

Abstract
An important adaptive feature of heat acclimation (HA) is the induction of cross tolerance against novel stressors (HACT) Reprogramming of gene expression leading to enhanced innate cytoprotective features by attenuating damage and/or enhancing the response of “help” signals plays a pivotal role. Hypoxia-inducible factor-1α (HIF-1α), constitutively upregulated by HA (1 mo, 34°C), is a crucial transcription factor in this program, although its specific role is as yet unknown. By using a rat HA model, we studied the impact of disrupting HIF-1α transcriptional activation [HIF-1α:HIF-1β dimerization blockade by intraperitoneal acriflavine (4 mg/kg)] on its mitochondrial gene targets [phosphoinositide-dependent kinase-1 (PDK1), LON, and cyclooxygenase 4 (COX4) isoforms] in the HA rat heart. Physiological measures of cardiac HACT were infarct size after ischemia-reperfusion and time to rigor contracture during hypoxia in cardiomyocytes. We show that HACT requires transcriptional activation of HIF-1α throughout the course of HA and that this activation is accompanied by two metabolic switches: 1) profound upregulation of PDK1, which reduces pyruvate entry into the mitochondria, consequently increasing glycolytic lactate production; 2) remodeling of the COX4 isoform ratio, inducing hypoxic-tolerant COX4.2 dominance, and optimizing electron transfer and possibly ATP production during the ischemic and hypoxic insults. LON and COX4.2 transcript upregulation accompanied this shift. Loss of HACT despite elevated expression of the cytoprotective protein heat shock protein-72 concomitantly with disrupted HIF-1α dimerization suggests that HIF-1α is essential for HACT. The role of a PDK1 metabolic switch is well known in hypoxia acclimation but not in the HA model and its ischemic setting. Remodeling of COX4 isoforms by environmental acclimation is a novel finding.
Keywords: HIF-1α, PDK1, COX4 isoforms, heat acclimation-mediated cross tolerance, heart, acriflavine
heat acclimation (HA) is a reversible “within lifetime” phenotypic adaptation to high ambient temperatures (15, 17). The primary physiological signature of HA is increased heat tolerance. An additional, equally important feature of the acclimated phenotype is acquired cytoprotection to novel stressors via cross-tolerance mechanisms [heat acclimation-mediated cross tolerance (HACT)]. The induction of HA involves short (3–5 days) and long acclimatory phases (25–30 days), but cross tolerance is apparent only following long-term HA, when acclimatory homeostasis is achieved (2, 48, 51). It stems from enhancement of innate cytoprotective pathways, which confer thermotolerance (previously linked solely to the heat shock response; Ref. 7). Based on a sharing principle, these cytoprotective pathways produce “on call” molecules to combat the consequences of several other forms of stress (3, 16, 17, 20). The first evidence of the importance of cellular protein reserves was the profound elevation of cardiac heat shock protein-72 (HSP72) levels following long-term HA (16, 34) and that the extent of protection was related to the amount of protein accumulated. The observations that the HA-protected rat cardiophenotype shows 1) enhanced glycolytic capacity at a slower rate; 2) larger preischemic endogenous glycogen stores; and 3) upregulation of phosphofructo-2 kinase 2 transcript (PFK2) (a rate-limiting glycolytic enzyme in the normoxic HA rat heart; Ref. 8) and the regulation of these components by hypoxia-inducible factor-1α (HIF-1α) (39, 41) led us to hypothesize that HIF-1α, the master regulator of O2 homeostasis, is also an important mediator of HACT. This hypothesis was supported by investigations carried out on rat hearts studied during heat stress and ischemia-reperfusion (32, 34) and on frontal cortexes in mice after traumatic brain injury (43, 44). The transcription factor HIF-1α, the master regulator of oxygen homeostasis, activates transcription as HIF-1, a heterodimer of HIF-1α and HIF-1β [also called aryl hydrocarbon receptor nuclear translocator (ARNT)] subunits that dimerize and bind to DNA. Whereas HIF-1β is constitutively expressed, HIF-1α levels were initially discovered to increase during oxygen deprivation (9). We demonstrated that acclimation induces a twofold elevation of HIF-1α and transcriptional activation of its target genes (32, 45). The list of “acclimation-associated” transcriptionally activated HIF-1 target genes in the heart and brain is growing and now includes erythropoietin receptors (EpoRs), vascular endothelial growth factor (VEGF), the water channel aquaporin 4 (31, 32, 43, 47, 50), the small heat shock protein heme oxygenase (HO1) (32), GLUT1 (25), and several mitochondrial enzymes including cyclooxygenase 4 (COX4) isoforms (20). The literature available on HIF-1-induced hypoxic and ischemic-reperfusion protection (4, 5, 21, 37, 38) suggests that one important protective component is activation of orchestrated cellular networks controlling energy balance. The specific metabolic role of HIF-1 in the protected HA phenotype and the time required for its transcriptional activation to induce HACT remain undetermined. Recent findings suggest that important HIF-1-dependent metabolic switches in hypoxic mammalian cells suppress mitochondrial respiration by phosphoinositide-dependent kinase-1 (PDK1) activity (23) and alter the ratio between COX4.2-hypoxic-resistant and COX4.1-hypoxic-sensitive (1, 9, 22, 24) isoforms. Thus, upon hypoxia, the degradation of COX4.1 and elevation of COX4.2 optimize ATP production at any oxygen concentration (9). Notably, COX4.2 is a HIF-1 target, whereas COX4.1 is degraded by the HIF-1 target mitochondrial protease Lon, a key enzyme in the degradation of oxidized proteins within the mitochondrial matrix (35). Hence, the COX4.2-to-COX4.1 ratio (COX4.2/COX4.1) is regulated by HIF-1 (9). Based on our accumulated results of the HIF-1 acclimatome, we hypothesize that the involvement of HIF-1α in the regulation of mitochondrial oxidative electron transport chains (20, 24, 39, 41) signifies its contributions to HACT via regulating the balance between oxidative and glycolytic metabolism.
It is unclear whether HACT depends on continuous changes occurring during sustained HIF-1α transcriptional activation throughout acclimation or to activation of greater HIF-1α reserves once acclimatory homeostasis has been achieved (32, 45).
With the use of an established rat heart HACT model and examining the responses to the novel insults of hypoxia or ischemia-reperfusion following disruption of the transcriptional activation of HIF-1α, the aims of the present investigation were threefold: 1) to elucidate whether HACT depends on sustained HIF-1α transcriptional activation; 2) to deduce the role of HIF-1α-regulated mitochondrial genes in HACT by studying their expression profiles during an ischemic insult; and 3) to test whether HIF-1α is essential for HACT by measuring the HSP72 profile during the inhibition of HIF-1α dimerization.
MATERIALS AND METHODS
Male Rattus norvegicus (Sabra strain), initially weighing 80–90 g (3 wk old), fed Ambar laboratory chow with water ad libitum, and held under light-dark cycled conditions (12-12 h), were randomly assigned to HA for 30 days (HA 30) or to an untreated control (C) group with and without administration of a HIF-1α:HIF-1β dimerization blocker to abolish HIF-1α transcriptional activation. Each of the experimental groups was randomly divided into animals undergoing no further intervention and those in which the heart or cardiomyocytes were subjected to ischemia-reperfusion or hypoxic stress, respectively. This design allowed us to examine the effect of acclimation status on both basal acclimatory properties and on the superimposed (novel) ischemia-reperfusion or hypoxic stress responses. The use of the HIF-1α:HIF-1β dimerization blocker allowed us to study the impact of disrupted HIF-1α transcriptional activation on the HA process. C rats were held at an ambient temperature of 24 ± 1°C, whereas HA was attained by continuous exposure to 34 ± 1°C and 30–40% relative humidity, as previously described (13, 32). Body weight and colonic temperature (Tc) were monitored throughout the HA period (32). HIF-1α:HIF-1β dimerization blockade was achieved with acriflavine (ACF), a mixture of trypaflavin and proflavine, which does not appear to elicit any adverse side-effects when administered for extended periods of time under normoxic conditions (28, 42). Previously, Lee et al. (28, 42) showed that ACF binds directly to the PAS-B subdomain of HIF-1α and inhibits HIF-1α:HIF-1β dimerization (26). ACF was administered intraperitoneally (4 mg/1 kg) either acutely, 60–75 min before the (novel) insult (C-ACF1, HA-ACF1), or daily throughout the 30-day HA regimen (HA-ACF30) to assess the importance of sustained inhibition. We initially studied the loss or induction of HACT in the heart, using established physiological experimental paradigms. These experiments were followed by biochemical and molecular measurements to assess HIF-1α function in HACT. For HIF-1α mitochondrial target genes, pyruvate dehydrogenase kinase 1 (PDK1), cytochrome c oxygenase 4.2 (COX4.2), and mitochondrial Lon protease were studied. All experimental protocols were approved by the Ethics Committee for Animal Experimentation of The Hebrew University (Jerusalem, Israel) and complied with the guidelines of the National Research Council Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Physiological Measurements
Experiments on isolated perfused hearts.
