

Abstract
Prolonged decreases in urinary bladder blood flow are linked to overactive and underactive bladder pathologies. However, the mechanisms regulating bladder vascular reactivity are largely unknown. To investigate these mechanisms, we examined myogenic and vasoactive properties of mouse bladder feed arterioles (BFAs). Unlike similar-sized arterioles from other vascular beds, BFAs failed to constrict in response to increases in intraluminal pressure (5–80 mmHg). Consistent with this lack of myogenic tone, arteriolar smooth muscle cell membrane potential was hyperpolarized (−72.8 ± 1.4 mV) at 20 mmHg and unaffected by increasing pressure to 80 mmHg (−74.3 ± 2.2 mV). In contrast, BFAs constricted to the thromboxane analog U-46619 (100 nM), the adrenergic agonist phenylephrine (10 µM), and KCl (60 mM). Inhibition of nitric oxide synthase or intermediate- and small-conductance Ca2+-activated K+ channels did not alter arteriolar diameter, indicating that the dilated state of BFAs is not attributable to overactive endothelium-dependent dilatory influences. Myocytes isolated from BFAs exhibited BaCl2 (100 µM)-sensitive K+ currents consistent with strong inward-rectifier K+ (KIR) channels. Notably, block of these KIR channels “restored” pressure-induced constriction and membrane depolarization. This suggests that these channels, in part, account for hyperpolarization and associated absence of tone in BFAs. Furthermore, smooth muscle-specific knockout of KIR2.1 caused significant myogenic tone to develop at physiological pressures. This suggests that 1) the regulation of vascular tone in the bladder is independent of pressure, insofar as pressure-induced depolarizing conductances cannot overcome KIR2.1-mediated hyperpolarization; and 2) maintenance of bladder blood flow during bladder filling is likely controlled by neurohumoral influences.
Keywords: myogenic tone, potassium channels, urinary bladder, vasculature
the urinary bladder expands during filling to allow storage of urine. As the bladder fills, intravesical pressure remains relatively low and constant, allowing the bladder wall to markedly distend as the volume of stored urine increases (1). Maintaining adequate blood flow during bladder distension is a physiological challenge, but the bladder is highly vascularized with a specialized network of arterioles and venules capable of adapting to such expansion (20–22). These vessels are coiled within a specialized elastic sheath that allows them to elongate freely as the bladder wall thins and thus compensate for the longitudinal and axial stretch experienced during bladder filling (21, 27, 38). Even with these adaptations, however, bladder blood flow progressively decreases by nearly 50% during bladder filling, resulting in a state of transient ischemia in bladder tissue (50). Prolonging bladder ischemia, achieved experimentally by temporarily occluding the iliac artery, results in bladder overactivity, which eventually progresses to underactivity if the duration of ischemia is extended (2). These observations suggest the importance of minimizing the duration and magnitude of ischemia in maintaining proper bladder function, but provide few insights into the regulation of bladder blood flow per se. Ex vivo experiments have shown that the α1 antagonist prazosin blocks electrical field stimulation-induced constriction of submucosal arterioles, suggesting a role for sympathetic innervation of these vessels in regulating bladder arteriolar diameter, and by extension, bladder blood flow (18). Beyond this, very little is known about the properties of the bladder vasculature, and how these vessels regulate bladder blood flow before, during, and after micturition.
Under normal physiological conditions, most small arteries and arterioles in the body constrict in response to increases in intraluminal pressure. This behavior, termed the myogenic response, is a mechanism intrinsic to myocytes that maintains these vessels in a partially constricted state referred to as myogenic tone (56). The myogenic response, which increases in magnitude as vessel size decreases (5), contributes significantly to autoregulation of blood flow within specific organs, such as kidneys, retina, and the brain (9, 29, 54). This property helps to maintain consistent blood flow in the face of blood pressure fluctuations and prevents end-organ damage when blood pressure increases pathologically; as such, it is essential for the normal physiological function of many organs (36). Myogenic tone develops in response to intraluminal pressure-induced smooth muscle cell membrane depolarization, which activates voltage-dependent Ca2+ channels (VDCCs) and increases intracellular Ca2+, thereby resulting in constriction (16, 17, 25). In arteries that develop myogenic tone, smooth muscle cell resting membrane potential varies from −60 to −40 mV and is directly correlated with intraluminal pressure (19, 24, 44). This pressure-induced depolarization occurs even when arteries are fully dilated (25).
An important regulator of smooth muscle cell membrane potential, and thus smooth muscle excitability, is the strong inward-rectifier K+ (KIR) channel (7, 51, 52). KIR channels have two important characteristics that distinguish them from many other K+ channels: 1) they are activated by membrane potential hyperpolarization, and not depolarization; and 2) at a constant voltage, their open probability increases with extracellular K+, with half-activation corresponding to the K+ equilibrium potential (EK) (30, 35, 51). In many excitable cell types, the activity of KIR channels is responsible for maintaining a negative membrane potential near EK (35). However, the smooth muscle cells of pressurized small arteries with myogenic tone have a membrane potential of about −40 mV, which is quite positive of EK (−84.6 mV with 5.9 mM external K+). A depolarized resting potential relative to EK sets the stage for activation of KIR channels by external K+ to cause membrane potential hyperpolarization, provided that EK remains negative to the resting potential; EK would be at a resting potential of −40 mV when external K+ is ~25 mM (32, 44, 46). Thus changes in extracellular K+ concentration and membrane potential synergistically increase KIR channel conductance to hyperpolarize smooth muscle cells, ultimately causing vasodilation (33). This phenomenon is apparent in cerebral arteries, where members of the KIR2 subfamily, specifically KIR2.1, are expressed in smooth muscle and are involved in the regulation of arterial diameter and membrane potential (11, 33, 52).
In this study, we investigated the functional properties of urinary bladder feed arterioles and explored the mechanisms underlying the regulation of tone in this vascular bed. We discovered that, although bladder arterioles respond to multiple vasoconstrictors and vasodilators, they completely lack myogenic tone and remain hyperpolarized over a wide range of physiological pressures. Notably, blockade of KIR channels in intact arterioles caused pressure-dependent depolarization and constriction, suggesting that the lack of a myogenic response is attributable to the hyperpolarizing influence of KIR channels. Consistent with this conclusion, smooth muscle-specific knockout of the gene encoding the KIR2.1 isoform (KCNJ2) resulted in the development of significant myogenic tone. Collectively, these data suggest that pressure-induced depolarizing conductances are unable to overcome KIR channel-mediated membrane hyperpolarization and dilation, even as pressure increases. Because mechanical forces imposed on the distal vasculature by bladder distension and compression are predicted to produce increases in upstream luminal pressure, this myogenic-neutralizing mechanism prevents further pressure-induced restrictions in blood flow, and as such represents a novel biological strategy for maintaining blood flow during bladder filling.
METHODS
Mouse models and tissue preparation.
All procedures used in this study conform to institutional guidelines and were approved by the Institutional Animal Care and Use Committees of the University of Vermont. Wild-type male C57Bl/6 mice (12–20 wk old) and smooth muscle cell-specific KIR2.1 knockout mice (KIR2.1SMKO) were used for all experiments. KIR2.1SMKO mice were generated by crossing Kcnj2fl/fl mice [described previously (63)] with mice expressing tamoxifen-inducible Cre recombinase [CreER(T2)] under the control of the Myh11 (myosin, heavy polypeptide 11, smooth muscle) promoter (SMMHC-CreERT2 mice; courtesy of Dr. Stefan Offermanns) (53, 61). Adult (12–20 wk old) F1 progeny were injected with tamoxifen (30 mg/kg ip) once daily for 5 days to yield the KIR2.1SMKO mouse model.
