

Abstract
Liver and intestine are tightly linked through the venous system of the portal circulation. Consequently, the liver is the primary recipient of gut-derived products, most prominently dietary nutrients and microbial components. It functions as a secondary “firewall” and protects the body from intestinal pathogens and other microbial products that have crossed the primary barrier of the intestinal tract. Disruption of the intestinal barrier enhances microbial exposure of the liver, which can have detrimental or beneficial effects in the organ depending on the specific circumstances. Conversely, the liver also exerts influence over intestinal microbial communities via secretion of bile acids and IgA antibodies. This mini-review highlights key findings and concepts in the area of host-microbial interactions as pertinent to the bilateral communication between liver and gut and highlights the concept of the gut-liver axis.
a connection between intestine and liver was already postulated two millennia ago by Galen (129–c. 216 CE) in ancient Greece. He held that blood was continually formed by the liver from digested food, brought to it via the portal vein, and was subsequently passed through systemic veins to the periphery where it was consumed as nutrient or transformed into flesh. Seminal discoveries in the 16th and 17th century by Harvey (1578–1657 CE) and others demonstrated that blood is, in fact, not generated in the adult liver but continuously circulates in a stable volume through the systemic and pulmonary vascular systems. However, the ancient concept that nutrients travel from the gut through the portal vein to the liver where they are consumed for metabolic purposes has remained remarkably intact in modern times. Harvey also described the essential elements of the portal blood circulation as a parallel venous system that is fed by mesenteric arteries and drains through the liver directly into the vena cava (13). It is now well established that the liver receives ~70% of its blood supply from the portal vein, the direct outflow of the intestine. Therefore, the liver is the first and principal organ outside the intestine that is exposed to gut-derived products, i.e., ingested nutrients and the products of bacterial metabolism.
The close functional and vascular association between gut and liver has been termed the “gut-liver axis.” This now fashionable term has been applied to many functional connections between different organs and systems, such as the gut-brain axis, liver-lung axis, hypothalamic-pituitary-adrenal axis, and so on, and could be readily expanded to all organs, raising the question whether the underlying axis concepts are physiologically meaningful or simply used to elevate the importance of any particular observation. Nonetheless, the connections between gut and liver are indeed more intimate, direct, and extensive than those found in most other reported organ axes. A special association between these two digestive organs is also underlined by the common developmental origin of hepatocytes and intestinal epithelium from the ventral foregut endoderm (74).
In addition to being the primary recipient of gut-derived products, most prominently dietary nutrients, the liver also modulates intestinal functions by producing bile for release into the small intestine. Bile acids, the primary component of bile, have an important role in the absorption of lipids and lipid-soluble vitamins (56). In addition, bile acids, as well as liver-derived IgA transported in the bile, affect the intestinal microbiota and help to defend the intestine against microbial pathogens (31). Together, these bidirectional interactions of gut and liver can be viewed as centered around two major entities, nutrients and microbes. While nutrient absorption has been extensively studied for many decades, explorations of the interactions between host and intestinal microbiota are an emerging field of research. This mini-review highlights key findings and concepts in the area of host-microbial interactions as pertinent to the gut-liver axis.
Intestinal Integrity and Microbial Exposure of the Liver
The direct venous connection of gut and liver permits the rapid appearance of passively absorbed or actively translocated bacteria and their products from gut to liver. As a consequence, the quantity and composition of the intestinal microbiota influence the microbial exposure of the liver. In healthy individuals, intact bacteria are rarely detectable in the liver (2), presumably because the intestinal barrier is normally sufficient to prevent live bacteria from entering the portal system. Although the liver is devoid of intact bacteria under physiological conditions, bacterial mRNA and LPS are readily detectable in trace amounts in the liver (7, 20) and peripheral blood (19), suggesting that the liver and other organs are constantly exposed to and functionally influenced by bacterial products even under healthy conditions. Other important bacterial products such as lipoteichoic acid or flagellin have not yet been carefully investigated in the liver, probably because of the lack of sensitive assays, but it would not come as a surprise if these and other bacterial products will also be found at low levels in the liver under physiological conditions.
Absorption of bacterial products from the intestine into the portal and systemic circulation is influenced by dietary factors. For example, a high-fat diet was shown to increase plasma LPS concentrations in mice by a moderate but functionally relevant degree that attenuates insulin actions, leading to the concept of “metabolic endotoxemia” for this condition (8). Similarly, diets rich in saturated fats cause postprandial endotoxemia in humans (19, 39, 44). In vitro studies have confirmed that fatty acids can promote transepithelial LPS absorption (39), perhaps by inducing endoplasmic reticulum stress in epithelial cells and inhibiting their ability to form tight junctions and secrete protective mucus (26) (Fig. 1). Host factors are also involved in mediating dietary effects on epithelial permeability. For example, a high-fat diet promotes the production of endocannabinoids, bioactive lipids such as arachidonoylglycerol (2-AG), and N-arachidonoylethanolamine (AEA) that are derived from host membrane precursors or dietary fatty acids. AEA can diminish epithelial barrier functions in vitro and in vivo through the cannabinoid receptor 1 (CB1) (50), while 2-AG is thought to be barrier protective (1), indicating that the balance of different endocannabinoids is likely to determine their overall impact on intestinal barrier permeability and translocation of microbial products into the portal and systemic circulation (9).