Two experimental series were performed.
infarct size after ischemia-reperfusion injury in isolated rat hearts.
Rats were euthanized by ketamine-xylazine (8.5 mg/100 g body wt ketamine in 0.5% xylazine ip) followed by cervical dislocation. The heart was then removed and mounted on a Langendorff perfusion system and retrogradely perfused at a pressure of 100 cmH2O, as previously described (8). Briefly, for perfusion, Krebs-Henseleit buffer (KHB) was used containing the following (in mM): 120 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 1.25 CaCl2, 25 NaHCO3, and 11 glucose, at pH 7.4 and aerated with a mixture of 95% O2-5% CO2 at 37°C. After 10 min of equilibration, the hearts underwent global ischemia for 30 min, followed by 40 min of reperfusion. Immediately after, a 10% 2,3,5-triphenyltetrazolium chloride solution was infused until the coronary vasculature stained dark red. The hearts were removed, blotted dry and stored at −80°C. One-millimeter-wide slices were fixed in 10% paraformaldehyde for 72 h and then photographed. Total slice area vs. infarct area was calculated using Adobe Photoshop (32).
rigor contracture in cardiomyocytes.
Isolated hearts were mounted on a Langendorff perfusion system as described above. Cardiomyocytes were isolated using a modification of the technique described by Griffith et al. (11). Briefly, the hearts were retrogradely perfused for 5 min with KHB, then for 5 min with calcium-free KHB, and then for 10–20 min with calcium-free KHB containing 0.17–0.2 mg/ml collagenase type 2 (Worthington) and 0.008 mg/ml protease type XIV (Sigma). The left ventricle was dissociated, and the myocytes were resuspended twice in calcium-free KB containing the following (in mM): 70 KOH,40 KCl, 20 KH2PO4, 50 glutamic acid, 20 taurine, 10 HEPES free, 3 MgCl2, and 0.5 EGTA). The myocytes were then suspended in Tyrode buffer containing the following (in mM): 135 NaCl, 10 HEPES, 4.2 KCl, 1.2 MgCl2, 1.2 KH2PO4, and 1.2 CaCl2 and placed on the bottom of an anoxic chamber (46) on an inverted microscope stage attached to a videomotion unit and Felix data acquisition software (PTI). During anoxia, an upward laminar flow of ultrapure argon was maintained to exclude virtually all oxygen (Po2 < 0.02 Torr). The time from the onset of anoxia to rigor contracture for individual myocytes (an abrupt shrinkage to an inert rectangular form, which is the rigor state) visualized on the screen of the system was measured using a stopwatch (6, 46, 48).
Protein and mRNA Measurements
Tissue sampling.
Isolated perfused hearts were mounted on the Langendorff system as described above. The hearts were subjected to either global or 75% ischemia for 10 or 30 min, and the left ventricle was then separated and stored at −80°C until analysis.
Western blot analysis.
Proteins were extracted from the left ventricles of the hearts (32). Aliquots of the lysates were loaded onto 9 or 12% polyacrylamide gels under denaturing conditions (27), transferred onto nitrocellulose membranes, blocked for 1 h in phosphate-buffered saline containing 5% dried skimmed milk powder, and probed overnight at 4°C with primary antibody followed by incubation for 1 h with horseradish peroxidase-conjugated goat anti-rabbit IgG (Jackson Laboratory, West Grove, PA) at room temperature. Specific antibody binding was detected using enhanced chemiluminescence (Beit Haemek Biological Industries) and visualized by exposing an X-ray film to the membrane. The densities of the scanned protein bands were calculated using TINA software (Raytest, Straubenhardt, Germany). The antibodies used were the following: rabbit polyclonal anti-PDK1 (1:1,000; NOVUS Biologicals, Littleton, CO), rabbit polyclonal anti-HIF-1 α (1:500), rabbit polyclonal anti-HIF-1 β (1:2,000), rabbit polyclonal anti-HSP72 (1:10,000; Abcam), rabbit polyclonal COX4.2 and goat polyclonal COX4.1 (1:1,000; Sigma-Aldrich, St. Louis, MO). Rabbit polyclonal β-actin (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA) was used as a loading control. For further details, see Refs. 32, 48.
mRNA analyses.
Total RNA was extracted using Tri-reagent (Molecular Research Center) and was reverse transcribed according to the manufacturer’s instructions (USB, Cleveland, OH) (32). mRNAs were measured using quantitative real-time RT-PCR (ABI Prism 7000 Sequence Detection System; Applied Biosystems). Reaction volumes of 20 μl contained 10 μl of SYBR Green Master Mix (Applied Biosystems), 500 nM each of the forward and reverse primer, and 5 μl of diluted cDNA. The thermal profile for SYBR Green real-time RT-PCR was 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. The primers for real-time RT-PCR (COX4.2, LON, and β-actin) were designed using Primer Express software (Applied Biosystems) and are listed in Table 1.
Table 1.
Real-time PCR primers
| Gene Name | Gene ID | Forward | Reverse |
|---|---|---|---|
| LONp1 | NM_133404.1 | GAGATGGTGACTGAGGCTACCAG | ATCTCTGCAGTCAGGGCCTTC |
| COX4-2 | NM_053472.1 | CGGATGAGCCTTACTGCACA | GGAACTGGAGCCGGTACAAG |
| β-Actin | NM_031144.3 | TGTGGCATCCATGAAACTAC | ATTTGCGGTGCACGATGGAG |
COX4.2, cyclooxygenase 4.2.
Coimmunoprecipitation.
For assessment of HIF-1α:HIF-1β dimerization: 500 μg of protein extracts, (described above) together with 2 μl, 1:50 of monoclonal anti-HIF-1β antibody (Abcam) in immunoprecipitation buffer [20 mM Tris·HCl, pH 7.5, 1.5 mM MgCl2, 1 mM EDTA, 20% glycerol, 5 mM DTT, 0.25 M sucrose, 1 mM PMSF, 1 mM Na3VO4, and protease inhibitor cocktail (Roche)] were incubated for 2 h at 4°C with agitation. Protein A beads (KPL, Washington, DC) in immunoprecipitation buffer (50 μl) were added and incubated overnight. The beads were then precipitated by centrifugation for 1 min at 10,000 g at 4°C. After being washed, HIF-1 immunoprecipitation by anti-HIF-1α antibody was detected by immunoblot assay as described above.
PDH Activity
The functional performance of PDK1, PDH activity, determined by NADH reduction, was measured in C and HA hearts using the commercial MSP18 kit (MitoScience, Eugene, OR) according to the manufacturer’s instructions.
Statistics
Commercially available software (SigmaStat 2.03) was used for statistical analysis. Treatments were taken as the independent categorical variables, and individual animals or hearts were considered as random samples from the population. Repeated-measures one-way ANOVA was used to compare continuous online Tc and body weight measures. Tukey’s tests were applied, unless otherwise specified, for post hoc pairwise comparisons between the results obtained in control (nonacclimated) and acclimated animals and those in the controls and acclimated animals that received ACF throughout the 30-day HA period. Two-way ANOVA was used to test the impact of the combined environmental/pharmacological and acute stressors. Tukey’s test, unless otherwise specified, was applied for post hoc pairwise comparisons, data are expressed as means ± SE. Values of P < 0.05 were considered statistically significant.