All animals were euthanized with an injection of pentobarbital sodium (150 mg/kg ip) followed by decapitation. Urinary bladder, ureters, and urethra were harvested together and placed in chilled (4°C) HEPES-buffered saline containing (in mM) 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, and 7 glucose (pH 7.4). Bladder feed arterioles were isolated, beginning immediately proximal to where they enter the bladder wall and ending distal of the ureters at the bladder trigone. Arterioles (1–2 mm long, ~40 μm in diameter) were cleaned of connective tissue and isolated for further experimentation.
Chemicals.
All chemicals were acquired from Sigma-Aldrich (St. Louis, MO), except for U-46619 (EMD Millipore, Danvers, MA), charybdotoxin (EMD Millipore), and GSK2193974 (Tocris Biosciences, Bristol, UK).
Diameter measurements of isolated bladder arterioles.
Vessels were placed in a pressure arteriograph chamber (Instrumentation and Modeling Facility, University of Vermont, Burlington, VT) and attached to similar-sized glass cannulae using 12-0 monofilament suture. The proximal cannula was connected to a servo-controlled pressure-regulating device (Living Systems; St. Albans, VT), and the distal cannula was closed. Arteries were pressurized to 5 mmHg for ~15 min in aerated (20% O2, 75% N2, 5% CO2), warmed (37°C) physiological salt solution (PSS) containing (in mM) 119 NaCl, 24 NaHCO3, 4.7 KCl, 1.2 KH2PO4, 1.2 MgCl2, 2 CaCl2, 7 glucose, and 0.023 EDTA (pH 7.4). Internal diameter was monitored continuously with a CCD camera and edge-detection software (IonOptix, Milton, MA). All compounds were added to the superfusate (PSS), which was continuously aerated and recirculated through the chamber. Arteries were pressurized to 80 mmHg and exposed to Ca2+-free PSS with 5 mM EGTA at the conclusion of each experiment to obtain maximal diameter. In pressure experiments, intraluminal pressure was increased from 20 to 80 mmHg in 20-mmHg increments. In agonist/antagonist experiments, pressure was held steady at 60 mmHg.
Membrane potential recordings.
Vessels were cannulated in a pressure arteriograph chamber and pressurized as described above. Individual smooth muscle cells were impaled with a glass electrode containing 0.5 M KCl (tip resistance, 180–200 MΩ), and membrane potential was recorded using an AxoClamp-2A digital amplifier with an HS-2 headstage (Molecular Devices, Sunnyvale, CA). Successful cell penetration was determined by satisfaction of each of the following criteria: 1) sharp negative membrane potential deflection on entry, 2) stable potential for at least 30 s after entry, and 3) sharp return to ~0 mV upon removal of the electrode from the cell. In each vessel, 2–3 cells were impaled at each pressure. In some experiments, membrane potential was also measured in the presence of 60 mM KCl or 100 µM BaCl2.
Bladder arteriolar myocyte isolation.
After dissection, urinary bladders were placed in chilled (4°C) dissociation solution consisting of (in mM) 60 NaCl, 85 Na-glutamate, 5.6 KCl, 2 MgCl2, 10 HEPES, 10 glucose, 7 mannitol, and 1 mg/ml BSA (pH 7.4). Vessels were then cleaned of connective tissue and incubated for 20 min at 37°C in dissociation solution containing papain (0.5 mg/ml) and dithioerythritol (DTE; 1 mg/ml). Tissues were then transferred to dissociation solution containing collagenase (Type F; 1 mg/ml) and CaCl2 (100 μM), and incubated for 5 min at 37°C. Individual smooth muscle cells were obtained by transferring vessels to dissociation solution (free of enzymes, DTE, and CaCl2) and mechanically triturating using a fire-polished glass Pasteur pipette.
Electrophysiology.
Whole cell KIR currents in isolated bladder arteriolar myocytes were measured as described previously (26, 32, 52). Briefly, K+ currents elicited by a voltage-ramp protocol (−140 mV to +50 mV over a period of 200 ms) from a holding potential of −40 mV were measured using the perforated-patch configuration of the patch-clamp technique. The pipette solution contained 100 mM K+-aspartate, 30 mM KCl, 10 mM HEPES, 10 mM NaCl, 1 mM MgCl2, and 0.3 mg/ml amphotericin B (pH 7.2). The bath solution contained (in mM) 134 NaCl, 6 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 7 glucose (pH 7.4). KIR currents were defined as the component of total inward K+ current that was abolished in the presence of 100 μM Ba2+.
Data analysis and statistics.
Diameter data were analyzed using LabChart 7 Pro software (ADInstruments, Colorado Springs, CO). Membrane potential data were analyzed using ClampEx 9.2 software (Molecular Devices). Electrophysiological data were analyzed using pCLAMP 9 software (Molecular Devices). Statistical significance of differences between two groups of equal variance was determined using two-tailed, unpaired or paired Student’s t-tests, where appropriate (α = 0.05). For multiple sample comparisons, one- and two-way ANOVA were used followed by Bonferroni’s post hoc analysis to compare individual means. P values ≤ 0.05 were considered significant. Calculations were performed using Microsoft Excel (Microsoft) and GraphPad Prism (GraphPad Software).
RESULTS
Bladder feed arterioles lack myogenic tone.
To investigate the functional properties of bladder arterioles, we first assessed myogenic reactivity by measuring changes in the diameter of isolated, pressurized urinary bladder feed arterioles to increases in intraluminal pressure (Fig. 1). As intraluminal pressure increased from 20 to 80 mmHg, diameter increased in a stepwise fashion, remaining unchanged at constant pressure (Fig. 1A) and showing no hint of constriction to pressure (Fig. 1B). Chelation of extracellular Ca2+ by exposing arterioles to Ca2+-free PSS containing 5 mM EGTA did not affect diameter, consistent with a lack of active constriction (Fig. 1C).
Fig. 1.

Mouse bladder feed arterioles lack myogenic tone. Representative tracing (A) and summary graph (B) of responses to increasing intraluminal pressure in isolated, cannulated bladder feed arterioles. Myogenic tone was absent at all pressures. C: exposure of pressurized (at 80 mmHg) arterioles to Ca2+-free PSS containing 5 mM EGTA did not induce significant vasodilation, providing additional evidence for a lack of myogenic tone. N = 12.
Bladder feed arterioles respond to several vasoactive substances.
The absence of myogenic tone could reflect a loss of arteriolar viability. To address this possibility, we tested the effects of several standard vasoactive substances on the diameter of vessels held at a constant pressure (60 mmHg). Exposure to the thromboxane analog U-46619 (100 nM), phenylephrine (10 µM), or KCl (60 mM) caused stable and significant decreases in diameter (Fig. 2A), indicating that arterioles were viable and capable of contracting in response to vasoconstrictor agonists and depolarization. Constrictions induced by high K+ were largely abrogated by preincubation with the VDCC inhibitor, diltiazem (50 µM), as expected given the role of VDCCs in mediating depolarization-induced constriction (Fig. 2B). To test the effects of vasodilators, we first contracted arterioles with a submaximal concentration of U-46619 (25 nM) and then exposed them to the endothelium-dependent vasodilators carbachol (1 μM), a muscarinic receptor agonist, and NS-309 (1 μM), a selective activator of small- and intermediate-conductance Ca2+-sensitive K+ (SK and IK, respectively) channels (Fig. 2C). Carbachol and NS-309 caused near-maximal vasodilation, indicating that endothelial function in bladder arterioles remained intact. Thus the absence of myogenic tone is not caused by a general defect in the ability of the arterioles to constrict or dilate. Instead, the lack of myogenic tone in this type of arteriole reflects a specific alteration in their response to pressure.
Fig. 2.
