

We describe potential mechanisms contributing to aerobic exercise-conferred protection against high intravascular pressure. Subcutaneous adipose microvessels from exercise mice express less NADPH oxidase (NOX) II and more superoxide dismutase (SOD) and demonstrate less sensitivity to ANG II. In microvascular endothelial cells, shear stress reduced NOX II but did not influence SOD expression.
Keywords: hypertension, endothelium, microcirculation, exercise, oxidative stress
Abstract
High blood pressure has been shown to elicit impaired dilation in the vasculature. The purpose of this investigation was to elucidate the mechanisms through which high pressure may elicit vascular dysfunction and determine the mechanisms through which regular aerobic exercise protects arteries against high pressure. Male C57BL/6J mice were subjected to 2 wk of voluntary running (~6 km/day) for comparison with sedentary controls. Hindlimb adipose resistance arteries were dissected from mice for measurements of flow-induced dilation (FID; with or without high intraluminal pressure exposure) or protein expression of NADPH oxidase II (NOX II) and superoxide dismutase (SOD). Microvascular endothelial cells were subjected to high physiological laminar shear stress (20 dyn/cm2) or static condition and treated with ANG II + pharmacological inhibitors. Cells were analyzed for the detection of ROS or collected for Western blot determination of NOX II and SOD. Resistance arteries from exercised mice demonstrated preserved FID after high pressure exposure, whereas FID was impaired in control mouse arteries. Inhibition of ANG II or NOX II restored impaired FID in control mouse arteries. High pressure increased superoxide levels in control mouse arteries but not in exercise mouse arteries, which exhibited greater ability to convert superoxide to H2O2. Arteries from exercised mice exhibited less NOX II protein expression, more SOD isoform expression, and less sensitivity to ANG II. Endothelial cells subjected to laminar shear stress exhibited less NOX II subunit expression. In conclusion, aerobic exercise prevents high pressure-induced vascular dysfunction through an improved redox environment in the adipose microvasculature.
NEW & NOTEWORTHY We describe potential mechanisms contributing to aerobic exercise-conferred protection against high intravascular pressure. Subcutaneous adipose microvessels from exercise mice express less NADPH oxidase (NOX) II and more superoxide dismutase (SOD) and demonstrate less sensitivity to ANG II. In microvascular endothelial cells, shear stress reduced NOX II but did not influence SOD expression.
Listen to this article’s corresponding podcast at https://ajpheart.podbean.com/e/exercise-averts-high-pressure-induced-vascular-dysfunction/.
cardiovascular disease (CVD) is the number one cause of death in the United States (35). Hypertension is the number one risk factor for CVD (35), and endothelial dysfunction also precedes the development of CVD (23). However, the temporal relation between high blood pressure and endothelial dysfunction has not been not fully elucidated (7, 38, 42). Regular aerobic exercise lowers the risk of CVD and is beneficial for blood pressure and endothelial cell health (45). However, acute exercise bouts paradoxically increase the risk of cardiovascular events in sedentary individuals, in part through a transient increase in blood pressure and endothelial dysfunction (2, 9, 15). Extensive work characterizing subcutaneous adipose resistance arteries has been done in humans (22, 24, 40), demonstrating that endothelium-dependent vasodilation in these arteries correlates with clinical measures of peripheral vascular function (6), that high intraluminal pressure elicits endothelial dysfunction in these arteries (9, 40), and that regular exercise may be protective against said endothelial dysfunction (16, 40).
High blood pressure is associated with excessive oxidative stress in the endothelium. Primary sources of oxidative stress include NADPH oxidase II (NOX II) and mitochondrial ROS. The local renin-angiotensin system (RAS) plays a role in both of these pathways (8, 11, 46, 47). In adipose resistance arteries, blockade of the local RAS through angiotensin type 1 receptor (AT1R) blockade or angiotensin-converting enzyme (ACE) inhibition prevents both increased superoxide () levels and impairment of endothelium-dependent dilation after high pressure (11). Uncoupled endothelial nitric oxide synthase (eNOS) in the presence of tetrahydrobiopterin oxidation is also a source of superoxide (28). Recent findings suggest that exercise leads to an increase in antioxidant mechanisms that prevent oxidative stress and endothelial dysfunction in the face of high pressure (9, 40).
Isolated adipose resistance arteries from exercise-trained individuals produce less after high intraluminal pressure compared with resistance arteries from sedentary individuals and are protected from high pressure-induced vasodilator dysfunction (9, 40). Furthermore, H2O2 appears to be the mediator of preserved vasodilation in adipose resistance arteries from exercise-trained adults (9, 40). This is of physiological relevance because reduces nitric oxide (NO·) bioavailability and contributes to eNOS uncoupling. However, is also converted to H2O2 via spontaneous dismutation or enzymatic conversion by SOD, whereby H2O2 can act as an alternative vasodilator in times of reduced NO· bioavailability (43). In agreement, regular exercise has been shown to increase SOD in the aortic endothelium (41) and vascular smooth muscle (17). Exogenous SOD has been shown to rescue resistance artery flow-induced dilation (FID) after high intraluminal pressure (25). Thus, regular exercise-induced increases in SOD expression would seem to be a likely mechanism for reduced and improved H2O2-mediated vasodilator function in response to high intraluminal pressure after exercise training. Therefore, we hypothesized that regular exercise would preserve vascular function after exposure to high intraluminal pressure through increased SOD expression and improved H2O2-mediated dilation. We also hypothesized exercise would lead to a reduction in NOX II expression and that shear stress is the primary mediator of chronic exercise-related adaptations in regard to NOX II and SOD expression. This investigation is important in that the findings will elucidate the mechanisms through which regular exercise may protect the microvascular endothelium from high pressure, thus facilitating maintenance of microvascular perfusion.
METHODS
Exercise protocol.
Twelve- to sixteen-week-old male C57BL/6J mice (obtained from Jackson Laboratory, Bar Harbor, ME) were used for this study. Mice ran on an exercise wheel (12.7-cm diameter) placed inside their cage for 2 wk. The wheels were instrumented with a tachometer, composed of a ring magnet on the wheel axle and a Hall effect sensor (magnetic flux sensor) on the wheel apparatus base to count the number of revolutions. The analog signal from the Hall effect sensor was acquired continuously using an analog-to-digital converter and data logger (DI-710-ULS, Dataq Instruments, Akron, OH). WinDaq Data Acquisition and Playback Software was used to determine distance ran based on wheel revolutions and wheel diameter. One revolution corresponds to 40 cm; thus, the revolution count was converted to running distance by multiplying by 40 cm.