Fig. 1.

Importance of intestinal barrier in preventing liver exposure to intestinal microbes and their products. The intestinal epithelium serves as a physical and functional barrier that protects the liver from exposure to intestinal bacteria and their products. Multiple mechanisms are involved in protection, including a thick mucus layer (e.g., Muc-2), antimicrobial molecules (e.g., Reg3b) and tight junction molecules (e.g., JAM-A). These protective mechanisms can be compromised by dietary factors, injurious agents, and endogenous factors such as TNF or endocannabinoids. As a consequence, intestinal bacteria and their products can translocate into the portal vein and reach the liver.
While dietary factors can alter epithelial permeability to bacterial products, extensive physical or functional disruption of the intestinal barrier allows whole bacteria to enter the portal vein and liver in greatly increased numbers (Fig. 1). In illustration, when mice are fed with the irritant dextran sulfate sodium, a sulfated polysaccharide that disrupts the epithelium and leads to mucosal inflammation, live bacteria are readily detected in the liver (2). Similarly, patients with Crohn’s disease, which is characterized by chronic remitting-relapsing intestinal inflammation and epithelial barrier disruption, display increased levels of bacterial colonization in portal blood, liver, and peritoneum (38), further underlining that intestinal barrier loss intensifies liver exposure to intestinal microbes. The physical integrity of the intestinal barrier is also actively regulated, so its dysregulation can lead to bacterial translocation into the liver (11). As one example, TNF is a major regulator of tight junctions in the intestine (Fig. 1). Elevated TNF levels, secondary to alcohol administration to mice and men, lead to tight junction disruption and increased LPS levels in plasma and presumably liver (4). Deficiency in two critical signaling mediators of this effect, TNF receptor 1 and myosin light chain kinase, in gene-targeted mice reverses the TNF effects and is consequently associated with reduced plasma LPS levels (11).
Beyond constituting a physical barrier, the intestinal surface is protected by functional defenses. One of these is the mucus layer overlaying the epithelium. It is composed of an inner and an outer layer of highly glycosylated mucin molecules. Whereas the outer layer is rich in bacteria, the inner layer is largely devoid of bacteria (34). The most abundant mucus molecule is mucin-2 (Muc-2), which is selectively secreted by goblet cells in the intestine but not found in the liver. Loss of Muc-2 in gene-targeted mice leads to loss of the mucus barrier and can cause spontaneous colitis depending on the genetic background (63, 66). Importantly, upon oral challenge of these mice with intestinal pathogens, such as Citrobacter rodentium (3) and Salmonella enterica serovar Typhimurium (72), bacterial loads increase in intestine and liver, indicating that the Muc-2-dependent mucus barrier prevents intestinal pathogen expansion and dissemination to the liver.
Another active defense of the intestinal epithelium is production of antimicrobial molecules. Paneth cells at the bottom of the small intestinal crypts secrete several antimicrobial molecules including defensins (termed cryptdins in mice), cathelicidin, lysozyme, and C-type lectins (53). Other epithelial cells also produce antimicrobial peptides in the intestine (22, 30). These defense proteins target bacteria by attacking common surface molecules such as peptidoglycan of Gram-positive bacteria and the outer membrane of Gram-negative bacteria (51). For example, deficiency in Reg3b, a C-type lectin primarily expressed in intestine and pancreas but not the liver, leads to increased bacterial burden in colon and liver upon oral infection of knockout mice with S. Typhimurium, indicating that loss of a single antimicrobial molecule in the intestine can increase bacterial translocation into the liver (62). Further disruption of the epithelial barrier in these mice by alcohol feeding increases the number of mucosa-associated and liver bacteria, and this increase could be reversed by transgenic overexpression of Reg3b in intestinal epithelial cells, emphasizing the importance of intestinal epithelial defenses for protecting the liver against bacterial colonization (67).
Taken together, these findings clearly demonstrate that disturbance of the intestinal barrier results in increased influx of whole bacteria and their products into the portal system and liver, where they can impact a range of normal and pathological processes.
Fate of Enteric Microbes and Their Products in the Liver
The liver not only is a passive recipient of intestinal bacteria and their products that arrive through the portal system but also actively controls their numbers in the organ and their access to the systemic circulation. Kupffer cells, the resident macrophages of the liver, play a prominent role in the clearance of whole bacteria (57). Similarly, hepatic neutrophils, whose numbers in the liver are low under physiological conditions but can increase dramatically during acute inflammation, are effective pathogen killers in the liver (14, 33).
