

Abstract
Key points
Chronic perinatal nicotine exposure causes abnormal auditory brainstem responses and auditory processing deficits in children and animal models.
The effect of perinatal nicotine exposure on synaptic maturation in the auditory brainstem was investigated in granule cells in the ventral nucleus of the lateral lemniscus, which receive a single calyx‐like input from the cochlear nucleus.
Perinatal nicotine exposure caused a massive reduction in the amplitude of the excitatory input current.
This caused a profound decrease in the number and temporal precision of spikes in these neurons.
Perinatal nicotine exposure delayed the developmental downregulation of functional nicotinic acetylcholine receptors on these neurons.
Abstract
Maternal smoking causes chronic nicotine exposure during early development and results in auditory processing deficits including delayed speech development and learning difficulties. Using a mouse model of chronic, perinatal nicotine exposure we explored to what extent synaptic inputs to granule cells in the ventral nucleus of the lateral lemniscus are affected by developmental nicotine treatment. These neurons receive one large calyx‐like input from octopus cells in the cochlear nucleus and play a role in sound pattern analysis, including speech sounds. In addition, they exhibit high levels of α7 nicotinic acetylcholine receptors, especially during early development. Our whole‐cell patch‐clamp experiments show that perinatal nicotine exposure causes a profound reduction in synaptic input amplitude. In contrast, the number of inputs innervating each neuron and synaptic release properties of this calyx‐like synapse remained unaltered. Spike number and spiking precision in response to synaptic stimulation were greatly diminished, especially for later stimuli during a stimulus train. Moreover, chronic nicotine exposure delayed the developmental downregulation of functional nicotinic acetylcholine receptors on these neurons, indicating a direct action of nicotine in this brain area. This presumably direct effect of perinatal nicotine exposure on synaptic maturation in the auditory brainstem might be one of the underlying causes for auditory processing difficulties in children of heavy smoking mothers.
Keywords: auditory brainstem, excitatory synaptic transmission, nicotinic receptor
Key points
Chronic perinatal nicotine exposure causes abnormal auditory brainstem responses and auditory processing deficits in children and animal models.
The effect of perinatal nicotine exposure on synaptic maturation in the auditory brainstem was investigated in granule cells in the ventral nucleus of the lateral lemniscus, which receive a single calyx‐like input from the cochlear nucleus.
Perinatal nicotine exposure caused a massive reduction in the amplitude of the excitatory input current.
This caused a profound decrease in the number and temporal precision of spikes in these neurons.
Perinatal nicotine exposure delayed the developmental downregulation of functional nicotinic acetylcholine receptors on these neurons.
Abbreviations
- ACh
acetylcholine
- ACSF
artificial cerebral spinal fluid
- AP
action potential
- EPSC
excitatory postsynaptic current
- MEC
mecamylamine
- MLA
methyllycaconitine
- nAChR
nicotinic acetylcholine receptor
- VNLL
ventral nucleus of the lateral lemniscus
Introduction
Maternal smoking during pregnancy causes auditory processing deficits in babies and children throughout adolescence resulting in difficulties in speech perception and learning (McCartney et al. 1994). Perinatally nicotine exposed newborns and babies exhibit decreased orientation behaviour in response to auditory stimuli and distorted auditory brainstem responses (Key et al. 2007; Kable et al. 2009; Peck et al. 2010). Similarly, auditory processing deficits have been observed in neonatally nicotine exposed rats (Sun et al. 2008). Although millions of children are affected by nicotine exposure during embryonic development, the question how brain circuits in the ascending auditory pathway are altered by it has received little attention.
Nicotinic acetylcholine receptors (nAChRs), the main binding site of nicotine, are present throughout the ascending auditory pathway. Functional nAChRs mostly of the α9 and α10 subtype are expressed around birth on inner and outer hair cells and on the descending olivocochlear fibres to the hair cells of the inner ear (Roux et al. 2011; Simmons & Morley, 2011), where they promote synaptic maturation and stabilization during early development (Vetter et al. 1999; Murthy et al. 2009). Nicotinic acetylcholine receptors also exist throughout the auditory brainstem, midbrain and cortex. Particularly during early pre‐ and postnatal development the highly Ca2+ permeable α7 nAChR subtype is expressed throughout the ascending auditory pathway (Happe & Morley, 2004). This receptor subtype has been implicated in the maturation of neural circuits during early brain development (Liu et al. 2007). During this period postsynaptic α7 nAChR activity leads to glutamate receptor accumulation and stabilization in the membrane (Lozada et al. 2012; Halff et al. 2014) thereby contributing to map formation (Morishita et al. 2010).
The ventral nucleus of the lateral lemniscus (VNLL), an auditory brainstem nucleus which faithfully encodes the temporal pattern of sounds, contains one of the highest expression levels of α7 nAChRs during early postnatal development (Happe & Morley, 2004). The globular cells in the VNLL, the most abundant cell type in the ventral part of the VNLL, receive a calyx‐like single and powerful glutamatergic input (Berger et al. 2014; Caspari et al. 2015) that originates from octopus cells in the cochlear nucleus (Glendenning et al. 1981; Schofield & Cant, 1997). Octopus cells initiate action potentials with extreme temporal fidelity (Golding et al. 1995; Oertel & Wickesberg, 2002). Hence, these VNLL neurons provide a strong and well‐timed inhibition to neurons in the auditory midbrain (Saint Marie & Baker, 1990; Saint Marie et al. 1997; Riquelme et al. 2001) and have been suggested to be important for sound pattern analysis including speech perception (Adams, 1997; Oertel & Wickesberg, 2002; Spencer et al. 2015). These neurons are thus likely to contribute to the observed temporal processing deficits in response to chronic developmental nicotine* exposure.
The present study explores the effect of perinatal nicotine exposure on synaptic maturation of the calyx‐like auditory brainstem synapse in globular cells of the VNLL. These neurons developmentally express high levels of nAChRs (Happe & Morley, 2004), which are likely to regulate the maturation of glutamatergic inputs. We also addressed the question to what extent nicotine exposure alters the input–output relationship of these neurons. To obtain a more complete view of the physiological mechanisms underlying these changes, we analysed the functional distribution of nAChRs and characterized developmental changes in nAChR function induced by nicotine exposure in these neurons.
Methods
Ethical approval
All experiments were performed in accordance with German animal welfare legislation and approved by the Landesamt für Gesundheit und Soziales (LAGESO, Berlin, Germany). The authors understand the ethical principles under which The Journal of Physiology operates and the experiments conform to the principles and regulations outlined in The Journal (Grundy, 2015).
Animals and chronic nicotine treatment
Experiments were performed on C57/Bl6 mice bred in our departmental animal facility. Mice were exposed to 100 μg ml−1 nicotine (free base in 2% saccharin) via the drinking water for 3 weeks prior to mating, during pregnancy, and until weaning. Control groups included mice receiving 2% saccharin in the drinking water or plain drinking water. Drinking solutions were changed twice a week. All animals had ad libitum access to standard laboratory food pellets and drinking water at all times. The nicotine dose was chosen based on other studies showing that oral doses between 10 μg ml−1 and 200 μg ml−1 have effects on sensory processing and behaviour in rodents (Sparks & Pauly, 1999; Pauly et al. 2004; Huang et al. 2008; Horst et al. 2012; Zhu et al. 2012). However, perinatal nicotine exposure to similar doses had no or only little effect on hearing thresholds, pup survival and body weight (Liang et al. 2006; Sun et al. 2008; Zhu et al. 2012).
In the experiments with nicotine exposure, we used no more than two offspring from any given litter to minimize the contribution of litter effects to the data. We used 3–5 litters and 3–7 mice from each experimental group to achieve an n value of 9–21 neurons for each type of analysis. For the analysis of short‐term depression, only 2–4 mice from 2 litters were used for each condition to achieve an n value of 5 to 6.
Slice preparation
All agents were purchased from Sigma‐Aldrich (Germany) and Biotrend (Germany) unless otherwisew indicated.
Slices were obtained from male and female pups of nicotine‐treated (nic), saccharin‐treated (sac) and untreated (ctr) C57/Bl6 dams. Patch‐clamp recordings were performed from neurons of pups at postnatal day 8/9 (P8), 14/15 (P14) and 22/23 (P22). Animals were decapitated under isoflurane inhalation anaesthesia (5%). Brains were removed in ice‐cold oxygenated (95% O2–5% CO2) sucrose replacement solution (in mm: 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 6 MgCl2, 25 glucose and 200 sucrose, pH 7.4). Transverse brainstem slices (180 μm) were cut with a vibratome (VT1200S; Leica, Germany), incubated at 32°C for 15 min in oxygenated artificial cerebrospinal fluid (ACSF) containing (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2 and 25 glucose, and then maintained at room temperature. For recordings, slices were transferred to a recording chamber, which was perfused continuously with oxygenated ACSF at 32°C, and visualized with an upright microscope (Axioscope, Zeiss, Germany) using infrared‐differential interference contrast optics.
Electrophysiology
Current and voltage clamp recordings were made from visually identified VNLL neurons using a Multiclamp 700A amplifier (Axon Instruments, USA). All experiments were performed at a near‐physiological temperature of 32°C. Patch pipettes were pulled from borosilicate glass capillaries (BioMedical Instruments, Germany) on a DMZ Universal Puller (Zeitz Instruments, Germany). When filled with electrode solution, patch pipettes had a resistance of 2–4 MΩ.