Mice ran an average of 6 km every 24 h, similar to previously reported values (5, 29). Mice were acclimatized to the running wheels for 48 h before being given permanent access to the unlocked wheels. Mice ran on the wheels during the nocturne (2000 to 0800 hours) and were on a 12:12-h light-dark light cycle. Mice were fed normal mouse chow ad libitum. Mice were anesthetized with isoflurane (5%) and, except for where otherwise stated, euthanized for experimentation within 2 h of their last exercise bout. Mice were not fasted at the time of death. Control mice weighed 29.4 ± 0.54 g, whereas exercised mice weighed 28.0 ± 0.71 g. Animal experiments were conducted in accordance with the University of Illinois at Chicago’s Animal Care and Use Committee (ACC no. 15-037) and National Institutes of Health (NIH) guidelines.
Resistance artery FID.
Resistance artery FID was performed as previously described (24, 40). For further detail, refer to Fig. 1. Briefly, bilateral hindlimb subcutaneous (inguinal) fat pads were removed from mice and placed in cold (4°C) HEPES buffer. After dissection of vessel arcades, single resistance arteries (internal diameter: 86 ± 5.45 µm) were cannulated on glass micropipettes (internal diameters of ~20 to 40 µm) in an organ chamber filled with Krebs solution. The remaining arteries were snap frozen in liquid nitrogen for molecular biology experiments described below. Resistance artery diameter was monitored by placing the organ chamber on the stage of an inverted microscope attached to a real time video-measuring apparatus (model VIA-100, Boeckeler). Arteries were maintained at 37°C, continuously perfused with warm Krebs buffer, and aerated with a gas mixture of 21% O2 and 5% CO2.
Fig. 1.

Schematic of the isolated vessel preparation used to generate flow-induced dilation (FID). Flow is generated by manipulating pressure gradients. Cannulated resistance arteries are equilibrated at 60 cmH2O (44 mmHg) intraluminal pressure for 30 min. Endothelin-1 (ET-1; ~120 pM) is applied into the external bathing solution in the organ chamber in which the resistance artery is cannulated. Pressure gradients are crated to generate flow. A: to generate flow, one of the Krebs-filled reservoirs is moved up (a) cm and the other Krebs-filled reservoir is simultaneously moved down (a') cm from the 60-cm position. The pressure gradient between the two reservoirs is equal to (a + a') cmH2O. a and a' are equal in magnitude and opposite in direction. Thus, the sum of b and c will always result in an average pressure of 60 cmH2O (b = 60 + a cm and c = 60 – a' cm). Furthermore, the total pressure within the system is constant, 120 cmH2O. B: table of the corresponding resistance artery diameters (means ± SE) and shear stress values at the different pressure gradients (Δ10, Δ20, Δ40, Δ60, and Δ100 cm H2O) Shear stress was calculated from the flow rate in the vessel lumen and the diameter of the vessels using the following equation described by Ahn et al. (1): τ = 4(μQ)/(πr3), where μ is viscosity (0.015 g·cm−1·s−1), Q is the volumetric flow rate {[ΔP × r4]/[8/π × l)], where l is length}, r is the internal radius of the vessel (in µm converted to cm), and ΔP is the change in the pressure gradient (in cmH2O converted to Pa).
After an intraluminal pressure of 60 cmH2O (44 mmHg) was maintained for 30 min, arteries were constricted 40–60% with endothelin-1 (ET-1; 125 ± 5 pmol for control mice and 115 ± 3 pmol for exercised mice, not significant, means ± SE). FID was produced using two Krebs solution-filled reservoirs to generate pressure gradients of Δ10, Δ20, Δ40, Δ60, and Δ100 cmH2O. Pressure gradients are generated by moving the reservoirs in equal and opposite directions. Diameters were taken after 3 min of exposure to each pressure gradient and then moved to create the next pressure gradient. For the high pressure model, high intraluminal pressure (HILP; 150 cmH2O or 110 mmHg) was administered for 45 min followed by 15 min of reequilibration at 60 cmH2O, similar to previously published protocols (2, 9, 11). FID was measured in the absence and presence of the NOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 100 μM), H2O2 scavenger polyethylene glycol-catalase (PEG-Cat; 500 U/ml), AT1R blocker losartan (50 μM), NOX II inhibitors VAS2870 (2 μM) and NSC23766 (10 μM), and SOD inhibitor zinc-diethyldithiocarbamate (Zn-DDC; 1 mM), which were added in randomized order either 30 min before preconstriction with ET-1 or coincubated during HILP. The maximal diameter of every vessel (endothelium-independent vasodilation) was determined with papaverine (100 μM) at the end of each round of FID. FID was calculated as the percent change from ET-1-constricted diameter relative to the maximal diameter measured at rest before ET-1 constriction. Three control male C57BL/6J mice were used for experiments to determine if impaired FID occurred in another vascular bed. Six resistance arteries (n = 6) were obtained, one from each gracilis muscle of the three mice. The skeletal muscle resistance arteries were 63.9 ± 9.7 μm (means ± SD). In agreement with our previous work, we noted that skeletal muscle resistance arteries (32, 37) were smaller than adipose resistance arteries (11, 24, 40). VAS2870 was obtained from Enzo Life Sciences (East Farmingdale, NY). NSC23766 was obtained from Tocris Bioscience (Bristol, UK). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Resistance artery ANG II dose responses.
Resistance artery vasoconstriction to ANG II doses was performed as previously described (37). Resistance arteries maintained at an intraluminal pressure of 60 cmH2O (44 mmHg) were exposed to incremental doses of ANG II (10−10−10−6 M) for 3 min each. Minimal internal diameters were recorded for each dose. After the dose response, vessels were maximally dilated using papaverine (100 μM).
Resistance artery fluorescence.
Superoxide and H2O2 levels were measured as previously described (24, 40). Briefly, arteries were maintained at 37°C in 20-ml aerated organ chambers bathed in Krebs solution at an equilibration pressure of 60 cmH2O (44 mmHg) for 30 min or exposed to HILP (110 mmHg), as described above. Superoxide levels were determined using 5 μM dihydroethidium (DHE). H2O2 levels were assessed using 1 µM 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA). Both dyes were purchased from Thermo Fisher Scientific (Waltham, MA). After incubation with dyes, vessels were exposed to flow (Δ60 cmH2O) 30 min. Prepared vessels were then excised from the glass micropipettes and subsequently mounted on slides with Dako fluorescent mounting medium (Dako, Carpinteria, CA). Mounted vessels were then immediately examined via fluorescent microscopy (Eclipse 80i, Nikon). Acquired images were analyzed for fluorescence intensity in arbitrary units using NIH ImageJ software. Fluorescence was measured three times along different lengths of each vessel. Background intensities were subtracted, and fluorescence was normalized to vessel size to account for autofluorescence (34). Buthionine sulfoxamine (BOS; 1 mM) was used as a positive control to induce and H2O2 production in resistance arteries. A combination treatment of the SOD mimetic tiron (1 mM) and PEG-Cat (500 U/ml) was used for the negative control.
