

Abstract
Damage to endothelial cells contributes to acute kidney injury (AKI) by leading to impaired perfusion. Endothelial colony-forming cells (ECFC) are endothelial precursor cells with high proliferative capacity, pro-angiogenic activity, and in vivo vessel forming potential. We hypothesized that ECFC may ameliorate the degree of AKI and/or promote repair of the renal vasculature following ischemia-reperfusion (I/R). Rat pulmonary microvascular endothelial cells (PMVEC) with high proliferative potential were compared with pulmonary artery endothelial cells (PAEC) with low proliferative potential in rats subjected to renal I/R. PMVEC administration reduced renal injury and hastened recovery as indicated by serum creatinine and tubular injury scores, while PAEC did not. Vehicle-treated control animals showed consistent reductions in renal medullary blood flow (MBF) within 2 h of reperfusion, while PMVEC protected against loss in MBF as measured by laser Doppler. Interestingly, PMVEC mediated protection occurred in the absence of homing to the kidney. Conditioned medium (CM) from human cultured cord blood ECFC also conveyed beneficial effects against I/R injury and loss of MBF. Moreover, ECFC-CM significantly reduced the expression of ICAM-1 and decreased the number of differentiated lymphocytes typically recruited into the kidney following renal ischemia. Taken together, these data suggest that ECFC secrete factors that preserve renal function post ischemia, in part, by preserving microvascular function.
Keywords: angiogenesis, endothelial progenitor, hemodynamics, regeneration
renal ischemia-reperfusion (I/R) impairs vascular function by reducing renal blood flow and glomerular filtration while promoting parenchymal cell damage and sustained injury. It has become increasingly clear that the renal endothelium is a critically important target in the injury process as factors geared toward preserving endothelial function lessen the severity of I/R-induced renal damage (6). This endothelial damage may compromise renal blood flow by imparting changes in vascular tone and/or increasing inflammatory responses. In recent years, there has been a substantial number of reports suggesting that proangiogenic cell-based therapies provide protective effects against renal I/R (14–16, 36, 47, 50, 52, 54–56, 60). For example, mesenchymal stem cells (MSCs) from bone marrow and adipose tissue produce paracrine vasculotrophic factors thought to be important in the protection against renal I/R (42, 46, 50, 52, 54–56). Similarly, bone marrow-derived pro-angiogenic cells can significantly attenuate the degree of injury following I/R (29, 39). These cells provide beneficial effects against the development of injury, but do not incorporate into regenerating kidney to influence repair processes (14, 29). Other investigations have sought to directly replace endothelial function via transplantation of human umbilical vein endothelial cells (HUVEC) to athymic rats, which preserved postischemic microvascular flow in an endothelial nitric oxide synthase-dependent fashion and protected against subsequent renal damage (13).
In addition to acute endothelial dysfunction, there is a significant reduction in peritubular capillary density following acute kidney injury (AKI) (7). This reduction in peritubular capillary density is characterized by low endothelial cell proliferation and propensity to undergo endothelial-to-mesenchymal transition (5). Since the endogenous renal vasculature demonstrates little reparative capacity (5), strategies directed toward vascular protection or revascularization may be considered an important goal in maintaining short-term or long-term function in response to renal injury.
Yoder and colleagues have described non-hematopoietic endothelial progenitor cells referred to as endothelial colony-forming cells (ECFC), which can be derived from peripheral blood or tissues (2). ECFC with high proliferative potential (HPP) in single-cell colony-forming assays form perfused vessels in vivo and are considered true endothelial progenitor cells (64). It has been shown that human cord blood as well as HUVEC represent a rich source of human HPP-ECFC (31, 63, 69). However, the proliferative potential of ECFC varies depending on species, age, and tissue source. For example, Alvarez et al. (3) showed that rat pulmonary microvascular endothelial cells (PMVEC) have a substantially greater proportion of HPP-ECFC compared with rat pulmonary artery-derived cells (PAEC). Since HPP-ECFC are bone fide vasculoreparative cells, we hypothesized that supplementation with HPP-ECFC may influence the course of renal injury, and potentially engraft into the damaged kidney. To that end, we sought to investigate the effect of endothelial transplantation based upon the proliferative potential of ECFC following renal I/R.
METHODS
Animals.
Male Sprague-Dawley rats (initial weight ~250 g) were utilized in all studies. Rats were given free access to standard rat chow and water throughout our studies. Experiments were conducted in accordance with National Institutes of Health guidelines and were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee.
Cells.
Rat pulmonary microvascular endothelial cells (PMVEC) and rat pulmonary artery cells (PAEC) were isolated and expanded as described previously (3). These primary cultures were derived from Sprague Dawley rats and utilized between passages 5 and 7. The endothelial nature of PMVEC and PAEC was previously characterized by Alvarez et al. (3) and cells were validated according to their expression of CD31, KDR, and vWF, but were negative for CD45 and CD133. PMVEC have a significantly faster proliferation rate and a greater percentage of high proliferative potential HPP-ECFC than PAEC (3). PMVEC and PAEC were maintained in EGM-2 supplemented with 10% FBS (Hyclone) and grown on T75 flasks. On the day of transplant studies, cells were harvested by trypsin digestion, washed with PBS. In some studies, the cells were labeled with CMTPX (i.e., Cell tracker red, Invitrogen), according to the manufacturer’s instructions. The cells were then washed and resuspended in serum-free culture medium and maintained on ice until the time of transplant.
Human ECFC were derived from human cord blood according the protocol described previously by Yoder et al. (64). Human ECFC were maintained in T-225 flasks in EGM2 (Invitrogen) with 10% FBS. Fifty milliliters of conditioned serum-free medium was derived from 50 to 75% confluent human ECFC, corresponding to ~8–12 million cells following 2 days of incubation and concentrated by centrifugation using Centricon filters (3000 M.W. cutoff) to achieve an enrichment of ~10-fold. Therefore, 1 ml of CM results from the contribution of ~1.6–2.4 million cells.
Surgeries.
Acute kidney injury was induced by bilateral ischemia reperfusion injury to the kidneys by clamping both renal pedicles for 40 min using a surgical approach that has been described previously under anesthesia induced with ketamine (100 mg/kg) and pentobarbital (25–50 mg/kg) (40) or ketamine (100 mg/kg) and xylazine (5 mg/kg). The first cocktail was used in the initial series of experiments in which rat ECFC were tested; while the second anesthetic cocktail was used in studies of human ECFC derived conditioned media. The reason for the change was due to limited availability of pentobarbital which occurred between the times of the two studies. These two anesthetic regimens yielded consistent levels of renal injury.
For endothelial cell administration, we utilized an approach similar to that described by Brodsky et al., for the administration of HUVEC (13). The left carotid artery was cannulated with a PE-50 tubing filled with heparinized sterile saline, inserted toward the heart, while the artery distal to the insertion site was ligated with a silk-suture to prevent backleak. This catheter was utilized for the administration of cells (5 × 106 PMVEC or PAEC in 0.5 ml of vehicle) in a retrograde fashion immediately following the release of the clamps. The catheter was then slowly withdrawn and the carotid artery was immediately ligated proximal to the insertion site to prevent bleeding. In studies using ECFC-conditioned media, a volume of 0.5 ml of 10× concentrated conditioned media or “mock”-conditioned media from human ECFC was administered to the suprarenal aorta at the time of reperfusion using a 31-gauge needle.
