

Abstract
Key points
Visual input to the suprachiasmatic nucleus circadian clock is critical for animals to adapt their physiology and behaviour in line with the solar day.
In addition to direct retinal projections, the clock receives input from the visual thalamus, although the role of this geniculohypothalamic pathway in circadian photoreception is poorly understood.
In the present study, we develop a novel brain slice preparation that preserves the geniculohypothalamic pathway to show that GABAergic thalamic neurons inhibit retinally‐driven activity in the central clock in a circadian time‐dependent manner.
We also show that in vivo manipulation of thalamic signalling adjusts specific features of the hypothalamic light response, indicating that the geniculohypothalamic pathway is primarily activated by crossed retinal inputs.
Our data provide a mechanism by which geniculohypothalamic signals can adjust the magnitude of circadian and more acute hypothalamic light responses according to time‐of‐day and establish an important new model for future investigations of the circadian visual system.
Abstract
Sensory input to the master mammalian circadian clock, the suprachiasmatic nucleus (SCN), is vital in allowing animals to optimize physiology and behaviour alongside daily changes in the environment. Retinal inputs encoding changes in external illumination provide the principle source of such information. The SCN also receives input from other retinorecipient brain regions, primarily via the geniculohypothalamic tract (GHT), although the contribution of these indirect projections to circadian photoreception is currently poorly understood. To address this deficit, in the present study, we established an in vitro mouse brain slice preparation that retains connectivity across the extended circadian system. Using multi‐electrode recordings, we first confirm that this preparation retains intact optic projections to the SCN, thalamus and pretectum and a functional GHT. We next show that optogenetic activation of GHT neurons selectively suppresses SCN responses to retinal input, and also that this effect exhibits a pronounced day/night variation and involves a GABAergic mechanism. This inhibitory action was not associated with overt circadian rhythmicity in GHT output, indicating modulation at the SCN level. Finally, we use in vivo electrophysiological recordings alongside pharmacological inactivation or optogenetic excitation to show that GHT signalling actively modulates specific features of the SCN light response, indicating that GHT cells are primarily activated by crossed retinal projections. Taken together, our data establish a new model for studying network communication in the extended circadian system and provide novel insight into the roles of GHT‐signalling, revealing a mechanism by which thalamic activity can help gate retinal input to the SCN according to time of day.
Keywords: circadian, electrophysiology, intergeniculate leaflet, mouse, channelrhodopsin
Key points
Visual input to the suprachiasmatic nucleus circadian clock is critical for animals to adapt their physiology and behaviour in line with the solar day.
In addition to direct retinal projections, the clock receives input from the visual thalamus, although the role of this geniculohypothalamic pathway in circadian photoreception is poorly understood.
In the present study, we develop a novel brain slice preparation that preserves the geniculohypothalamic pathway to show that GABAergic thalamic neurons inhibit retinally‐driven activity in the central clock in a circadian time‐dependent manner.
We also show that in vivo manipulation of thalamic signalling adjusts specific features of the hypothalamic light response, indicating that the geniculohypothalamic pathway is primarily activated by crossed retinal inputs.
Our data provide a mechanism by which geniculohypothalamic signals can adjust the magnitude of circadian and more acute hypothalamic light responses according to time‐of‐day and establish an important new model for future investigations of the circadian visual system.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- BIC
(+)‐bicuculline
- CGP
CGP55845 hydrochloride
- ChR2
channelrhodopsin‐2
- CNQX
6‐cyano‐7‐nitroquinoxaline‐2,3‐dione disodium
- d‐AP5
d‐(–)‐2‐amino‐5‐phosphonopentanoic acid
- GAD2
glutamate decarboxylase 2
- GHT
geniculohypothalamic tract
- IGL
intergeniculate leaflet
- ipRGCs
ntrinsically photosensitive retinal ganglion cells
- NPY
neuropeptide Y
- OC
optic chiasm
- OT
optic tract
- PBS‐T
Triton‐X 100 in PBS
- PCA
principle component analysis
- PFA
paraformaldehyde
- pMEA
perforated multi‐electrode array
- PON
pretectal olivary nucleus
- RM
repeated measures
- SCN
suprachiasmatic nucleus
- vLGN
ventral lateral geniculate nucleus
- vSPZ
ventral subparaventricular zone
- ZT
Zeitgeber time
Introduction
Visual input to the suprachiasmatic nucleus (SCN) circadian clock plays an essential role, allowing mammals to optimize physiology and behaviour across the solar day. This function is supported by direct retinal projections to the SCN from intrinsically photosensitive, melanopsin‐expressing, retinal ganglion cells (ipRGCs) (Hattar et al. 2006). Although ipRGCs are thus essential for circadian photoentrainment (Güler et al. 2007; Chen et al. 2011), the SCN is also reciprocally connected to several other ipRGC target regions: the thalamic intergeniculate leaflet/ventral lateral geniculate nucleus (IGL/vLGN) and the pretectal olivary nucleus (PON) (Watts et al. 1987; Mikkelsen, 1992; Kalsbeek et al. 1993; Morin et al. 1994; Horvath, 1998; Moore et al. 2000).
So far, the IGL/vLGN projection to the SCN (the geniculohypothalamic tract; GHT) is the only component of this extended circadian system that has been investigated in detail. IGL/vLGN cells are primarily GABAergic and express a variety of peptide co‐transmitters, with cells expressing neuropeptide Y (NPY) providing a major (but not exclusive) contribution to the GHT (Moore & Card, 1994; Morin & Blanchard, 1995, 2001). In line with the presence of substantial input to the IGL/vLGN from the major brain arousal systems (Moore & Card, 1994), at present, the best‐defined role for the GHT is in mediating circadian responses to arousal‐inducing (‘non‐photic’) stimuli (Harrington, 1997; Morin & Allen, 2006). Hence, circadian responses to non‐photic stimuli (which alter the timing of rodent behavioural rhythms in a manner essentially opposite to light) are blocked by lesions of the GHT (Johnson et al. 1988; Janik & Mrosovsky, 1994; Wickland & Turek, 1994; Marchant et al. 1997) and can be mimicked by electrical stimulation of the GHT (Rusak et al. 1989), in vivo/in vitro application of NPY (Shibata & Moore, 1993; Huhman & Albers, 1994; Huhman et al. 1996) or GABA agonists (Smith et al. 1989; Gillespie et al. 1996; Huhman et al. 1997; Novak et al. 2004).
By contrast, despite the substantial retinal input to the IGL/vLGN (and its connections to the visual centres in the pretectum), the contribution of the GHT to circadian photoreception remains uncertain. Data from studies using pharmacological manipulation of either GABA (Gillespie et al. 1996, 1997; Novak & Albers, 2004) or NPY signalling suggest that neurochemicals released from the GHT could actively inhibit light‐driven phase shifting (Yannielli & Harrington, 2000; Lall & Biello, 2002, 2003; Yannielli et al. 2004). The extent to which these reported effects reflect the physiological contribution of the GHT to photic phase‐resetting or whether GABA and/or NPY released from the GHT directly modulates light‐evoked firing within the SCN remains uncertain. This is especially true in the case of GABA signalling, where the pharmacological approaches used above are unable to specifically distinguish effects arising as a result of GHT‐derived GABA release from those as a result of the action of local SCN neurons or other long‐range inputs.
Moreover, although lesion studies are equivocal regarding how the GHT influences circadian photoreception, on balance, these appear to favour the view that circadian light responses are diminished, rather than enhanced, without a functional GHT (Harrington & Rusak, 1986; Pickard et al. 1987; Johnson et al. 1989; Maywood et al. 1997; Edelstein & Amir, 1999a,b; Morin & Pace, 2002; Muscat & Morin, 2006; Evans et al. 2012). Similarly, existing attempts to define the nature of the visual signals conveyed by the GHT provide no clear consensus as to whether these are primarily activated, suppressed or unresponsive to increases in illumination (Harrington & Rusak, 1989; Zhang & Rusak, 1989; Thankachan & Rusak, 2005; Blasiak & Lewandowski, 2013).
In the present study, we set out to resolve these questions regarding how the GHT influences SCN visual processing. We first generate and validate an in vitro model that retains an intact extended circadian system, including a functional GHT. We next use this model, alongside optogenetic manipulations, to define the impact of GHT signals on SCN responses to optic input. Finally, we perform in vivo recordings coupled with local pharmacological inhibition or optogenetic excitation to reveal the physiological impact of the GHT on light‐evoked neural activity within the SCN.
Methods
Animals
All animal use was conducted in accordance with the Animals, Scientific Procedures, Act of 1986 (UK) and received institutional ethics committee and UK Home Office approval. In vitro electrophysiological experiments were performed on adult (40–130 day old) male and female Opn4+/tau‐lacZ mice and their wild‐type littermates (Hattar et al. 2006). For optogenetic control of GABAergic neurons, glutamate decarboxylase 2 (GAD2)‐IRES‐Cre mice (Taniguchi et al. 2011) were crossed with a second line (Ai32) exhibiting Cre recombinase‐dependent channelrhodopsin‐2/EYFP (ChR2/EYFP) fusion protein expression (Madisen et al. 2012). The resulting double heterozygous offspring thus exhibited ChR2/EYFP expression restricted to GABAergic (GAD‐2 expressing) neurons. In vivo electrophysiological experiments were performed on adult male C57/Bl6 wild‐type or GAD2‐cre; Ai32 mice under urethane anaesthesia. Animals were housed under a 12:12 h light/dark cycle at 22°C with food and water available ad libitum. Zeitgeber time (ZT) was used to define the projected time of the SCN circadian clock, with lights‐off defined as ZT 12.
