

Abstract
Pulmonary hypertension (PH) is characterized by increased pulmonary vascular resistance, pulmonary vascular remodeling, and increased pulmonary vascular pressures that often result in right ventricular dysfunction, leading to right heart failure. Evidence suggests that reactive oxygen species (ROS) contribute to PH pathogenesis by altering pulmonary vascular cell proliferation and intracellular signaling pathways. However, the role of mitochondrial antioxidants and oxidant-derived stress signaling in the development of hypoxia-induced PH is largely unknown. Therefore, we examined the role of the major mitochondrial redox regulator thioredoxin 2 (Trx2). Levels of Trx2 mRNA and protein were examined in human pulmonary arterial endothelial cells (HPAECs) and smooth muscle cells (HPASMCs) exposed to hypoxia, a common stimulus for PH, for 72 h. Hypoxia decreased Trx2 mRNA and protein levels. In vitro overexpression of Trx2 reduced hypoxia-induced H2O2 production. The effects of increased Trx2 protein level were examined in transgenic mice expressing human Trx2 (TghTrx2) that were exposed to hypoxia (10% O2) for 3 wk. TghTrx2 mice exposed to hypoxia had exacerbated increases in right ventricular systolic pressures, right ventricular hypertrophy, and increased ROS in the lung tissue. Trx2 overexpression did not attenuate hypoxia-induced increases in Trx2 oxidation or Nox4 expression. Expression of a dominant negative C93S Trx2 mutant that mimics Trx2 oxidation exacerbated hypoxia-induced increases in HPASMC H2O2 levels and cell proliferation. In conclusion, Trx2 overexpression failed to attenuate hypoxia-induced HPASMC proliferation in vitro or hypoxia-induced PH in vivo. These findings indicate that strategies to enhance Trx2 expression are unlikely to exert therapeutic effects in PH pathogenesis.
Keywords: human pulmonary artery smooth muscle cell, reactive oxygen species, hydrogen peroxide, pulmonary hypertension
pulmonary hypertension (PH) is associated with increased proliferation of vascular wall cells, pulmonary vascular remodeling, increased right ventricular systolic pressures (RVSP), and right ventricular hypertrophy (RVH) leading eventually to heart failure (3). Numerous studies suggest that increased production of reactive oxygen species (ROS) in excess of the endogenous antioxidant capacity results in an environment of oxidative stress that may contribute to PH pathogenesis (1, 3, 4, 20, 22). Excess ROS, which can be produced through various cellular mechanisms, such as mitochondrial oxidative phosphorylation or NAD(P)H oxidase (Nox) activity may play a vital role in PH pathogenesis (39). Mitochondria act as cellular O2 sensors, adjusting redox-signaling pathways to regulate vascular tone and regulate hypoxia-induced redox signaling (10, 33, 37). Therefore, changes in mitochondrial antioxidants may regulate proliferative responses of pulmonary vascular wall cells to hypoxia and thereby modulate PH.
There are many enzymatic and nonenzymatic mechanisms to protect cells from ROS. This study focused on the thioredoxin (Trx) family because Trxs interact directly with intracellular signaling molecules and selectively activate the DNA binding of a number of transcription factors that directly influence vascular cell growth and proliferation. There are three Trx isozymes expressed in mammalian cells: cytosolic (Trx1), mitochondrial (Trx2), and spermatozoan (spTrx). Mammalian Trxs reduce disulfides to dithiols as a result of two conserved cysteine residues in the active site (9, 24). In combination with cofactors including NAD(P)H, peroxiredoxin (Prx), and thioredoxin reductase (TrxR), Trxs reduce exposed protein disulfides (an S-S bond) to dithiols (two –SH groups) in oxidized proteins (31, 36). Peroxiredoxins are nonheme peroxidases that reduce endogenously generated H2O2. TrxRs reduce Trxs, and Trxs reduce Prxs (9, 42). Human Trx2 is a 166 amino acid, 18.2-kDa enzyme localized to the mitochondria. Trx2 is essential for normal development in mice and plays a crucial role in regulation of mitochondrial apoptosis signaling pathways (8).
ROS impact vascular tone, vascular cell proliferation, and apoptosis (7, 29). ROS derived from Nox2 and Nox4 are involved in long-term responses of the pulmonary vasculature to hypoxia. Nox4 expression is increased in pulmonary artery endothelial cells isolated from patients with idiopathic pulmonary arterial hypertension (IPAH) and in pulmonary and vascular tissue from hypoxia-induced PH models (1, 13, 27, 34, 38). Hypoxia-induced Nox4 activation likely contributes to vasoconstriction and cell proliferation, and recent studies indicate that Nox4 is responsible for basal H2O2 production in the vasculature (6, 30, 32). In human pulmonary arterial smooth muscle cells (HPASMCs), PEG-catalase prevented HPASMC proliferation, ERK 1/2 and NF-κB activation, and Nox4 expression, indicating that H2O2 participates in feed-forward activation of proliferative signaling (2). In addition, our recent study establishes that mitochondria-derived H2O2 drives the expression of Nox2 and Nox4 to promote PH (1).
Since Trx2 is one of the primary lines of defense against mitochondria-derived ROS, this study examines how this antioxidant system is affected by hypoxia and how overexpression of Trx2 affects ROS production and other pathobiological hallmarks of PH. To clarify the role of Trx2 in PH, human pulmonary arterial endothelial cells (HPAECs), HPASMCs, and transgenic human Trx2-overexpressing (TghTrx2) and littermate control (Lit Ctrl) mice were exposed to hypoxia using protocols previously shown to promote proliferation and PH, respectively (1, 13, 23, 27). Due to the function of Trx2 in regulating ROS levels, we hypothesized that overexpression of Trx2 would attenuate hypoxia-induced mitochondrial ROS levels, thereby preventing elevations in hypoxia-induced PH markers, vascular remodeling, and hypertrophy of the right ventricle. However, our findings demonstrate that Trx2 overexpression fails to prevent HPASMC proliferation in vitro or PH and vascular remodeling in vivo. These findings provide novel evidence that targeting mitochondrial redox status with Trx2 overexpression does not impact pathobiological changes in the pulmonary vasculature that lead to PH.