Kupffer cells, in particular, have been most extensively investigated for their importance and mechanisms of bacterial capture. For example, LPS has been detected in Kupffer cell vacuoles, suggesting that these cells contribute to endotoxin clearance (23), although the underlying mechanisms are poorly understood. In mouse models of Listeria monocytogenes infection, Kupffer cells efficiently phagocytose the bacteria (6), and cell depletion dramatically increases mortality after infection (18). This so-called “fast track” route of bacterial capture and killing in the liver is mediated in part by scavenger receptors on Kupffer cells (6). Nonopsonized, unbound bacteria can be removed via this rapid route, suggesting that this innate organ defense is active immediately after bacterial exposure without a need for antibacterial antibodies. In addition to scavenger receptors, which recognize both Gram-positive and Gram-negative bacteria, another innate receptor, the complement receptor of the immunoglobulin superfamily, contributes to the capture of Gram-positive bacteria, including L. monocytogenes and Staphylococcus aureus (73). Surprisingly, the interaction between lipoteichoic acid on Gram-positive bacteria and the complement receptor is independent of complement proteins and opsonization (73). The combination of these different receptors for bacterial capture might help to explain the ability of Kupffer cells to capture bacteria under flow conditions resembling sinusoidal blood movement, which is in contrast to other tissue macrophages that can only take up bacteria under static conditions with minimal shear forces (33, 43).
Despite the importance of clearing bacteria and their products rapidly and effectively from the liver to minimize organ damage, a need also exists to retain some fraction of bacteria to initiate adaptive immune responses. In this context, the liver has evolved a “slow track” of bacterial handling, which allows a small percentage of opsonized and platelet-bound bacteria to induce antibacterial, T-cell-mediated immunity via activation of CD8α dendritic cells (6). This is of particular importance for developing lasting immunity, as illustrated in listeriosis by the observation that reinfection can only be prevented by Listeria-specific CD8 T cells (25).
Functional Impact of Microbial Exposure of the Liver
Bacteria and their products in the liver have multi-faceted effects, which can be detrimental or beneficial depending on the circumstances (Fig. 2). LPS, the best-studied bacterial product in regard to liver impact (57), first binds to soluble LPS-binding protein in the serum, and this complex subsequently attaches to myeloid differentiation factor 2 (MD2) and CD14 on the cell surface of responsive cells. Together they activate Toll-like receptor (TLR) 4, resulting in the production of proinflammatory cytokines and chemokines. Consequently, high levels of LPS in the liver under disease conditions can activate the recruitment of inflammatory cells, which can potentially destroy the liver parenchyma (27). Interestingly, recognition of LPS and other microbial molecules may not only occur in Kupffer cells (61), but possibly also directly in hepatocytes, as suggested by the expression of functional TLR2 in these cells (46).
Fig. 2.
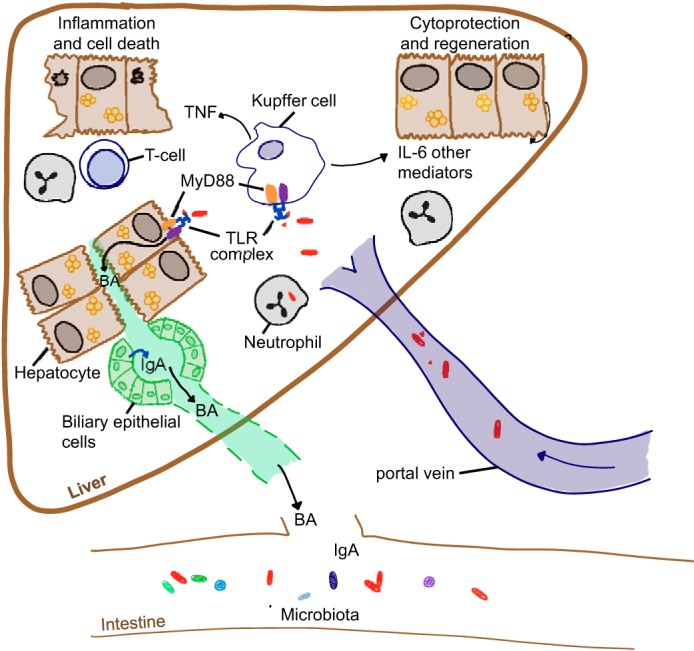
Beneficial and detrimental effects of bacterial products on liver function. Bacterial products reaching the liver can have beneficial or detrimental functions depending on the physiological circumstances. Stimulation of Kupffer and other liver cells via TLR-Myd88 leads to production of proinflammatory cytokines and chemokines, resulting in the recruitment of inflammatory cells and injury and death of hepatocytes. MyD88-dependent mechanisms can also alter the bile acid (BA) profile and together with other factors (e.g., IgA) modulate microbial composition in the intestine. Cytokines (e.g., IL-6) released by bacterially stimulated Kupffer cells activate cytoprotective mechanisms and promote liver regeneration.
LPS can synergize with other noxious stimuli in the liver. In alcoholic liver disease, increased levels of bacterial products such as LPS and peptidoglycan, which are presumably derived from the intestine, are found in the liver and blood (5, 24, 52). Activation of the respective TLRs on Kupffer and other liver cells promotes a cascade of events leading to the production and release of inflammatory cytokines (i.e., TNF-α, IL-1β) and subsequently injury, inflammation, and hepatocyte death (37). In another example, mice with a deletion of the tight junction molecule, JAM-A, in intestinal epithelial cells, display increased intestinal permeability and bacterial translocation to the liver (40). These mice develop more severe steatohepatitis when fed a diet high in saturated fat, fructose or cholesterol (54). Interestingly, colon biopsies from patients with nonalcoholic fatty liver disease show decreased levels of JAM-A and increased mucosal inflammation in the colon, suggesting that defects that primarily impact the intestine may contribute to liver disease (54).