X‐gal labelling and slice reconstruction
To trace ipRGC projections to the extended circadian system, Opn4+/tau‐lacZ mice were killed with an overdose of urethane (i.p. injection: 0.3 ml of 20% urethane) and were then transcardially perfused with 0.1 m PBS (pH 7.4) followed by cold, 4% paraformaldehyde (PFA) in PBS for 10 min. Brains were removed, post‐fixed in 4% PFA solution and then cryoprotected in 30% sucrose in PBS overnight. Coronal sections (60 μm thick) were obtained using a freezing sledge microtome (8000 Sledge; Bright Instruments, Luton, UK). To visualize β‐galactosidase expression, X‐gal staining was then performed as described previously (Sakhi et al. 2014). In brief, sections were washed twice for 10 min in Buffer B (0.1 m PBS at pH 7.4, 2 mm MgCl2, 0.01% Na‐desoxycholate and 0.02% [octylphenoxyl] polyethoxyethanol) and then incubated for 18 h in staining solution [Buffer B with 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide and X‐gal (1 mg ml−1); Bioline Reagents Ltd, London, UK] at 37ᵒC in the dark. Following X‐gal staining, sections were washed twice for 5 min in 0.1 m PBS and mounted onto glass slides. Nuclear fast red counterstain (Sigma‐Aldrich, Poole, UK) was applied to the sections for 5 min, followed by blot‐drying, dehydration steps (in 50%, 70% and 100% ethanol: 5 min each) and, finally, a clearing agent was added for 5 min (HistoChoice; Sigma‐Aldrich). Slides were then mounted with DPX (Sigma‐Aldrich) and coverslipped. Images of serial sections were acquired with a Plan‐Apochromat (20×/0.80) objective using a Pannoramic 250 Flash II slide scanner (3DHISTECH, Budapest, Hungary). Resulting images of each brain section were then manually aligned and imported into MATLAB (MathWorks Inc., Natick, MA, USA) where RGB colour ratios were used to distinguish X‐gal staining from background (fast red) and produce a 3‐D map of relative ipRGC fibre/terminal density.
Immunofluorescence processing
For visualization of ChR2 expression, GAD2‐Cre; Ai32 mice were killed and brains were cryoprotected and sectioned as indicated above for X‐gal labelling studies (except in this case at 50 μm thick). The resulting brain sections were washed three times in 1% Triton‐X 100 in PBS (PBS‐T) for 5 min each and then incubated in blocking solution (10% normal donkey serum, 0.2% PBS‐T) for 1 h at room temperature (21ᵒC). Sections were subsequently incubated in chicken anti‐GFP primary antibody (dilution 1:1000, diluted in blocking solution; ab13970; Abcam, Cambridge, MA, USA) overnight at 4˚C. After the incubation period, sections were washed three times in 0.2% PBS‐T for 15 min each and then incubated in Alexa Fluor 488‐AffiniPure donkey anti‐chicken secondary antibody (dilution 1:5000, diluted in blocking solution; 703‐545‐155‐JIR; Stratech, Singapore, Singapore) for 1 h at room temperature. Sections were washed three times in PBS for 15 min, mounted onto glass slides with DAPI‐containing Vectashield HardSet (H‐1500; Vector Laboratories, Inc., Burlingame, CA, USA) and coverslipped. Epifluorescence images of immunostaining were then acquired using a 250 Flash II slide scanner (Pannoramic; 3DHISTECH) as described above.
In vitro multi‐electrode recordings
Mice were killed by cervical dislocation followed by decapitation (ZT2‐4). Brains were then rapidly removed and placed into a custom‐made adult mouse brain matrix (Zivic Instruments, Pittsburgh, PA, USA). The base of the brains (just rostral to the cerebellum) were cut at 30° off the coronal plane and immediately mounted onto a metal stage and sectioned using a 7000 smz‐2 vibrating microtome (Campden Instrument, Loughborough, UK) in ice‐cold (4°C) sucrose‐based slicing solution composed of (in mm): 189 sucrose, 10 D‐glucose, 26 NaHCO3, 3 KCl, 5 MgSO4, 0.1 CaCl2 and 1.25 NaH2PO4, oxygenated with 95% O2 / 5% CO2 mixture. Once the optic chiasm (OC)/rostral pole of the SCN became visible, we then cut a 600 μm thick section that contained the SCN, IGL/vLGN and PON. Only one slice was produced per animal. The resulting slice was then transferred into a Petri dish containing oxygenated artificial cerebrospinal fluid (aCSF) composed of (in mm): 124 NaCl, 3 KCl, 24 NaHCO3, 1.25 NaH2PO4, 1 MgSO4, 10 glucose and 2 CaCl2; slices were then left to rest for 1 h at room temperature (22°C).
Multi‐electrode array recordings from acute brain slices were performed as described previously (Walmsley et al. 2015). Slices were placed, recording side down, onto 60pMEA100/30iR‐Ti‐gr perforated multi‐electrode arrays (pMEAs; Multi Channel Systems, MCS GmbH, Reutlingen, Germany), comprising 59 electrodes (site area: 707 μm2) spaced 100 μm apart with one dedicated low impedance reference electrode. Perforated arrays were chosen as a result of their improved slice perfusion rate, low signal‐to‐noise ratio and long‐term recording stability (Reinhard et al. 2014). Confirmation of optimal slice placement over the pMEA electrode sites was confirmed by overlaying images taken with a GXCAM−1.3 camera (GX Optical, Haverhill, UK). Where relevant, anatomical placement was also confirmed post hoc with X‐gal staining (for Opn4+/tau‐lacZ) or endogenous EYFP fluorescence (for GAD2‐Cre; Ai32), as discussed below.
During recordings, slices were held in place by both the bottom flow suction (via the MEA perforations) driven by a constant vacuum pump (MCS GmbH) and a weighted grid. The pMEA recording chamber was continuously perfused with pre‐warmed oxygenated aCSF (34 ± 1°C) to both slice surfaces at a combined rate of 2.5–3 ml min−1. For long‐term recordings (>26 h), 0.001% gentamicin (Sigma‐Aldrich) was added directly to the aCSF. Neural signals were acquired with MC_Rack software as time‐stamped action potential waveforms using a USB‐ME64 system and MEA1060UP‐BC amplifier (MCS GmbH, Germany). Signals were sampled at 50 kHz (12.5 kHz for long‐term recordings), high‐pass filtered at 200 Hz (second order Butterworth) and multi‐unit spikes crossing a threshold (usually set at −16.5 μV) were extracted for further analysis. In some cases, we also isolated single unit activity from these multi‐unit recordings, using principle components analysis (PCA)‐based spike sorting (Offline sorter V3; Plexon, Dallas, TX, USA). Reliable single unit isolation was confirmed by reference to MANOVA F statistics, J3 and Davies–Bouldin validity metrics (offline sorter) and the presence of a distinct refractory period (>20 ms for SCN neurons) in the interspike interval distribution.
For post hoc histological analysis, slices were submerged in 4% PFA at room temperature for 40 min and then kept overnight at 4ᵒC in 0.1 m PBS. In the case of X‐gal staining, sections were then additionally washed twice for 15 min in Buffer B and incubated overnight in staining solution at 37ᵒC in darkness (see solution details for X‐gal labelling above). All sections were subsequently washed twice for 5 min in 0.1 m PBS and mounted onto glass slides with Fluoromount (Sigma‐Aldrich). Images of X‐gal staining were acquired with ZEN digital imaging software (Carl Zeiss, Oberkochen, Germany) and EYFP fluorescence was captured using an BX51 upright microscope (4×/0.30 Plan Fln) (Olympus, Tokyo, Japan) using a Coolsnap ES camera (Photometrics, Huntington Beach, CA, USA) with MetaVue Software (Molecular Devices, Sunnyvale, CA, USA).
Electrical and optogenetic stimulation
To assess responses to optic input, retinal projections were activated by electrical stimulation of the OC using two methods: either biphasic stimulation via a neighbouring pair of pMEA electrodes visually confirmed to be situated within the OC (2500 mV, 100 μs duration, 0.5 Hz) or via an independently positioned stimulating concentric bipolar electrode (0.3–1.5 mA, 200 μs duration, 0.5 Hz) (FHC, Bowdoin, ME, USA). Similar stimulation parameters were used to activate the GHT [in this case with the stimulating electrode positioned within the IGL‐region at least ∼200 μm lateral to the optic tract (OT)]. Stimuli were applied as single pulses (0.5 Hz) or as a train of five stimuli, separated at 20 or 40 Hz.
For optogenetic stimulation, blue light flashes (5–10 ms duration, 0.5 Hz) were delivered by a 200 μm core fibre (465 nm LED, ∼800 mW mm−2 at the fibre tip; PlexBright; Plexon) placed 100–200 μm above the slice surface over the IGL or SCN region. Based on the numerical aperture of the fibre (0.66), this stimulus illuminates a ∼500 μm diameter portion of the slice (although, as a result of light scatter, the effective stimulated region will be larger). To assess functional convergence of retinal and thalamic inputs in the SCN and GHT signalling mechanisms, optical stimulation of the IGL/vLGN was synchronously delivered, or preceded pMEA electrical stimulation (2500 mV, 100 μs duration) of the OC at various latencies (25 ms to 1 s, 10–60 s between each OC/GHT pair: five or six different latencies were tested per experiment and delivered in an interleaved fashion). For long‐term recordings of thalamic evoked and spontaneous activity, we alternately stimulated optic input via a concentric electrode placed in the OC (0.6–1.2 mA, 200 μs duration, 0.02 Hz) and directly activated GHT GABAergic neurons via the optical fibre as above (5 ms duration, 0.02 Hz).