METHODS
Cell Culture
HPAECs (Clonetics, San Diego, CA) were grown at 37°C in 5% CO2 in endothelial cell growth medium (EGM; Lonza, Walkersville, MD) supplemented with provided growth factors and 10% fetal bovine serum (FBS) (13, 27, 41). HPAECs were used at passages lower than 7, and characteristic “cobblestone” endothelial cell morphology was confirmed with phase contrast microscopy before use.
HPASMCs (ScienCell Research Laboratories, Carlsbad, CA) were grown at 37°C in 5% CO2 in smooth muscle cell growth medium (SmGM-2; Lonza) supplemented with growth factors and 5% FBS. HPAECs or HPASMCs were plated in 10-cm cell culture dishes in 10% serum or serum free media overnight, respectively. Smooth muscle cells were used between passages 4 to 8 and smooth muscle cell elongated spindle morphology was confirmed with phase contrast microscopy before use. HPASMCs were then switched to media with 5% serum for hypoxia exposure. HPAECs or HPASMCs were exposed to normoxic (21% O2) or hypoxic (1% O2 or 10% O2) conditions for 72 h at 37°C in 5% CO2 as we have previously reported (13). Cell lysates were then extracted under hypoxic conditions in a C-Shuttle Glove Box (BioSpherix, Lacona, NY).
The genetic backgrounds of HPAECs and HPASMCs were characterized by STR profiling (Biosynthesis DNA Identity Testing Center, Lewisville, TX). Each HPAEC line used was derived from different male patients. HPASMCs were derived from four different male patients. All cell lines are established lines; however, other characterizing information was unavailable.
In vitro Trx2 Overexpression
Adenoviral vectors expressing a wild-type human Trx2 (WT Trx2) and an active site mutant of Trx2 (C93S Trx2) were purchased from Applied Biological Materials (Richmond, BC, Canada). A green fluorescent protein (GFP) adenovirus was used as a control for in vitro Amplex Red studies. Adenoviral vectors were used to infect HPASMCs at a multiplicity of infection of 2.
Measuring Relative Cell Number
The MultiTox-Glo Multiplex Cytotoxicity Assay (Promega, Madison, WI) was used according to the manufacturer’s instructions. In brief, HPASMC were plated in 96-well clear bottom black cell culture plates (Corning, Corning, NY). Cells were infected with adenovirus, as described above and then grown under normoxic or hypoxic conditions. Fifty microliters diluted glycyl-phenylalanyl-aminofluorocoumarin (GF-AFC) substrate were added to each well and incubated for 30 min at 37°C then measured with a Wallac Victor 3 fluorimeter (PerkinElmer, Waltham, MA) at 400 nm excitation/505 nm emission to detect live cells. The GF-AFC substrate can enter intact viable cells where it is cleaved by live-cell proteases to release the fluorogenic AFC. The fluorescent signal is directly proportional to the number of live cells.
Assessment of PH In Vivo
All animal studies were approved by the Atlanta Veterans Affairs Medical Center Institutional Animal Care and Use Committee and were performed in accordance with National Institutes of Health guidelines outlined in the Guide for the Care and Use of Laboratory Animals. Male, C57/BL6J mice (Jackson Laboratories), ages 6–9 wk, were either housed in ambient air (normoxia, 21% O2) or hypoxic conditions (10% O2) using the ProOx 110 (BioSpherix) for 3 wk (27) or 3 days to asses hypoxic effect on Trx2 expression.
In vivo Trx2 overexpression.
Human Trx2 was expressed in C57/BL6J mice under the CAG promoter using the Cre-LoxP system. The transgene contained a V5 epitope tag at the COOH terminus (17). TghTrx2 protein was localized to the mitochondria but not the cytoplasm, and there were no differences in Trx2 redox potential in mitochondria isolated from TghTrx2 or littermate control (Lit Ctrl) animals (17). TghTrx2 mice had no observed phenotype. Mice ages 6–9 wk, were utilized for these studies. To assess the effects of Trx2 overexpression on PH development, mice were exposed to normoxia or hypoxia as described above.
Several complementary methods were employed to assess the severity of hypoxia-induced PH in mouse models. Right ventricular systolic pressure (RVSP) was assessed using a 0.8-F microtip pressure transducer as previously reported (27). In brief, mice were lightly anesthetized with isoflurane, and the microtip pressure transducer (Transonic, Ithaca, NY) was inserted into the right jugular vein and advanced to the right ventricle. RVSP was monitored for 10 min, and data were analyzed using the Powerlab system (ADInstruments, Denver, CO) (27). Right ventricular hypertrophy (RVH) was assessed by calculating the ratio of the right ventricle weight to the left ventricle + septum weight (Fulton Index). Mouse hearts were removed, and the right ventricle was separated from the left ventricle and septum weighed (1, 27).
Pulmonary vascular remodeling was assessed with immunohistochemical and morphometric analyses (1, 13, 27). Lungs were perfused blood-free with calcium-free PBS + 1 mM EDTA at a perfusion pressure comparable to the measured RVSP to avoid confusing sustained vasoconstriction for morphological evidence of remodeling of the vascular wall (16, 26) and then lungs were placed in formalin overnight. Lung tissue was then paraffin embedded. Lung sections (5 μm) were fixed in 4% formaldehyde and washed three times in PBS, and endogenous peroxidase activity was quenched with 3% H2O2 in PBS. Sections were permeabilized with 0.05% Tween-20 (PBS-T), blocked with 5% donkey serum, and incubated overnight at 4°C with rabbit anti-α-smooth muscle actin (α-SMA) antibody (LabVision, Fremont, CA). Sections were incubated with biotinylated donkey anti-rabbit secondary antibody (Jackson Immunoresearch Laboratories, West Grove, PA) followed by horseradish peroxidase-streptavidin (Vectastain kit; Vector Laboratories, Burlingame, CA) as previously reported (1, 13). Ten to twenty arterioles (<100-µm diameter) per sample were assessed for α-SMA staining.