For current clamp recordings the pipette solution contained (in mm): 125 potassium gluconate, 5 KCl, 10 Hepes, 1 EGTA, 2 Na2ATP, 2 MgATP, 0.3 Na2GTP and 10 sodium phosphocreatine, pH 7.25 with KOH. To record synaptic currents patch electrodes were filled with (in mm): 99 CsMeSO4, 41 CsCl, 10 Hepes, 10 EGTA, 2 Na2ATP, 2 MgATP, 0.3 Na2GTP, 5 TEA‐Cl and 1 CaCl2, and 5 QX314, pH 7.25 with CsOH.
During current clamp experiments, the bridge balance was adjusted to compensate for artifacts arising from electrode resistance. During voltage clamp recordings, whole‐cell capacitance was compensated. The series resistance (<10 MΩ) was compensated to a residual of 2–2.5 MΩ and was not allowed to change more than 20% during recordings.
Neurons were selected to be ventrolateral VNLL neurons (Caspari et al. 2015) based on the following criteria: visual guided identification of area in the ventral VNLL lateral of the lemniscal fibre bundle with medium‐sized, round neurons, extremely fast rise (<0.3 ms) and decay kinetics (<0.8 ms) of excitatory postsynaptic currents (EPSCs; voltage clamp condition), and onset response pattern to depolarizing current step injections (current clamp condition) and little hyperpolarization‐activated cation current (I h) activation upon hyperpolarization.
Synaptic currents were evoked stimulating the ascending fibres with a glass electrode filled with 2 m NaCl. Stimulation electrodes were placed in the fibre bundle about 20–50 μm ventral to the recorded neuron. EPSCs were isolated by addition of 1 μm strychnine and 10 μm SR95531. EPSCs were evoked by brief biphasic pulses (100 μs, stimulation intensities 5–100 V) triggered by an analogue stimulus isolation unit (BSI‐950, Dagan Corporation, USA). Threshold for synaptic responses was usually around 20 V. For the analysis of input number, the stimulation intensity was gradually increased in increments of 5 V up to 50 V and thereafter in increments of 10 V up to 100 V. The number of input fibres was determined by visually identifying groups of similar EPSC amplitudes. Each group presumably reflects the activation of an individual input fibre (Kim & Kandler, 2003; Walcher et al. 2011). 10–90% rise time and tau decay (fitting a double‐exponential function for P8 VNLL neurons or a single‐exponential function for P14 and P22 VNLL neurons) of synaptic currents were assessed from responses evoked by minimal stimulation which was defined as the stimulation strength where a stepwise increase of 2 V resulted in the first measureable EPSC. In about 20% of the neurons recorded under both experimental conditions no EPSC could be evoked, which was most likely due to cut fibres in the brain slice. These neurons were excluded from the analysis. To estimate short‐term plastic changes of excitatory inputs to repetitive stimulations (50, 100, 200 and 300 Hz, 15 pulses, 10 repetitions), EPSC amplitudes were normalized to the first amplitude. Steady state depression was obtained by calculating the mean value of the last three normalized EPSC amplitudes.
The membrane resting potential was corrected by the junction potential of −10.5 mV. Input resistance was assessed from the peak hyperpolarization triggered by −100 pA current injection according to Ohm's law. Membrane time constants were calculated from the voltage deflection in response to −100 pA current injection using a single exponential fit.
Puff application experiments
Membrane potential changes and inward currents mediated by nAChRs were evoked by locally applying 1 mm acetylcholine (ACh) under constant perfusion of the recording chamber with ACSF. Atropine, but no other receptor blockers, was applied throughout the experiments to block muscarinic AChRs. ACh was applied with short pressure pulses (200 ms, ∼7 psi, 10 s interval) through a micropipette identical to the patch pipette using a pneumatic drug ejection system (PDES‐2L, npi electronic GmbH, Tamm, Germany). The tip of the puff pipette was positioned within 10–20 μm of the somata of the neurons. Mecamylamine (MEC) and methyllycaconitine (MLA) were used as specific nAChR antagonists. Stock solutions of all drugs were prepared in distilled water. Stock solutions were diluted to their final concentration in ACSF on the day of the experiment. The antagonists were applied by bath perfusion. The ACh‐induced depolarization of the membrane was calculated as the peak of voltage deflection from the average of three consecutive traces. ACh‐induced currents were determined as the peak of the current deflection from the average of five consecutive traces. To distinguish between the fast‐activating, MLA‐sensitive and the slow‐activating MEC‐sensitive peak current we set a time window. The initial MLA‐sensitive current amplitude was calculated at the peak between 0 and 0.2 s. The residual MEC‐sensitive current amplitude was determined at the peak of the remaining current after application of the MLA. The ACh‐induced charge was calculated as the area under the curve.
Data acquisition and statistical analysis
Stimulus generation and recordings were done with pCLAMP (Version 10.2, Axon Instruments). Both voltage and current signals were low‐pass filtered at 10 kHz with a four‐pole Bessel filter and sampled at a rate of 20 kHz. Traces were digitally filtered at 2 kHz. All electrophysiological data were analysed in IGOR Pro (Version 6.22, Wavemetrics, USA) using the custom‐written package Neuromatic (Jason Rothman, UCL, London, UK) or in Clampfit (Version 10.0, Axon Instruments).
Results are expressed as means ± standard error of the mean (SEM). Statistical significance was determined by the ANOVA test statistic followed by the appropriate post hoc test or by applying Student's paired or unpaired two‐tailed t test with significance thresholds of P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***). For non‐parametric distributions the Mann‐Whitney U test and Kruskal‐Wallis test followed by Games Howell or Dunn's multiple comparison were used.
Results
Perinatal nicotine exposure drastically reduces excitatory synaptic transmission in VNLL neurons
We investigated the effect of perinatal nicotine exposure on synaptic maturation in the VNLL, a nucleus that expresses a large number of nAChRs during development and plays a prominent role in the processing of the temporal acoustic pattern (Oertel, 1999; Spencer et al. 2015). Using whole‐cell electrophysiology in acute auditory brainstem slices we analysed excitatory postsynaptic currents (EPSCs) in neurons of the ventrolateral VNLL from untreated (ctr) and nicotine‐treated (nic) animals at postnatal day 22 (P22). We focused on globular neurons in the ventrolateral VNLL, since this group of neurons display distinct physiological properties (Caspari et al. 2015), and respond with high temporal precision to the temporal pattern of sounds (Recio‐Spinoso & Joris, 2014). To estimate the number of fibre inputs (step number) and the strength of the excitatory inputs we systematically increased the stimulation intensity (see Methods). As previously reported (Caspari et al. 2015) almost all VNLL neurons exhibited all‐or‐none EPSC responses with extremely large amplitudes irrespective of stimulation strength in both control and nicotine‐treated animals (Fig. 1 A and B). This implies that under both experimental conditions in most cases only one excitatory fibre input is contacting each postsynaptic neuron (ctr 1.20 ± 0.11, n = 15, nic 1.32 ± 0.11, n = 19, Mann‐Whitney U test, P = 0.567, Fig. 1 C). However, in nicotine‐treated animals the EPSC amplitude from one input fibre (min. EPSC) was almost 50% smaller than in control mice (min. EPSC amplitude: ctr 8.9 ± 1.2 nA, n = 17; nic 4.6 ± 0.9 nA, n = 19, Mann‐Whitney U test, P = 0.012, Fig. 1 C). Similarly, the maximal EPSC amplitude, the excitatory current evoked by all inputs, was greatly reduced in nicotine‐treated animals (max. EPSC amplitude: ctr 10.0 ± 1.2 nA, n = 15; nic 6.2 ± 1.0 nA, n = 19, Student's unpaired t test, P = 0.023, Fig. 1 C). In contrast, EPSC kinetics was only minimally affected by the nicotine treatment (tau decay: ctr 0.30 ± 0.01 ms, n = 17, nic 0.38 ± 0.05 ms, n = 19, Mann‐Whitney U test, P =0.384, Fig. 1 D, right; 10–90% rise time: ctr 0.18 ± 0.004 ms, n = 17, nic 0.21 ± 0.01 ms, n = 18; Mann‐Whitney U test, P = 0.025, Fig. 1 D, left). Saccharine‐only treatment had only minor effects on the synaptic input properties of VNLL neurons during the entire developmental period investigated (Fig. 2). We therefore compared the nicotine‐treated VNLL neurons only with the control VNLL neurons.
Figure 1. Excitatory postsynaptic currents are smaller in VNLL neurons of nicotine‐treated mice at P22.
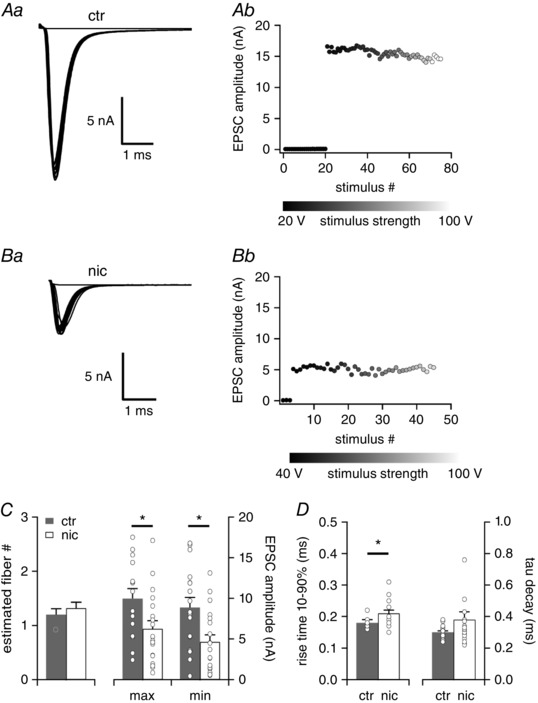
Aa and b, EPSC traces evoked by increasing stimulus strength representative for P22 neurons of untreated (ctr) mice (a) and the corresponding EPSC amplitudes as a function of stimulus number (b). Ba and b, EPSC traces evoked by increasing stimulus strength representative for P22 neurons of nicotine‐treated (nic) mice (a) and the corresponding EPSC amplitudes as a function of stimulus number (b). All traces represent the average of at least three consecutive recordings. C, the estimated number of excitatory input fibres, the total maximal EPSC amplitude and the minimal EPSC amplitude are shown for ctr (n = 15, grey bars) and nic (n = 19, white bars) VNLL neurons. * P < 0.05, Student's unpaired t test. D, rise time 10–90% and tau decay of the average EPSC evoked by minimal stimulation for ctr (n = 17, grey bars) and nic (n = 19, white bars) mice. * P < 0.05, Mann‐Whitney U test.