RESULTS
Colonic Temperature and Growth Rate During HA
Basal Tc and body weights of the rats from the various experimental groups are presented in Table 2. Tc and body weight were similar in the C and the C-ACF30-administered animals. Decreased growth rate and elevated Tc are consensus markers of successful acclimation in rats (13), and, similar to previous studies (12, 31, 44), Tc was higher and body weight lower in the HA rats than in the C rats. HA-ACF30-treated rats resembled the acclimated group, except that in the fourth week of the HA regimen, the growth rate of the HA-ACF30 group was significantly lower than that of the HA group.
Table 2.
Colonic temperature and body weight in the nonacclimated group and the acclimated groups in absence and presence of acriflavine
| Days |
Weeks |
||||||
|---|---|---|---|---|---|---|---|
| Group | Basal | 2 | 3 | 1 | 2 | 3 | 4 |
| Tc, °C | |||||||
| C | 36.8 ± 0.1 | 37.3 ± 0.09 | 36.8 ± 0.04† | 37.2 ± 0.04† | 36.7 ± 0.09† | 36.6 ± 0.05† | 36.7 ± 0.06† |
| C-ACF30 | 36.9 ± 01 | 37.2 ± 0.1 | 36.9 ± 0.09 | 37 ± 0.1 | 36.6 ± 0.08† | 36.7 ± 0.08† | 36.6 ± 0.08† |
| $HA | 37 ± 0.1 | 38 ± 0.1 | 37.5 ± 0.09† | 37.7±.08*‡ | 38 ± 0.1*‡ | 38 ± 0.1*‡ | 38 ± 0.1*‡ |
| &HA-ACF30 | 37 ± 0.13 | 37.6 ± 0.1 | 38.2 ± 0.1 | 37.7 ± 0.07 | 37.5 ± 0.08‡ | 38.1 ± 0.1 | 38.1 ± 0.1 |
| Body weight %change, g | |||||||
| C | 87.0 ± 1.6 | 103.0 ± 3.6 | 107.7 ± 2.7 | 133.0 ± 1.4 | 158.0 ± 5.3 | 218.9 ± 4.3 | 308.3 ± 4.0 |
| C-ACF30 | 86.4 ± 1.2 | 103.0 ± 1.3 | 107.8 ± 0.9 | 135.0 ± 1.6 | 159.6 ± 3.5 | 220.7 ± 4.7 | 310.5 ± 5.0 |
| $HA | 94.3 ± 2.1 | 111.0 ± 2.2 | 117.9 ± 2.6 | 127.1 ± 2.7 | 163.7 ± 2.9 | 207.0 ± 2.2* | 246.7 ± 2.0*†‡ |
| &HA-CF30 | 89.0 ± 3.0 | 101.9 ± 1.2 | 104.5 ± 2.8 | 129.2 ± 4.7 | 161.4 ± 5.8 | 199.2 ± 7.2 | 215.0 ± 6.8 |
Data are presented as means ± SE; n = 5. Tc, colonic temperature; HA, heat acclimated; C, nonacclimated; ACF, acriflavine. C-ACF, control ACF; ACF30, ACF administered for 30 days.
P < 0.001 vs. C by Tukey’s test for time-matched groups.
P < 0.001 vs. HA-ACF30 by Tukey’s test for time-matched groups.
P < 0.001 vs. C-ACF30 by Tukey’s test for time-matched groups.
P < 0.001: $HA vs. C; &HA-ACF30 vs. C-ACF30: HA vs. HA-ACF30 by repeated-measures ANOVA.
Evidence of HACT: Continuous But Not Acute HIF-1α Dimerization-Inhibition Abolishes HACT
Figure 1 presents infarct area (Fig. 1A) and time to rigor contracture (Fig. 1B) for the hearts that underwent global ischemia followed by reperfusion and for the hypoxic cardiomyocytes, respectively. Similar to previous studies (32, 48) HA conferred HACT in both preparations. The infarct size of HA rats was significantly smaller than that of C hearts (by 74%, P < 0.001) and time to rigor contracture profoundly longer (Δ 67% P < 0.001). ACF administration throughout the course of acclimation (HA-ACF30) profoundly diminished HACT, and infarct size was significantly larger than that of the HA hearts (P < 0.001). The hearts of the HA-ACF30 group, however, remained partially protected as infarct size was smaller than that of the C hearts (HA-ACF30: 36% vs. C: 46%; (P < 0.001). The time to rigor contracture for the cardiomyocytes from the HA-ACF30 animals did not differ from that in the C group. In contrast, in the bolus ACF administered animals (HA-ACF1), the HA phenotype was preserved. Both infarct size and time to rigor contracture did not differ from those of the HA animals. Infarct size was significantly smaller and time to rigor contracture significantly longer than in the C and the HA-ACF30 groups.
Fig. 1.
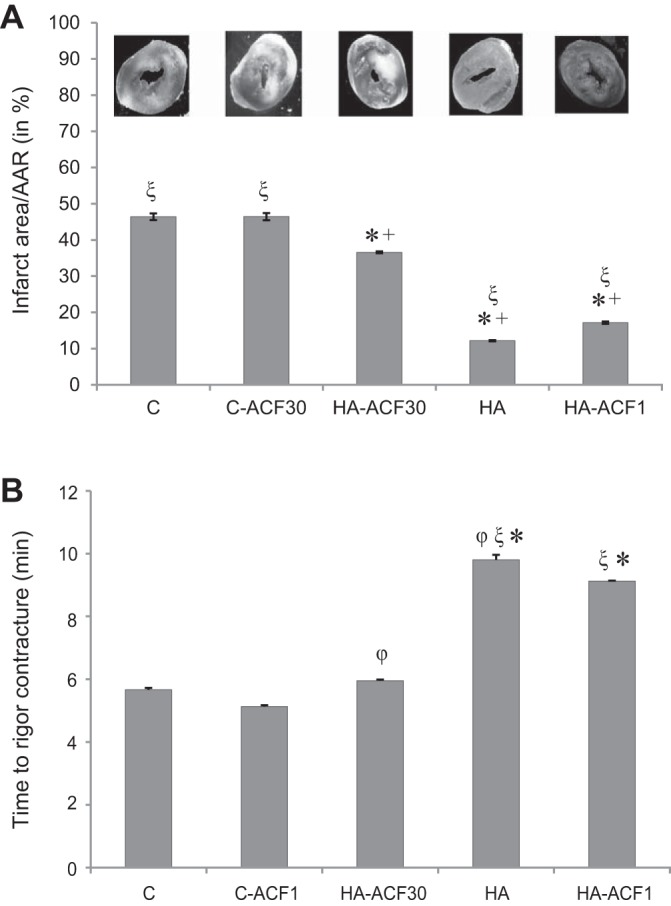
A and B: physiological markers of heat acclimation-mediated cross tolerance (HACT) in heat-acclimated (HA) hearts. A: infarct size, expressed as the percentage of infarct size/total slice area (AAR) in isolated hearts subjected to global ischemia for 30 min followed by 40 min of reperfusion. B: the time (in min) from the onset of anoxia to rigor contracture in individual cardiomyocytes. Experiments were conducted on HA hearts in the absence and presence of hypoxia-inducible factor (HIF)-1α:HIF-1β dimerization disruption by acriflavine (ACF) either during the 30-day HA period or acutely, 1 h before the test. Infarct size of HA rats was significantly smaller and time to rigor contracture upon hypoxia markedly longer than in the nonacclimated control (C) hearts, signifying cross tolerance. ACF administration during the course of acclimation (HA-ACF30) abolished HACT. Two-way ANOVA and Tukey’s post hoc test for pairwise comparisons in the different treatment groups were performed. *P < 0.001 vs. C; ξP < 0.001 vs. HA-ACF30; +P < 0.001 vs. C-ACF30; φP < 0.03 vs. HA-ACF1; **P < 0.01 vs. C.
Mitochondrial HIF-1α-Controlled Pathways in HACT
HIF-1α and HIF-1β levels and attenuation of their dimerization by ACF.