mRNA and Protein Analysis
Quantitative real-time PCR was employed to measure mRNA levels of Trx2 (human: forward: AGG GAG GGC TAG GCT GTG; reverse: ACT GAC CCT GAG AGG GCT TC) (mouse: forward: CAC ACA GAC CTT GCC ATT GA; reverse: CAC GTC CCC GTT CTT GAT). Total RNA was isolated from HPAECs and HPASMCs with Trizol according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA). Total RNA was reverse transcribed to synthesize cDNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Target cDNA was amplified using SYBR Green (Applied Biosystems, Hercules, CA). All data were normalized to GAPDH content of the same sample. Western blot analysis was employed to detect protein levels of Trx2, Nox4, β-actin, and GAPDH in vitro and in vivo. Cell and lung lysates were resolved on 10% or 4–12% Bis-Tris gels (Invitrogen), transferred onto nitrocellulose membranes (Millipore, Billerica, MA), and probed with primary antibodies specific for Trx2 1:100 [Go et al. (12)], Nox4 1:500 (Abcam), Nox1 (Santa Cruz Biotechnology, Santa Cruz, CA), and Nox2 (Santa Cruz Biotechnology), V5 1:250 epitope (Invitrogen), β-actin (Santa Cruz Biotechnology), or GAPDH 1:1000 (Sigma, St. Louis, MO). After being washed, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories). Immunodetection was performed using a near-infrared fluorescent method (Li-Cor, Lincoln, NE).
Measurement of Trx2 Redox Potential
Tissue samples were prepared as described (11). All steps were completed at 4°C unless stated. In short, tissue samples were homogenized in 10% trichloroacetic acid then centrifuged for 10 min at 12,000 g. The tissue pellet was resuspended in 100% acetone then centrifuged to pellet protein. The protein pellet was then sonicated in lysis/derivatization buffer (20 mM Tris·HCl, pH 8, 15 mM 4-acetoamido-4-maleimidylstilbene-2,2-disulphonic acid) with protease inhibitors then equilibrated in the dark at room temperature for 3 h. A fully reduced sample was prepared by treatment with 1,000 U PEG-catalase. After addition of nonreducing sample buffer (3.1 ml 1 M Tris·Cl, pH 6.8, and 1% wt/vol bromophenol blue), protein samples, including the reduced control (not shown), were separated on a freshly prepared 15% 1.0 mm polyacrylamide gel and then transferred to a nitrocellulose membrane. Immunodetection of reduced and oxidized Trx2 protein was performed as described above, and Trx2 redox potential was calculated using the Nernst equation, ETrx2 = −330 mV + 30 × ln(acceptor/donor).
Measurement of H2O2 Levels In Vitro and In Vivo
In vitro.
H2O2 released from treated HPASMCs was measured by horseradish peroxidase-catalyzed oxidation of the nonfluorescent molecule N-acetyl-3,7-dihydroxyphenoxazine (Amplex Red; Invitrogen) into the fluorescent molecule resorufin according to the manufacturer’s instructions. In brief, HPASMCs were preincubated with 200 μl of Krebs-Ringer Bicarbonate Buffer (KRPG) buffer for 3 h at 37°C in a 5% CO2 incubator. KRPG buffer from samples (50 μl) was collected and then loaded in duplicate into a clear-bottom 96-well plate. KRPG buffer from samples were then incubated with 50 μl of reaction mixture containing KRPG buffer, 100 μl/ml Amplex Red reagent, and 0.2 U/ml horseradish peroxidase (HRP) for 1 h at 37°C. Fluorescence was measured with a Wallac fluorimeter (Perkin Elmer) at excitation and emission wavelengths of 540 and 590 nm, respectively. Sample fluorescence was measured in duplicate, and H2O2 concentrations were calculated based on a H2O2 standard curve. H2O2 concentrations were normalized to BCA protein concentrations.
In vivo.
Lung tissue sections were incubated in KRPG buffer containing 100 µl/ml Amplex Red and 0.2 U/ml HRP for 1 h at 37°C. Menadione (0.5 M) was used as positive control, and 1,000 U/ml PEG-catalase was used as negative control to assess H2O2 production. Fluorescence was measured at excitation and emission wavelengths of 540 nm and 590 nm, respectively. Sample fluorescence was compared with that generated by H2O2 standard curve to calculate the concentrations of H2O2 released from tissue. H2O2 concentrations were normalized to wet tissue weight or total protein as determined by BCA assay (Invitrogen) (1, 13, 41).
Statistical Analysis
For all experiments, statistical analysis was performed using Graphpad Prism 6. Student’s t-test was used in all experiments comparing two groups. Two-way ANOVA was used in all experiments comparing four groups. Two-way ANOVA was followed by post hoc analysis with the Tukey test to detect differences between experimental groups. An α of P < 0.05 was considered statistically significant (27, 41). Error bars are means ± SE.
RESULTS
Effects of Hypoxia on Trx2
Mitochondrial ROS play a critical role in the development of multiple vascular complications including hypoxia-induced PH (1, 3, 10, 20). However, to our knowledge, previous studies have not examined the impact of hypoxia on Trx2 expression in pulmonary vascular wall cells. To determine if Trx2 is impacted by hypoxia exposure, we examined Trx2 in HPAECs and HPASMCs exposed to normoxia (21% O2) or hypoxia (1% O2) for 72 h (Fig. 1). Hypoxia exposure decreased Trx2 mRNA levels in HPAECs (Fig. 1A) and HPASMCs (Fig. 1B) in vitro and in lung tissue from mice exposed to hypoxia for 3 wk in vivo (Fig. 1C). Similarly, Trx2 protein levels were reduced by hypoxia exposure in HPAECs (Fig. 1D) and HPASMCs (Fig. 1E) in vitro. In contrast, Trx2 protein levels were not significantly reduced in lung tissue from mice exposed to hypoxia for 3 wk in vivo (Fig. 1F) or in lung tissue from rats exposed to the VEGFR inhibitor Su 5416 (data not shown). To further explore the relationship between oxygen level and Trx2 expression we compared Trx2 protein levels in HPASMCs exposed for 72 h to either 1 or 10% O2 (Fig. 2A). Oxygen levels similar to those used in the in vivo studies (10% O2) did not alter Trx2 expression. In contrast, exposing mice to 10% O2 in vivo for 72 h, rather than 3 wk, led to a significant increase in Trx2 expression (Fig. 2B).