Measurement of renal function.
At the indicated times, blood was obtained from rats under light isoflurane anesthesia via tail vein incisions. Blood was collected in 1.5-ml heparinized Eppendorf tubes, centrifuged at 3,000 g for 10 min. Serum creatinine was measured using a Point Scientific QT 180 Analyzer and creatinine reagent kit (Point Scientific, Canton, MI) according to the manufacturer’s specifications (57).
Evaluation of KIM-1 or ICAM-1 mRNA expression in the injured kidney.
Whole kidney mRNA was extracted from fresh-frozen tissue using a Direct-zol RNA extraction kit according to the manufacturer's instructions (Zymo, Irvine, CA). Kidney injury molecule-1 (KIM-1) mRNA expression was evaluated using predesigned Taqman primers (Life Technologies, Carlsbad, CA) with the 2−ΔΔCT analysis method (30).
Evaluation of renal hemodynamic response to I/R injury.
Rats were anesthetized with ketamine HCl (60 mg/kg), followed by Inactin (50–100 mg/kg) ip and placed on a heated surgical board to maintain body temperature at 37°C. The femoral vein was cannulated for intravenous infusion of 2% bovine serum albumin in 0.9% NaCl at a rate of 2 ml·h−1·100 g body wt−1. This catheter was also used for infusion of conditioned medium.
A midline abdominal incision was made, and a flow probe was placed around the renal artery for measurement of renal blood flow (RBF) via an ultrasonic Doppler flowmeter (model T206; Transonic Systems, Ithaca, NY). The left kidney was placed in a holder and an optical probe for laser Doppler flowmetry (Transonic) was implanted to a depth of ∼5.0 mm beneath the surface for measurements of renal outer medullary blood flow (MBF). Data were recorded using Biopac (Goleta, CA) data-acquisition software.
Following 30 min of equilibration, RBF and MBF values were measured for 30 min in 10-min time bins, with the final 10 min defined as baseline. Parameters were measured during ischemia and an additional 120 min of reperfusion. Values were normalized to each baseline value, and data are expressed as the average of these normalized values.
Evaluation of cell homing.
Prior to transplant, rat PMVEC were stained with cell tracker red CMTPX, as described above. Pilot studies indicated that tissue fixation impaired our detection of labeled cells. Therefore, cell fluorescence was examined in freshly harvested unfixed tissues. Kidneys, spleens, or lungs were removed from deeply anesthetized rats and immersed in ice-cold HEPES-Tyrode buffer (132 mM NaCl, 4 mM KCl, 1 mM CaCl2, 0.5 mM MgCl2, 10 mM HEPES, and 5 mM glucose, pH 7.4) that had been bubbled with 100% O2. Tissue slices were prepared using a hand microtome (Stadie Riggs Tissue Slicer) and stored in cold buffer and imaged within 1 h of tissue harvest. Images were obtained using a Zeiss LSM NLO confocal microscope equipped with AR and HeNe lasers and a ×40 water immersion lens, and a signal was obtained by 545 nm and detection at 565–615 nm.
Evaluation of infiltrating leukocytes.
Harvested kidneys were minced and digested in TL Liberase (2 µg/ml; Roche). The obtained cell suspension was filtered through a 100-μm filter mesh and washed with DMEM containing 10% fetal bovine serum (Cell Applications, San Diego, CA). The mononuclear cells were separated by Percoll (Sigma, St. Louis, MO) and counted by hemocytometer. To evaluate T lymphocytes, the cells were stained with antibodies against rat CD4 (PE-Cy7: BD Biolgend, San Diego, CA), CD8a (Alexa 647: BD Biolgend). To evaluate the cytokines secreted by T cells, the cells were stained for the CD4 surface marker, permeabilized using 0.1% saponin and stained with antibodies against rat IFN-γ (FITC: BD Biolgend) or IL-17 (FITC: BD Biolgend). Cells were scanned using flow cytometry (FACSCalibur, BD Biosciences), and scans were analyzed using Flowjo software (Tree Star, Ashland, OR). The gating strategy used for these analyses was exactly as we have previously described (32). The total numbers of the different T cell populations in the harvested kidney were calculated using the percentage of each cell type and the total cell number measured per gram of kidney.
Renal histology and immunohistochemistry.
Renal tubular damage was evaluated from formalin-fixed, paraffin-embedded samples stained using periodic acid-Schiff (PAS). Six random images (3 cortex, 3 outer medulla) were obtained using a Leica DMLB microscope (Scientific Instruments, Columbus, OH) using a ×20 objective. For each kidney, an average of 60 tubules were scored from images by an observer who was blinded to the treatments using a 1–4 scoring system described previously (8). Data presented are based on the average score per tubule corresponding to each animal.
Immunofluorescent analysis of ICAM-1.
Methanol-fixed 100-μm vibratome sections of kidneys were subjected to immunofluorescent staining using an ant-ICAM-1 antibody (BD Biosciences, San Jose, CA). ICAM-1-specific signals were developed using a tyramide-signal amplification kit (Invitrogen, Carlsbad, CA) as described previously (9). Confocal images were obtained using an Olympus FV 1000-MPE microscope using a ×20 objective (Center Valley, PA). Quantification of immunofluorescence was done with the aid of Fiji ImageJ. Data presented are based on the % total ICAM-1-stained area.
Statistical analysis.
Data are expressed as means ± SE. Differences in means were established by Student’s t-test or ANOVA as indicated. The 0.05 level of probability was utilized as the minimum criterion of significance. All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA).
RESULTS
The potential that ECFC may alter the course of renal dysfunction and/or repopulate the renal microvasculature as a function of proliferative potential was addressed by comparing the effect of administered rat PMVEC, which have a high percentage of HPP-ECFC, or rat PAEC, which have a low percentage of HPP-ECFC (3). Renal injury measured by increased serum creatinine was most prominent at 2 days of reperfusion. Relative to vehicle-treated control rats, PMVEC-treated rats had a lower peak creatinine level and a faster recovery of serum creatinine levels (Fig. 1A). In contrast, PAEC administration did not alter the course of renal injury relative to vehicle-treated rats. Despite evidence of recovery in all groups, the level of histological damage remained severe in post-ischemic, vehicle-treated animals at day 7 with evidence of sloughed cells and tubular dilatation in the outer medulla compared with PMVEC-treated rats (Fig. 1B). To further investigate the protective effect of PMVEC, additional animals were studied at 2 days following reperfusion. Similar to Fig. 1A, PMVEC-treated rats had lower peak serum creatinine levels (Fig. 1C) and reduced necrotic damage compared with vehicle-treated, postischemic rats (black arrows; Fig. 1, D and E).
Fig. 1.