Figure 2 shows HIF-1α levels and HIF-1α dimerization to HIF-1β using coimmunoprecipitation in the absence and after ACF administration. Similar to our previous observations, HIF-1α is significantly upregulated in the HA as compared with the C (32) and ACF groups (current observations, Fig. 2A). HIF-1α was further upregulated following 30 min of ischemia in the HA group (P < 0.002 vs. basal HA). HIF-1β increased slightly ~Δ10% (nonsignificant, data not shown) in the HA groups, both basal and following ischemia. HIF-1α:HIF-1β dimerization in the HA groups was significantly higher than in the C group (Fig. 2B), suggesting differences in the binding affinity of HIF-1α:HIF-1β between the C and the HA groups.
Fig. 2.

HIF-1α and HIF-1α:HIF-1β coimmunoprecipitation in the absence and presence of ACF in heat-acclimated (HA) and nonacclimated (C) groups. A: Western blot analysis of whole cell lysates from the left ventricle, showing marked elevation of HIF-1α in the HA group. B: HIF-1α:HIF-1β coimmunoprecipitation. HIF-1β was immunoprecipitated (IP) with protein A beads and separated on SDS-PAGE. Immunoblots (IB) of HIF-1α (in pixels) indicate HIF-1 subunit dimerization. HIF-1α:HIF-1β dimerization in the HA group was significantly higher than in C or ACF-treated groups. Each sample was tested 3 times in separate runs. Data in each bar represents means ± SE; n = 5. Representative protein bands are shown at the bottom: lane 1: C; lane 2: C-ACF1; lane 3: HA-ACF30; lane 4: HA; lane 5: HA-ACF1; a: basal; b: ischemia 10 min (Isc10); c: ischemia 30 min (Isc30). Two-way ANOVA and Tukey’s post hoc test for pairwise comparisons of the treatment groups were conducted. Between groups comparisons: *&Y@P < 0.001, significant difference of HA vs. C, C-ACF1, HA-ACF30, and HA-ACF1, respectively; φP < 0.001, significant difference of HA-ACF1 vs. HA-ACF30. Within group comparisons: #P < 0.001, HA Isc10 and HA Isc30 and HA-ACF1 vs. matched basal.
ACF significantly attenuated HIF-1α:HIF-1β dimerization. Attenuation was equal in the HA-ACF30 and HA-ACF1 groups under basal conditions, but less attenuation was detected in the ischemic HA-ACF1 group (Fig. 2B). The difference between HA-ACF30 and HA-ACF1 in the ischemic groups produced a “pseudo dose-response curve.”
Does HA affect PDK1?
PDK1 controls, via phosphorylation of the Ser 232 residue in the PDHE1α (a subunit in the PDH complex), the conversion of pyruvic acid to acetyl coenzyme A. Hence, PDK1 levels in C and HA hearts under basal and ischemic conditions were measured together with PDH activity. PDK1 was significantly upregulated in HA as compared with control rats under basal conditions. (~Δ72%, P < 0.001), but the rise accompanying ischemia was markedly greater in the nonacclimated group (Δ44% vs. Δ9%, respectively; Fig. 3). Concomitantly, PDH activity was lower in HA hearts than in the C hearts under basal conditions. PDH declined significantly faster during ischemia in the nonacclimated than in the HA rats (Fig. 3), highlighting the differences in functional activity of PDK1 in the HA and C groups. Both HA-ACF30 and HA-ACF1 administration abolished the ischemia-associated PDK1 elevation above the basal levels.
Fig. 3.
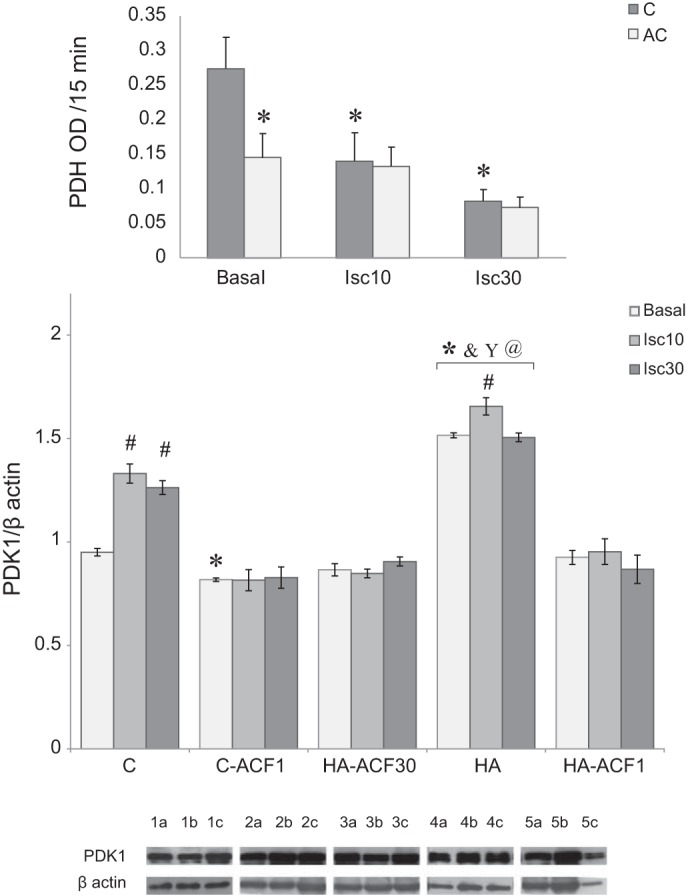
Phosphate dehydrogenase kinase 1 (PDK1) in the absence and presence of ACF in the heat-acclimated (HA) and nonacclimated (C) groups. A: Western blot analysis of whole cell lysates from the left ventricle. Each group represents basal PDK1 as well as levels after 10 and 30 min of global ischemia. HA significantly increased basal PDK1. Ischemia elevated PDK1 in both the HA and C groups with greater induction in the C hearts. ACF blocked PDK1 elevation. Each lysate was tested 3 times in separate runs. Data in each bar represent means ± SE; n = 5. Representative protein bands are shown at the bottom: lane 1: C; lane 2: C-ACF1; lane 3: HA-ACF30; lane 4: HA; lane 5: HA-ACF1; a: basal; b: Isc10; c: Isc30. In the inset, PDH activity in HA and C groups under basal or global ischemia (10 or 30 min) is presented. The C group demonstrated a profound decline in PDH activity following ischemia. For group abbreviations, statistics, and significance symbols, see Fig. 2 legend.
Does acclimation affect COX4.2 and LON mRNA?
To evaluate the impacts of HA and of inhibiting HIF-1α dimerization on transcriptional activation of the electron transport chain components, COX4.2 and LON transcripts were measured in the C group in the presence and absence of ACF and in the three HA groups, i.e., in the absence of ACF, in the one that received ACF throughout the 30-day HA period, and in the one that received bolus ACF on the day of exposure to the novel stressors. HA per se upregulated COX4.2 expression under both basal and ischemic conditions. This increase was abolished in the continued, daily ACF (HA-ACF30)-administered group. In the bolus ACF group (HA-ACF1), the COX4.2 transcript was lower than in the HA group but significantly higher than that in the HA-ACF30 group (P < 0.001, Fig. 4A).
Fig. 4.