Despite the ability of microbial products to induce or exacerbate liver disease, and the constant presence of at least low levels of LPS in the liver, the organ does not display overt inflammatory reactions under physiological conditions (33), suggesting that a threshold of activation exists for inflammatory responses to bacterial products. For instance, Kupffer cells are generally responsive to LPS, but possess mechanisms that attenuate the pro-inflammatory actions of LPS. The cells express lower levels of CD14 compared with peripheral blood monocytes, and exhibit decreased (but not absent) LPS responses (42). In addition, LPS-stimulated Kupffer cells secrete the anti-inflammatory cytokine IL-10, which downregulates pro-inflammatory cytokine responses (36).
Bacteria and their products are not only detrimental in the liver but also have beneficial effects. Mice treated with broad-spectrum antibiotics that markedly diminish the intestinal microbiota show impaired liver regeneration after partial hepatectomy (69). This and earlier work with LPS-hyporesponsive mice suggested that LPS is responsible for promoting liver regeneration after partial hepatectomy (15, 16). Studies in models of alcoholic liver disease and liver fibrosis also demonstrated a beneficial role of the commensal microbiota in protection against these diseases under specific circumstances (10, 47). Consistent with such beneficial effects of bacterial products in the liver, hepatocyte-specific deletion of the common TLR signaling adaptor, Myd88, predisposes to liver inflammation, steatosis, and insulin resistance (17). Another example of the liver-protective impact of intestinal microbes are Muc2-deficient mice, as these mice are protected from the development of fatty liver disease and alcoholic liver disease despite, and perhaps because of, their increased bacterial exposure in the liver (28, 29).
Although the mechanisms underlying liver protection and regeneration are not fully understood, production of IL-6 by liver macrophages promotes growth factor production and hepatoprotection, and has been proposed as a central cytokine responsible for liver regeneration (65). Consistent with this notion, mice deficient in MyD88 and TLR4 show impaired IL-6 production after partial hepatectomy (64). However, in spite of decreased IL-6 levels, complete liver restitution was observed in mice lacking one or more TLRs or MyD88 (59), suggesting that TLR- and MyD88-independent pathways exist that contribute to liver regeneration, possibly by mechanisms other than LPS recognition. Besides IL-6, production of IL-12 and IL-18 by hepatic natural killer cells in response to microbial ligands can induce IFN-γ, a cytokine that can also contribute to hepatic healing (57).
Role of the Liver in Controlling Intestinal Microbes
As the “distal” organ in the gut-liver axis, the liver receives microbial input from the intestine. However, the liver also exerts influence over intestinal microbes, commensurate with the two-way communication that is implied by the axis concept. Most importantly, the liver shapes microbial communities in the intestine via production and release of bile and IgA antibodies.
Bile acids, the primary component of bile, are synthesized in the liver from cholesterol. As amphipathic detergent-like molecules, they can exert direct effects on intestinal bacteria by causing membrane damage and disrupting protein and DNA functions, particularly in Gram-positive bacteria. In addition, bile acids are metabolized in the intestine by the gut microbiota to form secondary bile acids that can activate specific host receptors, particularly the nuclear farnesoid X receptor (FXR) and the G-protein-coupled bile acid receptor, Gpbar1 (also termed TGR5). These receptors regulate numerous immunological and metabolic pathways in the host, which can indirectly impact the intestinal microbiota. Bile acid composition may also be regulated indirectly by the microbiota, since deletion of the TLR signaling adaptor, Myd88, in hepatocytes altered the bile acid profile (17). Whether by direct or indirect mechanisms, feeding of bile acids or changes in bile acid composition can impact microbial composition in the intestine, such as reduction of Bacteroidetes and Actinobacteria, and expansion of Firmicutes at the phylum level (32), or decreases of Sutterella and Allobaculum at a lower taxonomic level (17). In another example, specific secondary bile acids can inhibit germination of Clostridium difficile, an opportunistic pathogen associated with antibiotics use (68). During infection, secondary bile acids are reduced, but successful fecal matter transplantation for treatment of C. difficile infection led to normalization of secondary bile acids, possibly providing a partial explanation for the efficacy of this intervention (68).
The liver is an important source of IgA, which is by far the most abundant immunoglobulin isotype in the intestinal lumen. In the intestine, IgA is produced by B cells and plasma cells in the lamina propria, and transported across intestinal epithelial cells via the polymeric immunoglobulin receptor (pIgR). In the liver, IgA producing plasma cells, originating from the Peyer’s patches, colonize portal regions and the submucosa of the biliary tract, and locally produce IgA, which together with serum IgA is transported across biliary epithelial cells (and possibly hepatocytes in some species but probably not humans) and secreted into the bile (49). The relative contributions of intestinal and hepatic production to luminal IgA levels varies by species, ranging from 5 to 10% in humans to >50% in some rodents (41). Furthermore, these contributions are presumably affected by the specific location in the gut and by meal status.