Rat pulmonary microvascular endothelial cells (PMVEC) protect against renal ischemia-reperfusion (I/R) injury and accelerate functional and structural recovery. A: serum creatinine (sCre) values for 7 days following I/R or sham surgery (n = 3) in rats treated with vehicle (n = 7), pulmonary artery endothelial cells (PAEC; n = 6), or PMVEC (n = 8). B: representative photomicrographs of periodic acid-Schiff (PAS)-stained kidney sections following 7 days of recovery from renal I/R. Dilated tubules are prominent in vehicle and PAEC-treated rats (black arrows), with some evidence of cell sloughing. Magnification is shown. C: results from a 2-day time course study comparing renal function in vehicle (n = 6)-treated vs. PMVEC-treated rats (n = 6) following I/R injury. D: corresponding photomicrographs of PAS-stained kidney sections following 2 days of recovery from renal I/R, indicating a greater degree of necrotic tubules (black arrows) in vehicle-treated rats. E: injury score corresponding to 2-day postischemic rats. Data are means ± SE. * and # indicate P < 0.05 in PMVEC-treated rats compared with PAEC- and vehicle-treated rats, respectively by Student’s t-test.
To investigate the potential mechanism of PMVEC-mediated protection, we investigated whether these cells influenced hemodynamic function in the early postischemic period by measuring total RBF and outer medullary blood flow (MBF) following reperfusion. Total RBF values rapidly recovered during the reperfusion phase and were similar to baseline values within 30–40 min. At 2 h of reperfusion, total RBF was ~90–95% of baseline in both vehicle-treated and PMVEC-treated animals (not significant; Fig. 2A). In contrast, MBF gradually declined over the course of 2 h following reperfusion in vehicle-treated rats. However, PMVEC-treated rats had significantly preserved MBF relative to vehicle-treated rats (Fig. 2B).
Fig. 2.
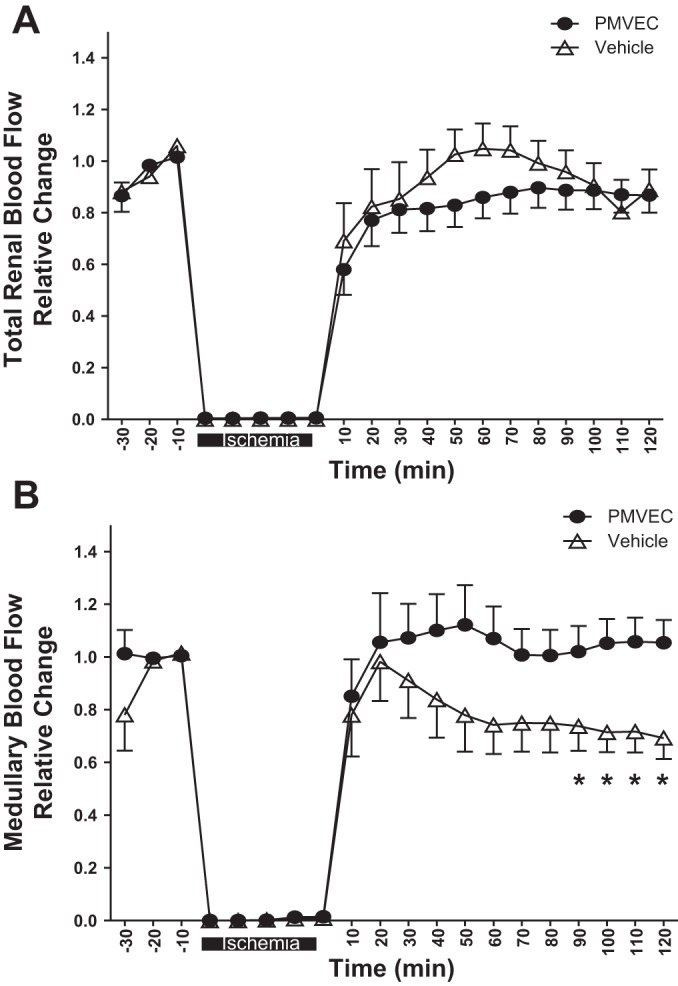
Rat PMVEC preserve medullary blood flow in the early postischemic period. Total renal blood (A) and medullary blood flow (B) were monitored for 30 min before ischemia and up to 120 min postreperfusion as labeled. Data are averaged in 10-min time bins normalized to the baseline values for each rat, and data are means ± SE *P < 0.05 in vehicle- relative to PMVEC-treated rats by ANOVA with repeated measures.
To determine whether transplanted PMVEC home to the postischemic kidney, cells were labeled with Celltracker red (CMTPX) just before administration and examined immediately following tissue harvest by confocal microscopy (Fig. 3A). There was no evidence of fluorescently labeled cells in postischemic kidneys at either 2 or 48 h following reperfusion (Fig. 3, B and C). In contrast, some fluorescently labeled cells were readily apparent in the spleen (white arrows, Fig. 3D) and lung (not shown).
Fig. 3.
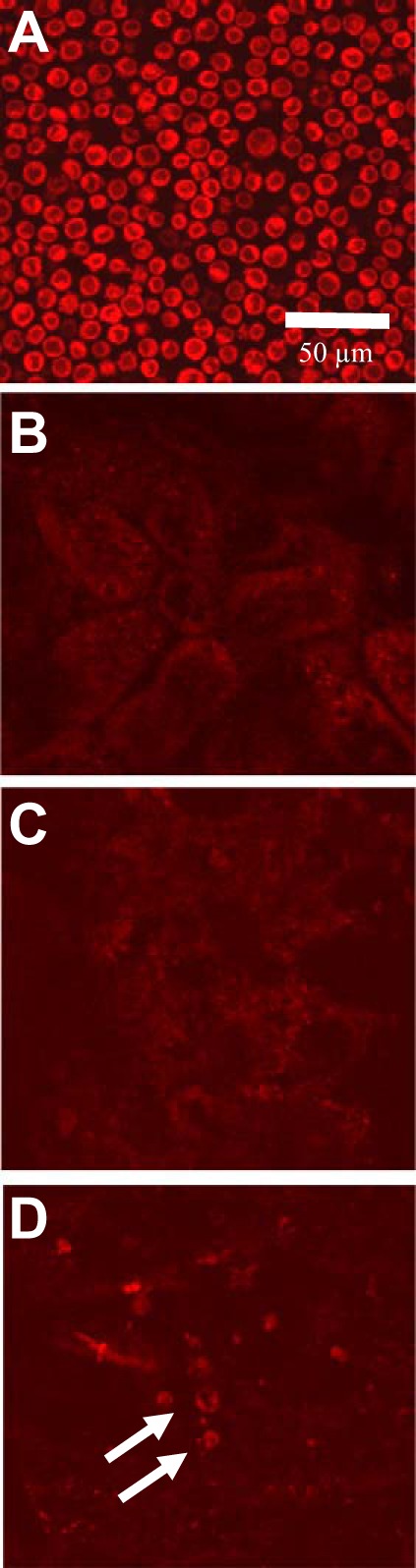
Rat PMVEC do not home to the kidney following transplantation. Shown are representative confocal images of freshly suspended PMVEC labeled with cell tracker red in vitro and imaged before transplantation (A). There were no labeled cellular structures in the kidney at either 2 h (B) or 2 days (C) posttransplantation. D: fluorescently labeled cells in spleen with a similar size and fluorescent intensity of preinfused PMVEC (white arrows). Similarly, labeled structures were readily apparent in lung (not shown). Magnification is shown in A.