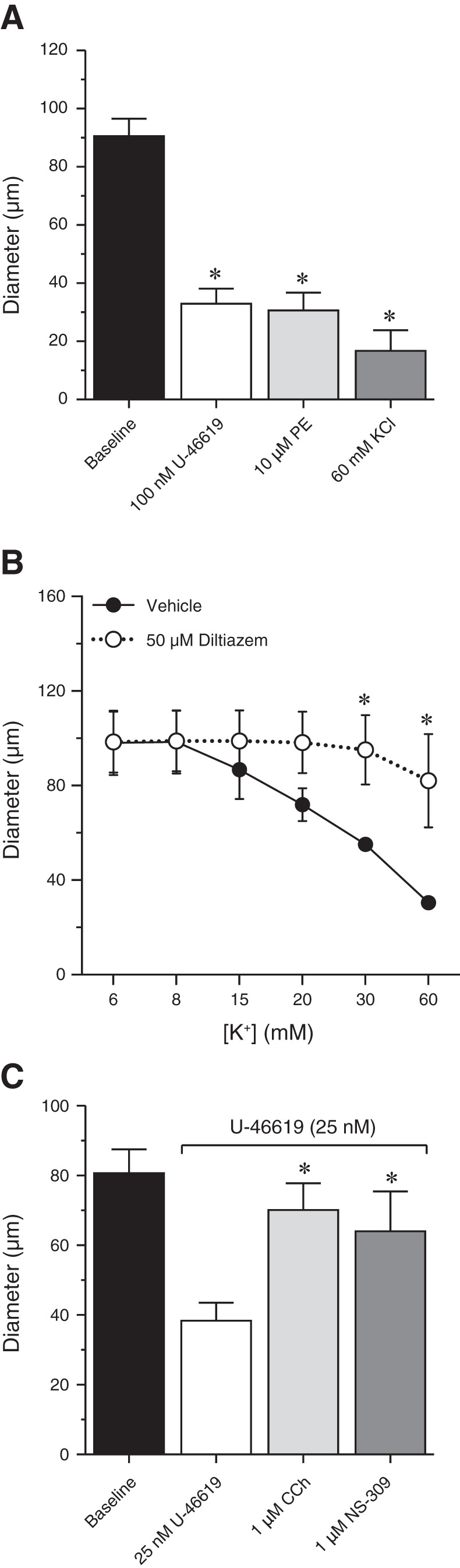
Mouse bladder feed arterioles respond to multiple vasodilators and vasoconstrictors. A: U-46619 (100 nM), phenylephrine (PE; 10 µM), and KCl (60 mM) elicited maximal contractile responses. B: KCl-induced contraction was inhibited by the L-type voltage-dependent calcium channel inhibitor diltiazem (50 µM). C: in vessels submaximally constricted with U-46619, both 1 µM CCh and 1 µM NS-309 caused maximal dilation. *P < 0.05 vs. baseline (A), vehicle (B), or U-46619 (C). N = 3–12.
The lack of myogenic tone is not caused by tonic vasodilatory influence from the endothelium.
In other vascular beds (e.g., cerebral arteries), pressure-induced constriction is opposed by tonic production of nitric oxide (NO) by the endothelium and by the tonic hyperpolarizing influence of endothelial IK and SK channels (15, 47). Small artery and arteriolar diameter is further regulated by activation of endothelial cell transient receptor potential vanilloid type 4 (TRPV4) channels, which are capable of causing near-maximal vasodilation (58). In addition, activation of large-conductance Ca2+-sensitive K+ channels in vascular smooth muscle opposes pressure-induced depolarization and constriction (43). To test whether the lack of myogenic tone in bladder feed arterioles was attributable to increased NO production, KCa channel activity, or TRPV4 channel activation, we exposed pressurized arterioles to the endothelial NO synthase (eNOS) inhibitor l-NNA (100 μM), a combination of KCa inhibitors (100 nM charybdotoxin + 300 nM apamin), or the potent TRPV4 channel antagonist GSK2193874 (10 nM) (Fig. 3). None of these interventions caused significant constriction, suggesting that the lack of myogenic tone is not attributable to the enhanced vasodilatory influence of endothelial NO production, TRPV4 channel activity, or KCa channel function.
Fig. 3.
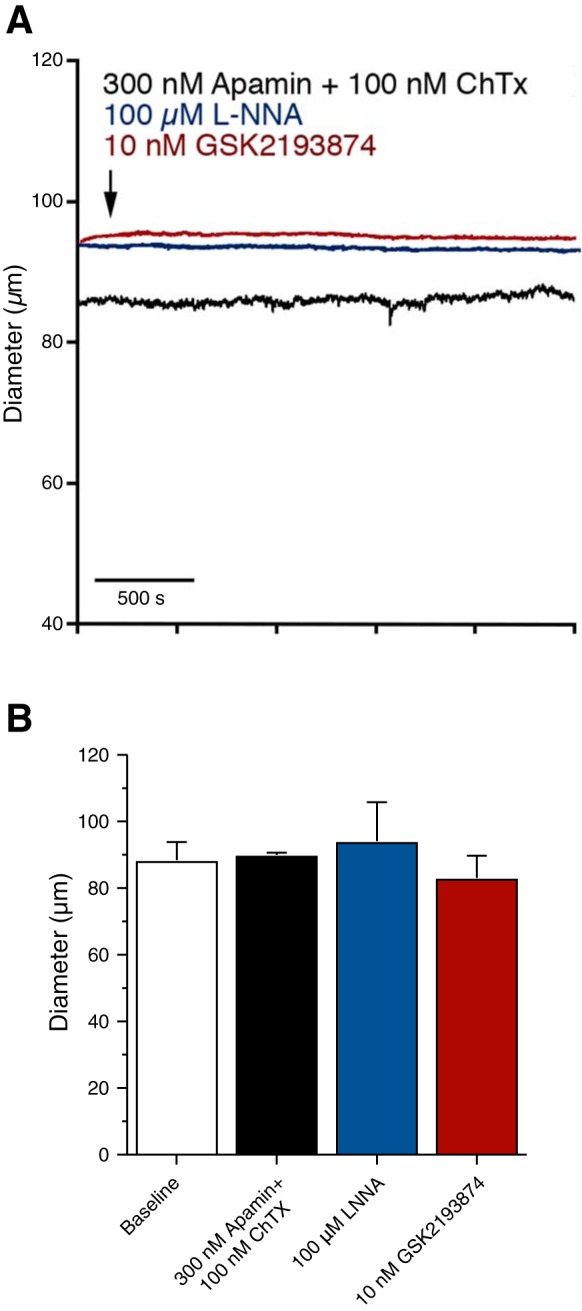
The lack of myogenic tone in mouse bladder feed arterioles is not due to endothelial NO production, increased TRPV4 channel activity, or KCa channels. A: representative tracings of bladder feed arteriole responses to 300 nM apamin + 100 nM charybdotoxin (ChTx) (black trace), 100 µM l-NNA (blue trace), or 10 nM GSK2193874 (red trace). B: inhibition of KCa channels (apamin/ChTx), eNOS (l-NNA), or TRPV4 channels (GSK2193874) had no effect on bladder feed arteriole diameter. N = 4–9.
Smooth muscle membrane potential is unaffected by pressure in bladder feed arterioles.
Elevation of intravascular pressure from low (20 mmHg) to physiological (80 mmHg) levels causes depolarization of smooth muscle cells from about −60 to −40 mV and constriction of small arteries (19, 24, 46). To determine if pressure depolarizes the membrane potential of smooth muscle cells in bladder feed arterioles, we measured membrane potential during increases in intraluminal pressure (Fig. 4). At 20 mmHg, bladder arteriole smooth muscle cell membrane potential was −72.8 ± 1.4 mV (Fig. 4A). Notably, increasing pressure to 80 mmHg did not cause significant depolarization (−74.3 ± 2.2 mV) (Fig. 4B). Addition of 60 mM KCl (at 80 mmHg) caused a depolarization to −22.7 ± 0.2 mV (Fig. 4C), a value close to the estimated EK (25, 42). These results indicate that pressure does not cause vasoconstriction because it does not depolarize the smooth muscle cells in the arteriole. Taken together, these data show that the membrane potential of bladder feed arteriolar smooth muscle cells is more hyperpolarized at low pressure than other arteries and is unaffected by pressure.