Figure 2. Maturation of excitatory postsynaptic inputs does not differ between VNLL neurons of saccharin‐treated and untreated mice.
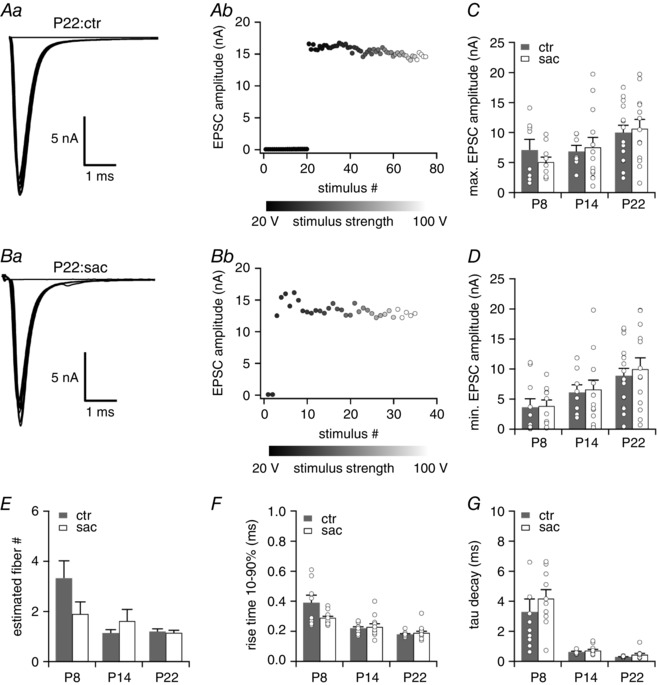
Aa and b, EPSC traces evoked by increasing stimulus strength representative for P22 neurons of untreated (ctr) mice (a) and the corresponding EPSC amplitudes as a function of stimulus number (b). Ba and b, EPSC traces evoked by increasing stimulus strength representative for P22 neurons of nicotine‐treated (nic) mice (a) and the corresponding EPSC amplitudes as a function of stimulus number (b). All traces represent the average of at least three consecutive recordings. C–E, the total maximal EPSC amplitude (C), the minimal EPSC amplitude (D) and the estimated number of excitatory input fibres (E) are shown for ctr and sac VNLL neurons of P8, P14 and P22 mice. P8, ctr n = 8, sac: n = 10; P14, ctr n = 7, sac: n = 13; P22, ctr n = 15, sac: n = 13. F and G, rise time 10–90% (F) and tau decay (G) of the average EPSC evoked by minimal stimulation for ctr and nic VNLL neurons of P8, P14 and P22 mice. P8, ctr n = 10, sac: n = 10; P14, ctr n = 8, sac: n = 12; P22, ctr n = 17, sac: n = 13.
To elucidate whether this nicotine‐induced prominent reduction in EPSC amplitude was due to a postsynaptic or a presynaptic effect we further studied short‐term plasticity of this large synapse with various stimulation frequencies and analysed spontaneous EPSCs. All excitatory inputs to VNLL neurons showed pronounced synaptic short‐term depression varying with stimulation frequency (Fig. 3 A and B). Nicotine exposure had no effect on the steady state depression for all stimulation frequencies tested (Fig. 3 C). To estimate the effect of nicotine treatment on the quantal size at this synapse we analysed spontaneous EPSCs (sEPSCs) in both control and nicotine‐treated mice. Again, nicotine treatment caused a significant reduction in sEPSC amplitude (ctr −60.5 ± 3.8 pA, n = 19, nic −49.7 ± 3.2 pA, n = 19; Mann‐Whitney U test, P = 0.034, Fig. 3 E). Nevertheless, the nicotine effect on sEPSC amplitude was much smaller compared to the nicotine‐induced reduction of evoked EPSCs. Again, sEPSC tau decay of both control and nicotine‐treated VNLL neurons remained similar (ctr 0.22 ± 0.01 ms, n = 19, nic 0.23 ± 0.01 ms, n = 19, Mann‐Whitney U test, P = 0.872, Fig. 3 F). Also, sEPSC frequency did not differ significantly between treatments (ctr 3.9 ± 0.8 Hz, n = 13, nic 2.7 ± 0.5 Hz, n = 13, Mann‐Whitney U test, P = 0.700, Fig. 3 G).
Figure 3. Increase of sEPSC frequency but no change in release probability in nicotine‐treated animals.
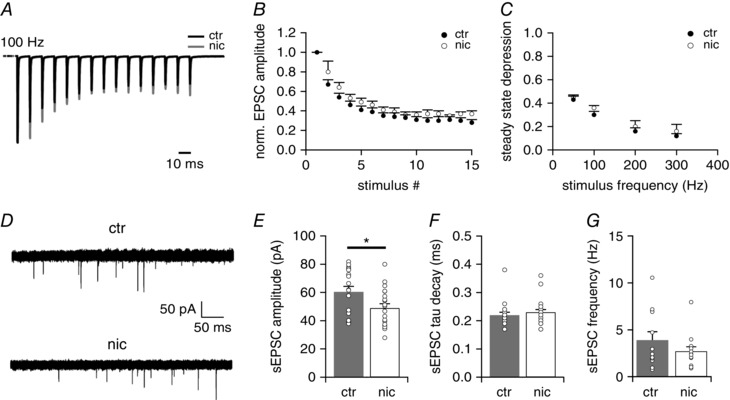
A, representative EPSC traces in response to 15 stimulations at 100 Hz for untreated (ctr; black trace) and nicotine‐treated (nic; grey trace) VNLL neurons. B, normalized EPSC amplitudes to 100 Hz stimulus frequency (ctr n = 6, nic n = 5). C, steady state depression of EPSCs in ctr and nic VNLL neurons as a function of stimulus frequency (ctr n = 6, nic n = 5). D, recording of sEPSCs from ctr (upper trace) and nic (bottom trace) VNLL neurons. E–G, sEPSC amplitude (E), sEPSC tau decay (F) and sEPSC frequency (G) of ctr and nic VNLL neurons (ctr n = 19, grey bars, nic n = 19, white bars). * P < 0.05, Mann‐Whitney U test.
Taken together, our data demonstrate that perinatal nicotine exposure leads to a profound reduction of evoked EPSC amplitudes and a smaller reduction of spontaneous EPSC amplitudes, whereas short‐term plasticity and sEPSC frequency remained unchanged. This indicates that perinatal nicotine treatment impairs synaptic transmission via a postsynaptic effect rather than a presynaptic effect.
Developmental remodelling of excitatory synaptic transmission is impaired in nicotine‐treated mice
To determine at what developmental stage nicotine exposure had its strongest effect we additionally recorded evoked EPSCs in control and nicotine‐treated pups just before (P8) and several days after hearing onset (P14). Just before hearing onset VNLL neurons of control animals received around three inputs. Within a few days after hearing onset (at P14) the number of inputs impinging on one VNLL neuron was reduced to one input in most cases (Fig. 4 A and D; Table 1). This developmental refinement was similar but slightly delayed in nicotine‐treated mice, where the pruning was only completed by P22 (Fig. 4 D). At P8 the single fibre EPSC amplitude (min. EPSC) did not statistically differ between control and nicotine‐treated VNLL neurons. Yet, at P22 this amplitude had increased more than twofold in control animals, whereas in nicotine‐treated mice this increase was much less pronounced and not statistically different to prehearing and juvenile animals (ctr, Kruskal‐Wallis test, P = 0.018; nic, Kruskal‐Wallis test, P = 0.069; Fig. 4 A and B; Table 1). Due to the reduction of inputs after hearing onset, the increase of the maximal EPSC, the current evoked by activating all incoming fibres, was less pronounced, and did not differ between control and nicotine‐treated animals (Fig. 4 C; Table 1) Also, perinatal nicotine exposure did not affect the pronounced developmental acceleration of evoked EPSCs, which also occurred during the first weeks after hearing onset (Fig. 4 E and F; Table 1).
Figure 4. Maturation of excitatory postsynaptic inputs is impaired in VNLL neurons of nicotine‐treated mice.