The lack of PMVEC homing suggests that soluble factors released from ECFC may provide protection against impaired renal blood flow following renal I/R. We conducted pilot studies to investigate whether soluble factors present in conditioned media of PMVEC may mediate protection from I/R injury. In one pilot study (n = 4), we utilized 5 ml of PMVEC-CM administered intraperitoneally and found that the increase in serum creatinine measured 24 h following reperfusion was significantly reduced by 44 ± 10% relative to mock CM-treated post-I/R rats (data not shown). However, to increase the translational relevance of this research, we sought to utilize CM from human cord blood ECFC, which have very high proliferative potential (63). In addition, we further modified our approach by concentrating hECFC-CM to facilitate a reasonable volume for intravascular administration. Relative to vehicle-injected control rats, hECFC-CM-treated rats manifested a significantly lower peak creatinine level following reperfusion (Fig. 4A). In addition, the level of histological damage was significantly less severe in ECFC-CM-treated rats compared with vehicle-treated animals at 2 days post-I/R (Fig. 4, B and C). To further assess renal injury, we evaluated KIM-1 mRNA expression and demonstrated that the expression of this marker for tubular injury was significantly reduced compared with vehicle-injected control rats (Fig. 4D).
Fig. 4.

Human endothelial colony-forming cells-conditioned medium (ECFC-CM) protects against renal I/R injury. A: serum creatinine for 2 days following I/R or sham surgery (n = 3) in vehicle (n = 7)- and ECFC-CM-treated rats (n = 7). B: representative photomicrographs of PAS-stained kidney sections following 2 days of recovery from renal I/R. Significant necrotic debris and tubular damage are present in vehicle-treated rat kidney 2 days postsurgery (black arrows). Magnification is shown. Tissue injury score is shown in C. D: KIM-1 mRNA expression in sham, vehicle-, or ECFC-CM-treated rats. Data are means ± SE. n.d., Not detectable. *P < 0.05 in ECFC-CM-treated relative to vehicle-treated rats by Student’s t-test.
To determine whether human ECFC-CM administration preserves hemodynamic function postischemia, we measured total RBF and outer MBF. Similar to studies described in Fig. 2A, total RBF values recovered to ~85% of control during the reperfusion phase and were not different between vehicle- and ECFC-CM-treated groups (Fig. 5A). In addition, MBF values returned toward control levels in hECFC-CM-treated animals, but remained significantly suppressed below baseline in vehicle-treated controls (Fig. 5B).
Fig. 5.
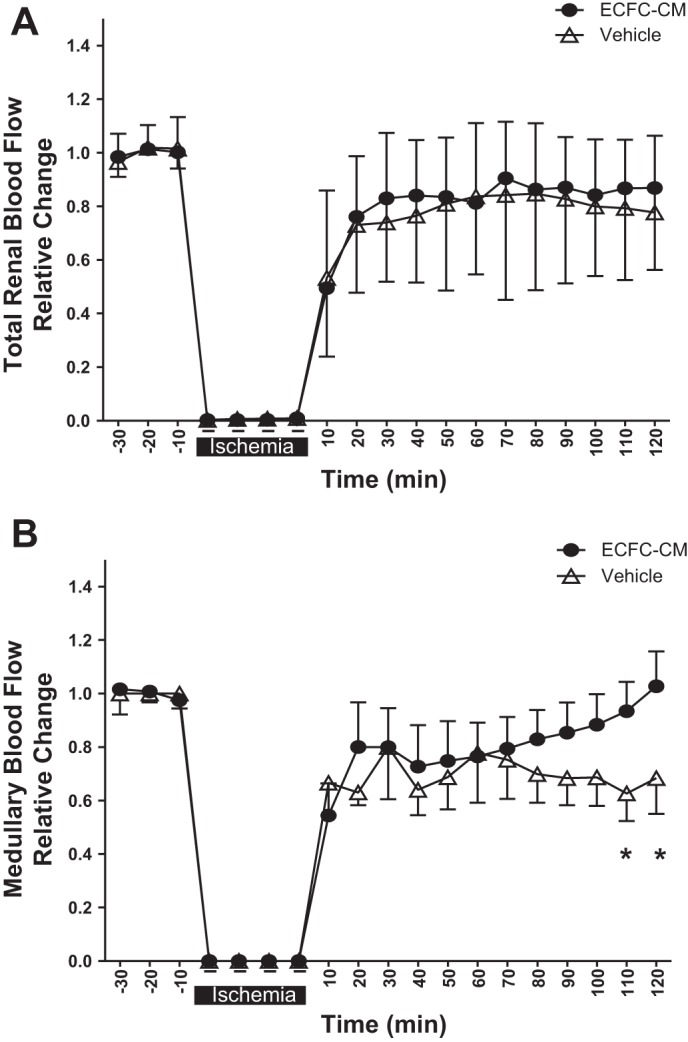
Human ECFC-CM preserves medullary blood flow in the early postischemic period. Total renal blood flow (A) and medullary blood flow (B) were monitored for 30 min before ischemia and up to 120 min postreperfusion as labeled. Data are averaged in 10-min time bins normalized to the baseline values for each rat and data are means ± SE. *P < 0.05 in vehicle- relative to ECFC-CM-treated rats by ANOVA with repeated measures.
It is has been suggested that endothelial cell dysfunction leads to increased leukocyte adhesion, which may contribute to the severity of renal damage in the post-ischemic state (5, 35). To determine whether hECFC-CM suppresses postischemic endothelial leukocyte adhesion, we first measured the mRNA expression of ICAM-1, an adhesion molecule known to be induced in endothelial cells in the early postischemic period and demonstrated that its expression was significantly increased within 5 h of reperfusion relative to sham. (Fig. 6A). Similarly, ICAM-1 protein was not detectable in kidneys of sham-operated rats while it was prominently induced in peritubular capillaries of postischemic rats as indicated by immunofluorescence (Fig. 6, B and C). Interestingly, both the mRNA expression of ICAM-1 (Fig. 6A) and the peritubular capillary protein expression of ICAM-1 (Fig. 6, B and C) were significantly attenuated by infusion of hECFC-CM.
Fig. 6.

Human ECFC-CM reduces adhesion molecular expression following recovery from I/R injury. Rats subjected to sham surgery or renal I/R and recovery for 5 h. Rats were treated with vehicle or ECFC-CM as labeled. A: mRNA expression levels for ICAM-1 derived from total RNA of whole kidney using real-time PCR. B and C: representative images of kidney ICAM-1 immunofluorescence in sham, vehicle-, and ECFC-CM-treated rats. C: quantitative analysis of % total area of ICAM-1 immunofluorescent-stained structures. Immunofluorescence data in C are % of total area compared with the mean value of sham-operated control rats. *P < 0.05 sham vs. I/R+vehicle by Student’s t-test. #P < 0.05 in I/R+vehicle- vs. I/R+ECFC-CM-treated rats.