Pharmacological manipulation
Pharmacological separation of the synaptic components of evoked responses was achieved by bath application of ionotropic NMDA (d‐(–)‐2‐amino‐5‐phosphonopentanoic acid; d‐AP5; 50 μm) and AMPA/kainate (6‐cyano‐7‐nitroquinoxaline‐2,3‐dione disodium; CNQX; 20 μm) glutamate receptor blockers. Contributions of GABAergic transmission were evaluated by bath perfusion with (+)‐bicuculline (BIC; 20 μm), an antagonist at ionotropic GABAA receptors, and CGP55845 hydrochloride (CGP; 3 μm), an antagonist at metabotropic GABAB receptors. At the end of all the experiments, slices were treated with bath application of NMDA (20 μm) to confirm maintained cell responsiveness, followed by TTX (1 μm) to confirm that acquired signals exclusively reflected Na+‐dependent action potentials. All drugs were purchased from Sigma‐Aldrich or Tocris (Bristol, UK), dissolved in ddH2O and kept as stock solutions at −20°C (with the exception of BIC and CGP, which were dissolved in dimethyl sulphoxide) and were diluted to their respective final concentrations in pre‐warmed, oxygenated aCSF.
Visual stimuli
Visual stimuli were delivered and quantified as described previously (Walmsley & Brown, 2015). Full field visual stimuli were generated via two LEDs (λmax 405 nm; half‐width: ±7 nm; Thorlabs, Newton, NJ, USA), controlled via LabVIEW (National Instruments, Austin, TX, USA) and neutral density filter wheels (Thorlabs). Light was supplied via flexible fibre optic light guides (7 mm diameter; Edmund Optics, York, UK) terminating in internally reflective plastic cones that fit snugly over the mouse's eye to prevent off‐target effects to stimulation of the opposing eye. Light measurements (at the level of the mouse eye) were performed using a calibrated spectroradiometer (Bentham Instruments, Reading, UK), with effective photoreceptor excitations calculated using established measures (Govardovskii et al. 2000; Jacobs & Williams, 2007; Walmsley et al. 2015). As a result of the choice of wavelength, the apparent brightness of our stimuli differed by no more than 0.2 log units for any of the known mouse opsins, with values reported in the presetn study reflecting effective irradiance for rod opsin.
In vivo multi‐electrode recordings
Mice were removed from their housing environment 1–2 h before lights off (ZT10‐11), anaesthetized by i.p. injection of urethane (1.55g kg−1 in 0.9% physiological saline) and then placed in a stereotaxic frame (SR‐15 M; Narishige International Ltd, London, UK), where their temperature was maintained at 37˚C using a homeothermic mat (Harvard Apparatus, Cambridge, UK). The surface of the skull was exposed and two holes were drilled using stereotaxic co‐ordinates (0.95 mm lateral to and 0.3 mm posterior to bregma for SCN recording electrode; 2.3 mm lateral to and 3.5 mm posterior to bregma for IGL probe) obtained from a mouse atlas (Paxinos & Franklin, 2001). Pupils were dilated with topical application of atropine (1% in saline) and a thin coating of mineral oil was then applied to prevent drying of the cornea (both sourced from Sigma‐Aldrich).
For SCN recordings 32‐channel electrodes were then coated with fluorescent dye (DiI; Invitrogen, Paisley, UK) and inserted into the brain 0.95 mm lateral and 0.3 mm caudal to bregma (angled at 9° relative to the dorsal‐ventral axis). We employed two different electrode configurations for these studies: Buszaki 32L (NeuroNexus, Ann Arbor, MI, USA) or 2 × 16Parallel (Cambridge Neurotech, Cambridge, UK), both of which produced equivalent data. Buszaki32L consisted of four shanks (200 μm spacing) each with eight closely spaced recordings sites in diamond formation (inter‐site distance of 22–42 μm; site area: 160 μm2). The 2 × 16 parallel electrodes had two shanks (250 μm spacing) each with two rows of eight recording sites (25 μm spacing; site area: 160 μm2). For manipulation of thalamic activity, a second neural probe (see below), again coated in DiI, was then inserted into the brain 2.3 mm lateral and 3.5 mm caudal to bregma (angled at 16° relative to the dorsal‐ventral axis) such that the tip of the electrode reached the IGL‐region.
In all cases, electrodes were then lowered into position using a fluid‐filled micromanipulator (MO‐10; Narishige International Ltd). After allowing at least 30 min for neural activity to stabilize following probe insertion, wideband neural signals were acquired using a Recorder64 system (Plexon), amplified (3000×) and digitized at 40 kHz. Timestamped multi‐unit spikes (1 ms segments of data) were then extracted from these recordings by threshold crossing (−40 μV) for subsequent analysis.
At the end of these in vivo experiments, brains were removed and post‐fixed in 4% PFA and then cryoprotected and sectioned (100 μm) as described above for X‐gal labelling studies. The DiI‐marked probe position (and where relevant endogenous GAD‐driven EYFP fluorescence) was then visualized and captured using a BX51 upright microscope (4×/0.30 Plan Fln) (Olympus) using a Coolsnap ES camera (Photometrics) with MetaVue Software (Molecular Devices).
Pharmacological inactivation of the visual thalamus
For these experiments, we implanted a 16‐site linear recording array (100 μm spacing; site area: 177 μm2; E16‐20 mm‐100‐177; NeuroNexus) with a drug cannula (outer diameter 165 μm; fused to the underside of the array) protruding 100 μm ventral to the probe tip into the IGL/vLGN region. To facilitate drug infusion into the visual thalamus, the cannula underlying the recording probe (100 μm aperture) was connected via flexible narrow bore tubing to a syringe pump (Harvard Apparatus) preloaded with muscimol (1 mm; Sigma).
After allowing neural activity to stabilize, we then evaluated baseline visual responsiveness across the SCN and visual thalamus. Mice were maintained in darkness and 5 s light steps were applied in an interleaved fashion to contralateral and/or ipsilateral eyes for a total of 10 repeats at logarithmically increasing intensities spanning 11.4–15.4 log photons cm−2 s−1 (interstimulus interval of 20 s).
Once we had collected this baseline data, we then began to infuse muscimol into the thalamus in 1 μl increments (delivered over 1 min) at the same time as monitoring responses to visual stimuli (5 s binocular light steps 15.4 log photons cm−2 s−1). Drug onset (observed after nominal injection volumes of 2–3 μl) was easily identifiable by a rapid loss of spontaneous and evoked neural activity, alongside a small increase in electrical noise. Following confirmation of abolition of thalamic activity by muscimol application, mice were then dark adapted (30 min) and the protocol above was repeated. In no case did we observe a reversal of drug effects during our recording sessions.
Optogenetic activation of the visual thalamus
These experiments were performed in GAD2‐cre; Ai32 mice. We implanted a 32‐site recording array (three parallel rows of 10–12 sites at 50 μm spacing; site area: 177 μm2; OALPPoly3; NeuroNexus) into the thalamus with an optical fibre (110 μm core; 0.66 numerical aperture) terminating 200 μm above the dorsal‐most recording site. For optogenetic stimulation, the optical fibre was connected to a 465 nm PlexBright LED (as described above), providing ∼630 mW mm−2 light energy at the fibre tip.
After allowing neural activity to stabilize, we first confirmed that we were able to manipulate GABAergic neuronal activity within the visual thalamus by monitoring multi‐unit responses to a train of 10 ms light flashes (100 repeats at 0.5 Hz). We then evaluated neural responses across the SCN and visual thalamus to bright light steps (15.4 log photons cm−2 s−1) targeting either the contralateral, ipsilateral or both eyes in the presence or absence of repeated optogenetic stimulation (5 s trains of 10 ms flashes at 5, 10 and 20 Hz). Each test stimulus was repeated 10 times in an interleaved fashion (interstimulus interval of 40 s).
Data analysis
Interpretation of multi‐unit data
Here, we evaluate neural responses to a range of stimulus conditions by monitoring ensemble activity detected across multiple recording electrodes within a single preparation. Multi‐unit recording has been used extensively to monitor SCN activity (Kubota et al. 1981; Tcheng & Gillette, 1996; Gribkoff et al. 1998; Yamazaki et al. 1998; Mrugala et al. 2000; Tousson & Meissl, 2004; Brown et al. 2006; VanderLeest et al. 2007; Nakamura et al. 2008) and provides a reliable indication of overall population activity within the nucleus (Schaap et al. 2003; Brown & Piggins, 2009) matching well with that attained using other single‐cell recording approaches (Green & Gillette, 1982; Groos & Hendriks, 1982; Shibata et al. 1982; Schaap et al. 1999; Pennartz et al. 2002; Itri et al. 2005; Nygård et al. 2005). Nonetheless, an important first consideration in interpreting the data from our multi‐electrode recordings is the size of the neural ensemble that such recordings represent and the extent to which signals detected at different electrodes can be considered independent.
Based on the typical peak SCN multi‐unit firing rates, we detect in vitro for recordings spanning a full circadian cycle (mean ± SD: 4.4 ± 6.4 spikes s−1 across each channel's most active 1 h epoch; n = 144 channels from five slices), relative to equivalent data obtained for single SCN cells (mean = 2.5 spikes s−1) (Brown & Piggins, 2009), and we consider it highly improbable that any single electrode samples from more than seven neurons. Given the small size and dense packing of SCN cells (diameter <10 μm; average inter‐soma distance ∼14 μm) (Abrahamson & Moore, 2001), this upper limit of seven neurons would be contained within a hemisphere with a radius of 17 μm relative to the centre of each recording site. Thus, we probably detect cells no further than 17 μm from each electrode (substantially smaller than the inter‐electrode distance of these arrays: 100 μm).