Fig. 1.

Trx2 expression is decreased in in vitro and in vivo models of hypoxia-induced pulmonary hypertension (PH). Trx2 mRNA normalized to GAPDH (fold change) in HPAECs (n = 3; A) and HPASMCs (n = 6; B) exposed to 72 h of hypoxia and mouse lung (n = 6; C) mRNA after 3 wk of hypoxia (10% O2) or normoxia (21% O2) exposure. Trx2 protein normalized to β-actin (fold change) in HPAECs (n = 5; D), HPASMCs (n = 5; E), and mouse lung (n = 5; F) protein. Trx2, thioredoxin 2; HPAECs, human pulmonary artery endothelial cells; HPASMCs, human pulmonary artery smooth muscle cells; mlung, mouse lung. Error bars = SE. *P < 0.05.
Fig. 2:

Trx2 expression is sensitive to fluctuations in oxygen levels. Trx2 protein normalized to β-actin (fold change) in HPASMCs (n = 6; A) exposed to either 1 or 10% O2 and mouse lung (n = 5; B) exposed to either 3 days or 3 wk of hypoxia (10% O2). Error bars = SE. *P < 0.05, compared with Normoxia.
Overexpression of Trx2 Prevents Hypoxia-Induced ROS Production but Not HPASMC Proliferation
Since thioredoxins play an important role in oxidative stress (21, 25), vascular remodeling (19), and hypoxia-induced proliferation (5), we hypothesized that Trx2 overexpression would prevent hypoxia-induced proliferation by attenuating hypoxia-induced increases in ROS. HPASMCs were infected with adenoviral wild-type Trx2 (WT Trx2) or a control GFP adenovirus (GFP) then incubated in normoxia (21% O2) or hypoxia (1% O2) for 72 h. Amplex Red was used to determine that WT Trx2 expression attenuated hypoxia-induced increases in extracellular H2O2 levels (Fig. 3A). However, WT Trx2 overexpression did not significantly impact hypoxia-induced increases in relative HPASMC cell number (Fig. 3B). Adenoviral infection alone had no impact on H2O2 levels or cell number (data not shown).
Fig. 3:

WT Trx2 overexpression in HPASMCs prevents hypoxia-induced ROS production but not proliferation. A; extracellular H2O2 levels in HPASMCs infected with either GFP control or wild-type Trx2 (WT Trx2) (n ≥ 4). B: cellular proliferation measured in HPASMCs infected with either GFP control or WT Trx2 (n = 3). ROS, reactive oxygen species; WT, wild type; GFP, green fluorescent protein. Error bars = SE. *P < 0.05, compared with Normoxia of same group; ƒP < 0.05, compared with Control Hypoxia).
Characterization of Trx2 in TghTrx2 Model
Since Trx2 overexpression reduced hypoxic increases in H2O2 but failed to attenuate hypoxic proliferation, we questioned if Trx2 overexpression would attenuate hypoxia-induced PH in vivo. A transgenic mouse model overexpressing human Trx2 was used to further examine the role of Trx2 in PH in vivo. This model (17, 40) expresses V5-tagged human Trx2 under the CAG promoter (Fig. 4A). Endogenous mouse and human Trx2 mRNA and protein levels were examined to confirm hTrx2 overexpression. Equivalent levels of mouse Trx2 mRNA were detected in the lungs of littermate control (Lit Ctrl) and TghTrx2 animals (Fig. 4B), while human Trx2 mRNA was only detected in the lungs of TghTrx2 mice (Fig. 4C). Human Trx2 protein was detected in TghTrx2 animals using the V5 epitope (Fig. 4D). Trx2 protein levels were detected using a Trx2 antibody (45) in Lit Ctrl and TghTrx2 animals. Endogenous Trx2 (mTrx2) migrated at 12 kDa and V5-tagged human Trx2 (hTrx2) migrated at ~15 kDa on the SDS-PAGE gel (Fig. 4E).
Fig. 4:

Human Trx2 overexpression in the TghTrx2 mouse model. A: schematic depicting Trx2 overexpression construct used to generate TghTrx2 mouse model. B: mouse Trx2 mRNA levels in Lit Ctrl or TghTrx2 (n = 6). Human Trx2 mRNA (n = 3; C) and protein (detected by V5 epitope) (n = 3; D) levels in Lit Ctrl and TghTrx2 animals. E: representative images of mouse (bottom) and human (top) Trx2 protein levels in Lit Ctrl and TghTrx2 animals. Lit Ctrl, littermate control. Error bars = SE.
Trx2 Overexpression Exacerbates Hypoxia-Induced PH In Vivo
Littermate control and TghTrx2 mice were exposed to 3 wk of normoxia (21% O2) or hypoxia (10% O2). As expected, chronic hypoxia exposure increased RVSP and RVH (measured by Fulton Index) (1, 27). Rather than attenuate these manifestations of PH, Trx2 overexpression exacerbated hypoxic increases in RVSP and RVH (Fig. 5, A and B). As expected, hypoxia also increased pulmonary vascular wall thickness, but Trx2 overexpression did not exacerbate vascular remodeling to a significant degree (Fig. 5, C and D). Since increases in H2O2 levels are observed in various models of PH (13, 27), we examined whether Trx2 overexpression modified H2O2 levels. Amplex Red was used to assess extracellular H2O2 levels in lung tissue from littermate control and TghTrx2 mice exposed to normoxia or hypoxia for 3 wk. Trx2 overexpression increased lung H2O2 levels in normoxia-exposed animals and exacerbated hypoxia-induced increases in lung H2O2 levels (Fig. 5A). Hypoxia-induced increases in lung Nox4 protein levels were also not altered by Trx2 overexpression (Fig. 6B). Similarly, Trx2 overexpression did not affect lung levels of Nox2 or Nox1 in mice (Fig. 6, C and D). There were no differences in body weight between Lit Ctrl or TghTrx2 mice and hypoxia-exposed Lit Ctrl (data not shown), and TghTrx2 mice showed similar increases in hematocrit, confirming hypoxia exposure (data not shown).