To determine whether hECFC-CM reduces postischemic inflammation, total and specific leukocyte populations were measured by FACS following 2 days of recovery from renal I/R (Fig. 7A). The total number of leukocytes, as well as the total number of CD4+ and CD8+ cells, were significantly elevated following renal I/R, but these were not influenced by the hECFC-CM (Fig. 7, B–D). Interestingly, alterations in specific populations were observed. For example, the total number of cells expressing the cytokine IL-17 (Fig. 7E) as well as T-helper 17 cells (i.e., CD4+/IL17+) was significantly attenuated in hECFC-CM-treated rats (Fig. 7F). Moreover, Th-1 cells, defined as CD4+/IFN-γ+, were also significantly attenuated in hECFC-CM-treated rats (Fig. 7G). These data suggest that reductions in specific anti-inflammatory cells may contribute to ECFC-mediated protection from I/R induced AKI.
Fig. 7.

Human ECFC-CM reduces infiltration of inflammatory cells in kidneys following I/R. Kidney resident monocytes were isolated from kidneys harvested 2 days postsurgery/treatment. A: gating strategy for FACS analysis. Lymphocytes were gated based on forward and side scatter plot. Total number of infiltrating monocytes (B), total number of CD4+ T cells (C), CD8+ T cells (D), total IL-17+ T cells (E), CD4+IL-17+ T cells (F), and IFN-γ+ cells (G) were quantified using fluorescence-activated cell sorting. Data are expressed as total number of each cell type per gram of tissue and are means ± SE. *P < 0.05 sham vs. I/R+vehicle by Student’s t-test. ϕP < 0.05 sham vs. I/R+ECFC-CM. #P < 0.05 I/R+vehicle vs. I/R+ ECFC-CM.
DISCUSSION
A long-term goal of cell-based therapy in the kidney seeks to protect renal perfusion via reducing the extent of vascular injury and/or promoting of vascular repair. Various cells of hematopoietic origin, often referred to as endothelial progenitor cells (EPC) or proangiogenic cells, have been identified which may promote vascular repair (4, 45) while numerous studies have demonstrated that a variety of different cell types can ameliorate the severity of AKI in preclinical models (14, 15, 36, 52, 54–56, 58, 61, 68).
ECFC are clonally distinct from hematopoietic CFU-ECs, have robust proliferative potential and form perfused vessels in vivo through a process of de novo vasculogenesis (64). In addition, these cells can be isolated from the vessel walls of a variety of tissue sources, suggesting that endothelial progenitors reside within the local vasculature (62). Alvarez et al. (3) demonstrated that PMVEC contain a high percentage of HPP-ECFC, from which >60% of the populations form colonies of >2,000 cells. In comparison, PAEC, are less proliferative containing only ~15% HPP-ECFC (3). In addition, we demonstrated that rat kidney contains little HPP-ECFC activity (10), and therefore PMVECs, rather than kidney-derived cells, were utilized in this study to evaluate the potential benefit of HPP-ECFC in the setting of AKI. The current study demonstrates that PMVEC have a powerful protective effect against I/R-induced renal injury, a property that was not shared by PAEC. These results are consistent with effects reported by Brodsky et al. (13) demonstrating HUVEC preserved early microvascular dysfunction and mitigated renal injury in athymic rats following I/R. It has been previously demonstrated that HUVEC contain a high level of HPP-ECFC (24).
Whether HPP-ECFC within PMVEC are responsible for mediating the protection is not yet clear. The isolation of HPP-ECFC has not been previously considered feasible based on expression of surface markers; however, endothelial cells have been shown to possess a side population based on differential Hoechst 33342 staining, which are enriched in colony-forming potential. These cells were shown to enhance the regeneration of endothelium in a hind limb ischemia model (37). Whether side population EC from PMVEC or other vascular beds can specifically contribute to renal protection will represent an important area of future investigation.
We hypothesized that ECFC, would home to the sites of renal injury, similar to CFU-ECs as described by Patschan et al. (39). However, no ECFC delivered by intravascular injection were detectable in postischemic kidneys, using confocal microscopy. While our search for cell homing was based on tracking of labeled cells using confocal microscopy, it is clearly reasonable to suggest that a small number of ECFC in kidney may not have been observed. Nevertheless, ECFCs were found primarily in lung and spleen. This appears similar to the dynamics of intravascular transplanted mesenchymal stem cells from various origins (53), which are thought to mediate their effects primarily via the production and release of soluble vasoactive/or anti-inflammatory compounds. To further evaluate the endocrine nature of ECFC activity, we also demonstrated that human ECFC-CM has protective activity against I/R injury (Fig. 4). These results are also consistent with recent studies from Burger et al. (14), who also demonstrated that human ECFC did not home to the kidneys of SCID mice following I/R, but did protect against AKI measured at either 24 or 72 h post-I/R. These investigators also demonstrated ECFC-mediated protection was characterized by significant attenuation of necrosis and apoptosis and a reduction in macrophage infiltration. Moreover, the attenuation of apoptosis by ECFC extended to both the tubular compartment as wells as the vascular compartment (14).
In viewing our results with respect to the hypothesis that endothelial dysfunction plays a primary role in the genesis of AKI (5, 35), it is noteworthy that the effects of rat PMVEC and human ECFC-CM on renal I/R appear to influence early alterations in renal hemodynamic function before the manifestation of significant renal tubular damage. Given that the renal medulla is hypoxic relative to the cortex and receives a smaller percentage of RBF, impaired perfusion in this region could exacerbate hypoxia and contribute toward cellular injury in this susceptible region (12). The early impairment of renal MBF can be observed within a few hours of reperfusion and is thought to correspond to the severity of the resulting injury (43). Reductions in MBF are exacerbated by l-NAME and ameliorated by the free radical scavenger N-acetyl cysteine (18). In addition, Regner et al. (44) demonstrated that the reduction in MBF in the first 2 h postischemia was abrogated by the 20-HETE analog 5,14, 20-HEDGE, which also mediated significant protection against tissue damage at 24 h. In addition, other agents such as volatile anesthetics (27), hydrogen sulfide, atrial natriuretic peptide, or the induction of heme-oxygenase-1 have all been shown to attenuate the immediate loss of MBF following I/R injury (43). Taken together, these observations indicate that the preservation of MBF represents an important mechanism by which either PMVEC or ECFC-CM protects against I/R-induced AKI.
The protective effects on MBF may be a function of modulating the renal inflammatory response. Inflammatory cell infiltration is a prominent early feature in the outer medullary vasa recta capillaries following injury characterized by the progressive influx of leukocytes, lymphocytes, dendritic cells, and macrophages into the interstitium (1, 22, 26, 28, 34, 66). It has been hypothesized that endothelial activation promoting cell infiltration contributes to the severity of injury either by sustaining reductions in perfusion and/or by promoting the liberation of cytokines which drive inflammation and injury (33).
In the current studies, we demonstrated that human ECFC-CM may influence early alterations in renal hemodynamics in part by influencing the expression of ICAM-1, which is known be enhanced on the surface of endothelial cells following I/R (11, 35, 70). Moreover, strategies using either antibodies or null mutant mice designed to inhibit the contribution of ICAM-1 or other adhesion molecule activity have been shown to reduce inflammation and the severity of AKI (23, 24, 26, 41, 48, 49).
