 2009. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 37, 2461–2470. (doi:10.1093/nar/gkp093) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tserovski L, Marchand V, Hauenschild R, Blanloeil-Oillo F, Helm M, Motorin Y. 2016. High-throughput sequencing for 1-methyladenosine (m(1)A) mapping in RNA. Methods 107, 110–121. (doi:10.1016/j.ymeth.2016.02.012) [DOI] [PubMed] [Google Scholar]
- 136.Birkedal U, Christensen Dalsgaard M, Krogh N, Sabarinathan R, Gorodkin J, Nielsen H. 2015. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. Engl. 54, 451–455. (doi:10.1002/anie.201408362) [DOI] [PubMed] [Google Scholar]
- 137.Grosjean H. 2009. DNA and RNA modification enzymes. Austin, TX: Landes Bioscience. [Google Scholar]
- 138.Chen K, et al. 2015. High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew. Chem. Int. Ed. Engl. 54, 1587–1590. (doi:10.1002/anie.201410647) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. 2015. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. (doi:10.1038/nmeth.3453) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Khoddami V, Cairns BR. 2014. Transcriptome-wide target profiling of RNA cytosine methyltransferases using the mechanism-based enrichment procedure Aza-IP. Nat. Protoc. 9, 337–361. (doi:10.1038/nprot.2014.014) [DOI] [PubMed] [Google Scholar]
- 141.Li X, Zhu P, Ma S, Song J, Bai J, Sun F, Yi C. 2015. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 11, 592–597. (doi:10.1038/nchembio.1836) [DOI] [PubMed] [Google Scholar]
- 142.Phelps K, Morris A, Beal PA. 2012. Novel modifications in RNA. ACS Chem. Biol. 7, 100–109. (doi:10.1021/cb200422t) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li X, Xiong X, Yi C. 2016. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods 14, 23–31. (doi:10.1038/nmeth.4110) [DOI] [PubMed] [Google Scholar]
- 144.Helm M, Motorin Y. 2017. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 23, 234 (doi:10.1038/nrg.2016.169) [DOI] [PubMed] [Google Scholar]
- 145.Schirmer M, D'Amore R, Ijaz UZ, Hall N, Quince C. 2016. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics 17, 125 (doi:10.1186/s12859-016-0976-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schaefer M. 2015. RNA 5-methylcytosine analysis by bisulfite sequencing. Meth. Enzymol. 560, 297–329. (doi:10.1016/bs.mie.2015.03.007) [DOI] [PubMed] [Google Scholar]
- 147.Conesa A, et al. 2016. A survey of best practices for RNA-seq data analysis. Genome Biol. 17, 13 (doi:10.1186/s13059-016-0881-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Baker M. 2015. Reproducibility crisis: blame it on the antibodies. Nature 521, 274–276. (doi:10.1038/521274a) [DOI] [PubMed] [Google Scholar]
- 149.Silva J, et al. 2003. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell 4, 481–495. (doi:10.1016/s1534-5807(03)00068-6) [DOI] [PubMed] [Google Scholar]
- 150.Egelhofer TA, et al. 2011. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 18, 91–93. (doi:10.1038/nsmb.1972) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Peach SE, Rudomin EL, Udeshi ND, Carr SA, Jaffe JD. 2012. Quantitative assessment of chromatin immunoprecipitation grade antibodies directed against histone modifications reveals patterns of co-occurring marks on histone protein molecules. Mol. Cell Proteomics 11, 128–137. (doi:10.1074/mcp.M111.015941) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Stollar BD. 1981. The antigenic potential and specificity of nucleic acids, nucleoproteins, and their modified derivatives. Arthritis Rheum. 24, 1010–1018. (doi:10.1002/art.1780240806) [DOI] [PubMed] [Google Scholar]
- 153.Seaman E, Van Vunakis H, Levine L. 1972. Serologic estimation of thymine dimers in the deoxyribonucleic acid of bacterial and mammalian cells following irradiation with ultraviolet light and postirradiation repair. J. Biol. Chem. 247, 5709–5715. [PubMed] [Google Scholar]
- 154.Munns TW, Oberst RJ, Sims HF, Liszewski MK. 1979. Antibody–nucleic acid complexes: immunospecific recognition of 7-methylguanine- and N6-methyladenine-containing 5'-terminal oligonucleotides of mRNA. J. Biol. Chem. 254, 4327–4330. [PubMed] [Google Scholar]