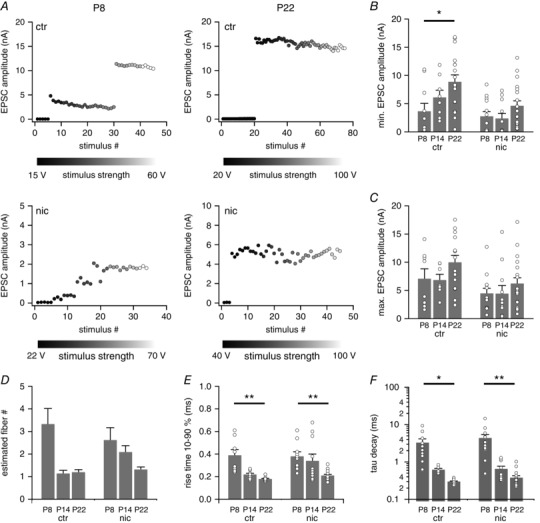
A, EPSC amplitudes as a function of stimulus number for untreated (ctr) and nicotine‐treated (nic) VNLL neurons of P8 and P22 mice. B–D, the total maximal EPSC amplitude (B), the minimal EPSC amplitude (C) and the estimated number of excitatory input fibres (D) are shown for ctr and nic VNLL neurons of P8, P14 and P22 mice. P8, ctr n = 8, nic n = 13; P14, ctr n = 7, nic n = 11; P22, ctr n = 15, nic n = 19. E and F, rise time 10–90% (E) and tau decay (F) of the average EPSC evoked by minimal stimulation for ctr and nic VNLL neurons of P8, P14 and P22 mice. P8, ctr n = 10, nic n = 13; P14, ctr n = 8, nic n = 11; P22, ctr n = 17, nic n = 19. At P8 there are no differences between ctr and nic VNLL neurons while developmental remodelling of excitatory inputs is delayed in nic mice. * P < 0.05, Students unpaired t test for parametric distributions and Mann‐Whitney U test for non‐parametric distributions.
Table 1.
Summary of excitatory postsynaptic properties of ctr and nic neurons
| Untreated (ctr) | Nicotine‐treated (nic) | |||||||
|---|---|---|---|---|---|---|---|---|
| P8 (n = 10/8) | P14 (n = 8/7) | P22 (n = 17/15) | P # | P8 (n = 13) | P14 (n = 11) | P22 (n = 19) | P # | |
| Min. EPSC amplitude (nA) | 3.6 ± 1.4 | 6.1 ± 1.3 | 8.9 ± 1.2 | 0.027 | 2.8 ± 0.8 | 2.4 ± 0.9 | 4.6 ± 0.9 | n.s. |
| Max. EPSC amplitude (nA) | 7.1 ± 1.7 | 6.8 ± 1.0 | 9.9 ± 1.2 | n.s. | 4.5 ± 0.9 | 4.5 ± 1.4 | 6.2 ± 1.0 | n.s. |
| Step number | 3.3 ± 0.7 | 1.1 ± 0.1 | 1.2 ± 0.1 | n.s. | 2.6 ± 0.6 | 2.1 ± 0.3 | 1.3 ± 0.1 | n.s. |
| Tau decay (ms) | 3.29 ± 0.87 | 0.62 ± 0.05 | 0.30 ± 0.01 | 0.004 | 4.37 ± 1.01 | 0.65 ± 0.12 | 0.44 ± 0.04 | 0.006 |
| 10–90% rise time (ms) | 0.39 ± 0.05 | 0.22 ± 0.01 | 0.18 ± 0.01 | 0.005 | 0.38 ± 0.04 | 0.34 ± 0.06 | 0.21 ± 0.01 | 0.004 |
Values are means ± SEM. The level of significance was determined using single‐factor ANOVA test followed by Scheffe's post hoc test or Kruskal‐Wallis test followed by Games Howell post hoc test. # P values for the post hoc comparison of P8 and P22 VNLL neurons from the respective mice.
Our data show that at P8, just before hearing onset, properties of excitatory inputs to VNLL neurons were rather similar in nicotine‐treated and control mice. However, the developmental refinement and the increase in total excitatory synaptic strength around and after hearing onset are affected by the nicotine treatment. This indicates that perinatal nicotine exposure disturbs the maturation of this excitatory synapse only around and after, but not before hearing onset.
Chronic nicotine exposure does not affect intrinsic membrane properties
Activation of nAChRs may also lead to changes in the ion channel composition of neurons. To test whether prenatal nicotine exposure affects the intrinsic membrane properties of neurons we applied depolarizing and hyperpolarizing current injections and recorded the voltage responses of VNLL neurons of control, saccharin‐ and nicotine‐treated animals just before (P8), a few days (P14) and several days after hearing onset (P22). Depolarization of the neurons resulted predominantly in an onset‐type firing pattern with one or only a few spikes at the onset of supra‐threshold depolarizing current injections. Hyperpolarization of the neurons resulted in a minor depolarizing voltage sag indicative of only little I h (Fig. 5 A). These results are in accordance with those of Caspari et al. (2015) showing a similar response pattern in adult‐like mice. Perinatal nicotine exposure did not change the firing pattern of the VNLL neurons at all developmental stages tested. Moreover, neither resting membrane potential, input resistance nor membrane time constants were affected by the nicotine treatment. Also, action potential half‐width, amplitude and latency were not changed (Fig. 5 B–G, Table 2). This shows that chronic activation of nAChRs in these neurons has no effect on the intrinsic membrane properties in VNLL neurons.
Figure 5. Nicotine exposure does not affect intrinsic membrane properties.
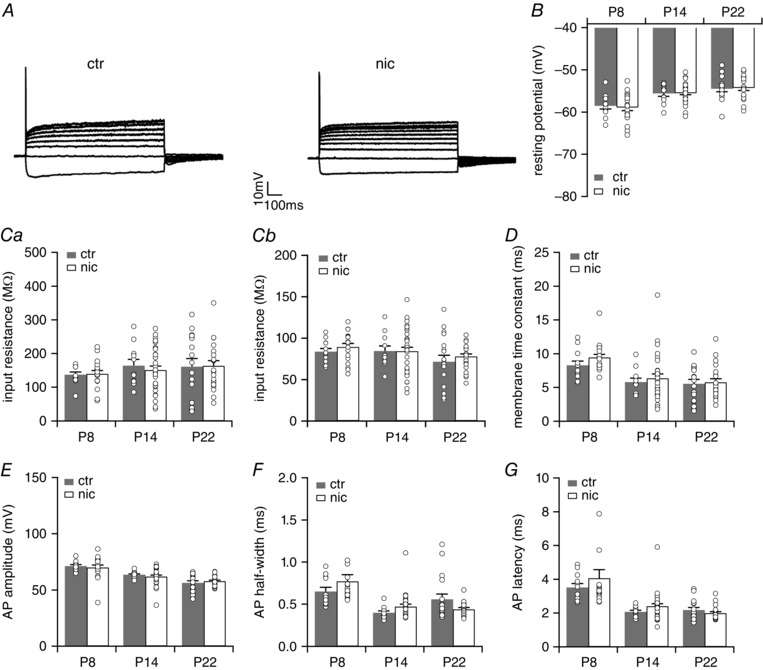
A, representative voltage traces recorded from neurons of untreated (ctr) and nicotine‐treated (nic) mice at P22 in the ventrolateral part of the VNLL. VNLL neurons displayed an onset‐type firing pattern to depolarizing current injections and a small voltage sage during hyperpolarizing current injections. B–D, resting membrane potential (B), peak input resistance, calculated at −100 pA current injection (Ca) and at +100 pA (Cb), and membrane time constant, calculated at −100 pA current injection (D), for ctr and nic VNLL neurons at P8, P14 and P22. E–G, action potential (AP) amplitude (E), AP half‐width (F) and AP latency (G) were evaluated using the first AP elicited by the lowest current injection. P8, ctr n = 12, nic n = 18; P14, ctr n = 11, nic n = 30; P22, ctr n = 17, nic n = 21.
Table 2.
Summary of membrane properties of ctr and nic neurons
| P8 | P22 | |||||
|---|---|---|---|---|---|---|
| Ctr (n = 12) | Nic (n = 18) | P # | Ctr (n = 17) | Nic (n = 21) | P # | |
| Passive membrane properties | ||||||
| Resting potential (mV) | −69.0 ± 0.8 | −69.4 ± 0.8 | n.s. | −65.0 ± 0.4 | −64.8 ± 1.2 | n.s. |
| Input resistance at −100 pA (MΩ) | 137 ± 8 | 139 ± 10 | n.s. | 161 ± 24 | 161 ± 32 | n.s. |
| Input resistance at +100 pA (MΩ) | 84 ± 4 | 89 ± 4 | n.s. | 71 ± 8 | 76 ± 3 | n.s. |
| Membrane time constant (ms) | 8.3 ± 0.6 | 9.4 ± 0.5 | n.s. | 5.5 ± 0.6 | 5.7 ± 0.5 | n.s. |
| Active membrane properties | ||||||
| Current threshold (pA) | 458 ± 23 | 511 ± 24 | n.s | 400 ± 46 | 395 ± 25 | n.s |
| Voltage threshold (mV) | −48.4 ± 0.4 | −48.2 ± 0.8 | n.s. | −45.8 ± 1.5 | −47.2 ± 0.7 | n.s. |
| AP amplitude (mV) | 71.3 ± 1.2 | 70.0 ± 2.3 | n.s. | 56.3 ± 1.8 | 57.8 ± 1.0 | n.s. |
| AP half‐width (ms) | 0.65 ± 0.05 | 0.77 ± 0.08 | n.s. | 0.56 ± 0.06 | 0.44 ± 0.02 | n.s. |
| AP latency (ms) | 3.5 ± 0.2 | 4.1 ± 0.5 | n.s. | 2.2 ± 0.2 | 2.0 ± 0.1 | n.s. |
| Cell capacitance (pF) | 25 ± 3 (6) | 28 ± 2 (15) | n.s | 15 ± 1 (17) | 14 ± 1 (20) | n.s |
Values are means ± SEM. Only the values for P8 and P22 are shown. The level of significance was determined using Student's unpaired t test or Mann‐Whitney U test. # P values for the comparison of age‐matched ctr and nic VNLL neurons.