Human adipose microvascular endothelial cells.
Experiments were performed on isolated human adipose microvascular endothelial cells (HAMECs) obtained from ScienCell (Cedro, Carlsbad, CA). HAMECs were grown in ScienCell endothelial cell growth medium (ECM-1007, PRF) supplemented with 5% FBS, EC growth supplement, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified incubator (5% CO2).
Shear stress experiments.
HAMECs were grown to 80–90% confluence. High physiological laminar shear stress (20 dyn/cm2 for 24 h) was generated using a previously described modified cone and plate design (3, 39). This level of shear stress has been used in several exercise mimetic models (14, 26). The cone used in these studies is designed to fit into a 20 × 100-mm tissue culture dish. Magnetism to rotate the cone is provided by magnets within a modified Thermo Scientific Super-Nuova multiplace stirrer (Thermo Fisher Scientific). The angle between the cone and plate is negligible (~0.5°), thus facilitating uniform flow over all HAMECs on the culture dish. HAMECs were switched to serum-free media before shear stress experiments to prevent bubbling. Control (static) HAMECs were also switched to serum-free media. All shear experiments were conducted under sterile conditions. Passages 5−8 were used for all experiments.
Cellular fluorescence.
HAMECs were grown to ~90% confluence on coverslips pretreated for 24 h with 2% bovine plasma fibronectin in basal media. Once the desired confluence was met, cells were incubated with the indicator DHE (5 μM) and cotreated with or without losartan (50 μM), NOX II inhibitors VAS2870 (2 μM) and NSC23766 (10 μM), or the SOD mimetic tiron (1 mM) for 30 min. For the final 15 min of incubation, ANG II (400 nM) was added, and for the final 10 min, the nucleic acid stain Hoechst 33342 (1 µM) was added. Cells were then washed twice using cold PBS and mounted on slides with DAKO fluorescent mounting medium. Mounted slips were then examined via fluorescent microscopy (Eclipse 80i, Nikon). Acquired images were analyzed for fluorescence intensity in arbitrary units using NIH ImageJ software. Random sequences of 10 cells were chosen from each treatment of each passage. This ensured that cases from no single passage disproportionately affected the data. These experiments were repeated four times.
Sample preparation for immunoblot analysis.
Resistance arteries were homogenized and sonicated (Qsonica Q55 Sonicator Ultrasonic Processor, Cole Palmer) in 60 μl RIPA buffer (Sigma) containing 3× protease and 3× phosphatase inhibitor cocktails (Thermo Fisher Scientific). After treatment with shear and pharmacological agents, cells were washed twice with cold PBS and lysed with 250 μl of 3× protease and phosphatase inhibitor cocktail. Lysates were then centrifuged at 17,000 rcf at 4°C for 20 min. Protein determination of tissue and cell lysates was determined using a bicinchoninic acid assay (Pierce, Thermo Fisher Scientific). Tissue and cell supernatants were subsequently heated at 95°C for 5 min at a 2:1 with a 5% β-mercaptoethanol 2× Laemmli sample buffer.
Western blot determination of protein expression.
For Western blot experiments, 20 μg of protein were loaded into each well of Bio-Rad 10% gels. Samples were processed by SDS-PAGE (time varied depending on molecular weight of target proteins at 200 V) and transferred to polyvinylidene difluoride membranes (0.22 μm, Bio-Rad). Proteins were wet transferred at 4°C from 70 to 90 min at 90-110 V depending on the molecular weight of the target protein. Membranes were blocked with Li-Cor blocking buffer (Lincoln, NE) for 2 h at room temperature. Membranes were incubated with primary antibodies for gp91phox and p47phox (1:1,000, Cell Signaling Technologies), GAPDH (1:5000, Cell Signaling Technologies), actin (1:2,000, Thermo Fisher), SOD I (1:3,300, R&D Systems), and SOD II and SOD III (1:1,000, Santa Cruz Biotechnology) in Li-Cor blocking buffer (0.2% Tween 20). Secondary antibodies (1:15,000) were incubated for 2 h in Li-Cor blocking buffer (0.2% Tween 20 and 0.01% SDS). Membranes were washed in Tris-buffered saline-Tween 20 (4 × 5 min) after antibody incubations. Membranes were transferred to Tris-buffered saline after the last wash and imaged within 1 h using a Li-Cor Odyssey scanner. Boxes were manually placed around each band of interest, which returned near-infrared fluorescent values of raw intensity with intralane background subtracted using Odyssey analytic software (27, 31).
Statistics.
All FID and ANG II dose-response comparisons were analyzed using two-way repeated-measures ANOVA using vessel diameter as a covariate followed by pairwise comparisons using Bonferroni’s adjustment (treatment × time). One-way ANOVA with Tukey’s post hoc test was used to compare HAMEC fluorescent data. Western blot and resistance artery fluorescence data were compared using t-tests. α was set at <0.05. With power (1 − β) set at 0.80 and α at <0.05 and assuming 20% differences in treatment conditions with 10% SD, it was calculated that five samples were needed for each condition allowing for two pairwise comparisons (adjusted α 0.025). In some cases, the effect size was adequately large that only four samples in each condition were needed to satisfy power (1 − β) set at 0.80.
RESULTS
Resistance artery FID and fluorescence.
Baseline resistance artery FID was similar in vessels obtained from control and exercise mice (Fig. 2, comparison not shown). After exposure to high intraluminal pressure, FID was reduced in control mouse arteries (P < 0.01 at 10, 20, 40, 60, and 100 cmH2O; Fig. 2A). FID was preserved after high intraluminal pressure in exercise mouse arteries (Fig. 2B). Vasodilation to papaverine was similar between resistance arteries from control and exercise mice, and there was no influence of high intraluminal pressure on the papaverine response (Fig. 2C).
Fig. 2.

High intraluminal pressure (HILP) impairs FID in resistance arteries from control mice but not exercised mice. A: high pressure (HILP) significantly reduced resistance artery FID compared with baseline (BSL) in control mouse arteries. *P < 0.01 at Δ10, 20, 40, 60, and 100 cmH2O. B: there was no difference in resistance artery FID after HILP compared with BSL in exercise mouse arteries. Vasodilation to papaverine was similar between resistance arteries from control and exercised mice. C: there was no influence of HILP on the papaverine response. Data are presented as means ± SE.