Fig. 4.

Bladder feed arteriolar smooth muscle cells do not depolarize in response to increases in intraluminal pressure. A–C: representative membrane potential recordings from mouse bladder feed arterioles pressurized to 20 mmHg (A) and 80 mmHg (B), and in the presence of 60 mM K+ (C). D: summary graph showing that smooth muscle cell membrane potential was unchanged as intraluminal pressure increased but was significantly depolarized by high (60 mM) extracellular K+. *P < 0.05 vs. 20 mmHg, †P < 0.05 vs. 80 mmHg; N = 3–4.
KIR channel activity is detected in bladder arteriolar myocytes.
BaCl2 acts as a relatively selective blocker of KIR channels at concentrations less than or equal to 100 µM (44, 59). Whole cell currents from isolated bladder arteriolar myocytes showed significant Ba2+-sensitive inward currents, indicating that arteriolar smooth muscle cells express functional KIR channels (Fig. 5, A and B). In pressurized vessel segments, addition of increasing concentrations of BaCl2 caused a concentration-dependent decrease in arteriolar diameter (Fig. 5, C and D). These data show that KIR channels are present and active in bladder feed arterioles, and may oppose the pressure-induced depolarizing conductances that would lead to the development of myogenic tone.
Fig. 5.

Inward-rectifier K+ (KIR) channel currents and functional role in isolated bladder arteriolar smooth muscle cells. A: representative traces of whole cell currents from isolated bladder arterioles in the absence (gray line) or presence (red line) of 100 µM BaCl2. Bladder arteriole myocytes exhibit a Ba2+-sensitive, inward-rectifying current (black line). B: summary graph of Ba2+-sensitive inward currents (at −140 mV and −100 mV membrane potentials) in isolated smooth muscle cells from bladder feed arterioles (n = 6 cells from N = 6 mice). C: representative tracings of bladder feed arteriolar responses to BaCl2 (100 µM). D: summary graph showing that BaCl2 caused a concentration-dependent constriction of bladder arterioles. N = 4–5.
As described above, a hallmark of arteries that possess KIR channels is that they dilate to small elevations of external K+ and constrict to higher concentrations (32, 44, 46). As shown in Fig. 2B, increasing K+ concentrations only caused constriction of bladder feed arterioles because no tone was present from which to dilate. To determine if bladder arterioles exhibit a biphasic response to increases in K+, we first submaximally contracted arterioles with 25 nM U-46619 and then exposed them to increasing concentrations of extracellular K+ (5.9–60 mM). Like other vessels that express KIR channels, bladder feed arterioles exposed to 8 mM K+ dilated, whereas those exposed to higher concentrations constricted (Fig. 6). Taken together, these findings demonstrate the presence of functional KIR channels and the potential for KIR-dependent regulation of vessel diameter and membrane potential in urinary bladder feed arterioles.
Fig. 6.
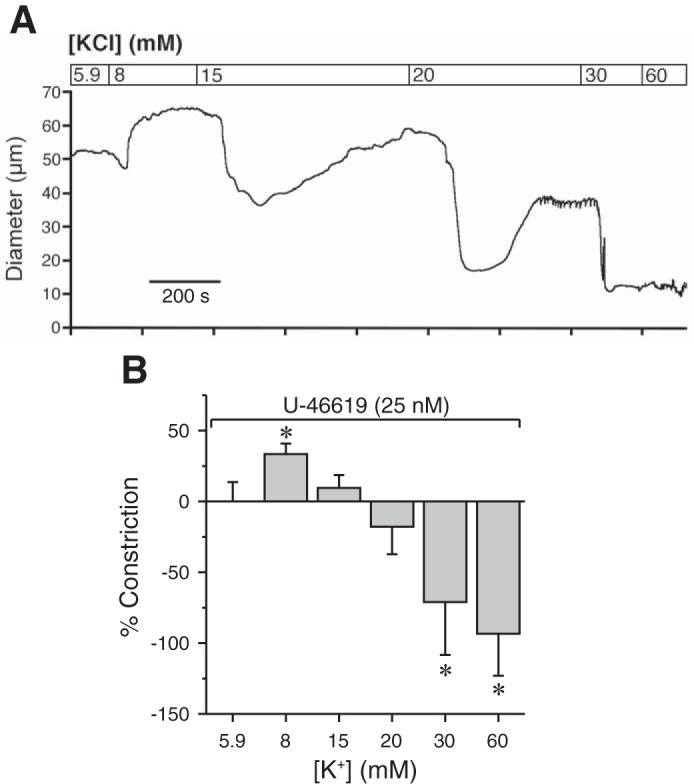
KIR channel activation or inhibition affects bladder arteriole diameter. A: representative tracing of bladder feed arteriole responses to increasing concentrations of K+. Vessels were first constricted with a submaximal concentration of U-46619 (25 nM). B: small increases in extracellular K+ caused relaxation that became constriction as concentration increased, indicative of KIR-mediated relaxation. *P < 0.05 vs. U-46619; N = 3.
KIR channel inhibition “restores” pressure-dependent smooth muscle membrane potential depolarization and vasoconstriction.
One possible explanation for the absence of myogenic tone in bladder feed arterioles is the absence of pressure-sensing, depolarizing ion channels. To investigate this, we measured membrane potential during increases in intraluminal pressure in the presence or absence of the KIR channel blocker BaCl2 (100 μM) (Fig. 7, A–E). In the absence of BaCl2, smooth muscle cell membrane potential was only slightly positive of EK but insensitive to changes in pressure (−70.4 ± 1.9 mV and −72.2 ± 0.7 mV at 20 mmHg and 80 mmHg, respectively) (Fig. 7, A and B). In marked contrast, with KIR channels blocked (100 μM BaCl2), pressurization to 20 and 80 mmHg caused membrane depolarization to −56.2 ± 3.1 and −43.3 ± 2.0 mV, respectively (Fig. 7, C and D). The prediction is that, under these conditions, the resultant pressure-induced depolarization should cause vasoconstriction. Indeed, in the presence of Ba2+, elevation of pressure to 60 and 80 mmHg led to constriction of these arterioles (Fig. 8, A–C). Together, these data suggest that the lack of myogenic tone in bladder feed arterioles is not attributable to the absence of pressure-induced depolarization mechanisms, but is rather due to the activity of KIR channels in bladder arteriolar smooth muscle and the inability of depolarizing conductances to overcome this hyperpolarizing influence.
Fig. 7.

Inhibiting KIR channels results in pressure-dependent depolarization of bladder arteriolar smooth muscle cells. A–D: representative membrane potential recordings from mouse bladder feed arterioles pressurized to 20 and 80 mmHg, in the absence (A, B) or presence (C, D) of the KIR channel inhibitor BaCl2 (100 µM). E: summary graph indicating that, in the absence of BaCl2, smooth muscle cell membrane potential was unchanged as intraluminal pressure increased. In the presence of BaCl2, increasing pressure from 20 to 80 mmHg caused a significant depolarization of smooth muscle cell membrane potential. *P < 0.05 vs. control, †P < 0.05 vs. 20 mmHg; N = 4.
Fig. 8.