Nicotine treatment decreases spiking fidelity and increases action potential jitter in VNLL neurons
Next, we were interested in discovering to what extent this considerable reduction in EPSC amplitude affects spiking behaviour in these neurons. A reduction in synaptic current amplitude may lead to spike failures and to a temporal imprecision in spike timing which decreases the information content of the spike response (Yang & Xu‐Friedman, 2015). To examine the reliability of AP firing at high stimulation frequencies, we performed current clamp recordings in ventrolateral VNLL neurons and stimulated the incoming fibres with regular trains at 50, 100, 200, 333 and 500 Hz (15 pulses, 10–20 repetitions). The stimulation intensity was chosen as minimal stimulation plus 15 V to reliably evoke action potentials in the presynaptic fibre. Experiments were conducted in the presence of 10 μm SR95531 and 1 μm strychnine to block inhibition. At P21–24 spikes are reliably produced at 50 Hz stimulation in both, control and nicotine treated animals. For higher frequencies (>100 Hz) the probability of spiking dropped considerably throughout the train in both control and nicotine‐treated VNLL neurons (Fig. 6 A). For frequencies of 200 Hz and 333 Hz the decrease in spike rate was more pronounced in VNLL neurons of nicotine‐treated animals compared to control animals (200 Hz: ctr 95.6 ± 2.7%, n = 9, nic 70.7 ± 11.0%, n = 9, Mann‐Whitney U test, P = 0.043; 333 Hz: ctr 72.8 ± 8.0%, n = 9, nic 42.1 ± 11.3%, n = 9, Student's unpaired t test, P = 0.042; Fig. 6 B ). To further examine the temporal precision of the APs, we analysed the distribution of the timing of the spikes (jitter) for the 100 Hz stimulation train as the number of failures was almost zero in both experimental groups under this condition. The timing of the APs was very similar for the first stimuli of the train but the jitter of the spiking response to later stimuli in the train was greatly enhanced in nicotine‐treated VNLL neurons compared to control neurons (Fig. 6 C and D). Interestingly, the jitter of EPSC peaks for 100 and 200 Hz train stimulation did not change after nicotine treatment (data not shown). Hence, there was a loss in temporal precision only of the spike signal after chronic nicotine treatment. This indicates a detrimental effect of perinatal nicotine exposure on spike timing and precision in auditory brainstem neurons renowned for their extremely low jittered spike response.
Figure 6. Reliability of AP generation in response to high frequency stimulus trains is decreased in nicotine‐treated mice.
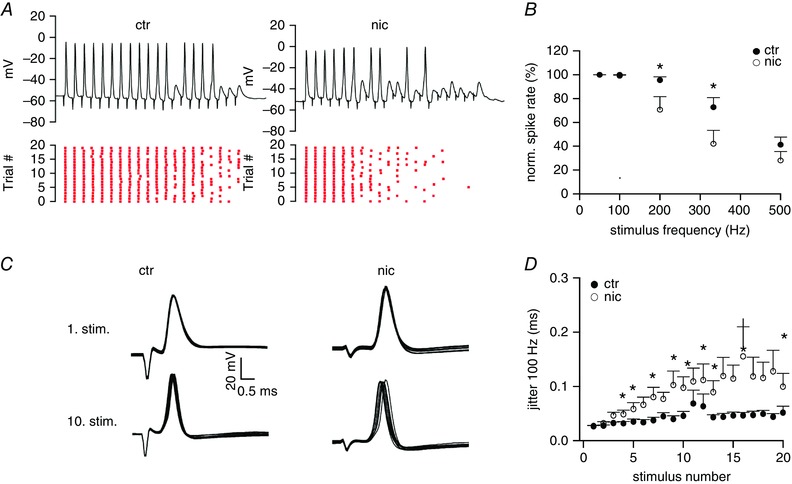
A, representative voltage traces recorded from VNLL neurons of untreated (ctr) and nicotine‐treated (nic) mice at P22 in response to 15 stimulations at 333 Hz (upper traces). The raster plots (bottom traces) show spiking over many similar trials. B, average spike rate showing a decrease in spike probability in nic mice for 200 Hz and 333 Hz stimulus trains. ctr n = 9, nic n = 9. * P < 0.05, Mann‐Whitney U test for 200 Hz and * P < 0.05, Student's unpaired t test for 333 Hz. C, presentative action potentials generated in response to the 1. and 10. stimulus of a 100 Hz train in control and nicotine treated mice. D, jitter of action potential timing agains stimulus number in control and nicotine‐treated mice for 100 Hz stimulation. Jitter is sginificantly increased in nicotine‐treated mice especially for later stimuli. ctr n = 9, nic n = 9. * P < 0.05. [Color figure can be viewed at wileyonlinelibrary.com]
Both α7‐type and non‐α7‐type nicotinic AChRs are present in VNLL neurons before hearing onset
Next, we were interested to discover whether ventrolateral VNLL neurons possess functional nicotinic acetylcholine receptors (nAChRs) during this period of synaptic remodelling. We therefore examined the effects of local application of acetylcholine (ACh) on VNLL neurons in slices obtained from mice before hearing onset at P8 while blocking muscarinic AChRs with atropine. The application of ACh (1 mm) with short pressure pulses (200 ms) to the somata of ventrolateral VNLL neurons resulted in a transient depolarization of the membrane potential ranging between 5 and 11 mV. The change in membrane potential consisted of a very fast rising depolarization followed by a slowly decaying repolarization lasting more than 1 s (Fig. 7 A). The mean voltage deflection elicited by puff application of ACh was 7.34 ± 0.84 mV (n = 7) (Fig. 7 B).
Figure 7. ACh‐induced nicotinic currents are mediated via α7‐type and non‐α7‐type nAChRs in VNLL neurons at P8.
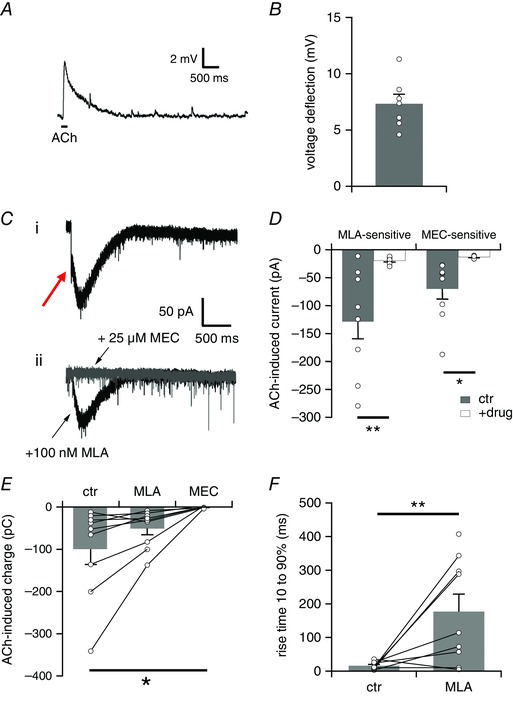
A, representative voltage trace showing the membrane potential change elicited by local application of ACh (1 mm) with a short pressure pulse (200 ms, indicated by the bar) in the presence of atropine (200 nm). The trace represents the average of three consecutive recordings. B, average voltage deflection induced by local application of ACh (1 mm, 200 ms) (n = 7). C, representative current traces recorded (i) under control conditions, and (ii) in the presence of MLA (100 nm, black trace) and subsequent additional application of MEC (25 μm, grey trace). Pressure application of ACh (1 mm, 200 ms) to the somata of VNLL neurons evoked a transient nicotinic inward current in the presence of atropine (200 nm) consisting of a fast, initial current component (indicated by the red arrow) and a slow residual current component. Holding potential was set to −60 mV. All traces represent the average of five consecutive recordings. D, left hand bars show average peak current amplitudes of the initial fast MLA‐sensitive current component in control recordings (ctr, n = 9, grey bars) and in the presence of MLA (+drug, n = 9, white bars). The initial fast current amplitude was determined as the negative peak between 0 and 200 ms of the trace. MLA completely blocked the initial current amplitude. Right hand bars show average peak current amplitudes of the slow MEC‐sensitive current component in the presence of MLA (ctr, n = 9, grey bars) and in the presence of MEC (+drug, n = 7, white bars). The slow current amplitude was determined as the negative peak after application of MLA. MEC completely blocked the slow current component. * P < 0.05, ** P < 0.01, Student's t test. E, nAChR‐mediated charge induced by local application of ACh (1 mm, 200 ms) in control recordings (ctr, n = 9), after addition of MLA (100 nm) (MLA, n = 9) and following additional application of MEC (25 μm) (MEC, n = 7). The MLA‐ and MEC‐sensitive components carried almost the same amount of charge flowing through the membrane. * P < 0.05, ANOVA followed by Scheffé’s post hoc test. F, rise time 10–90% of the ACh‐evoked current under control conditions (ctr, n = 8) and in the presence of MLA (MLA, n = 9). ** P < 0.01, Student's unpaired t test. [Color figure can be viewed at wileyonlinelibrary.com]
The majority of nAChRs in the brain are either α7 homomers or α4β2 heteromers, which differ in their kinetics and their calcium permeability and play different functional roles (Gotti et al. 2009). To characterize the subunit composition of the nAChRs whole‐cell patch clamp recordings were performed in voltage clamp mode in the presence of atropine to block mAChRs. Puff‐applying ACh onto the somata of ventrolateral VNLL neurons resulted in a small and transient inward current. Application of the α7‐type nAChR antagonist MLA, which binds to neuronal α‐bungarotoxin sites and is highly selective for the α7‐type nAChR (Alkondon et al. 1992), blocked the initial rapidly activating component of the inward current as indicated by a red arrow in Fig. 6 C (ctr −128.7 ± 30.2 pA, MLA: −19.7 ± 2.0 pA, n = 9, Student's paired t test, P = 0.007, Fig. 7 C and D). Nevertheless, a considerable current component was still left, indicating that the ACh‐induced current was also mediated via MLA‐insensitive nAChR subtypes. This residual current displayed a much slower time course. We therefore applied the general nAChR antagonist MEC (25 μm), a non‐specific blocker for all nAChR subtypes, which completely blocked the residual current in all cases (ctr −72.5 ± 22.0 pA, MEC: −13.3 ± 1.0 pA, n = 7, Student's paired t test, P = 0.040, Fig. 7 C and D). MLA almost completely eliminated the initial current but only abolished 49% of the charge flowing through the membrane (ctr −99.5 ± 36.5 pC, +MLA: −50.8 ± 15.0 pC, +MLA/MEC:−0.075 ± 0.94 pC, ANOVA, F (2,22) = 3.84, P = 0.037, Fig. 7 E). As MLA blocked the entire initial current, the rise time of the pre‐drug trace will give a good estimate for the rise time of the fast MLA‐sensitive current component. The rise time of the MLA‐sensitive fast current component differed significantly from the rise time of the slow residual current component (ctr 15.9 ± 4.0 ms, n = 8, residual: 189.3 ± 48.8 ms, n = 9, Student's unpaired t test, P = 0.007, Fig. 7 F). Taken together, our pharmacological study suggests that ventrolateral VNLL neurons possess functional α7‐ and non‐α7‐type nAChRs during this period of synaptic remodelling. Note, however, that an increase in presynaptic activity levels was never detected during our experiments, which suggests that functional nAChRs are missing on the presynaptic site at this synapse.