Vasoreactivity to ANG II was greater in control mouse arteries compared with exercise mouse arteries (P < 0.01 at 10-6−10-8 M; Fig. 3A). Impaired FID in control mouse arteries exposed to HILP was restored by AT1R blockade with losartan (P < 0.01 at 10, 20, 40, 60, and 100 cmH2O; Fig. 3B). FID in control mouse arteries exposed to HILP was also restored with NOX II inhibition via VAS2870 (P < 0.01 at 10, 20, 40, 60, and 100 cmH2O; Fig. 3C) and NSC23766 (P < 0.01 at 20, 40, 60, and 100 cmH2O; Fig. 3D). The addition of losartan or VAS2870 did not influence FID after high pressure exposure in exercise mouse arteries (Fig. 4, A and B). FID after high intraluminal pressure exposure was significantly reduced by the endogenous SOD inhibitor Zn-DDC in resistance arteries obtained from exercised mice (P < 0.01 at 20, 60, and 100 cmH2O; Fig. 4C).
Fig. 3.

Impaired FID after HILP in resistance arteries from control mice involves the local renin-angiotensin system (RAS) and NADPH oxidase II (NOX II). A: exercise mouse arteries were significantly less sensitive to ANG II-induced constriction compared with control (CON) mouse arteries. *P < 0.01 at 10−6−10−8 M. B: blockade of the angiotensin type 1 receptor (AT1R) with losartan (HILP + losartan) restored FID in control mouse arteries. *P < 0.01 at Δ10, 20, 40, 60, and 100 cmH2O. C: blockade of NOX II with VAS2870 (HILP + VAS2870) restored FID in control mouse arteries. *P < 0.01 at Δ10, 20, 40, 60, and 100 cmH2O. D: blockade of NOX II though inhibition of RAC1 guanine exchange factor (GEF) with NSC23766 (HILP + NSC23766) restored FID in control mouse arteries. *P < 0.01 at Δ 20, 40, 60, and 100 cmH2O. Data are presented as means ± SE.
Fig. 4.

Effect of ANG II, NOX II, and SOD blockade with HILP on FID in resistance arteries from exercised mice. A: there was no difference in resistance artery FID after HILP compared with HILP + blockade of AT1Rs (HILP + losartan) in exercise mouse resistance arteries. B: there was no difference in resistance artery FID after HILP compared with HILP + NOX II inhibition via VAS2870 (HILP + VAS2870) in exercise mouse resistance arteries. C: blockade of endogenous SOD with zinc-diethyldithiocarbamate (HILP + Zn-DDC) reduced FID compared with HILP alone in exercise mouse arteries. *P < 0.01 at Δ20, 60, and 100 cmH2O. Data are presented as means ± SE.
High pressure resulted in increased levels in control mice arteries compared with baseline pressure (P < 0.01; Fig. 5A). In contrast, exercise mouse arteries exposed to high pressure did not exhibit significantly greater levels compared with exercise mouse arteries maintained at baseline pressure (Fig. 5B). H2O2 levels were similar in control mouse arteries exposed to high pressure and control mouse arteries maintained at normal pressure (Fig. 5C). Arteries obtained from exercise mouse arteries exposed to high pressure exhibited significantly greater H2O2 levels compared with exercise mouse arteries maintained at 60 cmH2O (P < 0.01; Fig. 5D). The fluorescence control experiments indicated that the positive control BOS increased superoxide and H2O2 production in resistance arteries, whereas the application of tiron and PEG-Cat reduced the signal for superoxide and H2O2 (Fig. 5E).
Fig. 5.

Differential oxidative stress levels after HILP in resistance arteries from control and exercised mice. A: HILP evoked a significant increase in levels compared with BSL (60 cmH2O) in control mouse arteries. *P < 0.05. B: in exercise mouse arteries, HILP exposure did not evoke a significant increase in levels. C: in control mouse arteries, HILP did not evoke a significant increase in H2O2 levels compared with BSL. D: in exercise mouse arteries, HILP elicited a significant increase in H2O2 levels. *P < 0.05. E: data are presented as means ± SE. Buthionine sulfoxamine (BOS; 1 mM) increased and tiron (1 mM) + polyethylene glycol-catalase (PEG-Cat; 500 U/ml) decreased dihydroethidium (DHE) and 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) signals, indicating that these fluorophores are sensitive to increased ROS.
At baseline, FID in arteries from exercised mice was significantly reduced by both l-NAME and PEG-Cat, but the reduction via l-NAME was significantly greater than that of PEG-Cat (P < 0.01 at 20, 40, 60, and 100 cmH2O; Fig. 6A). In contrast, the preserved FID in exercise mouse arteries after exposure to HILP was also significantly reduced by PEG-Cat and l-NAME, but the reduction in the presence of PEG-Cat was significantly greater than that of l-NAME (P < 0.01 at 20, 40, 60, and 100 cmH2O; Fig. 6B). In the baseline condition, combination blockade of l-NAME + PEG-Cat resulted in a greater reduction in vasodilation than l-NAME alone (Fig. 6C, main effect for condition F = 3.13, P < 0.01). After high intraluminal pressure, combination blockade of l-NAME + PEG-Cat resulted in a greater reduction in vasodilation than PEG-Cat alone (Fig. 6D, main effect for condition F = 6.64, P < 0.05).
Fig. 6.

Preserved FID after HILP in resistance arteries from exercised mice involves a phenotypic switch from nitric oxide (NO·)-mediated dilation to H2O2-mediated dilation. A: in the basal condition, NG-nitro-l-arginine methyl ester (l-NAME) reduced FID significantly more so than PEG-Cat in exercise mouse arteries. *P < 0.01 at Δ20, 40, 60, and 100 cmH2O. B: after exposure to high pressure, PEG-Cat reduced FID significantly more so than l-NAME. *P < 0.01 at Δ20, 40, 60, and 100 cmH2O. C: in the BSL condition, a combination blockade of L-NAME + PEG-Cat resulted in a greater reduction in vasodilation than l-NAME alone. Significant main effect for condition: F = 3.13, P < 0.01. D: after HILP, a combination blockade of l-NAME + PEG-Cat resulted in a greater reduction in vasodilation than PEG-Cat alone. Significant main effect for condition: F = 6.64, P < 0.05. These findings indicate a switch from NO·-mediated dilation at rest to H2O2-mediated dilation after exposure to HILP. Data are presented as means ± SE.
Resistance artery protein expression.