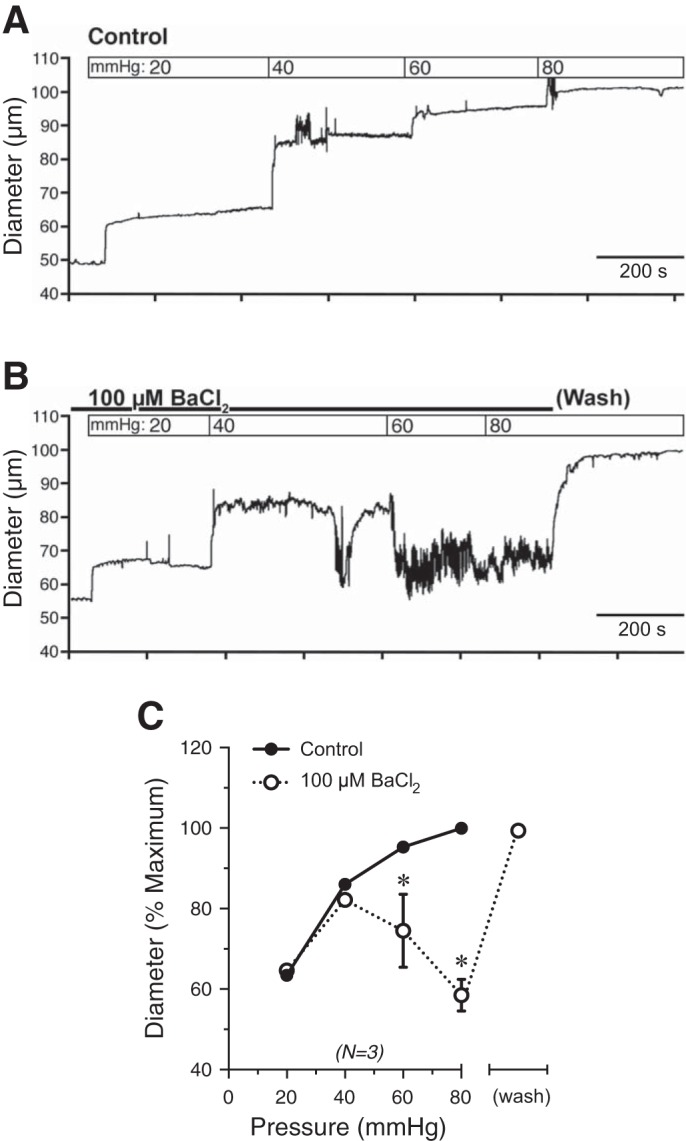
Inhibiting KIR channels results in pressure-dependent constriction of bladder feed arterioles. A, B: representative tracings of bladder feed arteriolar responses to increases in intraluminal pressure in the absence (A) or presence (B) of BaCl2 (100 µM). Vessels dilated to a diameter equivalent to controls after removal of BaCl2 at 80 mmHg. C: summary graph showing that BaCl2 resulted in pressure-induced constriction of bladder feed arterioles, indicating that KIR channel inhibition results in myogenic constriction. *P < 0.05 vs. control; N = 3.
Genetic ablation of smooth muscle KIR channels enables myogenic tone development.
In smooth muscle cells from small cerebral and coronary arterioles, activation of the KIR2.1 channel subtype causes membrane hyperpolarization, which leads to vasodilation (48, 64). Unfortunately, germline knockout of KIR2.1 causes cleft palate and neonatal lethality (64), making this model unsuitable for our studies. We circumvented this limitation by generating smooth muscle-specific, inducible KIR2.1-knockout mice (KIR2.1SMKO) (see methods). To validate that tamoxifen treatment reduced KIR2.1 function, we measured Ba2+-sensitive K+ currents in isolated bladder arteriolar smooth muscle cells from untreated and tamoxifen-treated KIR2.1SMKO mice (Fig. 9). Isolated smooth muscle myocytes from KIR2.1SMKO mice that did not receive tamoxifen showed no change in Ba2+-sensitive K+ currents compared with C57Bl/6 control mice. However, Ba2+-sensitive K+ currents were nearly abolished in KIR2.1SMKO mice treated with tamoxifen, indicating successful knockout of the KIR2.1 channel.
Fig. 9.
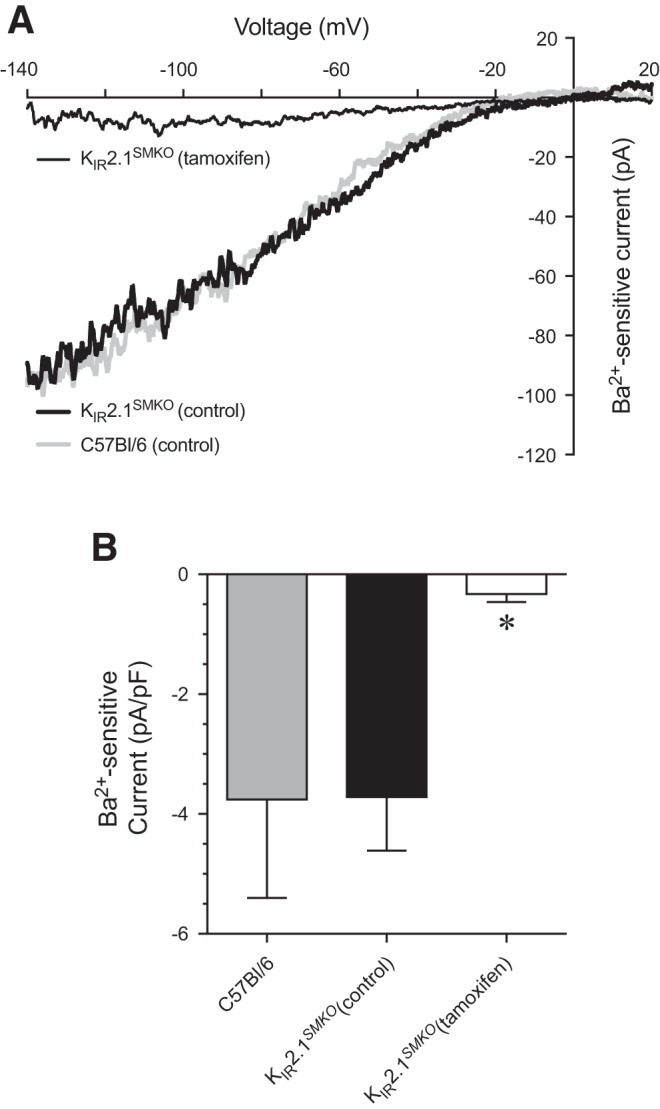
KIR channel currents in isolated bladder arteriolar smooth muscle cells from KIR2.1SMKO mice. A: without tamoxifen treatment, isolated bladder arteriole myocytes from KIR2.1SMKO mice exhibited Ba2+-sensitive inward-rectifying currents. With tamoxifen treatment, Ba2+-sensitive currents were nearly abolished. B: summary graph of Ba2+-sensitive inward current densities (at a membrane potential of −140 mV) from KIR2.1SMKO mice. In both A and B, currents from C57Bl/6 mice are shown for comparison (see Fig. 5). *P < 0.05; n = 6–11 cells from N = 2–3 mice.
To assess the contribution of smooth muscle KIR channels to the absence of pressure-induced constriction of urinary bladder feed arterioles, we examined arteriolar responses to changes in pressure in KIR2.1SMKO mice. Bladder arterioles from KIR2.1SMKO mice developed myogenic tone in response to increases in pressure, but exhibited profound vasomotion and contraction at higher pressures (Fig. 10A). At 40, 60, and 80 mmHg, the diameter of KIR2.1SMKO bladder arterioles was significantly reduced compared with that of control arterioles (Fig. 10B). These findings suggest that smooth muscle KIR channels are active in bladder feed arterioles, and that removal of KIR channels allows for pressure-induced depolarizing conductances to cause myogenic tone to develop.
Fig. 10.

Knockout of KIR2.1 channels from smooth muscle cells results in myogenic tone development in bladder arterioles. A: responses to increasing intraluminal pressure in isolated, cannulated bladder feed arterioles from KIR2.1SMKO mice. B: summary graph showing development of significant myogenic tone at 40, 60, and 80 mmHg in KIR2.1SMKO mice compared with control mice. *P < 0.05 vs. control; N = 9–13.