IgA production in the intestine and liver is largely dependent on the normal microbiota, as it is nearly absent in germ-free mice (45). Conversely, IgA is important for controlling intestinal microbial loads and protection of the mucosal-luminal interface. For example, an inability to class switch the immunoglobulin heavy chain to IgA, as observed in mice lacking activation-induced cytidine kinase, leads to a significant increase in the biomass of anaerobic microbes in the small intestine (21). In pIgR-deficient mice, the diversity of the cecal microbiota was also altered compared with wild-type mice, which might be responsible for the increased susceptibility of these mice to DSS-induced colitis because treatment with antibiotics attenuated weight loss and mortality (55). Similarly, IgA-deficient mice show increased susceptibility to intestinal injury in this model (48). The transition from the neonatal to the adult microbiota is also controlled by IgA. Thus, mice lacking IgA show persistent colonization with γ-proteobacteria, which are normally present in newborns but lost in adults. The continued presence of these bacteria can induce proinflammatory cytokines in the colon and enhance intestinal inflammation (48). These observations correlate with those in humans, as IgA-deficient individuals are more prone to developing inflammatory and autoimmune gastrointestinal disorder including ulcerative colitis and Crohn’s disease (71).
Outlook
Although the importance of the microbiota in the bidirectional communication between intestinal tract and liver is becoming ever more evident, much remains to be learned about the details and consequences of these interactions. What is the spectrum of intestinal bacteria that enter and persist in the liver under physiological and different disease conditions? How does it differ from the composition in different sites of the intestine? Does the microenvironment of the liver permit selective survival of some bacteria, but not others, and what are the functional consequences of this bacterial selection for liver functions? Are other bacterial products and metabolic functions beyond LPS important? For instance, flagellin is recognized by TLR5, whose potential role in the liver is only beginning to be appreciated (60, 70). Production of saturated long-chain fatty acids by the microbiota is diminished in alcoholic liver disease, and their dietary supplementation attenuated alcoholic liver injury (12). Surely, other bacterial metabolites exist with selective effects on the liver and other host organs. Another poorly understood aspect of the gut-liver axis and its role in controlling host-microbial interactions is the differentiation and migration of immune cells. For example, in conditions of extra-intestinal inflammation associated with inflammatory bowel disease such as primary sclerosing cholangitis, CD8 T cells are primed by dendritic cells in the intestine and subsequently migrate into the liver where they promote cholangitis (58). Recognition of bacterial ligands from enteric bacteria by TLR4 appears to be important in mediating this recruitment of CD8 T cells into the liver (35), but the importance of specific bacteria and their products in these processes and, more broadly, for different liver diseases remains to be established. Finally, the mechanisms that determine the balance of liver destruction versus protection by microbial products will be an important topic of future research.
GRANTS
This work was supported by National Institutes of Health Grants AA020864 and CA106802.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.B. and L.E. prepared figures; K.B. and L.E. drafted manuscript; K.B., V.K., and L.E. edited and revised manuscript; K.B., V.K., and L.E. approved final version of manuscript.
REFERENCES
- 1.Alhamoruni A, Lee AC, Wright KL, Larvin M, O’Sullivan SE. Pharmacological effects of cannabinoids on the Caco-2 cell culture model of intestinal permeability. J Pharmacol Exp Ther 335: 92–102, 2010. doi: 10.1124/jpet.110.168237. [DOI] [PubMed] [Google Scholar]
- 2.Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, Grieco A, Van Vlierberghe H, Fahrner R, Patuto N, Bernsmeier C, Ronchi F, Wyss M, Stroka D, Dickgreber N, Heim MH, McCoy KD, Macpherson AJ. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med 6: 237ra66, 2014. doi: 10.1126/scitranslmed.3008618. [DOI] [PubMed] [Google Scholar]
- 3.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog 6: e1000902, 2010. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bode C, Bode JC. Activation of the innate immune system and alcoholic liver disease: effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol Clin Exp Res 29, Suppl: 166S–171S, 2005. doi: 10.1097/01.alc.0000189280.19073.28. [DOI] [PubMed] [Google Scholar]
- 5.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol 4: 8–14, 1987. doi: 10.1016/S0168-8278(87)80003-X. [DOI] [PubMed] [Google Scholar]
- 6.Broadley SP, Plaumann A, Coletti R, Lehmann C, Wanisch A, Seidlmeier A, Esser K, Luo S, Rämer PC, Massberg S, Busch DH, van Lookeren Campagne M, Verschoor A. Dual-Track Clearance of Circulating Bacteria Balances Rapid Restoration of Blood Sterility with Induction of Adaptive Immunity. Cell Host Microbe 20: 36–48, 2016. doi: 10.1016/j.chom.2016.05.023. [DOI] [PubMed] [Google Scholar]