Similarly, comparing the typical light evoked multi‐unit activity we detect in vivo (mean ± SD: 14.1 ± 15.5 spikes s−1 for a 5 s, 405 nm light step providing 15.4 log photons cm−2 s−1 to both eyes; n = 295 channels from 20 mice) relative to typical single cell responses to an equivalent stimulus (mean = 9.7 spikes s−1) (Walmsley & Brown, 2015), we estimate that our multi‐unit recordings derive from no more than five cells per channel. This suggests an upper limit to the listening range of each electrode of ∼15 μm (i.e. half the inter‐electrode distance for these arrays: 22–42 μm).
To provide additional confidence that the multi‐unit signals we record at neighbouring electrodes are truly independent, we also calculated the incidence of synchronous spike detection (defined here as spikes occurring within ± 200 μs) at adjacent channels across subsets of our in vivo or in vitro SCN recordings. Because some SCN neurons are known to communicate using gap junctions (Long et al. 2005), the results of this analysis should be considered as an upper bound to the extent to which we may detect the same neuron at more than one electrode.
In the case of in vitro recordings, we analysed data from 794 pairs of horizontally or vertically adjacent channels that showed detectable neural activity (from five slices recorded over a full circadian cycle) and found the incidence of synchronous spikes to be very low (median = 1.2% of spikes at each channel, interquartile range = 0.4–2.9% of spikes). An equivalent analysis performed on data from in vivo recordings (560 pairs of adjacent channels from 10 recordings) revealed a broadly similar incidence of synchronous spikes (median = 2.1% of spikes at each channel, interquartile range = 1.2–3.7% of spikes). Thus, for both recording modalities, we consider the signals detected at individual channels as almost exclusively representing the activity of distinct groups of neurons.
Anatomical locations of recorded ensembles
As indicated above, placement of recording electrodes in all experiments was confirmed by overlaid reconstructions of the known recording array geometry and histological images (using ventricles and fibre tracts as landmarks and, where available, X‐gal staining or EYFP fluorescence). The anatomical extent of our recording arrays ensured that, in many experiments, some recording sites were located outside our regions of interest. For most of the analysis reported here, where electrode sites situated formally just outside these regions of interest exhibited functional responses to stimulation of retinal afferents pathways (see below), such data were included in our overall analysis. In particular, for hypothalamic recordings, retinally‐driven responses occasionally identified at recording sites located in the ventral subparaventricular zone (vSPZ), just dorsal to the SCN, were also included in our analysis. Exceptions to the rule above were our analysis of circadian rhythms in SCN, vSPZ and IGL/vLGN output (analysis presented in Figs 3 and 6), where we excluded data from sites located outside the borders of these regions evident in the anatomical images. Small errors in our anatomical classification of recording site location probably have not influenced our conclusions. Hence, including or excluding data from sites located at the projected anatomical borders of the regions in question would produce results that are essentially identical to those reported in the present.
Figure 3. Angled slices remain viable for more than 24 h and display circadian rhythms in electrical activity.
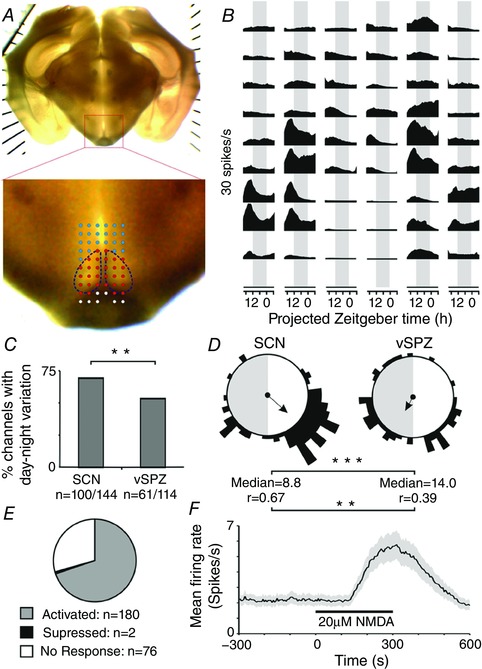
A, image of a representative angled slice preparation illustrating the distribution of pMEA recording sites within and surrounding the SCN. Red circles indicate sites categorized as being located in the SCN; blue circles indicate sites categorized as located in the vSPZ. B, multi‐unit firing rate profiles recorded across the 59 sites of the electrode array from slice in (A). Data were binned (60 s) and smoothed with a 2 h boxcar filter. C, bar chart illustrating the percentage of channels within the SCN and vSPZ considered rhythmic (based on data from five slices; for details of analysis, see Methods). Difference in the proportions of rhythmic channels was analysed by Fisher's exact test. D, Rayleigh vector plots displaying the timing of peak SCN firing activity detected at rhythmic channels in the SCN (n = 100) and vSPZ (n = 61). Arrows indicate the median phase and strength of clustering (r value). Differences in phase and clustering strength were analysed by the Mann–Whitney U test and Browne–Forsythe's test, respectively (analysis performed on linearized data; see Methods). E, proportion of channels across the SCN and vSPZ from the above experiments exhibiting significant changes in firing in response to a 5 min application of 20 μm NMDA, applied > 26 h after slice preparation. F, mean ± SEM firing rate across all NMDA‐activated channels (n = 180), illustrating robust changes in firing. Grey shaded areas in (B) and (D) correspond to the projected timing of the light/dark transition. ** P < 0.01 and *** P < 0.001, respectively.
Figure 6. Geniculohypothalamic modulation of SCN responses to optic input.
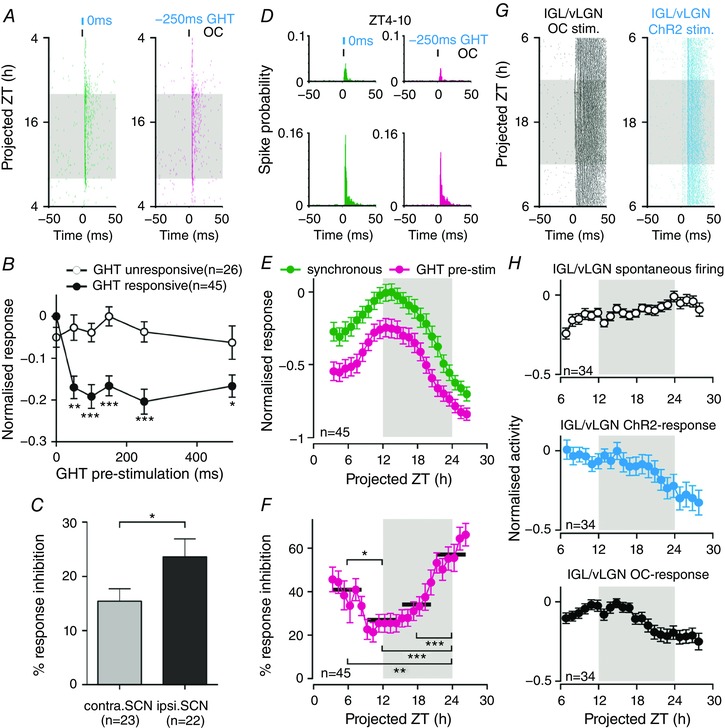
A, perievent raster of SCN multi‐unit responses to electrical stimulation of the OC; synchronous with (left), or 250 ms after (right), optogenetic activation of GABAergic GHT projection neurons (for recording set‐up, see Fig. 5
F). B, normalized mean ± SEM OC‐driven responses of SCN subpopulations as a function of GHT pre‐stimulation latency: at most recording sites (n = 45/71 OC‐responsive sites from three slices; for classification details, see Methods), GHT pre‐stimulation evoked long‐lasting suppression of OC‐driven responses (two‐way RM ANOVA: GHT pre‐stimulation, Group and interaction, all P < 0.001; asterisks indicate significance differences between groups in Sidak's post hoc tests). C, GHT‐driven inhibition of OC‐evoked responses was significantly greater ipsilateral to the optogenetically stimulated LGN (n = 22 vs. n = 23 contralateral sites; t test, P < 0.05). D, perievent histograms of SCN responses (perievent rasters are depicted in A) showing enhanced OC‐driven responses during the late day/early projected evening. E, normalized mean ± SEM response of GHT‐responsive sites (n = 45) to OC‐stimulation: synchronous with ( ), or subsequent to (
), or subsequent to ( ), optical activation of GABAergic GHT projection neurons. In both cases, an F test indicated a significantly better fit to a sinusoidal (24 h) vs. linear function (both P < 0.001 where a linear process generated the data). F, GHT‐driven inhibition of OC‐evoked responses (data from E, expressed as a percentage of the response observed under synchronous stimulation) varies significantly across the 24 h in vitro recording period (one‐way RM ANOVA of 6 h binned data; P < 0.001; asterisks indicate significance differences between timepoints in Tukey's post‐hoc tests). G, perievent raster of IGL/vLGN multi‐unit responses to OC and optical stimulation (as in Fig. 5
C). H, normalized mean ± SEM IGL/vLGN population activity (n = 34 responding sites from three slices) under basal conditions (top), following direct ChR2 (middle) or OC stimulation (bottom). In none of the above cases did we observe evidence of pronounced day‐night variation (F test, all P = 0.999; which were more probably generated by a linear vs. sinusoidal model). Shaded areas in (A) and (E–H) represent projected night. In (B) and (F): *
P < 0.05, **
P < 0.01 and ***
P < 0.001, respectively.