DISCUSSION
The role of the vasculature in bladder function has received relatively little research attention, despite the fact that prolonged decreases in bladder blood flow are linked to both overactive and underactive bladder (28, 45). In the present study, we investigated the properties of bladder feed arterioles to determine how these vessels might regulate bladder blood flow during normal bladder function. Unexpectedly, we found that bladder arterioles, unlike similarly sized vessels in most other vascular beds, lack myogenic tone. We further found that inhibition of the arteriolar smooth muscle KIR channel or ablation of the gene encoding it “restored” myogenic tone, producing smooth muscle depolarization and vascular diameter responses to pressure that were similar to those observed in vessels that normally exhibit myogenic tone (19, 44). These results indicate that the nonmyogenic phenotype of bladder feed arterioles is attributable to the activity of smooth muscle KIR channels. Thus, in the absence of other stimuli, KIR channel activity induces a hyperpolarizing influence that cannot be overcome by pressure-induced depolarization, thereby maintaining bladder feed arterioles in a state of maximal dilation.
Blood flow and bladder function.
The lack of myogenic tone in the bladder vasculature suggests that bladder arterioles do not autoregulate blood flow through direct responses to changes in intravascular pressure. Although this would be enormously detrimental in the cerebral or renal vasculature (5, 6, 60), it may be a specialized adaptation that serves to counteract the effects of bladder distension on vessel diameter. During normal bladder filling, distension of the bladder wall stretches the vasculature and imposes axial forces on it, causing vessel narrowing and compression, both of which are predicted to translate to higher intravascular pressure (per Poiseuille’s law) in upstream vessels, including feed arterioles. If bladder arterioles were capable of developing myogenic tone, this increase in pressure would cause these arterioles to constrict, further diminishing blood flow to the downstream vasculature and exacerbating the direct effects of vessel stretch and compression on blood flow. Thus the absence of myogenic tone is advantageous for maintaining blood flow and perfusion in the face of the forces imposed on the vasculature by expansion of the bladder wall. The forces imparted on the vasculature as the bladder wall distends can also lead to transient ischemia and acute hypoxia (31). Prolonged hypoxia, caused by inflammation, outlet obstruction, or vesical artery occlusion, is correlated with prolonged decreases in bladder blood flow, progressive bladder dysfunction, and, ultimately, bladder necrosis if blood flow is not restored (2–4, 37, 49). Vesical artery occlusion and decreased blood flow are also associated with significant bladder wall remodeling processes, including collagen deposition, urothelial barrier disruption, and production of proangiogenic factors and reactive oxygen species (2, 12, 31). Collectively, these observations suggest that the bladder is extremely sensitive to changes in blood flow, as even a modest reduction in blood flow leads to hypoxia, and prolonged changes in blood flow can cause bladder overactivity and remodeling.
Nonmyogenic regulation of the bladder vasculature.
An absolute lack of vasoregulatory capacity would be extremely detrimental in the context of an empty bladder, potentially causing capillary bed damage, tissue damage, and edema (36, 57). Our results show that, despite their myogenic incompetence, bladder feed arterioles remain sensitive to multiple endogenous vasoconstrictors that are capable of causing a prolonged reduction in vessel diameter in vivo. In the absence of a myogenic response, urinary bladder arteriolar diameter (and by extension bladder blood flow) are likely regulated through pressure-independent neurohumoral mechanisms. This regulation is likely attributable, at least in part, to nervous system innervation, as evidenced by our finding that the α-adrenergic agonist phenylephrine contracts bladder arterioles (Fig. 2). Electrical field stimulation-induced constriction of bladder submucosal arterioles is blocked by the α1 antagonist prazosin (18), indicating sympathetic innervation of these vessels. However, other nerves, including cholinergic, nitrergic, and primary afferent nerves, may also innervate these vessels, setting the stage for complex, heterogeneous neural regulation of bladder blood flow (28, 39).
It is also possible that bladder blood flow is regulated locally through the release of vasoactive substances from within the bladder wall. NO, acetylcholine, endothelin-1 (ET-1), and ATP are all released from the urothelium as the bladder fills (10, 14, 23, 40). Although detrusor smooth muscle appears to be insensitive to NO (8, 13, 34), changes in NO, ET-1, and ATP release from the urothelium are correlated with urinary bladder dysfunction (23, 41). This suggests that, in the absence of myogenic regulation of arteriolar diameter, a balance between neurohumoral input and vasoactive substances released from the urothelium may regulate bladder blood flow locally. Further support for this idea is provided by the observation that bladder blood flow significantly increases during micturition in an NO-dependent manner (49). Since myogenic tone is absent, an alternative vasoconstricting mechanism must be present to allow this relaxation to occur. Additional studies will be required to determine if, and how, locally released vasoactive substances and neurohumoral mechanisms regulate bladder blood flow, and whether dysfunction of these mechanisms influences normal bladder physiology.
Future directions: KIR channels and pressure-induced depolarization.
What remains unclear is whether the lack of myogenic tone in bladder feed arterioles is due to an increase in hyperpolarizing conductances or a decrease in depolarizing conductances activated by changes in intraluminal pressure. The fact that pressure does induce constriction in bladder feed arterioles when KIR channels are absent or inhibited indicates that these vessels possess a mechanism capable of causing myogenic tone development. One possibility is that KIR channel current density in urinary bladder feed arterioles is increased relative to smooth muscle cells from other vascular beds. However, it is difficult to accurately make this comparison owing to the wide range of reported current densities within and between vascular beds. Under comparable conditions (60 mM extracellular K+, −100 mV), smooth muscle cell KIR channel current density ranges from −1.6 pA/pF in hamster middle cerebral artery to −6.1 pA/pF in rat parenchymal arterioles and −9.0 pA/pF in the rat basilar artery (32, 55, 62). Nevertheless, it appears that the mechanisms that normally drive pressure-induced depolarization, although present, are not able to overcome the hyperpolarizing conductance through KIR channels. It is also possible that pressure-independent depolarizing conductances are present in these vessels. Although bladder arteriolar smooth muscle cell membrane potential is hyperpolarized compared with smooth muscle cells from other arteries, it is still positive of EK. This suggests some sort of mechanism is indeed present to oppose the hyperpolarizing influence of KIR channels. Future experiments are needed to seek to uncover the nature and identity of these depolarizing mechanisms, and determine how the balance between hyperpolarizing and depolarizing influences work together to regulate bladder arteriolar diameter before, during, and after bladder filling.
Conclusions.
Collectively, our findings show that urinary bladder feed arterioles fail to develop myogenic tone unless KIR channels are blocked or smooth muscle KIR2.1 channels are genetically ablated. This KIR channel-dependent neutralization of arteriolar myogenic tone represents a unique specialization of the bladder vasculature that likely evolved to prevent increases in feed arteriolar pressure produced by the recurrent distension and contraction of the bladder wall from inducing a myogenic response that would further restrict blood flow. Thus this adaptation can be viewed as a mechanism for maintaining adequate tissue perfusion during the micturition cycle. In the absence of an operational pressure-induced constriction response, bladder blood flow must be regulated by other means, presumably neurohumoral influences or local release of vasoconstrictors from the urothelium/bladder wall. Consistent with this supposition, urinary bladder feed arterioles retain the ability to constrict and dilate in response to multiple vasoactive compounds in a manner qualitatively similar to that of other arterioles of similar size.