Developmental decline of nicotinergic currents is delayed in nicotine‐treated mice
The expression of nAChRs especially of the α7‐subtype is developmentally regulated and drops profoundly during the first 3 postnatal weeks in auditory brainstem neurons (Happe & Morley, 2004). On the other hand, there is evidence that chronic nicotine exposure upregulates nAChRs (Molinari et al. 1998; Buisson & Bertrand, 2001; Kawai & Berg, 2001). We therefore determined the developmental profile of functional nAChRs in VNLL neurons of control and nicotine‐treated animals. At P8 puff application of ACh induced a small but pronounced inward current in both control and nicotine‐treated animals (ctr −74.8 ± 15.2 pA, n = 24, nic −64.5 ± 14.5 pA, n = 16, Kruskal‐Wallis test, P > 0.99, Fig. 8 A and B). For both experimental conditions this current consisted of two current components, a rapidly activating current component and a slowly activating current component. At P14 this current was greatly diminished in control animals, whereas in nicotine‐treated animals the ACh induced current persisted (ctr −19.8 ± 3.9 pA, n = 24, nic −76.3 ± 16.4 pA, n = 18, Kruskal‐Wallis test, P = 0.0014, Fig. 8 A and B). However, at P22 the Ach‐evoked current had almost disappeared for both experimental groups (ctr −21.7 ± 8.9 pA, n = 18, nic −22.4 ± 5.4 pA, n = 19; Kruskal‐Wallis test, P > 0.99, Fig. 8 A and B). Not only the current amplitude but also the proportion of neurons with responses to ACh decreased with age. Almost ∼80% of the neurons responded to ACh at P8, whereas at P14 only ∼30% of the neurons in control animals, but still more than ∼80% of the neurons in nicotine‐treated animals displayed ACh‐induced currents. At P22 ACh‐evoked inward currents could be elicited in ∼17% of the control VNLL neurons while ∼47% of nicotine‐treated VNLL neurons still responded to ACh with a small inward current (Fig. 8 B and C).
Figure 8. Decrease in nAChR‐mediated currents is delayed in nicotine‐treated mice.
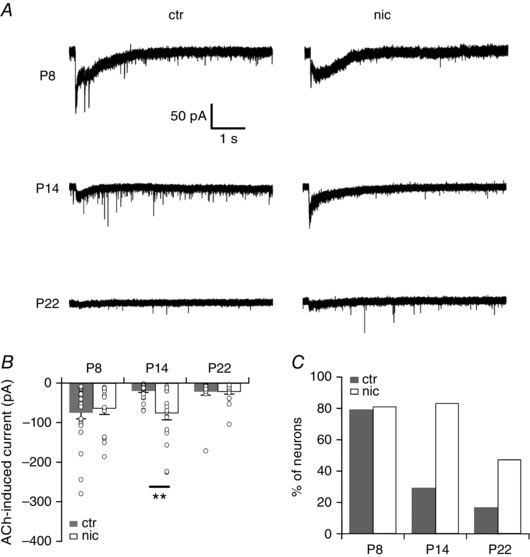
A, representative current traces recorded from VNLL neurons of untreated (ctr) and nicotine‐treated (nic) mice at P8, P14 and P22 to local application of ACh (1 mm, 200 ms) in the presence of atropine (200 nm). Holding potential was set to −60 mV. All traces represent the average of five consecutive recordings. B, initial nAChR‐mediated peak current amplitude for VNLL neurons ctr and nic mice at P8, P14 and P22. There is no decrease in current amplitude at P14 in nic VNLL neurons. P8, ctr n = 24, nic n = 16; P14, ctr n = 24, nic n = 18; P22, ctr n = 18, nic n = 19. ** P < 0.01, Kruskal‐Wallis test followed by Dunn's multiple comparison. C, number of neurons with responses to local application of ACh (1 mm, 200 ms) for the different ages. The proportion of neurons with responses to ACh is delayed in nic mice.
This parallel decline in functional nAChRs and synaptic remodelling indicates that the reduction in EPSC amplitude might be a direct effect of chronic activation of nAChRs on VNLL neurons and not an indirect affect by altering neural activity levels at the preceding neural circuits.
Discussion
Developmental chronic nicotine exposure causes deterioration of temporal auditory processing in humans and in animal models (McCartney et al. 1994; Sun et al. 2008; Kable et al. 2009; Peck et al. 2010). We therefore investigated the effect of perinatal nicotine exposure on synaptic transmission and integration in the VNLL, an auditory brainstem involved in the analysis of the temporal pattern of sounds. We show that excitatory synaptic strength in VNLL neurons is profoundly reduced by perinatal nicotine exposure. As a consequence, both efficacy and the timing of action potential initiation deteriorate, suggesting maladapted transmission of information during sound processing. In contrast, nicotine exposure had little or no effect on synaptic pruning and the maturation of intrinsic neuronal properties. We also show that these neurons express functional postsynaptic α7‐type and non‐α7‐type nAChRs only during this critical period. Nicotine exposure delays this developmental decline of nAChR expression, arguing in favour of a direct effect of nAChR activation. These results provide evidence for a prominent developmental effect of nAChR activation on synaptic maturation in the VNLL.
nAChR activation may directly modulate synaptic transmission and maturation in the VNLL
An important question arising during our study was whether nicotine acts directly on the synapse under investigation or whether neural circuits upstream of VNLL neurons are altered and mediate an activity‐dependent effect on the development of VNLL synapses. We showed that nAChRs of the α7 subtype are functional in postsynaptic VNLL neurons during early development and nicotine exposure prolongs the presence of these receptors. This is in agreement with anatomical studies, which show that the prominent labelling of the α subunit of the nAChR around hearing onset greatly diminishes thereafter (Happe & Morley, 2004). These receptors are highly permeable for calcium and activation of these receptors results in plastic changes of neural circuits and ultimately leads to the remodelling and refinement of input maps (Aramakis et al. 2000; Liu et al. 2007; Abreu‐Villaça et al. 2011). It is thus likely that excessive perinatal nicotine signalling modifies synaptic development directly in the VNLL.
But how do postsynaptic nAChRs directly interact with the development of the excitatory inputs? In our study the amplitude of excitatory synaptic transmission was greatly reduced by developmental nicotine exposure, while the kinetics of the synaptic currents remained unchanged. Excitatory synaptic transmission in VNLL granule cells of mature animals is mediated via a single calyx‐like synapse, which results in an all‐or‐nothing response to stimulation of the incoming ascending fibres (Berger et al. 2014; Caspari et al. 2015). Thus a reduction in input strength might be a reflection of fewer presynaptic release sites, a change in release probability or a decrease in postsynaptic receptor density. Since neither the frequency of sEPSCs nor short‐term synaptic depression was altered, but sEPSC amplitude was reduced after nicotine exposure, a decrease in AMPA receptor density at the postsynaptic membrane might at least partially explain this reduction in synaptic strength. Indeed, direct and acute activation of nAChRs increases the diffusion of GluR1 subunits of AMPA receptors to the postsynaptic site and strengthens AMPA receptor‐mediated currents by changing the phosphorylation state of the receptor (Halff et al. 2014; Tang et al. 2015). Both of these processes are dependent on Ca2+ influx and involve interaction with several other postsynaptic proteins. In contrast, chronic developmental activation of nAChRs may have more diverse effects on synaptic transmission. Previous studies show that developmental nicotine exposure may either increase or decrease the strength of synaptic transmission depending on brain area, age and receptor subtype investigated (Huang et al. 2008; Pilarski et al. 2011; Wang et al. 2011; Jaiswal et al. 2015). One possible explanation for these diverse effects is the strong and rapid desensitization of the α7‐containing nAChRs (Albuquerque et al. 2009), leading to diminished signalling through this receptor when nicotine is applied chronically. It is most likely that α7‐containing nAChRs in VNLL neurons are permanently desensitized during nicotine application and, as a consequence, these receptors are upregulated.