), optical activation of GABAergic GHT projection neurons. In both cases, an F test indicated a significantly better fit to a sinusoidal (24 h) vs. linear function (both P < 0.001 where a linear process generated the data). F, GHT‐driven inhibition of OC‐evoked responses (data from E, expressed as a percentage of the response observed under synchronous stimulation) varies significantly across the 24 h in vitro recording period (one‐way RM ANOVA of 6 h binned data; P < 0.001; asterisks indicate significance differences between timepoints in Tukey's post‐hoc tests). G, perievent raster of IGL/vLGN multi‐unit responses to OC and optical stimulation (as in Fig. 5
C). H, normalized mean ± SEM IGL/vLGN population activity (n = 34 responding sites from three slices) under basal conditions (top), following direct ChR2 (middle) or OC stimulation (bottom). In none of the above cases did we observe evidence of pronounced day‐night variation (F test, all P = 0.999; which were more probably generated by a linear vs. sinusoidal model). Shaded areas in (A) and (E–H) represent projected night. In (B) and (F): *
P < 0.05, **
P < 0.01 and ***
P < 0.001, respectively.
Identification of responding channels
Acute changes in neural activity evoked by electrical or optogenetic stimulations were classified as excitatory or inhibitory when the average spike counts across multiple stimulus repeats (typically substantially > 100), respectively, exceeded the upper or lower bounds of the 99% confidence limits for prestimulus spike counts (within at least one 10 ms bin < 100 ms after stimulus onset). For the majority of data reported in the present study (fast excitatory responses which are relatively time locked to the stimulus), representative multi‐unit data illustrating such responses are presented as perievent histograms reporting spike probability per unit time relative to stimulus onset. The exceptions are data for GHT‐driven inhibitory responses where perievent histograms are instead converted to average firing rates per unit time across trials.
Where relevant (analysis presented in Figs 6 and 7), channels identified as responding to electrical stimulation of the OC were further categorized as receiving modulatory input from the GHT. For this analysis, channels were considered ‘GHT responsive’ when the OC‐evoked response following prior optogenetic stimulation of the GHT (at any latency) was significantly different (P < 0.05) from the response evoked following synchronous stimulation (paired t tests with Bonferroni correction, based on responses to at least 120 trials at each latency).
Figure 7. Geniculohypothalamic‐driven inhibition of SCN responses to optic input involves GABAA and GABAB receptors.
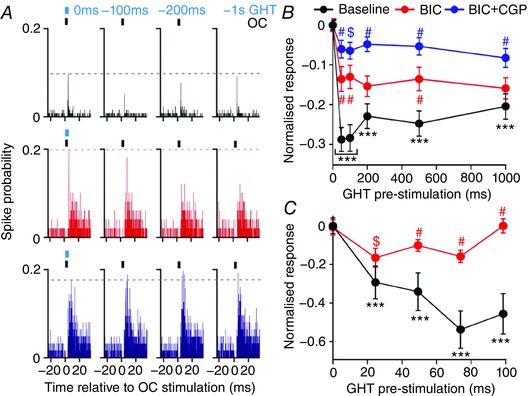
A, perievent histograms of representative SCN multi‐unit responses to electrical stimulation of the OC at varying latencies of GABAergic GHT neuron pre‐stimulation; under control conditions (top), following bath application of the GABAAR antagonist BIC (20 μm; middle), or co‐application of BIC (20 μm) and the GABABR antagonist CGP55845 (3 μm, CGP; bottom). B, normalized mean ± SEM population responses to OC‐stimulation as a function of GHT pre‐stimulation latency (n = 44 GHT‐responsive sites from a total of 81 OC‐responsive sites in three slices). Bath application of BIC substantially attenuated GHT‐driven inhibition at short latencies; residual responses under BIC were further attenuated when co‐applied with CGP. C, normalized mean ± SEM population responses to OC‐stimulation in a separate, second experiment investigating the effects of shorter GHT pre‐stimulation latencies (n = 14 GHT responsive/21 OC‐responsive sites from three slices): note that the initial components of the GHT‐evoked inhibition were almost completely attenuated by BIC application alone. Data in (B) and (C) were analysed by two‐way RM ANOVA with Sidak's post hoc tests (P < 0.01 for GHT pre‐stimulation, treatment and interaction in both cases). Black symbols in (B) and (C) represent significant differences relative to 0 ms delay (*** P < 0.001). Red symbols represent significant differences between BIC and baseline. Blue symbols (B) represent differences between BIC and BIC + CGP (# P < 0.01, $ P < 0.05).
For in vivo recordings, recorded ensembles at each site were considered light responsive when mean firing rates over the course of 5 s binocular light exposure (15.4 log photons cm−2 s−1) were significantly different from the firing rate in the 5 s epoch immediately prior to light exposure (paired t test from responses to 10 trials).
For analysis of responsiveness to pharmacological agents (NMDA) (Fig. 3), channels were considered responsive based on previously established criteria (Brown et al. 2008) and this was when the mean firing rate changed by more than 20% during the 3–8 min after the start of drug application relative to pre‐drug levels. To account for the possible confounding effects of baseline drift, pre‐drug firing was extrapolated based on a linear fit to the mean firing rates observed over the 5 min epoch immediately prior to the start of drug application.
Analysis of multi‐unit responses
Where we report average time‐course data for excitatory responses in the presence and absence of pharmacological blocking agents (Fig. 2.), we combined data from all channels identified (as above) as responding across each of the two to six slices tested. For this analysis, we first calculated, for each channel, perievent response histograms (5ms bin size) for the 6 min epochs immediately prior to drug application and 3–9 min immediately following the start of drug application (corresponding to 180 trials in either epoch). These histograms were then normalized by subtracting pre‐stimulus spike counts (averaged across the 50 ms preceding stimulation) and divided by the maximal response observed under baseline conditions. For comparison of pre‐ and post‐drug responses, the resulting datasets were then analysed by two‐way repeated measures (RM) ANOVA with Sidak's test for multiple comparisons (Prism; GraphPad Software Inc., San Diego, CA, USA).
Figure 2. Angled slice preparations retain OT connectivity across all major non‐image forming nuclei.

A, image of the recorded side (i.e. side placed facing recording sites) of an acute angled slice prepared from an Opn4+/tau‐lacZ reporter mouse before and (B) after X‐gal staining. C, schematic of the slice preparation showing stimulating electrode and pMEA placements used. D–F, electrical stimulation of the OC produces widespread excitatory responses across the pretectum (D), LGN (E) and SCN/peri‐SCN (F). Traces represent the probability of spike occurrence (from 600 trials) relative to stimulation onset (arrows) at each site on the 59‐electrode recording array for an illustrative slice. G–I, bath application of ionotropic glutamate receptor blockers CNQX (20 μm) and d‐AP5 (50 μm) completely abolished the evoked excitatory responses in the pretectum (G; n = 21 sites responding to OC stimulation from three slices), LGN (H; n = 45 sites from six slices) and SCN (I; n = 68 sites from two slices). Left: normalized responses to OC stimulation before (●) and after () drug application (see Methods). Shaded region indicates timepoints where the post‐drug response is significantly reduced (***
P < 0.001) relative to baseline (two‐way RM ANOVA with Sidak's multiple comparison test). Right: perievent rasters of multi‐unit activity from representative recording sites (green shading = 6 min drug application).
For quantification of the time‐course and magnitude of SCN responses to GHT‐stimulation, we produced perievent histograms (responses to at least 300 trials, bin size = 1 ms) and smoothed with a 10 ms boxcar filter. Latency was measured as time to the largest positive poststimulus deflection and response magnitude was measured as the percentage change in spike probability relative to the average spike probability during the 50 ms epoch preceding stimulation. The effects of pharmacological agents on this latter parameter were compared by paired t tests of equivalent analysis performed on responses occurring 0–10 min prior to drug application vs. 5–15 min after the start of drug perfusion.
For comparisons of activity evoked during combined GHT/OC stimulation (Figs 6 and 7), responses were quantified as the change in spike probability between the 25 ms epochs immediately before and after OC stimulation. For analysis, the corresponding data for each channel were then normalized according to the maximal response observed at any GHT‐OC stimulation delay. For experiments involving pharmacological blocking agents, this normalization was applied separately for baseline and each of the drug treatments (25 min epochs starting immediately prior to or 5 to 30 min after the start of each 30 min drug application; corresponding to 50 trials/epoch). Statistical significance was assessed using two‐way RM ANOVA and Sidak's test, as above.
For most analyses of in vivo visual response magnitude, the change in firing at each light responsive channel (relative to the 5 s epoch immediately prior to light stimulation) was normalized according to the largest value obtained for that site under all stimulus conditions tested (i.e. across each intensity/eye/degree of GHT manipulation). Changes in amplitude/sensitivity (Fig. 8), were then assessed by fitting irradiance response relationships to the normalized data using four‐parameter sigmoidal functions, with an F test (GraphPad) being used to determine whether the fitted parameters differed significantly between pre‐ and post‐muscimol application for each stimulus condition. Where this analysis indicated significant differences in the irradiance response relationship pre‐ vs. post‐muscimol for a given condition, we renormalized the two curves to the same maximal value and repeated the F test (to specifically assess whether there were differences in sensitivity). For assessment of changes in visual response amplitude as a function of GHT stimulation frequency (Fig. 9 F), data were normalized separately for each eye and analysed by two‐way RM ANOVA. Equivalent data for LGN recordings are analysed and presented in raw (not normalized) form.
Figure 8. Geniculohypothalamic signalling modulates specific features of the SCN light response in vivo .