GRANTS
This research was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants K01-DK-103840 (to N. R. Tykocki) and R37-DK-053832 (to M. T. Nelson) and American Heart Association Grant 14-POST-20480144 (to T. A. Longden).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.R.T., T.J.H., and M.T.N. conceived and designed research; N.R.T., A.D.B., and T.A.L. performed experiments; N.R.T., A.D.B., and T.A.L. analyzed data; N.R.T., A.D.B., and M.T.N. interpreted results of experiments; N.R.T. and A.D.B. prepared figures; N.R.T. drafted manuscript; N.R.T. and M.T.N. edited and revised manuscript; N.R.T., A.D.B., T.J.H., and M.T.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Stefan Offermanns for creating and providing the SMMHC-CreERT2 mice, and Dr. David Hill-Eubanks for input and editorial assistance.
REFERENCES
- 1.Andersson KE, Arner A. Urinary bladder contraction and relaxation: physiology and pathophysiology. Physiol Rev 84: 935–986, 2004. doi: 10.1152/physrev.00038.2003. [DOI] [PubMed] [Google Scholar]
- 2.Azadzoi KM, Tarcan T, Kozlowski R, Krane RJ, Siroky MB. Overactivity and structural changes in the chronically ischemic bladder. J Urol 162: 1768–1778, 1999. doi: 10.1016/S0022-5347(05)68236-5. [DOI] [PubMed] [Google Scholar]
- 3.Bajory Z, Hutter J, Krombach F, Messmer K. The role of endothelin-1 in ischemia-reperfusion induced acute inflammation of the bladder in rats. J Urol 168: 1222–1225, 2002. doi: 10.1016/S0022-5347(05)64629-0. [DOI] [PubMed] [Google Scholar]
- 4.Bajory Z, Hutter J, Krombach F, Messmer K. Microcirculation of the urinary bladder in a rat model of ischemia-reperfusion-induced cystitis. Urology 60: 1136–1140, 2002. doi: 10.1016/S0090-4295(02)01952-0. [DOI] [PubMed] [Google Scholar]
- 5.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 79: 387–423, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Davis MJ. Perspective: physiological role(s) of the vascular myogenic response. Microcirculation 19: 99–114, 2012. doi: 10.1111/j.1549-8719.2011.00131.x. [DOI] [PubMed] [Google Scholar]
- 7.Edwards FR, Hirst GD, Silverberg GD. Inward rectification in rat cerebral arterioles; involvement of potassium ions in autoregulation. J Physiol 404: 455–466, 1988. doi: 10.1113/jphysiol.1988.sp017299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrén I, Iversen H, Jansson O, Adolfsson J, Wiklund NP. Localization of nitric oxide synthase activity in the human lower urinary tract and its correlation with neuroeffector responses. Urology 44: 683–687, 1994. doi: 10.1016/S0090-4295(94)80206-8. [DOI] [PubMed] [Google Scholar]
- 9.Faraci FM, Baumbach GL, Heistad DD. Myogenic mechanisms in the cerebral circulation. J Hypertens Suppl 7: S61–S64, 1989. [PubMed] [Google Scholar]
- 10.Ferguson DR, Kennedy I, Burton TJ. ATP is released from rabbit urinary bladder epithelial cells by hydrostatic pressure changes--a possible sensory mechanism? J Physiol 505: 503–511, 1997. doi: 10.1111/j.1469-7793.1997.503bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 9: 1397–1403, 2006. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 12.Ghafar MA, Anastasiadis AG, Olsson LE, Chichester P, Kaplan SA, Buttyan R, Levin RM. Hypoxia and an angiogenic response in the partially obstructed rat bladder. Lab Invest 82: 903–909, 2002. doi: 10.1097/01.LAB.0000021135.87203.92. [DOI] [PubMed] [Google Scholar]
- 13.Gillespie JI. Phosphodiesterase-linked inhibition of nonmicturition activity in the isolated bladder. BJU Int 93: 1325–1332, 2004. doi: 10.1111/j.1464-410X.2004.04840.x. [DOI] [PubMed] [Google Scholar]
- 14.Hanna-Mitchell AT, Beckel JM, Barbadora S, Kanai AJ, de Groat WC, Birder LA. Non-neuronal acetylcholine and urinary bladder urothelium. Life Sci 80: 2298–2302, 2007. doi: 10.1016/j.lfs.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hannah RM, Dunn KM, Bonev AD, Nelson MT. Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab 31: 1175–1186, 2011. doi: 10.1038/jcbfm.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harder DR, Gilbert R, Lombard JH. Vascular muscle cell depolarization and activation in renal arteries on elevation of transmural pressure. Am J Physiol Renal Fluid Electrolyte Physiol 253: F778–F781, 1987. [DOI] [PubMed] [Google Scholar]
- 17.Harder DR. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circ Res 55: 197–202, 1984. doi: 10.1161/01.RES.55.2.197. [DOI] [PubMed] [Google Scholar]
- 18.Hashitani H, Takano H, Fujita K, Mitsui R, Suzuki H. Functional properties of suburothelial microvessels in the rat bladder. J Urol 185: 2382–2391, 2011. doi: 10.1016/j.juro.2011.02.046. [DOI] [PubMed] [Google Scholar]
- 19.Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol 3: a004549, 2011. doi: 10.1101/cshperspect.a004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hossler F, Kao R, Monson F. Microvasculature of the mammalian urinary bladder: a preliminary comparative study using corrosion casting. Microsc Microanal 11: 1204–1205, 2005. doi: 10.1017/S1431927605502356. [DOI] [Google Scholar]
- 21.Hossler FE, Monson FC. Evidence for a unique elastic sheath surrounding the vesicular arteries of the rabbit urinary bladder–studies of the microvasculature with microscopy and vascular corrosion casting. Anat Rec 252: 472–476, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 22.Hossler FE, Monson FC. Structure and blood supply of intrinsic lymph nodes in the wall of the rabbit urinary bladder--studies with light microscopy, electron microscopy, and vascular corrosion casting. Anat Rec 252: 477–484, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 23.Khan MA, Thompson CS, Dashwood MR, Mumtaz FH, Morgan RJ, Mikhailidis DP. Endothelin-1 and nitric oxide in the pathogenesis of urinary tract disorders secondary to bladder outlet obstruction. Curr Vasc Pharmacol 1: 27–31, 2003. doi: 10.2174/1570161033386600. [DOI] [PubMed] [Google Scholar]
- 24.Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol Heart Circ Physiol 269: H348–H355, 1995. [DOI] [PubMed] [Google Scholar]
- 25.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508: 199–209, 1998. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koide M, Bonev AD, Nelson MT, Wellman GC. Inversion of neurovascular coupling by subarachnoid blood depends on large-conductance Ca2+-activated K+ (BK) channels. Proc Natl Acad Sci USA 109: E1387–E1395, 2012. doi: 10.1073/pnas.1121359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowalewska PM, Burrows LL, Fox-Robichaud AE. Intravital microscopy of the murine urinary bladder microcirculation. Microcirculation 18: 613–622, 2011. doi: 10.1111/j.1549-8719.2011.00123.x. [DOI] [PubMed] [Google Scholar]
- 28.Kozlowski R, Siroky MB, Krane RJ, Azadzoi KM. Regulation of blood flow and microcirculation resistance in rabbit bladder. J Urol 168: 1608–1614, 2002. doi: 10.1016/S0022-5347(05)64529-6. [DOI] [PubMed] [Google Scholar]
- 29.Kur J, Bankhead P, Scholfield CN, Curtis TM, McGeown JG. Ca(2+) sparks promote myogenic tone in retinal arterioles. Br J Pharmacol 168: 1675–1686, 2013. doi: 10.1111/bph.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leech CA, Stanfield PR. Inward rectification in frog skeletal muscle fibres and its dependence on membrane potential and external potassium. J Physiol 319: 295–309, 1981. doi: 10.1113/jphysiol.1981.sp013909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin AT, Juan YS. Ischemia, hypoxia and oxidative stress in bladder outlet obstruction and bladder overdistention injury. Low Urin Tract Symptoms 4, Suppl 1: 27–31, 2012. doi: 10.1111/j.1757-5672.2011.00134.x. [DOI] [PubMed] [Google Scholar]