- 7.Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 292: G518–G525, 2007. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- 8.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772, 2007. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 9.Cani PD, Plovier H, Van Hul M, Geurts L, Delzenne NM, Druart C, Everard A. Endocannabinoids–at the crossroads between the gut microbiota and host metabolism. Nat Rev Endocrinol 12: 133–143, 2016. doi: 10.1038/nrendo.2015.211. [DOI] [PubMed] [Google Scholar]
- 10.Chen P, Miyamoto Y, Mazagova M, Lee KC, Eckmann L, Schnabl B. Microbiota Protects Mice Against Acute Alcohol-Induced Liver Injury. Alcohol Clin Exp Res 39: 2313–2323, 2015. doi: 10.1111/acer.12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen P, Stärkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 61: 883–894, 2015. doi: 10.1002/hep.27489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen P, Torralba M, Tan J, Embree M, Zengler K, Starkel P, van Pijkeren JP, DePew J, Loomba R, Ho SB, Bajaj JS, Mutlu EA, Keshavarzian A, Tsukamoto H, Nelson KE, Fouts DE, Schnabl B. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 148: 203–214, 2015. doi: 10.1053/j.gastro.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohn R. A brief history of the portal circulation. AMA Arch Intern Med 100: 848–852, 1957. doi: 10.1001/archinte.1957.00260110164023. [DOI] [PubMed] [Google Scholar]
- 14.Conlan JW, North RJ. Neutrophils are essential for early anti-Listeria defense in the liver, but not in the spleen or peritoneal cavity, as revealed by a granulocyte-depleting monoclonal antibody. J Exp Med 179: 259–268, 1994. doi: 10.1084/jem.179.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cornell RP. Restriction of gut-derived endotoxin impairs DNA synthesis for liver regeneration. Am J Physiol 249: R563–R569, 1985. [DOI] [PubMed] [Google Scholar]
- 16.Cornell RP, Liljequist BL, Bartizal KF. Depressed liver regeneration after partial hepatectomy of germ-free, athymic and lipopolysaccharide-resistant mice. Hepatology 11: 916–922, 1990. doi: 10.1002/hep.1840110603. [DOI] [PubMed] [Google Scholar]
- 17.Duparc T, Plovier H, Marrachelli VG, Van Hul M, Essaghir A, Stahlman M, Matamoros S, Geurts L, Pardo-Tendero MM, Druart C, Delzenne NM, Demoulin JB, van der Merwe SW, van Pelt J, Backhed F, Monleon D, Everard A, Cani PD. Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 66: 620–632, 2016. doi: 10.1136/gutjnl-2015-310904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebe Y, Hasegawa G, Takatsuka H, Umezu H, Mitsuyama M, Arakawa M, Mukaida N, Naito M. The role of Kupffer cells and regulation of neutrophil migration into the liver by macrophage inflammatory protein-2 in primary listeriosis in mice. Pathol Int 49: 519–532, 1999. doi: 10.1046/j.1440-1827.1999.00910.x. [DOI] [PubMed] [Google Scholar]
- 19.Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr 86: 1286–1292, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Etienne-Mesmin L, Vijay-Kumar M, Gewirtz AT, Chassaing B. Hepatocyte toll-like receptor 5 promotes bacterial clearance and protects mice against high-fat diet-induced liver disease. Cell Mol Gastroenterol Hepatol 2: 584–604, 2016. doi: 10.1016/j.jcmgh.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science 298: 1424–1427, 2002. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- 22.Forman RA, deSchoolmeester ML, Hurst RJ, Wright SH, Pemberton AD, Else KJ. The goblet cell is the cellular source of the anti-microbial angiogenin 4 in the large intestine post Trichuris muris infection. PLoS One 7: e42248, 2012. doi: 10.1371/journal.pone.0042248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fox ES, Thomas P, Broitman SA. Clearance of gut-derived endotoxins by the liver. Release and modification of 3H, 14C-lipopolysaccharide by isolated rat Kupffer cells. Gastroenterology 96: 456–461, 1989. doi: 10.1016/0016-5085(89)91571-0. [DOI] [PubMed] [Google Scholar]
- 24.Fujimoto M, Uemura M, Nakatani Y, Tsujita S, Hoppo K, Tamagawa T, Kitano H, Kikukawa M, Ann T, Ishii Y, Kojima H, Sakurai S, Tanaka R, Namisaki T, Noguchi R, Higashino T, Kikuchi E, Nishimura K, Takaya A, Fukui H. Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: relation to severity of liver disturbance. Alcohol Clin Exp Res 24, Suppl 4: 48S–54S, 2000. [PubMed] [Google Scholar]
- 25.Gregory SH, Liu CC. CD8+ T-cell-mediated response to Listeria monocytogenes taken up in the liver and replicating within hepatocytes. Immunol Rev 174: 112–122, 2000. doi: 10.1034/j.1600-0528.2002.017405.x. [DOI] [PubMed] [Google Scholar]