Fig. 5:

Overexpression of Trx2 does not ameliorate physiological markers of hypoxia-induced PH. RVSP (n ≥ 12; A), Fulton Index (n = 14; B), pulmonary vessel muscularization measured by smooth muscle actin staining of vessels smaller than 100 µm in Lit Ctrl and TghTrx2 mice exposed to Hypoxia (10% O2) or Normoxia (21% O2) for 3 wk (n = 3; C and D). RVSP, right ventricular systolic pressures; Fulton index, right ventricle hypertrophy. Error bars = SE. *P < 0.05, compared with Normoxia groups; #P < 0.05, compared with Lit Ctrl Hypoxia.
Fig. 6:

Trx2 Overexpression does not prevent hypoxia-induced increase in Nox4. H2O2 production (n ≥ 5; A), Nox 4 protein levels (n = 8; B), Nox 1 (n = 8; C), or Nox2 (n = 8; D) protein levels in Lit Ctrl and TghTrx2 mice exposed to Hypoxia (10% O2) or Normoxia (21% O2) for 3 wk. Nox4, NAD(P)H oxidase 4; Nox1, NAD(P)H oxidase 1; Nox2, NAD(P)H oxidase 2. Error bars = SE. *P < 0.05, compared with Normoxia groups; #P < 0.05, compared with Lit Ctrl Hypoxia.
Overexpression of hTrx2 Does Not Change Trx2 Redox Potential After Hypoxia Exposure
We used redox Western blot techniques (11) to compare reduced Trx2 (active) to oxidized Trx2 (inactive) protein levels in pulmonary tissue. The Nernst equation was used to compare the redox potential of Trx2 in littermate controls and TghTrx2 mice after exposure to 3 wk of hypoxia or normoxia. Hypoxia increased Trx2 redox potential in Lit Ctrl mice (−335.3 ± 1.6 to −312.3 ± 4.4), and Trx2 overexpression failed to alter these hypoxic reductions (−346.7 ± 12.7 to −316.1 ± 4.9) (Fig. 7).
Fig. 7:
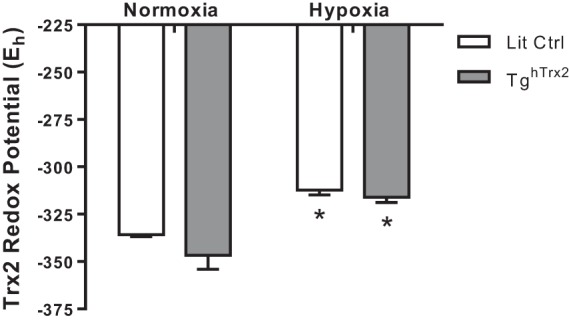
Hypoxia decreases Trx2 redox potential in vivo. Trx2 Redox state was calculated using the Nernst equation in conjunction with densitometry analysis of native gels. Error bars = SE. *P < 0.05, compared with Normoxia of same group.
Mutant Trx2 Overexpression Increases HPASMC H2O2 Levels and Proliferation
To further examine the role of Trx2 oxidation in pulmonary vascular cell proliferation, the effects of overexpression of a C93S mutant (corresponding to C36 in the mitochondrial processed form) (45) of Trx2 were examined. C93S Trx2 is unable to reduce the disulfide of target proteins and thereby mimics the inactive oxidized form of Trx2. In HeLa cells, overexpression of wild-type Trx2 (WT Trx2), but not the C93S Trx2 mutant, led to decreased TNF-α-induced oxidative stress and apoptosis (15). GFP or C93S Trx2 was overexpressed in HPASMCs, and H2O2 and proliferation responses were assessed in normoxic and hypoxic conditions. C93S Trx2 did not prevent hypoxia-induced increases in ROS production (Fig. 8A). Expression of C93S Trx2 significantly increased relative cell number under normoxic conditions and exacerbated hypoxia-induced proliferation (Fig. 8B) without affecting cell death (data not shown).
Fig. 8:

Trx2 activity is important for preventing hypoxia-induced ROS production and proliferation in HPASMC. A: extracellular H2O2 levels in HPASMC expressing either GFP control or C93S Trx2 (n ≥ 7). B: cellular proliferation measured in HPASMC expressing either GFP control or C93S Trx2 (n = 3). Error bars = SE. *P < 0.05, compared with normoxia of same group; #P < 0.05, compared with GFP Normoxia; ƒP < 0.05, compared with GFP Hypoxia.
DISCUSSION
To our knowledge, this study provides the first analysis of the effects of hypoxia on Trx2 in pulmonary vascular wall cells. Our results demonstrate that hypoxia exposure decreases Trx2 expression in HPAECs and HPASMCs in vitro and in mouse lung in vivo. Trx2 levels were dynamically regulated by both the level and duration of hypoxia. Overexpression of Trx is protective in many models of oxidative stress (19, 21, 25, 35), and we recently reported that mitochondria-targeted catalase overexpression attenuated hypoxia-induced PH in the mouse (1). Therefore, we initially hypothesized that overexpression of the mitochondrial Trx isoform Trx2 would attenuate hypoxia-induced PH by lowering mitochondrial H2O2 levels. However, while Trx2 overexpression in vitro attenuated hypoxia-induced increases in HPASMC H2O2 levels, it failed to attenuate hypoxia-induced HPASMC proliferation. Furthermore, Trx2 overexpression in vivo increased H2O2 levels in the lungs of normoxia-exposed mice and exacerbated hypoxia-induced increases in lung H2O2 levels. These increased lung levels of H2O2 were associated with exacerbation of hypoxia-induced increases in RVSP, RVH, and pulmonary vascular remodeling. These observations were explained by evidence that hypoxia oxidized and thereby increased the redox state of Trx2 in vivo, a derangement that was not reversed by Trx2 overexpression. The current study also provides novel evidence that Trx2 expression is dynamically regulated by both the duration and severity of hypoxia exposure.