- 32.Longden TA, Dabertrand F, Hill-Eubanks DC, Hammack SE, Nelson MT. Stress-induced glucocorticoid signaling remodels neurovascular coupling through impairment of cerebrovascular inwardly rectifying K+ channel function. Proc Natl Acad Sci USA 111: 7462–7467, 2014. doi: 10.1073/pnas.1401811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longden TA, Nelson MT. Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 22: 183–196, 2015. doi: 10.1111/micc.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longhurst PA, Briscoe JA, Rosenberg DJ, Leggett RE. The role of cyclic nucleotides in guinea-pig bladder contractility. Br J Pharmacol 121: 1665–1672, 1997. doi: 10.1038/sj.bjp.0701328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marrelli SP, Johnson TD, Khorovets A, Childres WF, Bryan RM Jr, Busija DW. Altered function of inward rectifier potassium channels in cerebrovascular smooth muscle after ischemia/reperfusion: Editorial Comment. Stroke 29: 1469–1474, 1998. doi: 10.1161/01.STR.29.7.1469. [DOI] [PubMed] [Google Scholar]
- 36.Mellander S. Functional aspects of myogenic vascular control. J Hypertens Suppl 7: S21–S30, 1989. [PubMed] [Google Scholar]
- 37.Mine S, Yamamoto T, Mizuno H, Endo K, Matsukawa Y, Funahashi Y, Kato M, Hattori R, Gotoh M. Effect of tamsulosin on bladder microcirculation in rat model of bladder outlet obstruction using pencil lens charge-coupled device microscopy system. Urology 81: 155–159, 2013. doi: 10.1016/j.urology.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Miodoński AJ, Litwin JA. Microvascular architecture of the human urinary bladder wall: a corrosion casting study. Anat Rec 254: 375–381, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 39.Mitsui R, Hashitani H. Immunohistochemical characteristics of suburothelial microvasculature in the mouse bladder. Histochem Cell Biol 140: 189–200, 2013. doi: 10.1007/s00418-012-1074-5. [DOI] [PubMed] [Google Scholar]
- 40.Mumtaz FH, Khan MA, Sullivan ME, Thompson CS, Mikhailidis DP, Morgan RJ, Dashwood MR. Potential role of endothelin and nitric oxide in physiology and pathophysiology of the lower urinary tract. Endothelium 7: 1–9, 1999. doi: 10.3109/10623329909165307. [DOI] [PubMed] [Google Scholar]
- 41.Munoz A, Smith CP, Boone TB, Somogyi GT. Overactive and underactive bladder dysfunction is reflected by alterations in urothelial ATP and NO release. Neurochem Int 58: 295–300, 2011. doi: 10.1016/j.neuint.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neild TO, Kotecha N. Relation between membrane potential and contractile force in smooth muscle of the rat tail artery during stimulation by norepinephrine, 5-hydroxytryptamine, and potassium. Circ Res 60: 791–795, 1987. doi: 10.1161/01.RES.60.5.791. [DOI] [PubMed] [Google Scholar]
- 43.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science 270: 633–637, 1995. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 44.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol 268: C799–C822, 1995. [DOI] [PubMed] [Google Scholar]
- 45.Nomiya M, Yamaguchi O, Akaihata H, Hata J, Sawada N, Kojima Y, Andersson KE. Progressive vascular damage may lead to bladder underactivity in rats. J Urol 191: 1462–1469, 2014. doi: 10.1016/j.juro.2013.10.097. [DOI] [PubMed] [Google Scholar]
- 46.Nystoriak MA, O’Connor KP, Sonkusare SK, Brayden JE, Nelson MT, Wellman GC. Fundamental increase in pressure-dependent constriction of brain parenchymal arterioles from subarachnoid hemorrhage model rats due to membrane depolarization. Am J Physiol Heart Circ Physiol 300: H803–H812, 2011. doi: 10.1152/ajpheart.00760.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524–526, 1987. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 48.Park WS, Han J, Earm YE. Physiological role of inward rectifier K(+) channels in vascular smooth muscle cells. Pflügers Arch 457: 137–147, 2008. doi: 10.1007/s00424-008-0512-7. [DOI] [PubMed] [Google Scholar]
- 49.Pontari MA, Hanno PM, Ruggieri MR. Comparison of bladder blood flow in patients with and without interstitial cystitis. J Urol 162: 330–334, 1999. doi: 10.1016/S0022-5347(05)68552-7. [DOI] [PubMed] [Google Scholar]
- 50.Pontari MA, Ruggieri MR. Sex differences and role of nitric oxide in blood flow of canine urinary bladder. Am J Physiol Regul Integr Comp Physiol 276: R407–R413, 1999. [DOI] [PubMed] [Google Scholar]
- 51.Quayle JM, Dart C, Standen NB. The properties and distribution of inward rectifier potassium currents in pig coronary arterial smooth muscle. J Physiol 494: 715–726, 1996. doi: 10.1113/jphysiol.1996.sp021527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quayle JM, McCarron JG, Brayden JE, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am J Physiol Cell Physiol 265: C1363–C1370, 1993. [DOI] [PubMed] [Google Scholar]
- 53.Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, Martins JR, Jodar M, Moro C, Offermanns S, Feng Y, Demolombe S, Patel A, Honoré E. Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Reports 13: 1161–1171, 2015. doi: 10.1016/j.celrep.2015.09.072. [DOI] [PubMed] [Google Scholar]
- 54.Roman RJ, Harder DR. Cellular and ionic signal transduction mechanisms for the mechanical activation of renal arterial vascular smooth muscle. J Am Soc Nephrol 4: 986–996, 1993. [DOI] [PubMed] [Google Scholar]
- 55.Sancho M, Samson NC, Hald BO, Hashad AM, Marrelli SP, Brett SE, Welsh DG. KIR channels tune electrical communication in cerebral arteries. J Cereb Blood Flow Metab 0271678X16662041, 2016. doi: 10.1177/0271678X16662041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schubert R, Mulvany MJ. The myogenic response: established facts and attractive hypotheses. Clin Sci (Lond) 96: 313–326, 1999. doi: 10.1042/cs0960313. [DOI] [PubMed] [Google Scholar]
- 57.Segal SS. Regulation of blood flow in the microcirculation. Microcirculation 12: 33–45, 2005. doi: 10.1080/10739680590895028. [DOI] [PubMed] [Google Scholar]
- 58.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336: 597–601, 2012. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sonkusare SK, Dalsgaard T, Bonev AD, Nelson MT. Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J Physiol 594: 3271–3285, 2016. doi: 10.1113/JP271652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan CO, Hamner JW, Taylor JA. The role of myogenic mechanisms in human cerebrovascular regulation. J Physiol 591: 5095–5105, 2013. doi: 10.1113/jphysiol.2013.259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horváth B, Maser-Gluth C, Greiner E, Lemmer B, Schütz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14: 64–68, 2008. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 62.Wu BN, Luykenaar KD, Brayden JE, Giles WR, Corteling RL, Wiehler WB, Welsh DG. Hyposmotic challenge inhibits inward rectifying K+ channels in cerebral arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 292: H1085–H1094, 2007. doi: 10.1152/ajpheart.00926.2006. [DOI] [PubMed] [Google Scholar]
- 63.Ye W, Chang RB, Bushman JD, Tu Y-H, Mulhall EM, Wilson CE, Cooper AJ, Chick WS, Hill-Eubanks DC, Nelson MT, Kinnamon SC, Liman ER. The K+ channel KIR2.1 functions in tandem with proton influx to mediate sour taste transduction. Proc Natl Acad Sci USA 113: E229–E238, 2016. doi: 10.1073/pnas.1514282112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res 87: 160–166, 2000. doi: 10.1161/01.RES.87.2.160. [DOI] [PubMed] [Google Scholar]