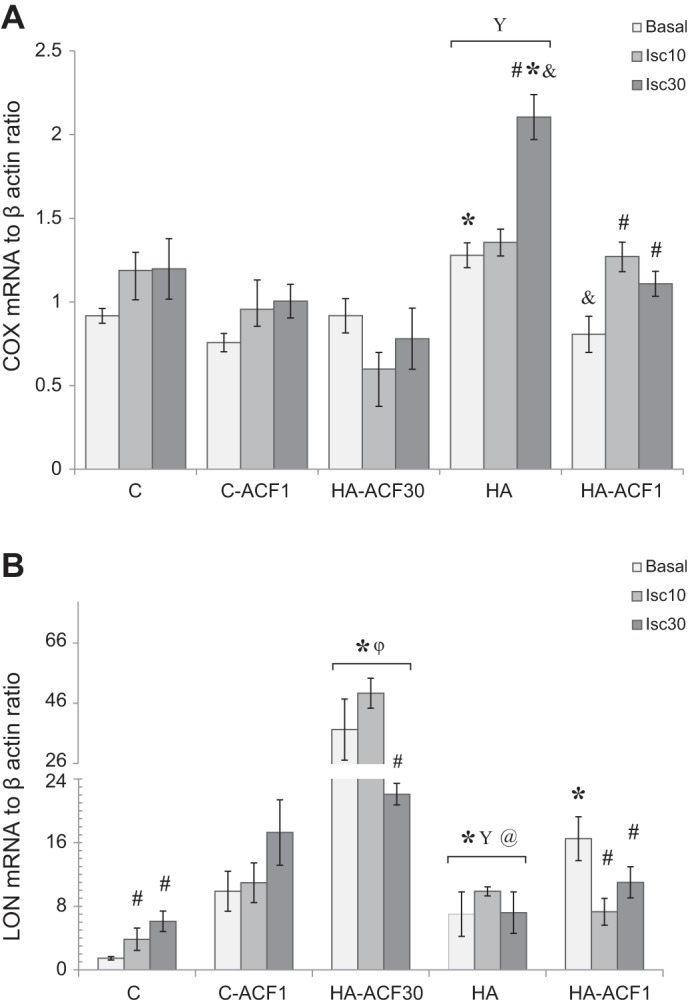
Quantitative RT-PCR analyses of cyclooxygenase 4.2 (COX4.2; A) and LON (B) transcripts in the left ventricle of HA and C hearts in the absence and presence ACF. HA increased both COX4.2 and LON expression. Daily ACF administration suppressed COX4.2 but induced marked LON expression. Hearts were analyzed under basal and global ischemic (10 or 30 min) conditions. mRNA from each animal was measured independently 3 times. Each bar represents means ± SE; n = 5. For group abbreviations, statistics, and significance symbols in A as well as between groups comparisons in B, see Fig. 2 legend. Within group comparisons vs. matched basal in B: #P = 0.001, C and HA-ACF30; #P = 0.02, HA-ACF1.
LON was upregulated by acclimation and following continuous ACF administration (Fig. 4B). This increase was much more pronounced than the COX4.2 inhibition in that group. In the group that received bolus administration of ACF, basal (preischemic) LON levels were significantly higher than those of the HA and C groups but much lower than those in the group that received ACF over the entire 30-day HA period. Otherwise, there were no significant differences among these groups.
Does HA affect COX4.2 and COX4.1 proteins?
Given the significant changes in the COX4.2 and LON transcript levels, COX4.2 and COX4.1 protein expression was also measured. HA significantly increased the COX4.2 subunit under both basal conditions and following 10 min of ischemia. The COX4.1 subunit showed an insignificant increase under similar conditions (not shown). Consequently, HA significantly increased the COX4.2/COX4.1 (Fig. 5). Bolus ACF administration (HA-ACF1) decreased the isoform ratio profoundly compared with HA alone, whereas daily ACF administration (HA-ACF30) resulted in a marked elevation of the COX4.2/COX4.1.
Fig. 5.
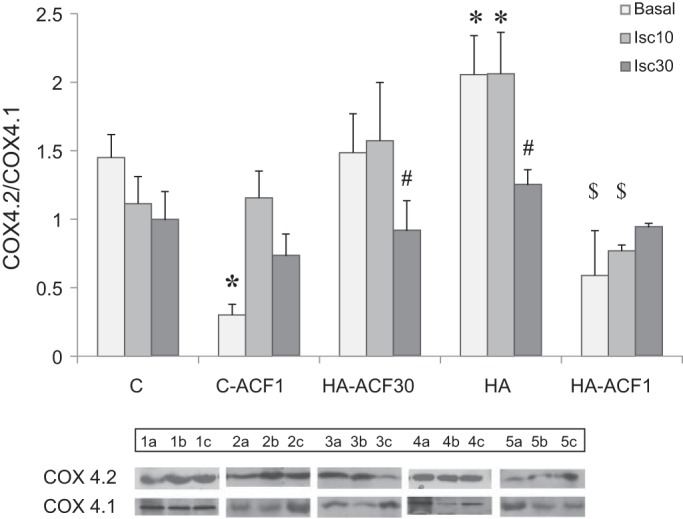
COX4.2-to-COX4.1 protein ratio (COX4.2/COX4.1) in left ventricles of HA and C hearts in the absence and presence of ACF. Hearts were tested under basal conditions and after ischemia (10 or 30 min). HA resulted in a marked elevation of the COX4.2/COX4.1. Protein expression was measured by Western blot analysis of whole cell lysates. Each lysate was tested 3 times in separate runs. Each bar represents means ± SE; n = 3–5. Representative protein bands are shown at the bottom: a: basal; b: Isc10; c: Isc30. For group abbreviations, see Fig. 2 legend. *P < 0.05 HA vs. matched C by two-way ANOVA, Dunnett’s test. $P < 0.001 HA-ACF1 vs. HA-ACF30; #P < 0.02 HA-ACF30 basal vs. Isc30.
Continuous and Acute HIF-1α Dimerization Blockade Do Not Affect HSP72 Induction
The results shown in Fig. 6 confirm our previous data that HA augments HSP72 reserves (33, 34) and demonstrate that ACF, administered either as a bolus or on a continuous daily basis, does not change HSP72 levels. HSP72 levels in the two HA-ACF-treated groups and in the HA group were significantly higher than those in the C and the C-ACF1 groups (two-way ANOVA, P < 0.001).
Fig. 6.
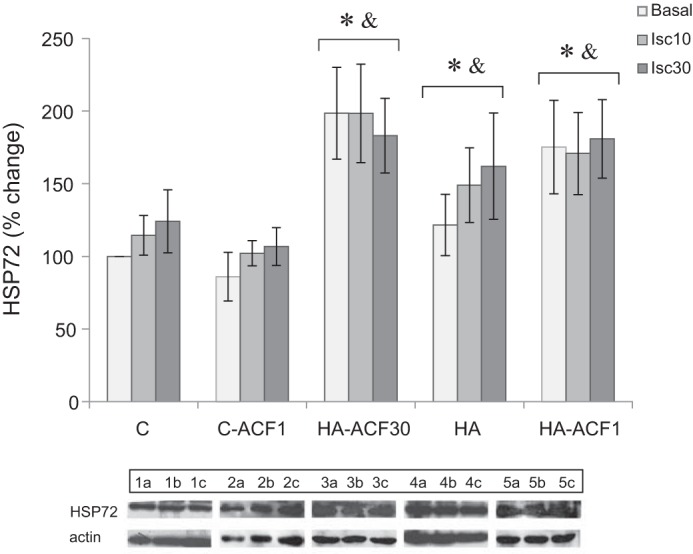
Heat shock protein-72 (HSP72) levels in the left ventricle of HA and C hearts. Protein expression was measured by Western blot analysis of whole cell lysates. Each group represents basal HSP72 levels as well as levels after 10 and 30 min of global ischemia in the absence and presence of ACF administration. HSP72 elevation was present in the HA hearts despite both daily and bolus ACF administration. Each lysate was tested 3 times in separate runs. Data in each bar represent means ± SE; n = 5. Representative protein bands are shown at the bottom: a: basal; b: Isc10; c: Isc30. For group abbreviations, see Fig. 2 legend. *P < 0.05 vs. C; &P < 0.05 vs. C-ACF1 by two-way ANOVA, Dunnett’s test.
DISCUSSION
With the use of an experimental rat heart model in which HIF-1 transcriptional activation is disrupted, this investigation is the first to show that the full spectrum of protection afforded by HIF-1α in the HACT mechanism requires transcriptional activation of HIF-1α throughout the HA period. We also demonstrate that HA induces two mitochondrial metabolic switches, one linked to PDK1 elevation and the other to an increase in the COX4.2-to-4.1 subunit ratio. The COX4.2-to-COX4.1 subunit ratio increases significantly after acclimation, thereby optimizing electron transfer reactions during times of decreased energy availability. Despite continuous inhibition of HIF-1α:HIF-1β dimerization, HACT was not completely abolished, indicating that continuous HIF-1α transcriptional activation is essential but by itself is insufficient to achieve the full spectrum of HACT associated with the HA phenotype.
The large cytoprotective HSP72 reserves attained by acclimation are still present in the blocked HIF-1α acclimation model and thus likely explain the partial protection evident when the responses to ischemia and hypoxic insults in this group are compared with those of the control, nonacclimated group. These issues are discussed below.