- 26.Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, Kang A, Schreiber V, Wong KY, Magor G, Denman S, Begun J, Florin TH, Perkins A, Cuív PO, McGuckin MA, Hasnain SZ. High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci Rep 6: 28990, 2016. doi: 10.1038/srep28990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamesch K, Borkham-Kamphorst E, Strnad P, Weiskirchen R. Lipopolysaccharide-induced inflammatory liver injury in mice. Lab Anim 49, Suppl 1: 37–46, 2015. doi: 10.1177/0023677215570087. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann P, Chen P, Wang HJ, Wang L, McCole DF, Brandl K, Stärkel P, Belzer C, Hellerbrand C, Tsukamoto H, Ho SB, Schnabl B. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 58: 108–119, 2013. doi: 10.1002/hep.26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartmann P, Seebauer CT, Mazagova M, Horvath A, Wang L, Llorente C, Varki NM, Brandl K, Ho SB, Schnabl B. Deficiency of intestinal mucin-2 protects mice from diet-induced fatty liver disease and obesity. Am J Physiol Gastrointest Liver Physiol 310: G310–G322, 2016. doi: 10.1152/ajpgi.00094.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hase K, Eckmann L, Leopard JD, Varki N, Kagnoff MF. Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun 70: 953–963, 2002. doi: 10.1128/IAI.70.2.953-963.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA 103: 3920–3925, 2006. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 141: 1773–1781, 2011. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 33.Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol 14: 996–1006, 2013. doi: 10.1038/ni.2691. [DOI] [PubMed] [Google Scholar]
- 34.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 105: 15064–15069, 2008. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.John B, Crispe IN. TLR-4 regulates CD8+ T cell trapping in the liver. J Immunol 175: 1643–1650, 2005. doi: 10.4049/jimmunol.175.3.1643. [DOI] [PubMed] [Google Scholar]
- 36.Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol 22: 226–229, 1995. doi: 10.1016/0168-8278(95)80433-1. [DOI] [PubMed] [Google Scholar]
- 37.Kudo H, Takahara T, Yata Y, Kawai K, Zhang W, Sugiyama T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol 51: 168–175, 2009. doi: 10.1016/j.jhep.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 38.Laffineur G, Lescut D, Vincent P, Quandalle P, Wurtz A, Colombel JF. [Bacterial translocation in Crohn disease]. Gastroenterol Clin Biol 16: 777–781, 1992. [PubMed] [Google Scholar]
- 39.Laugerette F, Vors C, Géloën A, Chauvin MA, Soulage C, Lambert-Porcheron S, Peretti N, Alligier M, Burcelin R, Laville M, Vidal H, Michalski MC. Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. J Nutr Biochem 22: 53–59, 2011. doi: 10.1016/j.jnutbio.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 40.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, Dermody TS, Nusrat A, Parkos CA. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 204: 3067–3076, 2007. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemaître-Coelho I, Jackson GD, Vaerman JP. Rat bile as a convenient source of secretory IgA and free secretory component. Eur J Immunol 7: 588–590, 1977. doi: 10.1002/eji.1830070818. [DOI] [PubMed] [Google Scholar]
- 42.Lichtman SN, Wang J, Lemasters JJ. LPS receptor CD14 participates in release of TNF-alpha in RAW 264.7 and peritoneal cells but not in kupffer cells. Am J Physiol 275: G39–G46, 1998. [DOI] [PubMed] [Google Scholar]
- 43.Llorente C, Schnabl B. Fast-track clearance of bacteria from the liver. Cell Host Microbe 20: 1–2, 2016. doi: 10.1016/j.chom.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 44.Lyte JM, Gabler NK, Hollis JH. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis 15: 186, 2016. doi: 10.1186/s12944-016-0357-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Macpherson AJ, Köller Y, McCoy KD. The bilateral responsiveness between intestinal microbes and IgA. Trends Immunol 36: 460–470, 2015. doi: 10.1016/j.it.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Matsumura T, Degawa T, Takii T, Hayashi H, Okamoto T, Inoue J, Onozaki K. TRAF6-NF-kappaB pathway is essential for interleukin-1-induced TLR2 expression and its functional response to TLR2 ligand in murine hepatocytes. Immunology 109: 127–136, 2003. doi: 10.1046/j.1365-2567.2003.01627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mazagova M, Wang L, Anfora AT, Wissmueller M, Lesley SA, Miyamoto Y, Eckmann L, Dhungana S, Pathmasiri W, Sumner S, Westwater C, Brenner DA, Schnabl B. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J 29: 1043–1055, 2015. doi: 10.1096/fj.14-259515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mirpuri J, Raetz M, Sturge CR, Wilhelm CL, Benson A, Savani RC, Hooper LV, Yarovinsky F. Proteobacteria-specific IgA regulates maturation of the intestinal microbiota. Gut Microbes 5: 28–39, 2014. doi: 10.4161/gmic.26489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moro-Sibilot L, Blanc P, Taillardet M, Bardel E, Couillault C, Boschetti G, Traverse-Glehen A, Defrance T, Kaiserlian D, Dubois B. Mouse and human liver contain immunoglobulin A-secreting cells originating from Peyer’s patches and directed against intestinal antigens. Gastroenterology 151: 311–323, 2016. doi: 10.1053/j.gastro.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Muccioli GG, Naslain D, Bäckhed F, Reigstad CS, Lambert DM, Delzenne NM, Cani PD. The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol 6: 392, 2010. doi: 10.1038/msb.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mukherjee S, Hooper LV. Antimicrobial defense of the intestine. Immunity 42: 28–39, 2015. doi: 10.1016/j.immuni.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 52.Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 32: 742–747, 2000. doi: 10.1016/S0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 53.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14: 141–153, 2014. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 54.Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, Smith T, Neish AS, Li H, Tan S, Wu P, Liu X, Yu Y, Farris AB, Nusrat A, Parkos CA, Anania FA. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 151: 733–746.e12, 2016. doi: 10.1053/j.gastro.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reikvam DH, Derrien M, Islam R, Erofeev A, Grcic V, Sandvik A, Gaustad P, Meza-Zepeda LA, Jahnsen FL, Smidt H, Johansen FE. Epithelial-microbial crosstalk in polymeric Ig receptor deficient mice. Eur J Immunol 42: 2959–2970, 2012. doi: 10.1002/eji.201242543. [DOI] [PubMed] [Google Scholar]