- 155.Munns TW, Liszewski MK, Hahn BH. 1984. Antibody–nucleic acid complexes: antigenic domains within nucleosides as defined by solid-phase immunoassay. Biochemistry 23, 2958–2964. (doi:10.1021/bi00308a017) [DOI] [PubMed] [Google Scholar]
- 156.Castleman H, Meredith RD, Erlanger BF. 1980. Fine structure mapping of an avian tumor virus RNA by immunoelectron microscopy. Nucleic Acids Res. 8, 4485–4499. (doi:10.1093/nar/8.19.4485) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ayub M, Hardwick SW, Luisi BF, Bayley H. 2013. Nanopore-based identification of individual nucleotides for direct RNA sequencing. Nano Lett. 13, 6144–6150. (doi:10.1021/nl403469r) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith AM, Abu-Shumays R, Akeson M, Bernick DL. 2014. Capture, unfolding, and detection of individual tRNA molecules using a nanopore device. Front. Bioeng. Biotechnol. 3, 91 (doi:10.3389/fbioe.2015.00091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Garalde DR, et al. 2016. Highly parallel direct RNA sequencing on an array of nanopores. bioRxiv (doi:10.1101/068809) [DOI] [PubMed] [Google Scholar]
- 160.Zaringhalam M, Papavasiliou FN. 2016. Pseudouridylation meets next-generation sequencing. Methods 107, 63–72. (doi:10.1016/j.ymeth.2016.03.001) [DOI] [PubMed] [Google Scholar]
- 161.Legrand C, Tuorto F, Hartmann M, Liebers R, Jacob D, Helm M, Lyko F. 2017. Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. bioRxiv. (doi:10.1101/130419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. 2013. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856. (doi:10.1261/rna.041178.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Molinie B, et al. 2016. m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat. Methods 13, 692–698. (doi:10.1038/nmeth.3898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sibbritt T, Patel HR, Preiss T. 2013. Mapping and significance of the mRNA methylome. Wiley Interdiscip. Rev. RNA 4, 397–422. (doi:10.1002/wrna.1166) [DOI] [PubMed] [Google Scholar]
- 165.Suzuki T, Suzuki T. 2014. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res 42, 7346–7357. (doi:10.1093/nar/gku390) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Miyauchi K, Ohara T, Suzuki T. 2007. Automated parallel isolation of multiple species of non-coding RNAs by the reciprocal circulating chromatography method. Nucleic Acids Res. 35, e24 (doi:10.1093/nar/gkl1129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Limbach PA, Paulines MJ. 2016. Going global: the new era of mapping modifications in RNA. Wiley Interdiscip. Rev. RNA 8, e1367 (doi:10.1002/wrna.1367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gonzalez C, Lopez-Rodriguez A, Srikumar D, Rosenthal JJC, Holmgren M. 2011. Editing of human K(V)1.1 channel mRNAs disrupts binding of the N-terminus tip at the intracellular cavity. Nat. Commun. 2, 436 (doi:10.1038/ncomms1446) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Colina C, Palavicini JP, Srikumar D, Holmgren M, Rosenthal JJC. 2010. Regulation of Na+/K+ ATPase transport velocity by RNA editing. PLoS Biol. 8, e1000540 (doi:10.1371/journal.pbio.1000540) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Seeburg PH, Higuchi M, Sprengel R. 1998. RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res. Brain Res. Rev. 26, 217–229. (doi:10.1016/S0165-0173(97)00062-3) [DOI] [PubMed] [Google Scholar]
- 171.Rula EY, Lagrange AH, Jacobs MM, Hu N, Macdonald RL, Emeson RB. 2008. Developmental modulation of GABA(A) receptor function by RNA editing. J. Neurosci. 28, 6196–6201. (doi:10.1523/JNEUROSCI.0443-08.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Morabito MV, Abbas AI, Hood JL, Kesterson RA, Jacobs MM, Kump DS, Hachey DL, Roth BL, Emeson RB. 2010. Mice with altered serotonin 2C receptor RNA editing display characteristics of Prader-Willi syndrome. Neurobiol. Dis. 39, 169–180. (doi:10.1016/j.nbd.2010.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu B, Liang X-H, Piekna-Przybylska D, Liu Q, Fournier MJ. 2008. Mis-targeted methylation in rRNA can severely impair ribosome synthesis and activity. RNA Biol. 5, 249–254. (doi:10.4161/rna.6916) [DOI] [PubMed] [Google Scholar]