Resistance arteries obtained from exercised animals expressed lower protein levels of the gp91phox NOX II subunit compared with arteries from control mice (P < 0.05; Fig. 7A). Resistance arteries obtained from exercised animals also expressed lower protein levels of the p47phox NOX II subunit compared with arteries from control mice (P < 0.01; Fig. 7B). In contrast, exercise mouse arteries expressed more SOD I (P < 0.01; Fig. 7C), SOD II (P < 0.01; Fig. 7D), and SOD III protein (P < 0.05; Fig. 7E).
Fig. 7.

Differential expression of NOX II subunits and SOD isoforms in resistance arteries from control and exercised mice. A: there was a significant reduction in gp91phox expression in exercise mouse arteries compared with control mouse arteries. *P < 0.05. B: there was a significant reduction in p47phox expression in exercise mouse arteries compared with control mouse arteries. *P < 0.01. C: there was a significant increase in SOD I expression in exercise mouse arteries compared with control mouse arteries. *P < 0.01. D: there was a significant increase in SOD II expression in resistance arteries from exercise mouse arteries compared with control mouse arteries. *P < 0.01. E: there was a significant increase in SOD III expression in in exercise mouse arteries compared with control mouse arteries. *P < 0.05. Data are presented as means ± SE.
HAMEC fluorescence and protein expression.
HAMECs exposed to ANG II exhibited significantly greater levels compared with HAMECs maintained in the control condition. AT1R blockade with losartan, NOX II inhibition with VAS2870 or NSC23766, and treatment with exogenous SOD mimetic tiron all prevented increased (P < 0.01; Fig. 8). In HAMECs exposed to high shear stress, there was less protein expression of the gp91phox and p47phox NOX II subunits than in static cells (P < 0.01 for both; Fig. 9, A and B). High shear stress did not influence SOD I or SOD II protein expression (Fig. 9, C and D). Endothelial cells do not express SOD III (19, 33). All blots were normalized to β-actin or GAPDH.
Fig. 8.

Effects of ANG II on superoxide levels in human adipose microvascular endothelial cells (HAMECs). In HAMECs, treatment with ANG II (400 nM) evoked a significant increase in levels. *P < 0.05. Combined treatment with ANG II + losartan (AT1R blocker), VAS2870 or NSC23766 (NOX II inhibitors), or tiron (SOD mimetic) significantly reduced the ANG II-induced increase in in HAMECS. †P < 0.05. Data are presented as means ± SE.
Fig. 9.

Expression of NOX II subunits and SOD isoforms in HAMECs exposed to laminar shear stress. A: exposure to high shear stress in HAMECs reduced gp91phox protein expression compared with HAMECs raised under static conditions. *P < 0.05. B: in HAMECs, exposure to high shear stress reduced p47phox expression compared with HAMECs raised under static conditions. *P < 0.01. C and D: there was no effect of high shear stress in HAMECs compared with cells grown under static conditions in regard to SOD I expression (C) or SOD II expression (D). Data are presented as means ± SE.
In additional FID experiments, skeletal muscle (gracilis) resistance arteries obtained from control mice displayed reduced FID after high pressure exposure (P < 0.01 at 20, 40, 60, and 100 cmH2O; Fig. 10). FID was preserved after high intraluminal pressure in adipose resistance arteries from a mouse that was regularly exercised but withheld from exercise for 24 h (Fig. 11).
Fig. 10.
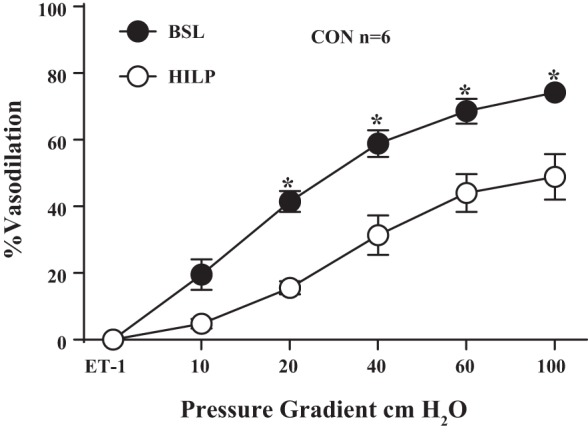
HILP impairs FID in skeletal muscles in resistance arteries from control mice. HILP significantly reduced FID compared with BSL in control mouse arteries. *P < 0.01 at Δ20, 40, 60, and 100 cmH2O. Data are presented as means ± SE.
Fig. 11.
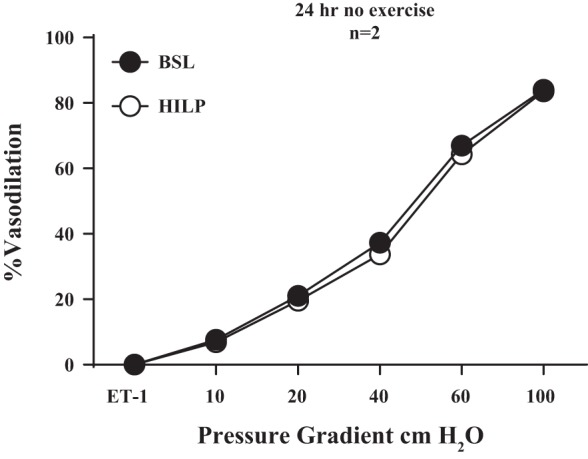
HILP does not impair FID in resistance arteries from exercised mice after 24 h of wheel removal. There was no difference in resistance artery FID after HILP compared with BSL in resistance arteries from exercised mice who did not run for 24 h. Data are presented as means ± SE.
DISCUSSION
The main findings from this study are that high intraluminal pressure induced resistance artery vasodilator dysfunction in a RAS/NOX II-dependent fashion in control mice, whereas arteries from exercised mice were protected from this occurrence. An illustrative summary of the primary findings is shown in Fig. 12.We have previously shown that ANG II plays a role in high pressure-induced vascular function (10, 11), and the local RAS has been well described elsewhere (4, 36). Yet, the molecular means through which regular exercise dampens local RAS-induced endothelial dysfunction remains poorly understood. Illustrating the role of local RAS losartan restored impaired FID in control mouse resistance arteries after high pressure exposure. This finding is in agreement with previous reports of impaired vasodilator function in ex vivo resistance arteries exposed to increased intraluminal pressure being rescued via ACE inhibitors or AT1R blockade (9, 11, 37). Control mouse arteries also displayed greater vasoconstrictive sensitivity to ANG II compared with exercise mouse arteries. In addition, exercise mouse arteries displayed protection against local activation of the RAS, as indicated by the preserved dilation after HILP and no impact of AT1R blockade (Fig. 4).
Fig. 12.