To our surprise the pruning of excitatory inputs to VNLL neurons was not diminished in nicotine‐treated mice. A direct link between nAChRs and synaptic remodelling has been suggested for a number of cortical neural circuits (Liu et al. 2007; Lozada et al. 2012; Ramanathan et al. 2015). However, these experiments mostly relied on the deletion of specific nAChR subtypes or on disruption of cholinergic inputs to the target cells which in both cases reduces cholinergic signalling. As pointed out above, we do not know whether cholinergic signalling is enhanced or diminished or just altered in timing during chronic nicotine administration. Thus we cannot rule out a possible contribution of cholinergic signalling to synaptic refinement in the VNLL at the moment.
Possible indirect effect of nicotine on VNLL neurons via activity‐dependent mechanisms
Since the VNLL synapse undergoes massive changes just after hearing onset, when activity levels surge, an activity‐dependent component in the maturation of this synapse cannot be ruled out. It is therefore possible that besides these direct effects of nAChRs activation on VNLL synapse maturation, indirect, nicotine‐induced changes in activity levels and timing upstream of this synapse might also play a role. For example, inner and outer hair cells receive prominent descending cholinergic inputs as early as embryonic day 16 (Simmons & Morley, 2011). Activation of the corresponding receptors, which mostly comprise the α9 and α10 subunits, regulate synapse stabilization and maturation of the cochlea (Vetter et al. 1999; Murthy et al. 2009; Turcan et al. 2010). It is also known that a deletion of the α9 subunit in the cochlea modifies the temporal pattern of spontaneous activity in the auditory nerve fibre and impairs synaptic refinement in an auditory brainstem projection before hearing onset (Clause et al. 2014). Nonetheless, to what extent perinatal nicotine exposure affects physiology and activity levels in the cochlea is unknown at the moment.
In the ascending auditory pathway, octopus cells of the cochlear nucleus, which provide the large calyx‐like input to the VNLL granule cells, are under cholinergic regulation during early postnatal development. Right around hearing onset, between P3 and P12, these neurons receive abundant cholinergic inputs, acetylcholine esterase levels are elevated and α7‐containing nAChRs are found in this region during the same time period (Morley et al. 2004). This indicates that nicotine might regulate activity levels in octopus cells which could lead to aberrant synaptic development at its output, the excitatory synapses in the VNLL (Happe & Morley, 2004).
Functional consequences of nicotine‐mediated impaired synaptic transmission in the VNLL
Our experiments show that the nicotine‐mediated reduction in synaptic strength also leads to spike failures and increased jitter in VNLL neurons. For two reasons these observed changes might be even larger in the in vivo situation compared to our recordings in acute brain slices. First, spontaneous spiking activity, which is generally missing in the acute brain slices preparation of the auditory brainstem, leads to a chronically depressed state of synapses in different parts of the auditory brainstem with generally much reduced synaptic currents (Hermann et al. 2007; Lorteije et al. 2009; Kuenzel et al. 2011). Since our experiments show that state spike failure and jitter was greatly augmented in nicotine‐treated animals, especially later during train stimulation when synaptic depression had reached steady state levels, spike failure and jitter might be more pronounced in vivo. Second, we pharmacologically blocked inhibitory inputs, which are prominent in these neurons (Caspari et al. 2015), since we could not reliably evoke coincident excitatory and inhibitory input activity. We also do not know whether sound stimulation evoked coincident excitatory and inhibitory input activity in these neurons. Nevertheless, coincident inhibitory and excitatory input activity might increase spike failures, especially later in the stimulus train as previously shown in cochlear nucleus neurons (Chanda & Xu‐Friedman, 2010).
But what would an increase in spike failure and deterioration of spike timing in VNLL neurons mean for the perception of acoustic signals? The granule neurons in the VNLL provide a powerful and short latency inhibitory input to neurons in the VNLL and the inferior colliculus (Nayagam et al. 2005, 2006; Spencer et al. 2015). This inhibition arrives a few milliseconds before the excitation in these neurons. Since octopus cells, the input to the VNLL neurons, and VNLL neurons themselves, respond best to broadband sound stimuli (Rhode et al. 1983; Rhode & Smith, 1986), a depression of spectral splatter that occurs at the start of a sharp onset sound was proposed as a function of this circuit during sound processing (Spencer et al. 2015). Other proposed functions of preceding inhibition are the shaping of selectivity of frequency modulations and the determination of first spike latency (Gittelman & Pollak, 2011; Pollak et al. 2011). In all cases, a maladapted preceding inhibition as observed after developmental nicotine exposure may degrade temporal processing of sounds including speech. This proposed mechanism of perinatal nicotine exposure on neuronal activity in the inferior colliculus is supported by measurements of auditory brainstem responses in infants of heavy‐smoking and non‐smoking mothers. In two independent studies, wave V latency, corresponding to inferior colliculus activity, was significantly shorter in infants of smoking mothers compared to non‐smoking mothers (Kable et al. 2009; Peck et al. 2010). Weaker onset inhibition in the inferior colliculus induced by unreliable spiking of VNLL neurons could be an underlying cause of this observation.
Since nAChRs are broadly expressed in the ascending auditory pathway, perinatal nicotine exposure may not exclusively affect the maturation of VNLL synapses but could lead to alterations in neural circuits throughout the ascending auditory system. Cortical circuits, including the auditory cortex, are especially affected by changes in nicotinic activity levels (Aramakis et al. 2000; Liang et al. 2006). Nonetheless, our study provides the first evidence for a profound effect of perinatal nicotine exposure on the development of neural circuits in the auditory brainstem possibly involved in the temporal sound analysis important for speech processing.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
V.B. and U.K. conceived and designed the study. V.B. and U.K. contributed to the acquisition, analysis and interpretation of the data in this study. V.B. and U.K. drafted and wrote the manuscript. Both authors have approved the final version of the manuscript. Both authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SPP1608, SFB885) to U.K.
Acknowledgements
We thank Karin Heufelder for maintaining the mouse colony and establishing the nicotine and saccharin administration.
This is an Editor's Choice article from the 1 June 2017 issue.
References
- Abreu‐Villaça Y, Filgueiras CC & Manhães AC (2011). Developmental aspects of the cholinergic system. Behav Brain Res 221, 367–378. [DOI] [PubMed] [Google Scholar]
- Adams JC (1997). Projections from octopus cells of the posteroventral cochlear nuceus to the ventral nucleus of the lateral lemniscus in cat and human. Aud Neurosci 3, 335–350. [Google Scholar]
- Albuquerque EX, Pereira EFR, Alkondon M & Rogers SW (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89, 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Wonnacott S & Albuquerque EX (1992). Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Mol Pharmacol 41, 802–808. [PubMed] [Google Scholar]
- Aramakis VB, Hsieh CY, Leslie FM & Metherate R (2000). A critical period for nicotine‐induced disruption of synaptic development in rat auditory cortex. J Neurosci 20, 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C, Meyer EMM, Ammer JJ & Felmy F (2014). Large somatic synapses on neurons in the ventral lateral lemniscus work in Pairs. J Neurosci 34, 3237–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B & Bertrand D (2001). Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J Neurosci 21, 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari F, Baumann VJ, Garcia‐Pino E & Koch U (2015). Heterogeneity of intrinsic and synaptic properties of neurons in the ventral and dorsal parts of the ventral nucleus of the lateral lemniscus. Front Neural Circuits 9, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S & Xu‐Friedman MA (2010). Neuromodulation by GABA converts a relay into a coincidence detector. J Neurophysiol 104, 2063–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clause A, Kim G, Sonntag M, Weisz CJC, Vetter DE, Rűbsamen R & Kandler K (2014). The precise temporal pattern of prehearing spontaneous activity is necessary for tonotopic map refinement. Neuron 82, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittelman JX & Pollak GD (2011). It's about time: how input timing is used and not used to create emergent properties in the auditory system. J Neurosci 31, 2576–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glendenning KK, Brusno‐Bechtold JK, Thompson GC & Masterton RB (1981). Ascending auditory afferents to the nuclei of the lateral leminscus. J Comp Neurol 197, 673–703. [DOI] [PubMed] [Google Scholar]
- Golding NL, Robertson D & Oertel D (1995). Recordings from slices indicate that octopus cells of the cochlear nucleus detect coincident firing of auditory nerve fibers with temporal precision. J Neurosci 15, 3138–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L & Zoli M (2009). Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol 78, 703–711. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halff AW, Gómez‐Varela D, John D & Berg DK (2014). A novel mechanism for nicotinic potentiation of glutamatergic synapses. J Neurosci 34, 2051–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happe HK & Morley BJ (2004). Distribution and postnatal development of α7 nicotinic acetylcholine receptors in the rodent lower auditory brainstem. Dev Brain Res 153, 29–37. [DOI] [PubMed] [Google Scholar]
- Hermann J, Pecka M, Gersdorff H von, Grothe B & Klug A (2007). Synaptic transmission at the calyx of held under in vivo‐like activity levels. J Neurophysiol 98, 807–820. [DOI] [PubMed] [Google Scholar]