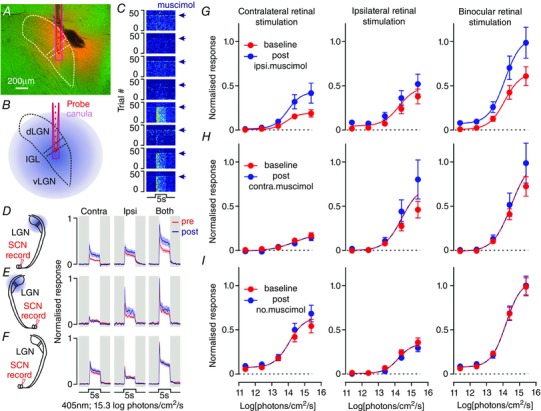
A, histological reconstruction of a DiI‐labelled recording electrode/drug cannula placement in the LGN (light microscopic image, pseudocoloured green). B, schematic showing recording electrode/drug cannula placement in LGN and projected spread of drug infusion. C, multi‐unit responses detected at level of probe in A–B, to 5 s light step (405 nm, 15.3 log photons cm−2 s−1) presented to both eyes before and after infusion of the GABAA receptor agonist muscimol (1 mm; 2–3 μl injection at 1 μl min−1) completely abolished spiking activity at recording sites up to 750 μm from cannula tip. D, normalized mean ± SEM SCN population responses to 5 s light steps (405 nm, 15.3 log photons cm−2 s−1) presented to the contralateral (left eye), ipsilateral (right eye) or both eyes, before and after infusion of muscimol into the ipsilateral LGN (n = 63 sites from five animals); schematic (left) shows recording electrode placement in the SCN and projected spread of drug infusion in the LGN. E and F, data as in (D) for responses following infusion of muscimol into the contralateral LGN (E; n = 33 sites from three mice) or under control conditions (F; identical protocol with no drug delivery, n = 127 sites from six mice); schematics of the recording set‐up are shown on the left. G–I, normalized mean (±SEM) SCN population responses to 5 s light steps of varying intensity presented to the contralateral (left), ipsilateral (right) or both eyes (left, middle and right, respectively), before () and after muscimol (or no drug) infusion ( ) into the ipsilateral LGN (G), contralateral LGN (H), or under control conditions (I). For (G–I) an F test was performed to determine whether a single sigmoidal function could explain the irradiance response relationship pre‐and post‐muscimol. This was the case (P > 0.05) in all but (G) contralateral and binocular (P = 0.001 and 0.002, respectively).
) into the ipsilateral LGN (G), contralateral LGN (H), or under control conditions (I). For (G–I) an F test was performed to determine whether a single sigmoidal function could explain the irradiance response relationship pre‐and post‐muscimol. This was the case (P > 0.05) in all but (G) contralateral and binocular (P = 0.001 and 0.002, respectively).
Figure 9. Optogenetic activation of the GHT inhibits specific features of the SCN light response in vivo .
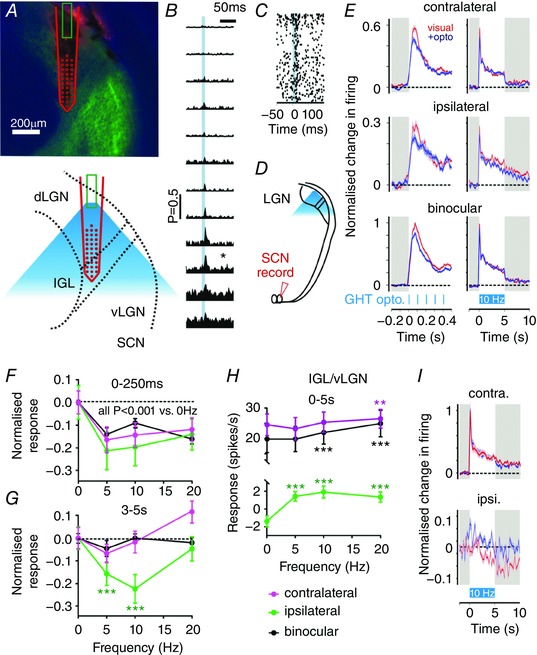
A, upper: histological reconstruction of a DiI‐labelled optrode placement in the LGN of a GAD2‐Cre; Ai32 mouse (DiI in red, ChR2‐EYFP in green, DAPI in blue); lower: schematic showing optrode placement in LGN and projected spread of optical illumination (based on 0.66 numerical aperture fibre). B, representative perievent histograms showing responses from optrode in (A) (responses to 100 trials; for clarity, responses shown only for central 12 sites) to 10 ms light flashes (465 nm LED; ∼630 mW mm−2 at fibre tip, represented by blue shading). C, perievent raster for optogenetic responses identified at the site in (B) indicated by an asterisk (*). D, schematic of recording set‐up for data in (E–G). E, normalized mean ± SEM SCN population responses to 5 s light steps (405 nm, 15.3 log photons cm−2 s−1) presented to the contralateral, ipsilateral or both eyes, in the absence (red) and presence (blue) of optogenetic stimulation of the GHT (for clarity, data for 10 Hz trains only are shown). Data are derived from 72 light responsive channels (from six mice). Left: initial components of the response (10 ms bin size, smoothed with a 50 ms boxcar filter); right: full light response (200 ms bin size). F and G, normalized mean ± SEM change in light‐evoked SCN activity as a function of GHT stimulation frequency for stimulation of contralateral, ipsilateral or both eyes, either during early (F; 0–250 ms) or later (G; 3–5 s after start of visual stimulation) components of the response. Data were analysed by two‐way RM ANOVA with Sidak's post hoc test. In (F), ANOVA revealed a significant effect of stimulation frequency (P < 0.001) but not eye or interaction, post hoc tests indicated all frequencies were significantly reduced compared to no GHT stimulation. In (G), ANOVA revealed a significant interaction between eye and stimulation frequency, coloured asterisks represent significant differences relative to the no‐GHT stimulation group (0 Hz) for that eye. H, mean ± SEM light‐evoked IGL/vLGN activity from experiments above. Data are based on changes across full 5 s stimulation (from 41 light and optically responsive sites from six mice), analysis performed on the early and later epochs (as in F and G; not shown) produced qualitatively similar results: two‐way RM ANOVA revealed significant interaction (eye × stimulation frequency; P < 0.001). Coloured asterisks represent significant differences relative to the no‐GHT stimulation group for that eye (Sidak's post hoc test). I, normalized mean ± SEM IGL/vLGN responses from dataset above for contralateral and ipsilateral visual stimuli in the absence (red) or presence (blue) of optogenetic stimulation (for clarity, data for 10 Hz trains only are shown). Grey shaded areas in (E) and (I) represent darkness. In (G) and (H): ** P < 0.01 and *** P < 0.001, respectively.
Analysis of circadian rhythmicity
For analysis of circadian rhythmicity in long‐term hypothalamic recordings (data presented in Fig. 3), data from each channel were considered to exhibit circadian variation when they could be better fit by a sinusoidal function (constrained to have a period between 20 and 28 h) vs. a first‐order polynomial (calculated using custom MATLAB routines). Data from a small number of channels where we did not detect multi‐unit activity (n = 37/295 from from slices; primarily channels located in the OT or over the third ventricle) were excluded from this analysis. Of the remaining channels, the identified best fit functions described the data well (median ± SD of Pearson's r values = 0.81 ± 0.16, n = 258). For those channels considered to exhibit circadian variation (median ± SD of Pearson's r values for sinusoidal fit = 0.83 ± 0.15, n = 168), we next calculated the timing of peak firing. For the analysis reported in the present study, multi‐unit time series were binned (60 s) and smoothed (2 h boxcar filter), with the ZT of bin exhibiting maximal values taken as the timing of the peak.
The phase and co‐ordination of the daily peak in electrical activity was then calculated by Rayleigh analysis using custom MATLAB scripts as described previously (Walmsley et al. 2015). Comparable analysis performed using peak ZTs estimated directly from the sinusoidal fits produced essentially identical results. Differences in clustering strength and phasing were assessed by first linearizing the data such that, within each dataset, each observation was no more than 12 h displaced from the overall circular median (i.e. subtracting 24 h from those observations that occurred > 12 h later and adding 24 h to those observations occurring > 12 h earlier than the median). We then applied conventional analysis to identify differences in variance (Browne–Forsythe's test) and median (Mann–Witney U test).
For other analysis of circadian variation reported in the present study (Fig. 6), spontaneous or evoked responses at each responding recording site within the region of interest were 1 h‐binned and then normalized according to the peak value observed across all bins. We then tested whether the resulting normalized population datasets were better fit by a sinusoidal function (with 24 h periodicity) or a first‐order polynomial (via an F test; GraphPad). Where data were better fit by a sinusoidal function, we rebinned/normalized the data into 6 h blocks and performed one‐way RM ANOVA with Tukey's post hoc tests.
Results
Generation and validation of a functional in vitro model of the extended circadian system
To better understand the physiological roles of GHT signals in modulating SCN responses to retinal input, we first set out to develop an in vitro model that preserved functional connectivity across the extended circadian system. Because projections between the SCN, IGL/vLGN and PON all run via the OT (Watts et al. 1987; Mikkelsen, 1992; Kalsbeek et al. 1993; Morin et al. 1994; Horvath, 1998; Moore et al. 2000), we initially aimed to determine what angle and thickness of brain section would be required to preserve OT‐projections to all of these nuclei largely intact. Accordingly, we exploited the fact that M1‐type ipRGC projections specifically identify nuclei of the extended circadian system in the mouse brain (Hattar et al. 2006). Using a selective reporter mouse line, Opn4tau‐LacZ, we were thus able to readily visualize retinal projections to all three nuclei in X‐gal stained coronal serial sections (Fig. 1 A). After aligning the resulting sections, we then generated a 3‐D map of M1‐ipRGC central projections. Based on a flattened sagittal plane view of this projection map (Fig. 1 B), we determined that a 600 μm thick brain section cut at 30° off the coronal plane would preserve OT projections and large portions of the SCN, IGL/vLGN and PON intact (for coronal plane reconstruction of the resulting slice, see Fig. 1 C).
Figure 1. Modelling the non‐image forming visual projection.

A, central targets of M1‐type ipRGCs visualized via X‐gal stained sections from Opn4+/tau‐LacZ mice. B, Sagittal reconstruction of forebrain M1‐type ipRGC axon/terminal labelling intensity, indicating chosen slice angle and thickness: 30° off the coronal plane, 600 μm thickness. C, projected ‘coronal’ view of slice depicted in (B) flattened across the z‐axis.