Schematic of high pressure/ANG II-induced vascular oxidative stress and exercise-induced adaptation. A: high pressure increases ANG II, which acts on AT1R (1). AT1R stimulation results in the assembly of the NOX II complex, which is composed of several subunits including membrane-bound gp91phox and p22phox and cytosolic p47 phox, p67 phox, and the GTPase Rac1 (2). Active NOX II produces (3), scavenges the vasodilator NO· to form peroxynitrite (4), or SOD converts to H2O2, which can serve as an alternative vasodilator in place of NO· (5). The deleterious effects of ANG II-induced oxidative stress were inhibited by administration of pharmacological agents including the AT1R blocker losartan (6), NOX II inhibitor VAS2870 (7), and the RAC GEF inhibitor NSC23766 (8). Endogenous SOD activity was blocked with Zn-DDC in resistance arteries from exercised mice (9). B: regular exercise reduced expression of NOX II subunits. C: regular exercise increased the expression of SOD isoforms.
The role of NOX II-induced oxidative stress was indicated by high pressure resulting in increased O2− levels in control mouse arteries and restoration of FID by treatment with NOX II inhibition. In contrast, exercise mouse arteries were not impacted by NOX II blockade after HILP (Fig. 4). Furthermore, exercise mouse arteries did not exhibit increased (Fig. 6) and were also found to express significantly less protein for NOX II subunits gp91phox and p47phox (Fig. 7), which have been shown to play a critical role in generation in endothelial cells (21, 30). Collectively, these findings may partially explain the preserved FID after high pressure exposure in exercise mouse arteries. We did not assess NO· due to limited tissue availability. However, the finding that l-NAME reduced vasodilation after high intraluminal pressure, coupled with reduced NOX II expression and levels, suggests that exercise mouse arteries had greater relative NO· bioavailability. However, NO· was not the predominate vasodilator after high pressure in resistance arteries from exercised mice.
The preserved dilation in resistance arteries from exercised mice was accompanied by a phenotypic switch from primarily NO·-dependent to H2O2-dependent dilation. This was indicated by the reduced FID after H2O2 scavenging with PEG-Cat, by the reduced FID after inhibition of endogenous SOD with Zn-DDC, and, finally, by the increased H2O2 fluorescence after high pressure exposure. While the reduction in preserved dilation with catalase in exercise mouse arteries supports previous findings (9, 40), we show here, for the first time, that the blockade of endogenous SOD also abrogates the preserved FID. These findings suggest that exercise mouse arteries maintained FID after high pressure due to greater SOD activity/expression and a resultant increase in the conversion of to H2O2. In agreement, exercise mouse arteries were found to express significantly more SOD protein compared with control mouse arteries. This finding is also similar to previously reported regular exercise-driven adaptations in other microvascular depots (41, 44).
We previously reported PEG-Cat reactivity was largely unaffected, whereas the response to l-NAME was largely reduced, after high pressure exposure in resistance arteries obtained from sedentary humans. These findings suggest impaired dilation in sedentary humans is due to impaired NO· bioavailability without a concomitant phenotypic switch to rescue vasodilation (9, 40). We did not denude vessels in this particular study to determine if our findings were solely attributable to the endothelium, although our group has done this in the past (9). We were also not able to distinguish if the molecular changes made in the vascular wall were specific to the endothelium as isolating the endothelium and quantifying protein expression solely from this vascular tunic was not feasible. As a substitution, we used HAMECs. We hypothesized that ANG II is the primary mediator of high pressure-induced oxidative stress and endothelial dysfunction. To directly assess the effect of ANG II on the endothelium, we used ANG II treatment in HAMECs. Laminar shear stress was used to mimic exercise.
Treatment with ANG II in HAMECs resulted in increased levels. Treatment with losartan prevented the ANG II-mediated increase in . These findings provide additional evidence suggesting that the local RAS is an upstream mediator of oxidative stress in response to high pressure. NOX II inhibition also reduced the ANG II-mediated increases in , indicating that NOX II is a primary downstream mediator of local RAS. The finding that HAMECs exposed to shear stress expressed significantly lower protein of the gp91phox and p47phox NOX II subunits indicates that increased shear stress during exercise bouts could be the primary stimulus for reduced NOX II-dependent . Contrary to our initial hypothesis, SOD protein expression in HAMECs was unaffected by laminar shear stress. This finding could be due to several reasons, including that SOD changes in resistance arteries were primarily due to changes in vascular smooth muscle. Alternatively, paracrine factors released during whole body acute exercise may play a role in regulating endothelial SOD expression in the adipose microvasculature (10, 18, 48). Nonetheless, treatment with exogenous tiron also prevented ANG II-mediated increases in levels in HAMECs, suggesting that exercise-induced increases in SOD could play a role in combating oxidative stress after local activation of the RAS in the adipose microcirculation. A limitation to the HAMEC model was that static cells were used as a comparison with high physiological shear, whereas a low shear stress model may have served as a more appropriate control to model in vivo conditions.
Additional experiments (n = 2) were performed on arteries obtained from an exercised mouse whose wheel was removed for 24 h before death. Endothelium-dependent dilation was preserved after high pressure exposure, despite no exercise being performed at least 24 h before the artery was obtained (Fig. 11). These findings are in agreement with our previous human studies (4, 18). Taken together, these findings suggest preserved endothelium-dependent dilation after high pressure in exercisers is due to chronic exercise adaptations. The exact time course for this regular exercise-conferred protection remains to be elucidated. Additional experiments performed in skeletal muscle (gracilis m.) resistance arteries suggest impaired FID after high intraluminal pressure may be systemic (Fig. 10). However, our findings are of particular relevance to the adipose tissue microcirculation. Impaired adipose tissue perfusion may contribute to CVD risk by promoting inflammation through the secretion of proinflammatory adipokines (20). Prior studies have shown that inflammation modulates endothelial dysfunction in human adipose resistance arteries (12, 13). Our findings suggest regular exercise may play a role in preventing this impaired perfusion after high pressure exposure.
Clinical implications.
The collective findings presented here elucidate a damaging pathway elicited by high pressure involving local activation of the local RAS and activation of NOX II, which results in excess oxidative stress in the vascular wall. Our results suggest regular exercise protects against high pressure-induced microvascular dysfunction by creating a favorable vascular redox environment in the adipose microcirculation. Regular exercise may protect against high pressure by creating a favorable redox environment, thus lessening the risk for cardiovascular events.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants 2NIH-R01-HL-070187-11 (to T. Fukai), R01-HL-116976 (to T. T. Fukai and M. Ushio-Fukai), K23-HL-085614 (to S. A. Phillips), R01-HL-095701 (to S. A. Phillips), and R01-HL-095701- 03S1 (to A. T. Robinson); Department of Veterans Affairs Merit Review Grant 2I01BX001232-05 (to T. Fukai), and American Heart Association Scientist Development Grant 15SDG25700406 (to V. Sudhahar).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.T.R. conceived and designed research; A.T.R., V.S., M.C., A.M.M., and M.A. performed experiments; A.T.R. analyzed data; A.T.R., I.S.F., J.T.B., M.D.B., T.F., and S.A.P. interpreted results of experiments; A.T.R. prepared figures; A.T.R. drafted manuscript; A.T.R., I.S.F., V.S., J.T.B., M.C., A.M.M., M.A., M.U.-F., M.D.B., T.F., and S.A.P. edited and revised manuscript; A.T.R., I.S.F., V.S., J.T.B., M.C., A.M.M., M.A., M.U.-F., M.D.B., T.F., and S.A.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Sang Joon Ahn and Heather Grimm for technical assistance.