- Horst NK, Heath CJ, Neugebauer NM, Kimchi EY, Laubach M & Picciotto MR (2012). Impaired auditory discrimination learning following perinatal nicotine exposure or β2 nicotinic acetylcholine receptor subunit deletion. Behav Brain Res 231, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y‐Y, Kandel ER & Levine A (2008). Chronic nicotine exposure induces a long‐lasting and pathway‐specific facilitation of LTP in the amygdala. Learn Mem 15, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal SJ, Buls Wollman L, Harrison CM, Pilarski JQ & Fregosi RF (2015). Developmental nicotine exposure enhances inhibitory synaptic transmission in motor neurons and interneurons critical for normal breathing. Dev Neurobiol 76, 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kable JA, Coles CD, Lynch ME & Carroll J (2009). The impact of maternal smoking on fast auditory brainstem responses. Neurotoxicol Teratol 31, 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H & Berg DK (2001). Nicotinic acetylcholine receptors containing α7 subunits on rat cortical neurons do not undergo long‐lasting inactivation even when up‐regulated by chronic nicotine exposure. J Neurochem 78, 1367–1378. [DOI] [PubMed] [Google Scholar]
- Key APF, Ferguson M, Molfese DL, Peach K, Lehman C & Molfese VJ (2007). Smoking during pregnancy affects speech‐processing ability in newborn infants. Environ Health Perspect 115, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G & Kandler K (2003). Elimination and strengthening of glycinergic/GABAergic connections during tonotopic map formation. Nat Neurosci 6, 282–290. [DOI] [PubMed] [Google Scholar]
- Kuenzel T, Borst JGG & Heijden M van der (2011). Factors controlling the input–output relationship of spherical bushy cells in the gerbil cochlear nucleus. J Neurosci 31, 4260–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Poytress BS, Chen Y, Leslie FM, Weinberger NM & Metherate R (2006). Neonatal nicotine exposure impairs nicotinic enhancement of central auditory processing and auditory learning in adult rats. Eur J Neurosci 24, 857–866. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhang J & Berg DK (2007). Role of endogenous nicotinic signaling in guiding neuronal development. Biochem Pharmacol 74, 1112–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorteije JAM, Rusu SI, Kushmerick C & Borst JGG (2009). Reliability and precision of the mouse calyx of held synapse. J Neurosci 29, 13770–13784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z & Berg DK (2012). Glutamatergic synapse formation is promoted by α7‐containing nicotinic acetylcholine receptors. J Neurosci 32, 7651–7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney JS, Fried PA & Watkinson B (1994). Central auditory processing in school‐age children prenatally exposed to cigarette smoke. Neurotoxicol Teratol 16, 269–276. [DOI] [PubMed] [Google Scholar]
- Molinari EJ, Delbono O, Messi ML, Renganathan M, Arneric SP, Sullivan JP & Gopalakrishnan M (1998). Up‐regulation of human α7 nicotinic receptors by chronic treatment with activator and antagonist ligands. Eur J Pharmacol 347, 131–139. [DOI] [PubMed] [Google Scholar]
- Morishita H, Miwa JM, Heintz N & Hensch TK (2010). Lynx1, a cholinergic brake limits plasticity in adult visual cortex. Science 330, 1238–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley BJ, Warr WB & Rodriguez–Sierra J (2004). Transient expression of acetylcholinesterase in the posterior ventral cochlear nucleus of rat brain. J Assoc Res Otolaryngol 5, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V, Taranda J, Elgoyhen AB & Vetter DE (2009). Activity of nAChRs containing α9 subunits modulates synapse stabilization via bidirectional signaling programs. Dev Neurobiol 69, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayagam DAX, Clarey JC & Paolini AG (2005). Powerful, onset inhibition in the ventral nucleus of the lateral lemniscus. J Neurophysiol 94, 1651–1654. [DOI] [PubMed] [Google Scholar]
- Nayagam DAX, Clarey JC & Paolini AG (2006). Intracellular responses and morphology of rat ventral complex of the lateral lemniscus neurons in vivo . J Comp Neurol 498, 295–315. [DOI] [PubMed] [Google Scholar]
- Oertel D (1999). The role of timing in the brain stem auditory nuclei of vertebrates. Annu Rev Physiol 61, 497–519. [DOI] [PubMed] [Google Scholar]
- Oertel D & Wickesberg RE (2002). Ascending pathways through ventral nuclei of the lateral lemniscus and their possible role in pattern recognition in natural sounds In Integrative Functions in the Mammalian Auditory Pathway, eds Oertel D, Popper AN. & Fay RR, pp. 207–237. Springer, New York. [Google Scholar]
- Pauly JR, Sparks JA, Hauser KF & Pauly TH (2004). In utero nicotine exposure causes persistent, gender‐dependant changes in locomotor activity and sensitivity to nicotine in C57Bl/6 mice. Int J Dev Neurosci 22, 329–337. [DOI] [PubMed] [Google Scholar]
- Peck JD, Neas B, Robledo C, Saffer E, Beebe L & Wild RA (2010). Intrauterine tobacco exposure may alter auditory brainstem responses in newborns. Acta Obstet Gynecol Scand 89, 592–596. [DOI] [PubMed] [Google Scholar]
- Pilarski JQ, Wakefield HE, Fuglevand AJ, Levine RB & Fregosi RF (2011). Developmental nicotine exposure alters neurotransmission and excitability in hypoglossal motoneurons. J Neurophysiol 105, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak GD, Gittelman JX, Li N & Xie R (2011). Inhibitory projections from the ventral nucleus of the lateral lemniscus and superior paraolivary nucleus create directional selectivity of frequency modulations in the inferior colliculus: a comparison of bats with other mammals. Hear Res 273, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan DS, Conner JM, Anilkumar AA & Tuszynski MH (2015). Cholinergic systems are essential for late‐stage maturation and refinement of motor cortical circuits. J Neurophysiol 113, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recio‐Spinoso A & Joris PX (2014). Temporal properties of responses to sound in the ventral nucleus of the lateral lemniscus. J Neurophysiol 111, 817–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhode WS, Oertel D & Smith PH (1983). Physiological response properties of cells labeled intracellularly with horseradish peroxidase in cat ventral cochlear nucleus. J Comp Neurol 213, 448–463. [DOI] [PubMed] [Google Scholar]
- Rhode WS & Smith PH (1986). Encoding timing and intensity in the ventral cochlear nucleus of the cat. J Neurophysiol 56, 261–286. [DOI] [PubMed] [Google Scholar]
- Riquelme R, Saldaña E, Osen KK, Ottersen OP & Merchán MA (2001). Colocalization of GABA and glycine in the ventral nucleus of the lateral lemniscus in rat: an in situ hybridization and semiquantitative immunocytochemical study. J Comp Neurol 432, 409–424. [DOI] [PubMed] [Google Scholar]
- Roux I, Wersinger E, McIntosh JM, Fuchs PA & Glowatzki E (2011). Onset of cholinergic efferent synaptic function in sensory hair cells of the rat cochlea. J Neurosci 31, 15092–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint Marie RL & Baker RA (1990). Neurotransmitter‐specific uptake and retrograde transport of [3H]glycine from the inferior colliculus by ipsilateral projections of the superior olivary complex and nuclei of the lateral lemniscus. Brain Res 524, 244–253. [DOI] [PubMed] [Google Scholar]
- Saint Marie RL, Shneiderman A & Stanforth DA (1997). Patterns of gamma‐aminobutyric acid and glycine immunoreactivities reflect structural and functional differences of the cat lateral lemniscal nuclei. J Comp Neurol 389, 264–276. [DOI] [PubMed] [Google Scholar]
- Schofield BR & Cant NB (1997). Ventral nucleus of the lateral lemniscus in guinea pigs: cytoarchitecture and inputs from the cochlear nucleus. J Comp Neurol 379, 363–385. [DOI] [PubMed] [Google Scholar]
- Simmons DD & Morley BJ (2011). Spatial and temporal expression patterns of nicotinic acetylcholine alpha 9 and alpha 10 subunits in the embryonic and early postnatal inner ear. Neuroscience 194, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JA & Pauly JR (1999). Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmacology 141, 145–153. [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Nayagam DAX, Clarey JC, Paolini AG, Meffin H, Burkitt AN & Grayden DB (2015). Broadband onset inhibition can suppress spectral splatter in the auditory brainstem. PLoS One 10, e0126500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Hansen A, Zhang L, Lu J, Stolzberg D & Kraus KS (2008). Neonatal nicotine exposure impairs development of auditory temporal processing. Hear Res 245, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Luo D, Yang J, Xu X‐Y, Zhu B‐L, Wang X‐F, Yan Z & Chen G‐J (2015). Modulation of AMPA receptor mediated current by nicotinic acetylcholine receptor in layer I neurons of rat prefrontal cortex. Sci Rep 5, 14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Slonim DK & Vetter DE (2010). Lack of nAChR activity depresses cochlear maturation and up‐regulates gaba system components: temporal profiling of gene expression in α9 null mice. PLoS One 5, e9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter DE, Liberman MC, Mann J, Barhanin J, Boulter J, Brown MC, Saffiote‐Kolman J, Heinemann SF & Elgoyhen AB (1999). Role of α9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron 23, 93–103. [DOI] [PubMed] [Google Scholar]
- Walcher J, Hassfurth B, Grothe B & Koch U (2011). Comparative posthearing development of inhibitory inputs to the lateral superior olive in gerbils and mice. J Neurophysiol 106, 1443–1453. [DOI] [PubMed] [Google Scholar]
- Wang H, Dávila‐García MI, Yarl W & Gondré‐Lewis MC (2011). Gestational nicotine exposure regulates expression of AMPA and NMDA receptors and their signaling apparatus in developing and adult rat hippocampus. Neuroscience 188, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H & Xu‐Friedman MA (2015). Skipped‐stimulus approach reveals that short‐term plasticity dominates synaptic strength during ongoing activity. J Neurosci 35, 8297–8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang X, Xu Y, Spencer TJ, Biederman J & Bhide PG (2012). Prenatal nicotine exposure mouse model showing hyperactivity, reduced cingulate cortex volume, reduced dopamine turnover, and responsiveness to oral methylphenidate treatment. J Neurosci 32, 9410–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]