To further confirm the impact of Trx2 oxidation on PASMC biology, in vitro overexpression of wild-type Trx2 was compared with a dominant negative Trx2 mutant. Wild-type Trx2 attenuated H2O2 production in HPASMC exposed to hypoxia but did not attenuate hypoxia-induced proliferation. C93S Trx2, an active site mutant that mimics inactive oxidized Trx2, increased proliferation in normoxia, exacerbated proliferation in hypoxia, and exacerbated hypoxia-induced H2O2 levels. These findings indicate that Trx2 oxidation itself may significantly stimulate proliferation. To our knowledge, these findings are the first to provide evidence that oxidative modification of Trx2 may contribute to increased H2O2 production and the proliferative PASMC phenotype that contributes to PH pathogenesis. These findings are also consistent with previous findings in which superoxide dismutase 2 (SOD2) overexpression exacerbated hypoxia-induced increases in H2O2, proliferation and vascular muscularization, and RVSP (1).
While our current and previous findings (1) indicate that H2O2 plays a critical role in pulmonary vascular wall cell proliferation and PH, ROS other than H2O2, such as superoxide, likely also contribute to hypoxia-induced cell proliferation. These results suggest that Trx2 oxidation state plays an important role in maintaining vascular function and preventing vascular diseases.
The mechanisms by which hypoxia-induced Trx2 oxidation contributes to PASMC proliferation remain to be defined. We speculate that the failure of Trx2 overexpression to attenuate hypoxia-induced pulmonary vascular cell proliferation and PH may be due in part to previously reported Trx2 interactions with apoptosis signal-regulating kinase-1 (ASK-1). Trx2 binds to ASK-1 in the mitochondria, preventing ASK-1 translocation into the nucleus and subsequent stimulation of apoptotic pathways (18, 46). As a result, Trx2 overexpression could increase cell proliferation through inhibition of ASK-1 and proapoptotic signaling. Furthermore, our data are consistent with a previous report that indicates that decreasing Prx3, a Trx2 target, is required for hypoxia-inducible factor-1α (HIF-1α)-dependent proliferation in clear cell renal cell carcinoma (43). Reductions in Trx2 would be expected to reduce the availability of active Prx3, thereby promoting proliferation, potentially in a HIF-1α-dependent manner.
Trx2 also plays a key role in regulating mitochondrial redox state, the electrochemical gradient, and ATP generation (24) through the reduction of oxidized proteins. Evidence from our laboratory indicates that hypoxia-induced, mitochondria-derived H2O2 activates a signaling cascade that promotes proliferation (1). Additional evidence suggests that ROS generated from the mitochondria in vascular endothelial cells contribute to pulmonary disease (24). In vitro knockdown of Trx2 causes accumulation of ROS (24), and Trx2 protects against ROS-induced endothelial cell dysfunction (8). Similar to the results we have shown in vitro, Trx2 mRNA levels are decreased in the hearts of hypoxia-exposed rats (47). In contrast to the current study, Trx2 overexpression in mice attenuates angiotensin II-induced hypertension by reducing angiotensin II-induced ROS increase (40). Collectively these reports suggest that Trx2 may differentially regulate diverse ROS-mediated pathologies.
Hypoxia exposure has previously been shown to enhance ROS and Nox expression (28), impair antioxidant capacity in HPAECs, and promote Nox4 expression (13, 27). The current results confirm that hypoxia exposure elevated lung Nox4 and H2O2 levels in vivo and further demonstrate that increases in Trx2 levels are not effective at mitigating hypoxia-induced increases in Nox4 or Nox2 levels. We previously reported that hypoxia increased PASMC Nox4 expression, H2O2 production, and proliferation and that these hypoxic derangements were blocked by PEG-catalase treatment indicating a feed forward effect of H2O2 on Nox4 expression (1, 2). Taken together, these observations suggest that Trx2 overexpression failed to attenuate hypoxic increases in Nox4 expression because it failed to attenuate hypoxic increases in lung H2O2 or to restore hypoxic reductions in redox potential. Overexpression of Trx2R, which may maintain active Trx2 or a nonoxidizable form of Trx2, may be more effective in the targeted attenuation of mitochondrial H2O2 and therefore PH pathogenesis.
There are several limitations to the current study. 1) Our findings also demonstrate that the duration and severity of hypoxia exposure play an important role in Trx2 expression and activity. Because we have examined only a limited number of combinations of hypoxia duration and concentration, we cannot exclude the possibility that Trx2 overexpression might have greater impact in models with hypoxic conditions not examined in the present study. 2) Previous work in our laboratory established a vital role for mitochondrial H2O2 in PH development (1), and since H2O2 is freely diffusible, elevated H2O2 levels may exert detrimental effects without directly interacting with Trx2, Prx3, or other mitochondrial antioxidant proteins. Because Trx2 function is part of an integrated series of antioxidant molecules, we speculate that overexpression of additional components in the Trx2 pathway may be required to maintain optimal and sustained Trx2 activity. For example, in addition to peroxiredoxin and thioredoxin reductase, the glutathione and glutaredoxin (Grx) systems (14, 44, 45) can regulate Trx2. Studies designed to examine the interrelationships among these antioxidant systems during hypoxic conditions may identify additional strategies by which Trx2 can be therapeutically targeted. Finally, 3) hypoxia activates proliferative signaling pathways through redox- and H2O2-independent mechanisms that are insensitive to Trx2 expression and activity that were not examined in the current study.