REFERENCES
- 1.Ahn SJ, Fancher IS, Bian JT, Zhang CX, Schwab S, Gaffin R, Phillips SA, Levitan I. Inwardly rectifying K+ channels are major contributors to flow-induced vasodilatation in resistance arteries. J Physiol. In press. doi: 10.1113/JP273255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD. An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol Heart Circ Physiol 307: H1587–H1593, 2014. doi: 10.1152/ajpheart.00557.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. J Biol Chem 277: 3388–3396, 2002. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 4.Campbell DJ. Clinical relevance of local renin angiotensin systems. Front Endocrinol (Lausanne) 5: 113, 2014. doi: 10.3389/fendo.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chinsomboon J, Ruas J, Gupta RK, Thom R, Shoag J, Rowe GC, Sawada N, Raghuram S, Arany Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc Natl Acad Sci USA 106: 21401–21406, 2009. doi: 10.1073/pnas.0909131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dharmashankar K, Welsh A, Wang J, Kizhakekuttu TJ, Ying R, Gutterman DD, Widlansky ME. Nitric oxide synthase-dependent vasodilation of human subcutaneous arterioles correlates with noninvasive measurements of endothelial function. Am J Hypertens 25: 528–534, 2012. doi: 10.1038/ajh.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dharmashankar K, Widlansky ME. Vascular endothelial function and hypertension: insights and directions. Curr Hypertens Rep 12: 448–455, 2010. doi: 10.1007/s11906-010-0150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 102: 488–496, 2008. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 9.Durand MJ, Dharmashankar K, Bian JT, Das E, Vidovich M, Gutterman DD, Phillips SA. Acute exertion elicits a H2O2-dependent vasodilator mechanism in the microvasculature of exercise-trained but not sedentary adults. Hypertension 65: 140–145, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durand MJ, Lombard JH. Low-dose angiotensin II infusion restores vascular function in cerebral arteries of high salt-fed rats by increasing copper/zinc superoxide dimutase expression. Am J Hypertens 26: 739–747, 2013. doi: 10.1093/ajh/hpt015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durand MJ, Phillips SA, Widlansky ME, Otterson MF, Gutterman DD. The vascular renin-angiotensin system contributes to blunted vasodilation induced by transient high pressure in human adipose microvessels. Am J Physiol Heart Circ Physiol 307: H25–H32, 2014. doi: 10.1152/ajpheart.00055.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farb MG, Bigornia S, Mott M, Tanriverdi K, Morin KM, Freedman JE, Joseph L, Hess DT, Apovian CM, Vita JA, Gokce N. Reduced adipose tissue inflammation represents an intermediate cardiometabolic phenotype in obesity. J Am Coll Cardiol 58: 232–237, 2011. doi: 10.1016/j.jacc.2011.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farb MG, Tiwari S, Karki S, Ngo DT, Carmine B, Hess DT, Zuriaga MA, Walsh K, Fetterman JL, Hamburg NM, Vita JA, Apovian CM, Gokce N. Cyclooxygenase inhibition improves endothelial vasomotor dysfunction of visceral adipose arterioles in human obesity. Obesity (Silver Spring) 22: 349–355, 2014. doi: 10.1002/oby.20505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feairheller DL, Park JY, Rizzo V, Kim B, Brown MD. Racial differences in the responses to shear stress in human umbilical vein endothelial cells. Vasc Health Risk Manag 7: 425–431, 2011. doi: 10.2147/VHRM.S22435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franklin NC, Ali M, Goslawski M, Wang E, Phillips SA. Reduced vasodilator function following acute resistance exercise in obese women. Front Physiol 5: 253, 2014. doi: 10.3389/fphys.2014.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franklin NC, Robinson AT, Bian JT, Ali MM, Norkeviciute E, McGinty P, Phillips SA. Circuit resistance training attenuates acute exertion-induced reductions in arterial function but not inflammation in obese women. Metab Syndr Relat Disord 13: 227–234, 2015. doi: 10.1089/met.2014.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest 105: 1631–1639, 2000. doi: 10.1172/JCI9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukai T, Siegfried MR, Ushio-Fukai M, Griendling KK, Harrison DG. Modulation of extracellular superoxide dismutase expression by angiotensin II and hypertension. Circ Res 85: 23–28, 1999. doi: 10.1161/01.RES.85.1.23. [DOI] [PubMed] [Google Scholar]
- 19.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 15: 1583–1606, 2011. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res 118: 1786–1807, 2016. doi: 10.1161/CIRCRESAHA.115.306885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Görlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res 87: 26–32, 2000. doi: 10.1161/01.RES.87.1.26. [DOI] [PubMed] [Google Scholar]
- 22.Goslawski M, Piano MR, Bian JT, Church EC, Szczurek M, Phillips SA. Binge drinking impairs vascular function in young adults. J Am Coll Cardiol 62: 201–207, 2013. doi: 10.1016/j.jacc.2013.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green DJ, Jones H, Thijssen D, Cable NT, Atkinson G. Flow-mediated dilation and cardiovascular event prediction: does nitric oxide matter? Hypertension 57: 363–369, 2011. doi: 10.1161/HYPERTENSIONAHA.110.167015. [DOI] [PubMed] [Google Scholar]
- 24.Grizelj I, Cavka A, Bian JT, Szczurek M, Robinson A, Shinde S, Nguyen V, Braunschweig C, Wang E, Drenjancevic I, Phillips SA. Reduced flow-and acetylcholine-induced dilations in visceral compared with subcutaneous adipose arterioles in human morbid obesity. Microcirculation 22: 44–53, 2015. doi: 10.1111/micc.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang A, Sun D, Kaley G, Koller A. Superoxide released to high intra-arteriolar pressure reduces nitric oxide-mediated shear stress- and agonist-induced dilations. Circ Res 83: 960–965, 1998. doi: 10.1161/01.RES.83.9.960. [DOI] [PubMed] [Google Scholar]