Continuous But Not Acute Inhibition of HIF-1α Dimerization Markedly Attenuates HACT Protection
Models for studying the role of HIF-1α in cardioprotection and/or hypoxia adaptation, which upregulate expressed HIF-1α, used pharmacological or genetic tools to stabilize normoxic HIF-1α levels (37, 38). HA leads to an almost twofold increase in HIF-1α and an increase of 203% in the nuclear-to-cytosolic fraction ratio of HIF-1α (32). Here, the importance of upregulated HIF-1α was studied by disrupting HIF-1α:HIF-1β heterodimerization, either by continuous administration of ACF throughout the 30-day acclimation period (HA-ACF30) or by bolus ACF administration shortly before exposure to the insult (HA-ACF1). This approach was used to elucidate whether HACT requires sustained HIF-1α transcriptional activation. Consistent with the results of previous studies demonstrating that sustained ACF administration does not affect body weight (28, 36, 42), we showed that sustained ACF administration did not affect growth rate and Tc of the controls. The Tc of the HA-ACF30 was not affected; however, the growth rate of this group was ~12% lower than HA rats, particularly noticeable during the last week of acclimation. It is important to note that HA decreases both growth and metabolic rates (13, 40). Analysis of the control of body weight in ACF-acclimated rats was beyond the scope of the current investigation. Coimmunoprecipitation experiments validated attenuated dimerization of the HIFs in the ACF-treated groups, while HA alone enhanced dimerization. A seemingly “attenuated-dimerization” dose response curve was seen between the HA-ACF1 and HA-ACF30 groups. However, as discussed in the following sections, no dose-response effect was seen in the studied HIF-1 targets.
We demonstrated that HACT was markedly attenuated by sustained ACF administration, evidenced by the finding that the time to the development of rigor contracture in the HA-ACF30 animals did not differ from that of the C group while infarct size was markedly larger than that of the HA heart (Fig. 1). In contrast, following bolus ACF administration (HA-ACF1), both infarct size and the time to development of rigor contracture were similar to those of the fully protected HA phenotype. We propose that these results provide unequivocal physiological confirmation of our hypothesis that long-term HIF-1 transcriptional activation is required to achieve HACT.
HIF-1 Mitochondrial Targets
Three major mitochondrial HIF-1 target gene candidates for inducing an adaptive metabolic switch were studied. For PDK1, the HA phenotype demonstrates a profound elevation of PDK1 levels with a significant decrease in PDH activity, implicating enhanced glycolysis. The consequences/benefits of elevated PDK1 levels were defined by Papandreou (38) et al. and Kim et al. (23) as an adaptive hypoxic metabolic switch through regulating mitochondrial respiration via limiting entry of pyruvate to the tricarboxylic acid cycle thus limiting pyruvic ATP production by aerobic metabolism. Likewise, Kim et al. (23) showed that enforced PDK1 expression in hypoxic HIF-1α null decreases mitochondrial respiration, attenuates hypoxic ROS generation, decreases the ATP required for maintenance of mitochondrial membrane potential, and rescues these cells from hypoxia-induced apoptosis. Analogously, HA myocytes demonstrate a marked decrease in ROS under basal, ischemic, and heat stress conditions (19), and the heart shows increased glycolytic potential (18). During global ischemia, using 13C NMR (8) we demonstrated profoundly enhanced anaerobic metabolism in HA hearts. Concomitantly, the glycolytic flux was slower, allowing longer utilization of augmented preischemic glycogen levels and cardioprotection. In contrast, the abrupt increase of lactic acid in C hearts rapidly arrested glycolysis (8). These findings correlate with the PDK1 profiles in the ischemic HA and C hearts in the current investigation, namely moderate vs. greater elevation of PDK1 upon ischemic stress and stable PDH activity vs. a decrease in the C heart (Fig. 3, inset), respectively. Given the stable PDH activity in the ischemic HA heart, we hypothesize that there is some continued pyruvic-supported mitochondria function. With respect to this, Assayag et al. (3) showed a preserved ADP-to-O2 ratio and preserved mitochondrial membrane potential in ischemic HA mitochondria in contrast to C mitochondria. In nonacclimated rats, ADP-to-O2 ratio was less than 0.5 vs. 2.5 in the HA hearts (3). In this investigation, the marked upregulation of PDK1 in the HA hearts was suppressed in both the continuous and bolus ACF-treated groups, suggesting that acute interference of HIF-1α dimerization, similarly to daily ACF administration, has an effect on PDK1 levels in our model. However, only the continuous disruption of HIFs dimerization (HA-ACF30) interfered with HACT, suggesting that the constitutive interaction between PDK1-PDH is required to exert the PDK1-induced metabolic shift. We suggest that PDH is an important metabolic intersection in this process. HA-ACF1 hearts benefited from the impact of elevated PDK1 on glycolysis throughout acclimation.
An important HIF-1α-regulated mitochondrial pathway during ischemia is the metabolic switch caused by elevated levels of the COX4.2 regulatory subunit in cytochrome c oxidase (electron transport chain-complex IV), which optimizes COX activity (41). We hypothesized that the elevated COX4.2/COX4.1 is a mitochondrial modulatory change in acclimated hearts that is responsible, in part, for the protective features associated with HACT. This hypothesis was assessed in the acclimated phenotype by studying LON and COX4.2 transcript levels. HA induced a profound elevation in the COX4.2 transcript, under both normoxic and ischemic conditions. In the HA-ACF30 group, COX4.2 transcription was inhibited. HA-ACF1 suppressed COX4.2 transcript levels under normoxic conditions but within group comparisons demonstrated enhanced COX4.2 transcription following ischemia. LON transcript showed a similar profile in the transition from control to the HA state while bolus and daily ACF administration demonstrated further LON upregulation, particularly in the HA-ACF30 where the relative increase was approximately five times. LON has several potential binding sites for stress-responsive transcription factors (in addition to HIF-1α) and increased mRNA levels may partially compensate for decreased protein levels, i.e., attenuation of HIF-1 transcriptional activation. Previously we demonstrated augmented HSP72 mRNA maintains adequate HSP72 (14). The results indicate that during acclimation LON is upregulated favoring proteolytic impact of LON on COX4.1 to maintain a lower COX4.1 level (9) while the COX4.2 transcript is upregulated. To further validate the COX4.2 subunit switch, the COX4.2-to-COX4.1 protein ratio was measured. The results of this investigation showed a significant elevation of the COX4.2-to-COX4.1 protein ratio in HA hearts under normoxic and following 10-min ischemic conditions. In the HACT model, increased COX4.2 transcription appears to depend on the time of transcriptional activation and is likely to be one of the major components of the COX isoform switch.
Is HIF-1α Necessary and Sufficient in HACT?
In this investigation we showed that disruption of HIF-1α:HIF-1β dimerization during acclimation significantly diminishes the protective features of HACT (Fig. 1, A and B). We also showed that daily ACF administration does not inhibit the establishment of augmented HSP72 reserves. HSP72 is considered an important element in cardioprotection, essential but apparently insufficient to render ischemic cardioprotection in several models (10, 12, 29). In the HA hearts, inhibition of HSP72 induction by β-adrenergic blockade during acclimation abolished HACT (33). Recently, β-adrenergic blockade was shown to repress HIF-1α induction in hemangioma cells (30). In retrospect, therefore, blockade of both HIF-1α dimerization and HSP72 augmentation during HA could explain the marked attenuation of HACT by β-adrenergic blockade (33). Taken together, the current results favor interaction between HSP72 and HIF-1 in HACT but affirm the pivotal role of HIF-1α in HACT. The study of Umschweif et al. (50) on spontaneous recovery from traumatic brain injury corroborates this conclusion. In the current model, HIF-1α and HSP72 inductions seem to be independent. This conclusion gains support from experiments in the Caenorhabditis elegans HIF-1 loss of function mutant (49) as well.