While a variety of cells are known to invade the postischemic kidney, there is growing evidence the T cells play a critical role in the development of AKI (19, 21, 32, 65, 67). It is interesting that ECFC-CM influences the activation and differentiation of T cells in the postischemic kidney, as evidenced by the reduced number of cells expressing the cytokine IL-17, including Th-17 cells (CD4+/IL17+). Recent studies from our own laboratory have shown that Th17 cells represent the most prominent T-helper population induced by renal I/R in rats (32). Moreover, Th17 infiltration is enhanced in vitamin D-deficient rats, which exacerbates AKI (20), while Th17 cell infiltration is reduced by the administration of adipose-derived stromal cells which attenuate AKI (25). A potential role for Th17 cells in the pathogenesis of AKI is supported by studies in which mice lacking the IL-17A gene were protected against the development of AKI induced by cisplatin (17, 32). It is possible that IL-17 produced from T cells may directly impact renal hemodynamics in the setting of AKI, representing an additional mechanism by which PMVEC and hECFC-CM convey vascular protection following ischemic injury. The suggestion that IL-17 may contribute to vascular oxidant stress derives from studies indicating that reactive oxygen species generation induced by ANG II is ablated in mice lacking the IL-17A gene. In addition, Nguyen et al. (38) demonstrated that IL-17 activates RhoA-Rho-kinase, leading to endothelial dysfunction and hypertension in mice. We suggest that inhibition of adhesion molecule activity by ECFC-CM represents a potential mechanism by which postischemic Th17 cell infiltration was attenuated; however, our results cannot exclude the possibility that ECFC-CM may have a direct effect on lymphocyte activation.
The lack of homing and the similar effect of conditioned media suggest that factors released by ECFC may preserve renal vascular function in the setting of AKI, in part, by targeting endothelial-leukocyte interactions. It is plausible to suggest that factors derived from ECFC could be utilized clinically in high-risk patients to mitigate the development of AKI. At the present time, the biochemical nature of the factor(s) that mediate this protection is not clear. However, recent studies demonstrated that ECFC-derived exosomes contain a high level of miR-486-5p and that inhibition of miR-486-5p attenuated the protective effects of exosomes in hypoxia-challenged endothelial cells in vitro (59).
In summary, our results confirm previous studies that soluble factors produced by ECFC protect against the development of AKI, and extend these results by indicating that protection is mediated in part by preventing early loss in renal medullary perfusion. ECFC also reduce the expression of adhesion molecules and attenuate the degree of inflammation, in particular, the infiltration of Th17 cells. The identification and exploitation of these soluble factors will be of considerable interest in the future to determine whether the effects of ECFC are mediated directly at the renal endothelium and/or influence inflammatory cells activated in response to injury.
GRANTS
This work was supported by National Institutes of Health (NIH) grants DK-063114 (D. P. Basile), an ARRA supplement to this same grant. J. A. Collett was supported by NIH T32 5T32 HL-079995-10 and NIH/NCATS TL1 Postdoctoral Fellowship TL1TR001107 (A. Shekar, PI). Additional funding for this study was from Riley Children’s Foundation (M. C. Yoder), pilot funding from George M. O’Brien Grant P30 DK-79312 (Bruce Molitoris, Program Director), Fortune Fry, and Bridge Funding from the Indiana University Research Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.A.C., M.C.Y., and D.P.B. conceived and designed research; J.A.C., P.M., A.C., and W.C.S. performed experiments; J.A.C., P.M., A.C., and D.P.B. analyzed data; J.A.C., P.M., M.C.Y., and D.P.B. interpreted results of experiments; J.A.C. prepared figures; J.A.C. and D.P.B. drafted manuscript; J.A.C., P.M., M.C.Y., and D.P.B. edited and revised manuscript; J.A.C., P.M., A.C., W.C.S., M.C.Y., and D.P.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Ellen Leonard, Jessica Friedrich, Meridith Balbach, and Pingyu Zing for technical support. We thank Seth Winfree for assistance with quantitative analysis of immunofluorescence.
REFERENCES
- 1.Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm 2009: 1, 2009. doi: 10.1155/2009/137072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarado-Moreno JA, Hernandez-Lopez R, Chavez-Gonzalez A, Yoder MC, Rangel-Corona R, Isordia-Salas I, Hernandez-Juarez J, Cerbulo-Vazquez A, Gonzalez-Jimenez MA, Majluf-Cruz A. Endothelial colony-forming cells: Biological and functional abnormalities in patients with recurrent, unprovoked venous thromboembolic disease. Thromb Res 137: 157–168, 2016. doi: 10.1016/j.thromres.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez DF, Huang L, King JA, ElZarrad MK, Yoder MC, Stevens T. Lung microvascular endothelium is enriched with progenitor cells that exhibit vasculogenic capacity. Am J Physiol Lung Cell Mol Physiol 294: L419–L430, 2007. doi: 10.1152/ajplung.00314.2007. [DOI] [PubMed] [Google Scholar]
- 4.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res 85: 221–228, 1999. doi: 10.1161/01.RES.85.3.221. [DOI] [PubMed] [Google Scholar]
- 5.Basile DP, Yoder MC. Renal endothelial dysfunction in acute kidney ischemia reperfusion injury. Cardiovasc Hematol Disord Drug Targets 14: 3–14, 2014. doi: 10.2174/1871529X1401140724093505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int 72: 151–156, 2007. doi: 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 7.Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol 281: F887–F899, 2001. doi: 10.1152/ajprenal.0050.2001. [DOI] [PubMed] [Google Scholar]
- 8.Basile DP, Dwinell MR, Wang SJ, Shames BD, Donohoe DL, Chen S, Sreedharan R, Van Why SK. Chromosome substitution modulates resistance to ischemia reperfusion injury in Brown Norway rats. Kidney Int 83: 242–250, 2013. doi: 10.1038/ki.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol 300: F721–F733, 2011. doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basile DP, Zeng P, Friedrich JL, Leonard EC, Yoder MC. Low proliferative potential and impaired angiogenesis of cultured rat kidney endothelial cells. Microcirculation 19: 598–609, 2012. doi: 10.1111/j.1549-8719.2012.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boesen EI, Crislip GR, Sullivan JC. Use of ultrasound to assess renal reperfusion and P-selectin expression following unilateral renal ischemia. Am J Physiol Renal Physiol 303: F1333–F1340, 2012. doi: 10.1152/ajprenal.00406.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brezis M, Rosen S. Hypoxia of the renal medulla–its implications for disease. N Engl J Med 332: 647–655, 1995. doi: 10.1056/NEJM199503093321006. [DOI] [PubMed] [Google Scholar]