- 26.Kim JS, Kim B, Lee H, Thakkar S, Babbitt DM, Eguchi S, Brown MD, Park JY. Shear stress-induced mitochondrial biogenesis decreases the release of microparticles from endothelial cells. Am J Physiol Heart Circ Physiol 309: H425−H433, 2015. doi: 10.1152/ajpheart.00438.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinoshita-Kikuta E, Kinoshita E, Matsuda A, Koike T. Tips on improving the efficiency of electrotransfer of target proteins from Phos-tag SDS-PAGE gel. Proteomics 14: 2437–2442, 2014. doi: 10.1002/pmic.201400380. [DOI] [PubMed] [Google Scholar]
- 28.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201–1209, 2003. doi: 10.1172/JCI200314172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laufs U, Werner N, Link A, Endres M, Wassmann S, Jürgens K, Miche E, Böhm M, Nickenig G. Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation 109: 220–226, 2004. doi: 10.1161/01.CIR.0000109141.48980.37. [DOI] [PubMed] [Google Scholar]
- 30.Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J Biol Chem 278: 12094–12100, 2003. doi: 10.1074/jbc.M209793200. [DOI] [PubMed] [Google Scholar]
- 31.Mahmood T, Yang PC. Western blot: technique, theory, and trouble shooting. N Am J Med Sci 4: 429–434, 2012. doi: 10.4103/1947-2714.100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahmoud AM, Szczurek MR, Blackburn BK, Mey JT, Chen Z, Robinson AT, Bian JT, Unterman TG, Minshall RD, Brown MD, Kirwan JP, Phillips SA, Haus JM. Hyperinsulinemia augments endothelin-1 protein expression and impairs vasodilation of human skeletal muscle arterioles. Physiol Rep 4: e12895, 2016. doi: 10.14814/phy2.12895. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=27796268&dopt=Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marklund SL. Expression of extracellular superoxide dismutase by human cell lines. Biochem J 266: 213–219, 1990. doi: 10.1042/bj2660213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monici M. Cell and tissue autofluorescence research and diagnostic applications. Biotechnol Annu Rev 11: 227–256, 2005. doi: 10.1016/S1387-2656(05)11007-2. [DOI] [PubMed] [Google Scholar]
- 35.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation 131: e29–e322, 2015. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 36.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 86: 747–803, 2006. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 37.Phillips SA, Pechman KR, Leonard EC, Friedrich JL, Bian JT, Beal AG, Basile DP. Increased ANG II sensitivity following recovery from acute kidney injury: role of oxidant stress in skeletal muscle resistance arteries. Am J Physiol Regul Integr Comp Physiol 298: R1682–R1691, 2010. doi: 10.1152/ajpregu.00448.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puddu P, Puddu GM, Zaca F, Muscari A. Endothelial dysfunction in hypertension. Acta Cardiol 55: 221–232, 2000. doi: 10.2143/AC.55.4.2005744. [DOI] [PubMed] [Google Scholar]
- 39.Reinhart-King CA, Fujiwara K, Berk BC. Physiologic stress-mediated signaling in the endothelium. Methods Enzymol 443: 25–44, 2008. doi: 10.1016/S0076-6879(08)02002-8. [DOI] [PubMed] [Google Scholar]
- 40.Robinson AT, Franklin NC, Norkeviciute E, Bian JT, Babana JC, Szczurek MR, Phillips SA. Improved arterial flow-mediated dilation after exertion involves hydrogen peroxide in overweight and obese adults following aerobic exercise training. J Hypertens 34: 1309–1316, 2016. doi: 10.1097/HJH.0000000000000946. [DOI] [PubMed] [Google Scholar]
- 41.Rush JW, Turk JR, Laughlin MH. Exercise training regulates SOD-1 and oxidative stress in porcine aortic endothelium. Am J Physiol Heart Circ Physiol 284: H1378–H1387, 2003. doi: 10.1152/ajpheart.00190.2002. [DOI] [PubMed] [Google Scholar]
- 42.Shimbo D, Muntner P, Mann D, Viera AJ, Homma S, Polak JF, Barr RG, Herrington D, Shea S. Endothelial dysfunction and the risk of hypertension: the multi-ethnic study of atherosclerosis. Hypertension 55: 1210–1216, 2010. doi: 10.1161/HYPERTENSIONAHA.109.143123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimokawa H. Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pflugers Arch 459: 915–922, 2010. doi: 10.1007/s00424-010-0790-8. [DOI] [PubMed] [Google Scholar]
- 44.Thengchaisri N, Shipley R, Ren Y, Parker J, Kuo L. Exercise training restores coronary arteriolar dilation to NOS activation distal to coronary artery occlusion: role of hydrogen peroxide. Arterioscler Thromb Vasc Biol 27: 791–798, 2007. doi: 10.1161/01.ATV.0000258416.47953.9a. [DOI] [PubMed] [Google Scholar]
- 45.Thompson PD, Franklin BA, Balady GJ, Blair SN, Corrado D, Estes NA III, Fulton JE, Gordon NF, Haskell WL, Link MS, Maron BJ, Mittleman MA, Pelliccia A, Wenger NK, Willich SN, Costa F; American Heart Association Council on Nutrition, Physical Activity, and Metabolism; American Heart Association Council on Clinical Cardiology; American College of Sports Medicine . Exercise and acute cardiovascular events placing the risks into perspective: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism and the Council on Clinical Cardiology. Circulation 115: 2358–2368, 2007. doi: 10.1161/CIRCULATIONAHA.107.181485. [DOI] [PubMed] [Google Scholar]
- 46.Vecchione C, Carnevale D, Di Pardo A, Gentile MT, Damato A, Cocozza G, Antenucci G, Mascio G, Bettarini U, Landolfi A, Iorio L, Maffei A, Lembo G. Pressure-induced vascular oxidative stress is mediated through activation of integrin-linked kinase 1/betaPIX/Rac-1 pathway. Hypertension 54: 1028–1034, 2009. doi: 10.1161/HYPERTENSIONAHA.109.136572. [DOI] [PubMed] [Google Scholar]
- 47.Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, Bauersachs J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of thioredoxin 2. Hypertension 54: 338–344, 2009. doi: 10.1161/HYPERTENSIONAHA.108.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woods D, Sanders J, Jones A, Hawe E, Gohlke P, Humphries SE, Payne J, Montgomery H. The serum angiotensin-converting enzyme and angiotensin II response to altered posture and acute exercise, and the influence of ACE genotype. Eur J Appl Physiol 91: 342–348, 2004. doi: 10.1007/s00421-003-0993-1. [DOI] [PubMed] [Google Scholar]