Perspectives and Significance
HACT depends on transcriptional activation and long-term changes in “on call” cytoprotective molecules and is achieved by two main mechanisms 1) attenuation of induced damage and 2) enhancement of acute innate help signals. Our results, that continuous inhibition of HIF-1α transcriptional activation during acclimation, but not acute transcriptional disruption, nullifies HACT, highlight the remodeling of two metabolic pathways initiated by upstream control of HIF-1α on mitochondrial target genes. These, shown in Fig. 7, follow the first strategy of HACT noted above: constitutive upregulation of PDK1 and, in turn, decreased PDH activity and enhanced reliance on glycolysis as reported by Eynan et al. (8), elevated LON levels, and increased COX4.2 expression resulting in a complex IV COX isoform switch, namely an increased COX4.2/COX4.1, which improves electron transfer chain efficiency. Collectively, this scenario protects the cardiomyocyte from excessive ROS production as demonstrated in our previous studies (19). Increased complex IV efficiency (COX4 isoforms ratio and the results from our earlier studies; Ref. 3) led us to hypothesize that pyruvate conversion to acetyl CoA is not completely abolished in the HA phenotype, and this helps maintain 30% ATP levels in the ischemic HA heart as shown in our P-NMR studies (17). To the best of our knowledge, remodeling of the COX4 isoforms has never been described in the heat acclimatory processes. Given that receptor remodeling during acclimation has been established (26) we propose that this strategy is evolutionarily conserved. The PDK1 metabolic switch reported here resembles the evolutionary conserved hypoxia adaptive response (38).
Fig. 7.

A major principle of HACT is attenuation of damage caused by the novel stressor by using the elevated reserves of cytoprotective elements and remodeled cytoprotective molecules. The scheme shows two metabolic switches activated by the reprograming of the expression of PDK1, COX4.2, and the LON protease. Frist, following 30 days of HA PDK1 is upregulated, suppressing PDH activity and causing decreased entry of pyruvic acid into the tricarboxylic acid cycle. During ischemia PDK1 is only slightly upregulated; however PDH activity is markedly downregulated (right). Eynan et al. (8), demonstrated that this is accompanied by a prolonged, slower glycolytic flux and lower levels of ROS production. In contrast, ischemic nonacclimated hearts demonstrate a profound elevation of PDK1 (left). Under these conditions glycolysis is rapidly arrested and ROS levels are significantly higher (8, 16). Second, increased LON transcript targeted COX4.1 and elevated COX4.2 transcript alters the COX4 isoform ratio, inducing hypoxic-tolerant COX4.2 dominance, and optimizing electron transfer in response to disrupted energetic conditions.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.A.-S., A.M., and E.S. performed experiments; R.A.-S., A.M., E.S., and M.H. analyzed data; R.A.-S., A.M., G.G., M.D.S., and M.H. interpreted results of experiments; R.A.-S., A.M., and M.H. prepared figures; R.A.-S., A.M., E.S., G.G., M.D.S., and M.H. approved final version of manuscript; G.G., M.D.S., and M.H. conceived and designed research; G.G., M.D.S., and M.H. edited and revised manuscript; M.H. drafted manuscript.
GRANTS
This study was supported by the U.S.-Israel Binational Fund BSF Grant 2009–499 and (in part) by the Intramural Program of the National Institute on Aging.
REFERENCES
- 1.Arnold S. The power of life–cytochrome c oxidase takes center stage in metabolic control, cell signalling and survival. Mitochondrion 12: 46–56, 2012. doi: 10.1016/j.mito.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Assayag M, Gerstenblith G, Stern MD, Horowitz M. Long- but not short-term heat acclimation produces an apoptosis-resistant cardiac phenotype: a lesson from heat stress and ischemic/reperfusion insults. Cell Stress Chaperones 15: 651–664, 2010. doi: 10.1007/s12192-010-0178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Assayag M, Saada A, Gerstenblith G, Canaana H, Shlomai R, Horowitz M. Mitochondrial performance in heat acclimation: a lesson from ischemia/reperfusion and calcium overload insults in the heart. Am J Physiol Regul Integr Comp Physiol 303: R870–R881, 2012. doi: 10.1152/ajpregu.00155.2012. [DOI] [PubMed] [Google Scholar]
- 4.Cai Z, Luo W, Zhan H, Semenza GL. Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc Natl Acad Sci USA 110: 17462–17467, 2013. doi: 10.1073/pnas.1317158110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerretelli P, Gelfi C. Energy metabolism in hypoxia: reinterpreting some features of muscle physiology on molecular grounds. Eur J Appl Physiol 111: 421–432, 2011. doi: 10.1007/s00421-010-1399-5. [DOI] [PubMed] [Google Scholar]
- 6.Cohen O, Kanana H, Zoizner R, Gross C, Meiri U, Stern MD, Gerstenblith G, Horowitz M. Altered Ca2+ handling and myofilament desensitization underlie cardiomyocyte performance in normothermic and hyperthermic heat-acclimated rat hearts. J Appl Physiol (1985) 103: 266–275, 2007. doi: 10.1152/japplphysiol.01351.2006. [DOI] [PubMed] [Google Scholar]
- 7.Commission for Thermal Physiology of the International Union of Physiological Sciences (IUPS Thermal Commission) Glossary of terms for thermal physiology. Third edition. Jpn J Physiol 51: 245–280, 2001. [Google Scholar]
- 8.Eynan M, Knubuvetz T, Meiri U, Navon G, Gerstenblith G, Bromberg Z, Hasin Y, Horowitz M. Heat acclimation-induced elevated glycogen, glycolysis, and low thyroxine improve heart ischemic tolerance. J Appl Physiol (1985) 93: 2095–2104, 2002. doi: 10.1152/japplphysiol.00304.2002. [DOI] [PubMed] [Google Scholar]
- 9.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129: 111–122, 2007. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 10.Gray CC, Amrani M, Yacoub MH. Heat stress proteins and myocardial protection: experimental model or potential clinical tool? Int J Biochem Cell Biol 31: 559–573, 1999. doi: 10.1016/S1357-2725(99)00004-7. [DOI] [PubMed] [Google Scholar]
- 11.Griffiths EJ, Wei SK, Haigney MC, Ocampo CJ, Stern MD, Silverman HS. Inhibition of mitochondrial calcium efflux by clonazepam in intact single rat cardiomyocytes and effects on NADH production. Cell Calcium 21: 321–329, 1997. doi: 10.1016/S0143-4160(97)90120-2. [DOI] [PubMed] [Google Scholar]
- 12.Honma Y, Tani M, Yamamura K, Takayama M, Hasegawa H. Preconditioning with heat shock further improved functional recovery in young adult but not in middle-aged rat hearts. Exp Gerontol 38: 299–306, 2003. doi: 10.1016/S0531-5565(02)00199-7. [DOI] [PubMed] [Google Scholar]
- 13.Horowitz M. Acclimatization of rats to moderate heat: body water distribution and adaptability of the submaxillary salivary gland. Pflugers Arch 366: 173–176, 1976. doi: 10.1007/BF00585874. [DOI] [PubMed] [Google Scholar]
- 14.Horowitz M. Epigenetics and cytoprotection with heat acclimation. J Appl Physiol (1985) 120: 702–710, 2015. doi: 10.1152/japplphysiol.00552.2015. [DOI] [PubMed] [Google Scholar]
- 15.Horowitz M. Genomics and proteomics of heat acclimation. Front Biosci (Schol Ed) 2: 1068–1080, 2010. doi: 10.2741/s118. [DOI] [PubMed] [Google Scholar]
- 16.Horowitz M. Heat acclimation and cross-tolerance against novel stressors: genomic–physiological linkage. Progr Brain Res 162: 373–392, 2007. doi: 10.1016/S0079-6123(06)62018-9. [DOI] [PubMed] [Google Scholar]
- 17.Horowitz M. Heat acclimation, epigenetics, and cytoprotection memory. Compr Physiol 4: 199–230, 2014. doi: 10.1002/cphy.c130025. [DOI] [PubMed] [Google Scholar]
- 18.Horowitz M. Matching the heart to heat-induced circulatory load: heat-acclimatory responses. News Physiol Sci 18: 215–221, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Horowitz M, Canaana H, Kohen R. ROS generated in the mitochondria may be linked with heat stress mediated HIF-1 transcriptional activation in the heart (Abstract 97). APS Intersociety Meeting Virginia Beach, VA: Am Physiol Soc, 2006. [Google Scholar]