- 13.Brodsky SV, Yamamoto T, Tada T, Kim B, Chen J, Kajiya F, Goligorsky MS. Endothelial dysfunction in ischemic acute renal failure: rescue by transplanted endothelial cells. Am J Physiol Renal Physiol 282: F1140–F1149, 2002. doi: 10.1152/ajprenal.00329.2001. [DOI] [PubMed] [Google Scholar]
- 14.Burger D, Viñas JL, Akbari S, Dehak H, Knoll W, Gutsol A, Carter A, Touyz RM, Allan DS, Burns KD. Human endothelial colony-forming cells protect against acute kidney injury: role of exosomes. Am J Pathol 185: 2309–2323, 2015. doi: 10.1016/j.ajpath.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 15.Burgos-Silva M, Semedo-Kuriki P, Donizetti-Oliveira C, Costa PB, Cenedeze MA, Hiyane MI, Pacheco-Silva A, Câmara NOS. Adipose tissue-derived stem cells reduce acute and chronic kidney damage in mice. PLoS One 10: e0142183, 2015. doi: 10.1371/journal.pone.0142183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC, Sordi A, Biancone L, Tetta C, Camussi G. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int 82: 412–427, 2012. doi: 10.1038/ki.2012.105. [DOI] [PubMed] [Google Scholar]
- 17.Chan AJ, Alikhan MA, Odobasic D, Gan PY, Khouri MB, Steinmetz OM, Mansell AS, Kitching AR, Holdsworth SR, Summers SA. Innate IL-17A-producing leukocytes promote acute kidney injury via inflammasome and Toll-like receptor activation. Am J Pathol 184: 1411–1418, 2014. doi: 10.1016/j.ajpath.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 18.Conesa EL, Valero F, Nadal JC, Fenoy FJ, López B, Arregui B, Salom MG. N-acetyl-L-cysteine improves renal medullary hypoperfusion in acute renal failure. Am J Physiol Regul Integr Comp Physiol 281: R730–R737, 2001. [DOI] [PubMed] [Google Scholar]
- 19.Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-gamma. J Immunol 176: 3108–3114, 2006. doi: 10.4049/jimmunol.176.5.3108. [DOI] [PubMed] [Google Scholar]
- 20.de Bragança AC, Volpini RA, Mehrotra P, Andrade L, Basile DP. Vitamin D deficiency contributes to vascular damage in sustained ischemic acute kidney injury. Physiol Rep 4: e12829, 2016. doi: 10.14814/phy2.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int 66: 486–491, 2004. doi: 10.1111/j.1523-1755.2004.761_3.x. [DOI] [PubMed] [Google Scholar]
- 22.Fuller TF, Sattler B, Binder L, Vetterlein F, Ringe B, Lorf T. Reduction of severe ischemia/reperfusion injury in rat kidney grafts by a soluble P-selectin glycoprotein ligand. Transplantation 72: 216–222, 2001. doi: 10.1097/00007890-200107270-00008. [DOI] [PubMed] [Google Scholar]
- 23.Haller H, Dragun D, Miethke A, Park JK, Weis A, Lippoldt A, Gross V, Luft FC. Antisense oligonucleotides for ICAM-1 attenuate reperfusion injury and renal failure in the rat. Kidney Int 50: 473–480, 1996. doi: 10.1038/ki.1996.338. [DOI] [PubMed] [Google Scholar]
- 24.Herter JM, Rossaint J, Spieker T, Zarbock A. Adhesion molecules involved in neutrophil recruitment during sepsis-induced acute kidney injury. J Innate Immun 6: 597–606, 2014. doi: 10.1159/000358238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collett JA, Mehrotra P, Crone A, Merfeld-Clauss S, March KL, Basile DP. Human adipose stromal cell therapy improves survival and reduces renal inflammation and capillary rarefaction in acute kidney injury. J Cell Mol Med 2017. doi: 10.1111/jcmm.13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly KJ, Williams WW Jr, Colvin RB, Bonventre JV. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc Natl Acad Sci USA 91: 812–816, 1994. doi: 10.1073/pnas.91.2.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim M, Ham A, Kim JY, Brown KM, D’Agati VD, Lee HT. The volatile anesthetic isoflurane induces ecto-5′-nucleotidase (CD73) to protect against renal ischemia and reperfusion injury. Kidney Int 84: 90–103, 2013. doi: 10.1038/ki.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi B-S, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li B, Cohen A, Hudson TE, Motlagh D, Amrani DL, Duffield JS. Mobilized human hematopoietic stem/progenitor cells promote kidney repair after ischemia/reperfusion injury. Circulation 121: 2211–2220, 2010. doi: 10.1161/CIRCULATIONAHA.109.928796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Mcniece IK, Bertoncello I, Kriegler AB, Quesenberry PJ. Colony-forming cells with high proliferative potential (HPP-CFC). Int J Cell Cloning 8: 146–160, 1990. doi: 10.1002/stem.5530080302. [DOI] [PubMed] [Google Scholar]
- 32.Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int 88: 776–784, 2015. doi: 10.1038/ki.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molitoris BA. Therapeutic translation in acute kidney injury: the epithelial/endothelial axis. J Clin Invest 124: 2355–2363, 2014. doi: 10.1172/JCI72269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molitoris BA, Sandoval R, Sutton TA. Endothelial injury and dysfunction in ischemic acute renal failure. Crit Care Med 30, Suppl: S235–S240, 2002. doi: 10.1097/00003246-200205001-00011. [DOI] [PubMed] [Google Scholar]
- 35.Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure. Kidney Int 66: 496–499, 2004. doi: 10.1111/j.1523-1755.2004.761_5.x. [DOI] [PubMed] [Google Scholar]
- 36.Morigi M, Introna M, Imberti B, Corna D, Abbate M, Rota C, Rottoli D, Benigni A, Perico N, Zoja C, Rambaldi A, Remuzzi A, Remuzzi G. Human bone marrow mesenchymal stem cells accelerate recovery of acute renal injury and prolong survival in mice. Stem Cells 26: 2075–2082, 2008. doi: 10.1634/stemcells.2007-0795. [DOI] [PubMed] [Google Scholar]
- 37.Naito H, Kidoya H, Sakimoto S, Wakabayashi T, Takakura N. Identification and characterization of a resident vascular stem/progenitor cell population in preexisting blood vessels. EMBO J 31: 842–855, 2012. doi: 10.1038/emboj.2011.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res 97: 696–704, 2013. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patschan D, Krupincza K, Patschan S, Zhang Z, Hamby C, Goligorsky MS. Dynamics of mobilization and homing of endothelial progenitor cells after acute renal ischemia: modulation by ischemic preconditioning. Am J Physiol Renal Physiol 291: F176–F185, 2006. doi: 10.1152/ajprenal.00454.2005. [DOI] [PubMed] [Google Scholar]
- 40.Phillips SA, Pechman KR, Leonard EC, Friedrich JL, Bian J-T, Beal AG, Basile DP. Increased ANG II sensitivity following recovery from acute kidney injury: role of oxidant stress in skeletal muscle resistance arteries. Am J Physiol Regul Integr Comp Physiol 298: R1682–R1691, 2010. doi: 10.1152/ajpregu.00448.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rabb H, Mendiola CC, Saba SR, Dietz JR, Smith CW, Bonventre JV, Ramirez G. Antibodies to ICAM-1 protect kidneys in severe ischemic reperfusion injury. Biochem Biophys Res Commun 211: 67–73, 1995. doi: 10.1006/bbrc.1995.1779. [DOI] [PubMed] [Google Scholar]