In summary, the current report provides novel evidence that sustained hypoxia decreases Trx2 expression and activity in the lung. Hypoxia regulates Trx2 in a dynamic fashion dependent on both the severity and duration of hypoxia. Overexpression of Trx2 fails to attenuate hypoxia-induced PASMC proliferation in vitro or hypoxia-induced increases in lung Nox4 expression and PH in vivo. These data indicate that the expression and redox state of Trx2 play an important role in the cellular response to hypoxia and suggest that future studies examining Trx2 should also consider broader antioxidant systems regulating the oxidation state of Trx2. This study highlights the complex pathobiology involved in cell and organ responses to hypoxia and the challenges inherent in simultaneously targeting hypoxia-induced redox and nonredox signaling in compartmentalized pathways.
GRANTS
This work was supported by National Research Service Award 1F31-HL-114386-01A1 (to S. E. Adesina), National Heart, Lung, and Blood Institute (NHLBI) Grant HL-102167 (CMH/RLS), Graduate Training in the Pharmacological Sciences 5T32GM008602 (to S. E. Adesina), National Institute of Environmental Health Sciences Graduate and Postdoctoral Training in Toxicology Public Health Service Grant 5T32ES12870-7 (to S. E. Adesina), Postdoctoral Training Grant in Academic Pulmonary Medicine NHLBI Grant T32-HL-076118 (to B. E. Wade), and Veterans Affairs Merit Review 1I01BX001910 (to C. M. Hart).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.E.A., B.E.W., K.M.B., B.-Y.K., C.R.W., and J.M. performed experiments; S.E.A., B.E.W., K.M.B., and R.L.S. analyzed data; S.E.A., B.E.W., Y.-M.G., and C.M.H. interpreted results of experiments; S.E.A., B.E.W., K.M.B., and R.L.S. prepared figures; S.E.A. and B.E.W. drafted manuscript; S.E.A., B.E.W., K.M.B., B.-Y.K., C.R.W., Y.-M.G., C.M.H., and R.L.S. edited and revised manuscript; S.E.A., B.E.W., K.M.B., B.-Y.K., C.R.W., J.M., Y.-M.G., C.M.H., and R.L.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Samantha Yeligar for assistance during the preparation of the manuscript. The contents reported herein do not represent the views of the Department of Veterans Affairs or the United States Government.
REFERENCES
- 1.Adesina SE, Kang BY, Bijli KM, Ma J, Cheng J, Murphy TC, Michael Hart C, Sutliff RL. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic Biol Med 87: 36–47, 2015. doi: 10.1016/j.freeradbiomed.2015.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bijli KM, Kleinhenz JM, Murphy TC, Kang BY, Adesina SE, Sutliff RL, Hart CM. Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation. Free Radic Biol Med 80: 111–120, 2015. doi: 10.1016/j.freeradbiomed.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 169: 764–769, 2004. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 4.Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res 92: 683–691, 2003. doi: 10.1161/01.RES.0000063424.28903.BB. [DOI] [PubMed] [Google Scholar]
- 5.Chen B, Nelin VE, Locy ML, Jin Y, Tipple TE. Thioredoxin-1 mediates hypoxia-induced pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 305: L389–L395, 2013. doi: 10.1152/ajplung.00432.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 45: 1340–1351, 2008. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunn LL, Buckle AM, Cooke JP, Ng MK. The emerging role of the thioredoxin system in angiogenesis. Arterioscler Thromb Vasc Biol 30: 2089–2098, 2010. doi: 10.1161/ATVBAHA.110.209643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebrahimian T, Touyz RM. Thioredoxin in vascular biology: role in hypertension. Antioxid Redox Signal 10: 1127–1136, 2008. doi: 10.1089/ars.2007.1985. [DOI] [PubMed] [Google Scholar]
- 10.Fuchs B, Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Grimminger F, Seeger W, Gudermann T, Weissmann N. Redox signaling and reactive oxygen species in hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol 174: 282–291, 2010. doi: 10.1016/j.resp.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Go YM, Jones DP. Thioredoxin redox Western analysis. Curr Protoc Toxicol 17: 12, 2009. doi: 10.1002/0471140856.tx1712s4. [DOI] [PubMed] [Google Scholar]
- 12.Go YM, Park H, Koval M, Orr M, Reed M, Liang Y, Smith D, Pohl J, Jones DP. A key role for mitochondria in endothelial signaling by plasma cysteine/cystine redox potential. Free Radic Biol Med 48: 275–283, 2010. doi: 10.1016/j.freeradbiomed.2009.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol 47: 718–726, 2012. doi: 10.1165/rcmb.2011-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanschmann EM, Lönn ME, Schütte LD, Funke M, Godoy JR, Eitner S, Hudemann C, Lillig CH. Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J Biol Chem 285: 40699–40705, 2010. doi: 10.1074/jbc.M110.185827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen JM, Zhang H, Jones DP. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-α-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol Sci 91: 643–650, 2006. doi: 10.1093/toxsci/kfj175. [DOI] [PubMed] [Google Scholar]
- 16.Hansen K, Nedergaard OA. Methodologic aspects of acetylcholine-evoked relaxation of rabbit aorta. J Pharmacol Toxicol Methods 41: 153–159, 1999. doi: 10.1016/S1056-8719(99)00035-0. [DOI] [PubMed] [Google Scholar]
- 17.He M, Cai J, Go YM, Johnson JM, Martin WD, Hansen JM, Jones DP. Identification of thioredoxin-2 as a regulator of the mitochondrial permeability transition. Toxicol Sci 105: 44–50, 2008. doi: 10.1093/toxsci/kfn116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, Min W. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 131: 1082–1097, 2015. doi: 10.1161/CIRCULATIONAHA.114.012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang YL, Chuang CY, Sung FC, Chen CY. Thioredoxin overexpression modulates remodeling factors in stress responses to cigarette smoke. J Toxicol Environ Health A 71: 1490–1498, 2008. doi: 10.1080/15287390802350030. [DOI] [PubMed] [Google Scholar]