- 20.Horowitz M, Assadi H. Heat acclimation-mediated cross-tolerance in cardioprotection: do HSP70 and HIF-1alpha play a role? Ann NY Acad Sci 1188: 199–206, 2010. doi: 10.1111/j.1749-6632.2009.05101.x. [DOI] [PubMed] [Google Scholar]
- 21.Jian B, Wang D, Chen D, Voss J, Chaudry I, Raju R. Hypoxia-induced alteration of mitochondrial genes in cardiomyocytes: role of Bnip3 and Pdk1. Shock 34: 169–175, 2010. doi: 10.1097/SHK.0b013e3181cffe7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadenbach B, Hüttemann M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 24: 64–76, 2015. doi: 10.1016/j.mito.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Kocha KM, Reilly K, Porplycia DSM, McDonald J, Snider T, Moyes CD. Evolution of the oxygen sensitivity of cytochrome c oxidase subunit 4. Am J Physiol Regul Integr Comp Physiol 308: R305–R320, 2015. doi: 10.1152/ajpregu.00281.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kodesh E, Levi E, Horowitz M. Combined heat-acclimation and exercise-training improves cardiac mechanical, metabolic performance and enhances cardioprotection. Med Sci Sports Exerc 42: 40–41, 2010. [Google Scholar]
- 26.Kodesh E, Nesher N, Simaan A, Hochner B, Beeri R, Gilon D, Stern MD, Gerstenblith G, Horowitz M. Heat acclimation and exercise training interact when combined in an overriding and trade-off manner: physiologic-genomic linkage. Am J Physiol Regul Integr Comp Physiol 301: R1786–R1797, 2011. doi: 10.1152/ajpregu.00465.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685, 1970. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA 106: 17910–17915, 2009. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Lennon SL, Quindry J, Hamilton KL, French J, Staib J, Mehta JL, Powers SK. Loss of exercise-induced cardioprotection after cessation of exercise. J Appl Physiol (1985) 96: 1299–1305, 2004. doi: 10.1152/japplphysiol.00920.2003. [DOI] [PubMed] [Google Scholar]
- 30.Li P, Guo Z, Gao Y, Pan W. Propranolol represses infantile hemangioma cell growth through the β2-adrenergic receptor in a HIF-1α-dependent manner. Oncol Rep 33: 3099–3107, 2015. doi: 10.3892/or.2015.3911. [DOI] [PubMed] [Google Scholar]
- 31.Maloyan A, Eli-Berchoer L, Semenza GL, Gerstenblith G, Stern MD, Horowitz M. HIF-1alpha-targeted pathways are activated by heat acclimation and contribute to acclimation-ischemic cross-tolerance in the heart. Physiol Genomics 23: 79–88, 2005. doi: 10.1152/physiolgenomics.00279.2004. [DOI] [PubMed] [Google Scholar]
- 32.Maloyan A, Eli-Berchoer L, Semenza GL, Gerstenblith G, Stern MD, Horowitz M. HIF-1α-targeted pathways are activated by heat acclimation and contribute to acclimation-ischemic cross-tolerance in the heart. Physiol Genomics 23: 79–88, 2005. doi: 10.1152/physiolgenomics.00279.2004. [DOI] [PubMed] [Google Scholar]
- 33.Maloyan A, Horowitz M. β-Adrenergic signaling and thyroid hormones affect HSP72 expression during heat acclimation. J Appl Physiol (1985) 93: 107–115, 2002. doi: 10.1152/japplphysiol.01122.2001. [DOI] [PubMed] [Google Scholar]
- 34.Maloyan A, Palmon A, Horowitz M. Heat acclimation increases the basal HSP72 level and alters its production dynamics during heat stress. Am J Physiol Regul Integr Comp Physiol 276: R1506–R1515, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Ngo JK, Pomatto LC, Davies KJ. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol 1: 258–264, 2013. doi: 10.1016/j.redox.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogle ME, Gu X, Espinera AR, Wei L. Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1α. Neurobiol Dis 45: 733–742, 2012. doi: 10.1016/j.nbd.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ong SG, Lee WH, Theodorou L, Kodo K, Lim SY, Shukla DH, Briston T, Kiriakidis S, Ashcroft M, Davidson SM, Maxwell PH, Yellon DM, Hausenloy DJ. HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc Res 104: 24–36, 2014. doi: 10.1093/cvr/cvu172. [DOI] [PubMed] [Google Scholar]
- 38.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: 187–197, 2006. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, Ordoñez A, Cuevas Y, Saez-Morales D, Garcia-Bermejo ML, Landazuri MO, Guinovart J, del Peso L. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS One 5: e9644, 2010. doi: 10.1371/journal.pone.0009644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ray DE. Roubicek CB, Hamidi M. Organ and gland weights of rats chronically exposed to 22° and 35°C. Growth 32: 1–12, 1968. [PubMed] [Google Scholar]
- 41.Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 1813: 1263–1268, 2011. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shay JES, Imtiyaz HZ, Sivanand S, Durham AC, Skuli N, Hsu S, Mucaj V, Eisinger-Mathason TK, Krock BL, Giannoukos DN, Simon MC. Inhibition of hypoxia-inducible factors limits tumor progression in a mouse model of colorectal cancer. Carcinogenesis 35: 1067–1077, 2014. doi: 10.1093/carcin/bgu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shein NA, Horowitz M, Alexandrovich AG, Tsenter J, Shohami E. Heat acclimation increases hypoxia-inducible factor 1alpha and erythropoietin receptor expression: implication for neuroprotection after closed head injury in mice. J Cereb Blood Flow Metab 25: 1456–1465, 2005. doi: 10.1038/sj.jcbfm.9600142. [DOI] [PubMed] [Google Scholar]
- 44.Shein NA, Horowitz M, Shohami E. Heat acclimation: a unique model of physiologically mediated global preconditioning against traumatic brain injury. Prog Brain Res 161: 353–363, 2007. doi: 10.1016/S0079-6123(06)61025-X. [DOI] [PubMed] [Google Scholar]
- 45.Shein NA, Horowitz M, Alexandrovich AG, Tsenter J, Shohami E. Heat acclimation increases hypoxia-inducible factor 1alpha and erythropoietin receptor expression: implication for neuroprotection after closed head injury in mice. J Cereb Blood Flow Metab 25: 1456–1465, 2005. doi: 10.1038/sj.jcbfm.9600142. [DOI] [PubMed] [Google Scholar]
- 46.Stern MD, Silverman HS, Houser SR, Josephson RA, Capogrossi MC, Nichols CG, Lederer WJ, Lakatta EG. Anoxic contractile failure in rat heart myocytes is caused by failure of intracellular calcium release due to alteration of the action potential. Proc Natl Acad Sci USA 85: 6954–6958, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugimoto N, Matsuzaki K, Ishibashi H, Tanaka M, Sawaki T, Fujita Y, Kawanami T, Masaki Y, Okazaki T, Sekine J, Koizumi S, Yachie A, Umehara H, Shido O. Upregulation of aquaporin expression in the salivary glands of heat-acclimated rats. Sci Rep 3: 1763, 2013. doi: 10.1038/srep01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tetievsky A, Cohen O, Eli-Berchoer L, Gerstenblith G, Stern MD, Wapinski I, Friedman N, Horowitz M. Physiological and molecular evidence of heat acclimation memory: a lesson from thermal responses and ischemic cross-tolerance in the heart. Physiol Genomics 34: 78–87, 2008. doi: 10.1152/physiolgenomics.00215.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Treinin M, Shliar J, Jiang H, Powell-Coffman JA, Bromberg Z, Horowitz M. HIF-1 is required for heat acclimation in the nematode Caenorhabditis elegans. Physiol Genomics 14: 17–24, 2003. doi: 10.1152/physiolgenomics.00179.2002. [DOI] [PubMed] [Google Scholar]
- 50.Umschweif G, Alexandrovich AG, Trembovler V, Horowitz M, Shohami E. Hypoxia-inducible factor 1 is essential for spontaneous recovery from traumatic brain injury and is a key mediator of heat acclimation induced neuroprotection. J Cereb Blood Flow Metab 33: 524–531, 2013. doi: 10.1038/jcbfm.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yacobi A, Stern-Bach Y, Horowitz M. The protective effect of heat acclimation from hypoxic damage in the brain involves changes in the expression of glutamate receptors. Temperature 1: 57–65, 2014. doi: 10.4161/temp.29719. [DOI] [PMC free article] [PubMed] [Google Scholar]