- 42.Rajashekhar G, Traktuev DO, Roell WC, Johnstone BH, Merfeld-Clauss S, Van Natta B, Rosen ED, March KL, Clauss M. IFATS collection: Adipose stromal cell differentiation is reduced by endothelial cell contact and paracrine communication: role of canonical Wnt signaling. Stem Cells 26: 2674–2681, 2008. doi: 10.1634/stemcells.2008-0277. [DOI] [PubMed] [Google Scholar]
- 43.Regner KR, Roman RJ. Role of medullary blood flow in the pathogenesis of renal ischemia-reperfusion injury. Curr Opin Nephrol Hypertens 21: 33–38, 2012. doi: 10.1097/MNH.0b013e32834d085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Regner KR, Zuk A, Van Why SK, Shames BD, Ryan RP, Falck JR, Manthati VL, McMullen ME, Ledbetter SR, Roman RJ. Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int 75: 511–517, 2009. doi: 10.1038/ki.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rehman J, Li J, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 107: 1164–1169, 2003. doi: 10.1161/01.CIR.0000058702.69484.A0. [DOI] [PubMed] [Google Scholar]
- 46.Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE, Pell CL, Johnstone BH, Considine RV, March KL. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation 109: 1292–1298, 2004. doi: 10.1161/01.CIR.0000121425.42966.F1. [DOI] [PubMed] [Google Scholar]
- 47.Shih YC, Lee PY, Cheng H, Tsai CH, Ma H, Tarng DC. Adipose-derived stem cells exhibit antioxidative and antiapoptotic properties to rescue ischemic acute kidney injury in rats. Plast Reconstr Surg 132: 940e–951e, 2013. doi: 10.1097/PRS.0b013e3182a806ce. [DOI] [PubMed] [Google Scholar]
- 48.Singbartl K, Forlow SB, Ley K. Platelet, but not endothelial, P-selectin is critical for neutrophil-mediated acute postischemic renal failure. FASEB J 15: 2337–2344, 2001. doi: 10.1096/fj.01-0199com. [DOI] [PubMed] [Google Scholar]
- 49.Singbartl K, Green SA, Ley K. Blocking P-selectin protects from ischemia/reperfusion-induced acute renal failure. FASEB J 14: 48–54, 2000. [DOI] [PubMed] [Google Scholar]
- 50.Tögel F, Cohen A, Zhang P, Yang Y, Hu Z, Westenfelder C. Autologous and allogeneic marrow stromal cells are safe and effective for the treatment of acute kidney injury. Stem Cells Dev 18: 475–486, 2009. doi: 10.1089/scd.2008.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tögel F, Westenfelder C. The role of multipotent marrow stromal cells (MSCs) in tissue regeneration. Organogenesis 7: 96–100, 2011. doi: 10.4161/org.7.2.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tögel F, Yang Y, Zhang P, Hu Z, Westenfelder C. Bioluminescence imaging to monitor the in vivo distribution of administered mesenchymal stem cells in acute kidney injury. Am J Physiol Renal Physiol 295: F315–F321, 2008. doi: 10.1152/ajprenal.00098.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tögel F, Zhang P, Hu Z, Westenfelder C. VEGF is a mediator of the renoprotective effects of multipotent marrow stromal cells in acute kidney injury. J Cell Mol Med 13, 8B: 2109–2114, 2009. doi: 10.1111/j.1582-4934.2008.00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tögel FE, Westenfelder C. Kidney protection and regeneration following acute injury: progress through stem cell therapy. Am J Kidney Dis 60: 1012–1022, 2012. doi: 10.1053/j.ajkd.2012.08.034. [DOI] [PubMed] [Google Scholar]
- 56.Tögel FE, Westenfelder C. Mesenchymal stem cells: a new therapeutic tool for AKI. Nat Rev Nephrol 6: 179–183, 2010. doi: 10.1038/nrneph.2009.229. [DOI] [PubMed] [Google Scholar]
- 57.Vella F. Textbook of Clinical Chemistry. Tietz NW, Editor. Philadelphia, PA: Saunders, 1986. [Google Scholar]
- 58.Villanueva S, Carreño JE, Salazar L, Vergara C, Strodthoff R, Fajre F, Céspedes C, Sáez PJ, Irarrázabal C, Bartolucci J, Figueroa F, Vio CP. Human mesenchymal stem cells derived from adipose tissue reduce functional and tissue damage in a rat model of chronic renal failure. Clin Sci (Lond) 125: 199–210, 2013. doi: 10.1042/CS20120644. [DOI] [PubMed] [Google Scholar]
- 59.Viñas JL, Burger D, Zimpelmann J, Haneef R, Knoll W, Campbell P, Gutsol A, Carter A, Allan DS, Burns KD. Transfer of microRNA-486-5p from human endothelial colony forming cell-derived exosomes reduces ischemic kidney injury. Kidney Int 90: 1238–1250, 2016. doi: 10.1016/j.kint.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 60.Westenfelder C, Togel FE. Protective actions of administered mesenchymal stem cells in acute kidney injury: relevance to clinical trials. Kidney Int Suppl 1, Suppl: 103–106, 2011. doi: 10.1038/kisup.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao W, Hu Q, Ma Y, Xiong W, Wu T, Cao J, Wu D. Human adipose-derived mesenchymal stem cells repair cisplatin-induced acute kidney injury through antiapoptotic pathways. Exp Ther Med 10: 468–476, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoder MC. Is endothelium the origin of endothelial progenitor cells? Arterioscler Thromb Vasc Biol 30: 1094–1103, 2010. doi: 10.1161/ATVBAHA.109.191635. [DOI] [PubMed] [Google Scholar]
- 63.Yoder MC, Du XX, Williams DA. High proliferative potential colony-forming cell heterogeneity identified using counterflow centrifugal elutriation. Blood 82: 385–391, 1993. [PubMed] [Google Scholar]
- 64.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R, Temm CJ, Prchal JT, Ingram DA. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 109: 1801–1809, 2007. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yokota N, Daniels F, Crosson J, Rabb H. Protective effect of T cell depletion in murine renal ischemia-reperfusion injury. Transplantation 74: 759–763, 2002. doi: 10.1097/00007890-200209270-00005. [DOI] [PubMed] [Google Scholar]
- 66.Ysebaert DK, De Greef KE, De Beuf A, Van Rompay AR, Vercauteren S, Persy VP, De Broe ME. T cells as mediators in renal ischemia/reperfusion injury. Kidney Int 66: 491–496, 2004. doi: 10.1111/j.1523-1755.2004.761_4.x. [DOI] [PubMed] [Google Scholar]
- 67.Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M, Verpooten GA, Eyskens EJ, De Broe ME. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol Dial Transplant 15: 1562–1574, 2000. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]
- 68.Zander AR, Lange C, Westenfelder C. Mesenchymal stromal cells: main factor or helper in regenerative medicine? Kidney Int Suppl 1: 74–76, 2011. doi: 10.1038/kisup.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Fisher N, Newey SE, Smythe J, Tatton L, Tsaknakis G, Forde SP, Carpenter L, Athanassopoulos T, Hale SJ, Ferguson DJ, Tyler MP, Watt SM. The impact of proliferative potential of umbilical cord-derived endothelial progenitor cells and hypoxia on vascular tubule formation in vitro. Stem Cells Dev 18: 359–376, 2009. doi: 10.1089/scd.2008.0071. [DOI] [PubMed] [Google Scholar]
- 70.Zizzi HC, Zibari GB, Granger DN, Singh I, Cruz LD, Abreo F, McDonald JC, Brown MF. Quantification of P-selectin expression after renal ischemia and reperfusion. J Pediatr Surg 32: 1010–1013, 1997. doi: 10.1016/S0022-3468(97)90388-2. [DOI] [PubMed] [Google Scholar]