- 174.Kane SE, Beemon K. 1987. Inhibition of methylation at two internal N6-methyladenosine sites caused by GAC to GAU mutations. J. Biol. Chem. 262, 3422–3427. [PubMed] [Google Scholar]
- 175.Narayan P, Ludwiczak RL, Goodwin EC, Rottman FM. 1994. Context effects on N6-adenosine methylation sites in prolactin mRNA. Nucleic Acids Res 22, 419–426. (doi:10.1093/nar/22.3.419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schwartz S, et al. 2013. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155, 1409–1421. (doi:10.1016/j.cell.2013.10.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Charpentier E, Marraffini LA. 2014. Harnessing CRISPR-Cas9 immunity for genetic engineering. Curr. Opin. Microbiol. 19, 114–119. (doi:10.1016/j.mib.2014.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Stepper P, Kungulovski G, Jurkowska RZ, Chandra T, Krueger F, Reinhardt R, Reik W, Jeltsch A, Jurkowski TP. 2016. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3 L methyltransferase. Nucleic Acids Res. 45, 1703–1713. (doi:10.1093/nar/gkw1112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nelles DA, Fang MY, O'Connell MR, Xu JL, Markmiller SJ, Doudna JA, Yeo GW. 2016. Programmable RNA tracking in live cells with CRISPR/Cas9. Cell 165, 488–496. (doi:10.1016/j.cell.2016.02.054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Garncarz W, Tariq A, Handl C, Pusch O, Jantsch MF. 2013. A high-throughput screen to identify enhancers of ADAR-mediated RNA-editing. RNA Biol. 10, 192–204. (doi:10.4161/rna.23208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Okamoto M, et al. 2012. Frequent increased gene copy number and high protein expression of tRNA (Cytosine-5-)-methyltransferase (NSUN2) in human cancers. DNA Cell Biol. 31, 660–671. (doi:10.1089/dna.2011.1446) [DOI] [PubMed] [Google Scholar]
- 182.Rouhani F, Kumasaka N, de Brito MC, Bradley A, Vallier L, Gaffney D. 2014. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 10, e1004432 (doi:10.1371/journal.pgen.1004432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. 2014. N(6)-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198. (doi:10.1038/ncb2902) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Batista PJ, et al. 2014. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. (doi:10.1016/j.stem.2014.09.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Geula S, et al. 2015. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347, 1002–1006. (doi:10.1126/science.1261417) [DOI] [PubMed] [Google Scholar]
- 186.Alon S, Garrett SC, Levanon EY, Olson S, Graveley BR, Rosenthal JJC, Eisenberg E, Guigó R. 2015. The majority of transcripts in the squid nervous system are extensively recoded by A-to-I RNA editing. eLife Sci. 4, e05198 (doi:10.7554/eLife.05198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Taliaferro JM, Wang ET, Burge CB. 2014. Genomic analysis of RNA localization. RNA Biol. 11, 1040–1050. (doi:10.4161/rna.32146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yoon YJ, Wu B, Buxbaum AR, Das S, Tsai A, English BP, Grimm JB, Lavis LD, Singer RH. 2016. Glutamate-induced RNA localization and translation in neurons. Proc. Natl Acad. Sci. USA 113, E6877–E6886. (doi:10.1073/pnas.1614267113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hoernes TP, Clementi N, Faserl K, Glasner H, Breuker K, Lindner H, Hüttenhofer A, Erlacher MD. 2016. Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code. Nucleic Acids Res. 44, 852–862. (doi:10.1093/nar/gkv1182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Licht K, Kapoor U, Mayrhofer E, Jantsch MF. 2016. Adenosine to inosine editing frequency controlled by splicing efficiency. Nucleic Acids Res 44, 6398–6408. (doi:10.1093/nar/gkw325) [DOI] [PMC free article] [PubMed] [Google Scholar]