- 20.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li GX, Hirabayashi Y, Yoon BI, Kawasaki Y, Tsuboi I, Kodama Y, Kurokawa Y, Yodoi J, Kanno J, Inoue T. Thioredoxin overexpression in mice, model of attenuation of oxidative stress, prevents benzene-induced hemato-lymphoid toxicity and thymic lymphoma. Exp Hematol 34: 1687–1697, 2006. doi: 10.1016/j.exphem.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Liu JQ, Folz RJ. Extracellular superoxide enhances 5-HT-induced murine pulmonary artery vasoconstriction. Am J Physiol Lung Cell Mol Physiol 287: L111–L118, 2004. doi: 10.1152/ajplung.00006.2004. [DOI] [PubMed] [Google Scholar]
- 23.Lu X, Murphy TC, Nanes MS, Hart CM. PPARgamma regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB. Am J Physiol Lung Cell Mol Physiol 299: L559–L566, 2010. doi: 10.1152/ajplung.00090.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Min W, Xu LK, Zhou HJ, Huang Q, Zhang H, He Y, Zhe X, Luo Y. Thioredoxin and redox signaling in vasculature-studies using Trx2 endothelium-specific transgenic mice. Methods Enzymol 474: 315–324, 2010. doi: 10.1016/S0076-6879(10)74019-2. [DOI] [PubMed] [Google Scholar]
- 25.Mitsui A, Hamuro J, Nakamura H, Kondo N, Hirabayashi Y, Ishizaki-Koizumi S, Hirakawa T, Inoue T, Yodoi J. Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxid Redox Signal 4: 693–696, 2002. doi: 10.1089/15230860260220201. [DOI] [PubMed] [Google Scholar]
- 26.Nakatsu K, Vandenberghe M, Kobus S, Kawamoto J, Brien JF, Marks GS. Endothelium-dependent relaxation of rabbit aorta by acetylcholine requires ethylenediaminetetraacetic acid. Can J Physiol Pharmacol 64: 1050–1052, 1986. doi: 10.1139/y86-179. [DOI] [PubMed] [Google Scholar]
- 27.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol 42: 482–490, 2010. doi: 10.1165/rcmb.2008-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, Fan TH, Mitchell PO, Sutliff RL, Hart CM. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol 40: 601–609, 2009. doi: 10.1165/2008-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care 31, Suppl 2: S170–S180, 2008. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 30.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol 31: 1368–1376, 2011. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 31.Roede JR, Hansen JM, Go YM, Jones DP. Maneb and paraquat-mediated neurotoxicity: involvement of peroxiredoxin/thioredoxin system. Toxicol Sci 121: 368–375, 2011. doi: 10.1093/toxsci/kfr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sedeek M, Hébert RL, Kennedy CR, Burns KD, Touyz RM. Molecular mechanisms of hypertension: role of Nox family NADPH oxidases. Curr Opin Nephrol Hypertens 18: 122–127, 2009. doi: 10.1097/MNH.0b013e32832923c3. [DOI] [PubMed] [Google Scholar]
- 33.Shimoda LA, Undem C. Interactions between calcium and reactive oxygen species in pulmonary arterial smooth muscle responses to hypoxia. Resp Physiol Neurobiol 174: 221–229, 2010. doi: 10.1016/j.resp.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sutliff RL, Kang BY, Hart CM. PPARgamma as a potential therapeutic target in pulmonary hypertension. Ther Adv Respir Dis 4: 143–160, 2010. doi: 10.1177/1753465809369619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Umekawa T, Sugiyama T, Kihira T, Murabayashi N, Zhang L, Nagao K, Kamimoto Y, Ma N, Yodoi J, Sagawa N. Overexpression of thioredoxin-1 reduces oxidative stress in the placenta of transgenic mice and promotes fetal growth via glucose metabolism. Endocrinology 149: 3980–3988, 2008. doi: 10.1210/en.2007-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watson WH, Yang X, Choi YE, Jones DP, Kehrer JP. Thioredoxin and its role in toxicology. Toxicol Sci 78: 3–14, 2004. doi: 10.1093/toxsci/kfh050. [DOI] [PubMed] [Google Scholar]
- 37.Waypa GB, Schumacker PT. Hypoxia-induced changes in pulmonary and systemic vascular resistance: where is the O2 sensor? Respir Physiol Neurobiol 174: 201–211, 2010. doi: 10.1016/j.resp.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wedgwood S, Lakshminrusimha S, Czech L, Schumacker PT, Steinhorn RH. Increased p22(phox)/Nox4 expression is involved in remodeling through hydrogen peroxide signaling in experimental persistent pulmonary hypertension of the newborn. Antioxid Redox Signal 18: 1765–1776, 2013. doi: 10.1089/ars.2012.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weir EK, Archer SL. The role of redox changes in oxygen sensing. Respir Physiol Neurobiol 174: 182–191, 2010. doi: 10.1016/j.resp.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, Bauersachs J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 54: 338–344, 2009. doi: 10.1161/HYPERTENSIONAHA.108.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams CR, Lu X, Sutliff RL, Hart CM. Rosiglitazone attenuates NF-κB-mediated Nox4 upregulation in hyperglycemia-activated endothelial cells. Am J Physiol Cell Physiol 303: C213–C223, 2012. doi: 10.1152/ajpcell.00227.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wood ZA, Schröder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci 28: 32–40, 2003. doi: 10.1016/S0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 43.Xi H, Gao YH, Han DY, Li QY, Feng LJ, Zhang W, Ji G, Xiao JC, Zhang HZ, Wei Q. Hypoxia inducible factor-1α suppresses Peroxiredoxin 3 expression to promote proliferation of CCRCC cells. FEBS Lett 588: 3390–3394, 2014. doi: 10.1016/j.febslet.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 44.Zhang H, Du Y, Zhang X, Lu J, Holmgren A. Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid Redox Signal 21: 669–681, 2014. doi: 10.1089/ars.2013.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Go YM, Jones DP. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch Biochem Biophys 465: 119–126, 2007. doi: 10.1016/j.abb.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res 94: 1483–1491, 2004. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]
- 47.Zhou W, Li S, Wan N, Zhang Z, Guo R, Chen B. Effects of various degrees of oxidative stress induced by intermittent hypoxia in rat myocardial tissues. Respirology 17: 821–829, 2012. doi: 10.1111/j.1440-1843.2012.02157.x. [DOI] [PubMed] [Google Scholar]