Using the results of this modelling, we next prepared live brain sections from Opn4+/tau‐LacZ mice, with the aid of a custom matrix that allowed us to accurately block the brain at the required angle for subsequent sectioning. From initial light microscopic visualization of the resulting slices, we could reliably (> 90% of slices) observe a continuous OT extending far beyond the thalamus and easily identifiable SCN, IGL/vLGN and pretectal regions (Fig. 2 A), as subsequently confirmed by post hoc X‐gal staining (Fig. 2 B). We next set out to confirm that OT connectivity across these visual nuclei remained functional in our slice preparation via pMEA recording.
As expected, electrical stimulation of the OC in these angled slices reliably evoked spike firing at subsets of recording sites located in the pretectum (n = 21 responding sites identified in three slices; Fig. 2 C and D), visual thalamus (n = 45 sites responding from six slices; Fig. 2 C and E) and SCN region (n = 68 sites from two slices; Fig. 2 C and F). To confirm that the OC‐evoked changes in spike rate that we recorded represented genuine synaptically‐driven responses, we then bath applied ionotropic glutamate receptor antagonists (CNQX, 20 μm and d‐AP5, 50 μm). This manipulation completely and reversibly abolished the evoked responses at all responding channels in the pretectum, thalamus and SCN/peri‐SCN region (two‐way RM ANOVA: Time × Treatment effects, all P < 0.001; Fig. 2 G and I). Taken together, these data confirm that our angled slice preparations retain a functional OT projection to all major subcortical visual nuclei.
Before using this model to investigate the impact of GHT signalling in more detail, however, we first aimed to confirm that the relatively thick (600 μm) slices required would remain viable for a sufficient length of time in vitro to facilitate analysis of circadian variation in circadian network function. We found that slice viability could be routinely maintained for longer than 24 h in vitro (Fig. 3 A). Hence, of 144 recording sites (from five slices) located within the SCN, 100 (∼70%) displayed evidence of circadian rhythmicity (Fig. 3 B and C; for analysis details, see Methods). Previous work indicates that ex vivo SCN firing rate rhythms (recorded using a wide variety of different methodologies) reliably report phasing of the SCN in vivo and are independent of the time of preparation, with peak firing consistently occurring during the mid‐late projected day (vanderLeest et al. 2009; for a detailed discussion of this point, see also Brown & Piggins, 2007). Consistent with this earlier work, we found that the timing of peak multi‐unit activity at SCN channels identified as rhythmic was strongly clustered around the mid‐late projected day (Fig. 3 D).
As expected for a major target of SCN output (Abrahamson & Moore, 2001), we also identified electrode sites located just dorsal to the SCN (in the vSPZ) exhibiting evidence of circadian variation in firing activity. Among this group, however, the proportion of sites identified as rhythmic was significantly lower than for SCN located electrodes (n = 61/114 vSPZ recording sites; Fisher's exact test: P < 0.01; Fig. 3 C). Moreover, the timing of peak firing for rhythmic vSPZ channels was significantly less well clustered and had a median phase significantly later than observed in the SCN (P < 0.01 and P < 0.001, respectively; for details of analysis, see Methods; Fig. 3 D).
To further evaluate the viability of these angled slices, at the end of these long‐term recordings (> 26 h after slice preparation), we bath applied NMDA (20 μm, 5 min application), a manipulation that should drive increases in firing in most SCN neurons (∼80% of cells in mice of a similar strain background; Cutler et al. 2003). As expected, this treatment drove robust changes in firing (see Methods) in > 70% of channels located across the SCN and vSPZ (n = 180/258 channels, no significant difference in responses between SCN and vSPZ; Fisher's exact test: P = 0.22; Fig. 3 E and F). Collectively, these data establish that our preparation remains viable for greater than one circadian cycle in vitro.
Impact of GHT signals on spontaneous SCN activity
Because our angled slice preparations retain intact OT projections across the extended circadian system, we expected that connections between the SCN, IGL/vLGN and PON (which follow the same trajectory) should be similarly preserved in this model. Here, we specifically focused on the GHT, although, in pilot experiments (six slices; data not shown), we were also able to readily detect communication between the IGL/vLGN and PON.
To test for the retention of a functional GHT, we next electrically stimulated the IGL/vLGN region via concentric electrodes at the same time as recording from the SCN and vSPZ (Fig. 4 A). Based on the predominately GABAergic nature of IGL/vLGN neurons (Moore & Card, 1994; Morin & Blanchard, 2001), to maximize our chances of observing inhibitory SCN responses to GHT stimulation, we performed these recordings across the mid‐late projected day when spontaneous SCN firing is highest (ZT 4–11). In line with data obtained following in vivo stimulation of the IGL/vLGN (Roig et al. 1997; González & Dyball, 2006), we found that single pulse stimulation of the GHT could evoke either inhibitory (Fig. 4 B) or excitatory responses (Fig. 4 C) across individual electrode sites within the SCN (n = 21 and n = 27 sites, respectively, from 12 slices).
Figure 4. Electrical stimulation of the GHT evokes inhibitory and excitatory responses in the SCN.

A, schematic of the slice preparation showing stimulation electrode and pMEA placement used. B, electrical stimulation (concentric electrode; 1.2 mA, 200 μs) of the IGL region evoked inhibitory responses at a subset of SCN recording sites (n = 21 sites from 12 slices; representative recording site shown). C, electrical stimulation of the GHT also evoked excitatory responses in a second group of SCN recording sites from these 12 slices (n = 27 sites; left: representative recording site shown), which could be abolished by bath application of ionotropic glutamate receptor blockers (CNQX, 20 μm and d‐AP5, 50 μm; right: representative recording site shown of four SCN recording sites tested). D, GHT‐evoked inhibitory responses (time to peak, 10 ms moving window) were typically slower and exhibited more variable timing than excitatory responses. E, where tested (n = 12 sites), short trains of pulses (five stimuli at 50 ms, i.e. 20 Hz, or five stimuli at 25 ms, 40 Hz) did not evoke significantly larger inhibitions than single pulse stimulation (one‐way ANOVA; P = 0.98). F, GHT‐evoked (1.2 mA, 200 μs; 5 × 40 Hz train) inhibitory responses (middle and bottom) but not excitatory responses (top) were blocked by bath application of the GABAAR antagonist, BIC (20 μm; representative SCN sites shown from a total of 23 excitatory and five inhibitory responses tested). G and H, quantification of the effect of BIC and CNQX/D‐AP5 relative to predrug values (black bars) for excitatory (G), and inhibitory responses (H). Data were analysed by a paired t test (except CNQX in G, where only four cells were tested). ** P < 0.01; n.s., P > 0.05.
Of note, we observed differences in the latency of increases vs. decreases in SCN firing following thalamic stimulation, with inhibitions typically displaying longer and more variable latency relative to the usually faster excitatory responses (Fig. 4 D; median ± SD time to peak: 53 ± 32 ms vs. 18 ± 10 ms, respectively; Mann–Whitney test: P < 0.001). These values represent the time to peak response rather than response onset (which is not always easy to accurately assess for inhibitory responses) and are equivalent those reported by previous in vivo studies (Roig et al. 1997; González & Dyball, 2006).
Because we observed a relatively low proportion of sites showing inhibitory responses to GHT stimulation in the experiments above, in some cases, we also evaluated the possibility that single pulse stimulation was simply insufficient to evoke a measureable inhibition in SCN firing. We found no evidence, however, that trains of GHT stimulation (five stimuli at 20 or 40 Hz), designed to approximate light‐driven burst firing seen in the visual thalamus (Brown et al. 2010; Howarth et al. 2014), resulted in a greater magnitude or proportion of sites exhibiting inhibitory responses (n = 12 sites; one‐way ANOVA; P > 0.05; Fig. 4 E).
In keeping with previous in vivo data then, thalamic stimulation in our angled slice preparation evokes a mixture of excitatory and inhibitory responses. Although the reported excitatory actions of GABA within the SCN (Wagner et al. 1997; Choi et al. 2008; Irwin & Allen, 2009; Farajnia et al. 2014) could, in principle, provide an explanation for the relatively high number of electrode sites showing excitatory responses to thalamic stimulation, our subsequent experiments rule this out as a primary explanation. Hence, where tested (n = 4 recording sites), excitatory responses were completely abolished by bath application of ionotropic glutamate receptor blockers (Fig. 4 C and G). By contrast, whereas inhibitory responses were reliably blocked by the GABAA receptor antagonist BIC (20 μm), excitatory responses were consistently unaffected (n = 5 and 23 sites tested, respectively; Fig. 4 F and H).
Although the presence of a very small number of glutamatergic cells in the mouse IGL‐region (experiment: 73818754) (Lein et al. 2007) provides one possible origin for the excitatory responses that we observed above, it is important to also note that many of the ipRGCs innervating the IGL send axon collaterals to the SCN (Pickard, 1985; Morin et al. 2003). Accordingly, it is more probable that antidromic activation of this ipRGC population accounts for the excitatory responses observed in some SCN neurons, as described previously for increased SCN Fos‐like immunoreactivity following electrical stimulation of the thalamus (Treep et al. 1995).
To circumvent this issue, we next turned to an optogenetics‐based approach to selectively activate GABAergic IGL/vLGN neurons in a temporally controlled manner. Accordingly, we employed a mouse line (GAD2‐cre; Ai32) where Cre‐recombinase specifically directs ChR2/EYFP expression to GABAergic (GAD2‐expressing) neurons (Taniguchi et al. 2011). As expected, the antibody enhanced fluorescence signal was highly enriched throughout the IGL/vLGN region (Fig. 5 A and B) and other predominantly GABAergic central nuclei (data not shown) in GAD2‐Cre; Ai32 mice, indicating robust expression of the ChR2/EYFP. Consistent with this anatomical localization, targeted optical stimulation (465 nm LED; ∼800 mW/mm2 at fibre tip, 10 ms flash; Fig. 5 B) over the IGL/vLGN (Fig. 5 C and E) evoked fast and reliable bursts of spike firing from neurons across the stimulated region.