- 56.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72: 137–174, 2003. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 57.Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology 130: 1886–1900, 2006. doi: 10.1053/j.gastro.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 58.Seidel D, Eickmeier I, Kühl AA, Hamann A, Loddenkemper C, Schott E. CD8 T cells primed in the gut-associated lymphoid tissue induce immune-mediated cholangitis in mice. Hepatology 59: 601–611, 2014. doi: 10.1002/hep.26702. [DOI] [PubMed] [Google Scholar]
- 59.Seki E, Tsutsui H, Iimuro Y, Naka T, Son G, Akira S, Kishimoto T, Nakanishi K, Fujimoto J. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology 41: 443–450, 2005. doi: 10.1002/hep.20603. [DOI] [PubMed] [Google Scholar]
- 60.Singh V, Chassaing B, Zhang L, San Yeoh B, Xiao X, Kumar M, Baker MT, Cai J, Walker R, Borkowski K, Harvatine KJ, Singh N, Shearer GC, Ntambi JM, Joe B, Patterson AD, Gewirtz AT, Vijay-Kumar M. Microbiota-dependent hepatic lipogenesis mediated by stearoyl CoA desaturase 1 (SCD1) Promotes Metabolic Syndrome in TLR5-Deficient Mice. Cell Metab 22: 983–996, 2015. doi: 10.1016/j.cmet.2015.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, Remick DG, Wang SC. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 31: 932–936, 2000. doi: 10.1053/he.2000.5634. [DOI] [PubMed] [Google Scholar]
- 62.van Ampting MT, Loonen LM, Schonewille AJ, Konings I, Vink C, Iovanna J, Chamaillard M, Dekker J, van der Meer R, Wells JM, Bovee-Oudenhoven IM. Intestinally secreted C-type lectin Reg3b attenuates salmonellosis but not listeriosis in mice. Infect Immun 80: 1115–1120, 2012. doi: 10.1128/IAI.06165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, Einerhand AW. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131: 117–129, 2006. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 64.Vaquero J, Campbell JS, Haque J, McMahan RS, Riehle KJ, Bauer RL, Fausto N. Toll-like receptor 4 and myeloid differentiation factor 88 provide mechanistic insights into the cause and effects of interleukin-6 activation in mouse liver regeneration. Hepatology 54: 597–608, 2011. doi: 10.1002/hep.24420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vaquero J, Riehle KJ, Fausto N, Campbell JS. Liver regeneration after partial hepatectomy is not impaired in mice with double deficiency of Myd88 and IFNAR genes. Gastroenterol Res Pract 2011: 727403, 2011. doi: 10.1155/2011/727403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, Kucherlapati R, Lipkin M, Yang K, Augenlicht L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 295: 1726–1729, 2002. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 67.Wang L, Fouts DE, Stärkel P, Hartmann P, Chen P, Llorente C, DePew J, Moncera K, Ho SB, Brenner DA, Hooper LV, Schnabl B. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe 19: 227–239, 2016. doi: 10.1016/j.chom.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weingarden AR, Dosa PI, DeWinter E, Steer CJ, Shaughnessy MK, Johnson JR, Khoruts A, Sadowsky MJ. Changes in colonic bile acid composition following fecal microbiota transplantation are sufficient to control clostridium difficile germination and growth. PLoS One 11: e0147210, 2016. doi: 10.1371/journal.pone.0147210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu X, Sun R, Chen Y, Zheng X, Bai L, Lian Z, Wei H, Tian Z. Oral ampicillin inhibits liver regeneration by breaking hepatic innate immune tolerance normally maintained by gut commensal bacteria. Hepatology 62: 253–264, 2015. doi: 10.1002/hep.27791. [DOI] [PubMed] [Google Scholar]
- 70.Xiao Y, Liu F, Yang J, Zhong M, Zhang E, Li Y, Zhou D, Cao Y, Li W, Yu J, Yang Y, Yan H. Over-activation of TLR5 signaling by high-dose flagellin induces liver injury in mice. Cell Mol Immunol 12: 729–742, 2015. doi: 10.1038/cmi.2014.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yel L. Selective IgA deficiency. J Clin Immunol 30: 10–16, 2010. doi: 10.1007/s10875-009-9357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A, Xia L, Vallance BA. The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar Typhimurium colitis. Infect Immun 81: 3672–3683, 2013. doi: 10.1128/IAI.00854-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zeng Z, Surewaard BG, Wong CHY, Geoghegan JA, Jenne CN, Kubes P. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe 20: 99–106, 2016. doi: 10.1016/j.chom.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 74.Zorn AM, Wells JM. Vertebrate endoderm development and organ formation. Annu Rev Cell Dev Biol 25: 221–251, 2009. doi: 10.1146/annurev.cellbio.042308.113344. [DOI] [PMC free article] [PubMed] [Google Scholar]