Figure 5. Optogenetics‐based selective activation of geniculohypothalamic signalling.
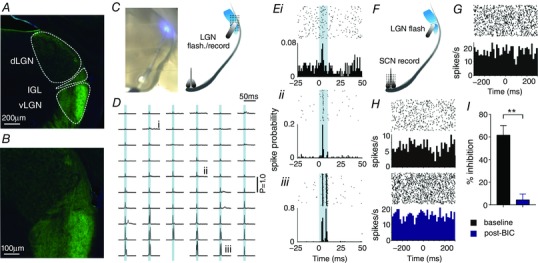
A, GFP‐immunostaining enhanced ChR2/EYFP expression in the thalamus of GAD2‐Cre; Ai32 mice, revealed robust expression in neural processes across the predominantly GABAergic IGL/vLGN region. B, central region of (A) showing EYFP signal across the dLGN and IGL/vLGN at higher magnification. C, live image of slice showing optical fibre positioning over IGL/LGN (left) and schematic of slice preparation with optical fibre and pMEA placements used (right). D and E, direct optical stimulation (465 nm LED; ∼800 mW mm−2 at fibre tip, 10 ms flash; represented by blue shaded area in traces) evoked robust and rapid excitatory responses across the IGL/vLGN and much weaker responses in the dLGN. Traces in (D) represent the probability of spike occurrence (from 200 trials) relative to stimulation onset at each site on the 59‐electrode recording array for the slice shown in (C). Data in (Ei, Eii and E iii) show perievent rasters and larger spike probability histograms for the traces indicated in (D). F, schematic of slice preparation showing optical fibre and pMEA placements used for investigation of GHT signalling. G, optical stimulation of GABAergic GHT projection neurons evoked modest inhibitory responses in a small subset of SCN recording sites (n = 12 sites from four slices; inhibition, mean ± SEM: 43.8 ± 5.5% of pre‐stimulation; latency, median ± SD: 60 ± 25 ms: representative SCN recording sites are shown in G and H). H and I, where tested (n = 6), optogenetic‐evoked inhibitory responses were abolished by bath application of the GABAAR antagonist, BIC (20 μm). H, data from a representative SCN recording site pre‐ and post‐BIC (upper and lower, respectively). I, mean ± SEM response magnitude pre‐ and post‐BIC (n = 6). Data were analysed by paired t test. ** P < 0.01.
Having established that our approach allows us to robustly activate IGL/vLGN neurons, we next evaluated the impact of GABAergic GHT projection neuron activity on spontaneous firing within the SCN region (Fig. 5 F). In line with our data above, although, in no case did we observe any excitatory responses in the SCN following selective stimulation of GABAergic GHT cells, we did find a small proportion of sites (n = 12 sites from four slices) that displayed statistically significant decreases in firing (Fig. 5 G and H; see Methods). The magnitude and timecourse of the resulting inhibitory responses were broadly similar to those following electrical stimulation (mean ± SEM inhibition: 43.8 ± 5.5%; time to peak, median ± SD: 60 ± 25 ms) and, where tested (n = 6 sites), these were abolished by the bath application of 20 μm BIC (paired t test P < 0.01; Fig. 5 I), consistent with their GABAergic origin. Taken together, our data suggest that, although GABAergic GHT projections are clearly intact in our angled slice preparation, they appear to have, at most, quite modest effects on spontaneous firing in the SCN.
Impact of GHT signals on SCN responses to optic input
Insofar as our ability to detect inhibitory GHT‐driven responses relies on the recipient cell to be firing action potentials, we speculated that the modest effects observed above reflected the fact that the primary role of this projection is to modulate responses to excitatory input from the retina. Indeed, anatomical studies suggest that GHT projections are concentrated within retinorecipient portions of the SCN (Mikkelsen, 1990; Moore et al. 2000; Abrahamson & Moore, 2001). To establish the influence of GHT signals on retinohypothalamic signalling, we next monitored SCN responses to electrical stimulation of the OC (simulating retinal input) concurrent with or following optical activation of the GABAergic GHT projection (Fig. 5 F). Given the relatively long latency of GHT‐driven inhibitory responses, we thus expected that activation of this pathway would only be able to modulate SCN responses if this occurred before (but not synchronous with) electrical stimulation of the OC. Moreover, to evaluate the possibility that the functional impact of GHT signals on the OC‐evoked response might change across the circadian day, we ran this protocol (1 OC pulse min−1) over a full 24 h recording epoch (Fig. 6 A).
We first calculated, for each independent recording site within the SCN, the average response to the several hundred stimulus presentations across the full 24 h recording epoch. We found that the majority (n = 45/71 sites from three slices) of hypothalamic recording sites at which we observed OC‐evoked spiking also showed a statistically significant reduction in this response when GHT stimulation occurred prior to OC stimulation (see Methods). The apparent magnitude of the effect at these ‘GHT‐responsive’ sites was relatively modest (∼20% reduction) but long‐lasting, being evident for all tested delays between 50 and 500 ms (two‐way RM ANOVA with Sidak's post hoc tests; Fig. 6 B). Consistent with our findings above, very few of these sites showed detectable changes in spontaneous firing following GHT stimulation (n = 4/45 sites passing criteria for responsiveness; for details, see Methods). Moreover, in line with anatomical studies indicating stronger GHT input to the ipsilateral vs. contralateral SCN (Pickard, 1982; Mikkelsen, 1990; Moore et al. 2000), we also found that the magnitude of the GHT‐driven inhibition of OC‐evoked responses (but not the proportion of GHT‐responsive sites) was significantly higher for sites located ipsilateral to the stimulated GHT (t test, P < 0.05; Fig. 6 C).
We also aimed to determine the extent to which the multi‐unit responses described above reflected the activity of individual units within the SCN. Accordingly, we performed PCA‐based spike sorting on data from channels were we observed OC‐evoked changes in firing. From these 71 channels, we were able to isolate 23 single units that exhibited clear excitatory responses to OC‐stimulation. More than half of these units (n = 12/23) exhibited a significant GHT‐driven modulation in the amplitude of OC‐evoked responses, a proportion that was not statistically different from the proportion of channels identified as GHT responsive (Fisher's exact test, P = 0.24). Importantly, the majority of these GHT‐responsive units (n = 10/12) were isolated from electrode sites where multi‐unit data were similarly modulated by GHT input, with only a few instances where single units isolated from GHT‐responsive channels lacked a significant modulation (n = 3 units from 13 channels) or vice versa (n = 2 units from 10 channels). Moreover, the magnitude of the GHT‐driven modulations in the responding single units was not significantly different from those that we report for multi‐unit data (two‐way RM ANOVA; P = 0.47 for population and P = 0.43 for interaction × GHT stimulation latency; analysis as in Fig 6 B). In sum, these data indicate that our analysis based on multi‐unit responses does not lead us to substantially underestimate the impact of GHT‐inputs on individual SCN neurons.
We next evaluated the extent to which these GHT‐driven modulations of hypothalamic responses to optic input varied across the circadian cycle. As expected, given the nocturnal increase in SCN sensitivity to light (Meijer et al. 1996, 1998; Brown et al. 2011; van Oosterhout et al. 2012), OC‐evoked multi‐unit responses themselves exhibited a very pronounced circadian variation, with peak activity occurring during the early projected night, both following synchronous activation of the GHT and following GHT pre‐stimulation (Fig. 6 A, D and E). Importantly, however, we could detect clear evidence of a circadian variation in the functional impact of GHT signals (one‐way RM ANOVA of 6 h binned data; P < 0.001; Fig. 6 F): thus, the percentage reduction in OC‐evoked responses was two‐fold greater during the projected day vs. the early‐mid projected night (Fig. 6 F). Based on these data, we next asked whether this variation in the functional impact of GHT stimulation reflected a change in the output/excitability of GABAergic neurons in the IGL/vLGN region or might instead reflect an SCN intrinsic mechanism. To this end, we directly recorded OC‐ and optically‐evoked responses from the IGL/vLGN region over a 24 h recording epoch (n = 34 sites from three slices; Fig. 6 G and H). Although, on average, we observed a slight decline in optically and electrically evoked activity towards the end of these recordings (Fig. 6 H), we saw no clear evidence for any pronounced circadian variation in either spontaneous or evoked activity across the IGL/vLGN (for analysis details, see Methods). In summary, we infer that daily variation in the functional impact of GHT signals on SCN responses involves a mechanism local to the SCN itself.
Focusing on the projected day, where GHT signals exert the most substantial effects on SCN response to OT input, we next aimed to better understand the properties and mechanistic basis of this influence. Hence, although by definition this mechanism probably involves GABA, many IGL cells co‐express NPY and/or other neuromodulators (Moore & Card, 1994; Morin & Blanchard, 1995, 2001). Accordingly, employing similar approaches to those described above, we investigated OC‐evoked responses in the SCN following optical stimulation of GHT GABAergic cells at variable latencies, in the presence and absence of GABA receptor blocking drugs (data analysed by two‐way RM ANOVA; Fig. 7 A).
As before, we observed a very long‐lasting inhibition of OC‐evoked responses at many sites (n = 44/81 OC‐responsive sites from three slices) following optical stimulation of the IGL/vLGN. This effect was maximal at GHT pre‐stimulation latencies of 50–100 ms and began to diminish at longer latencies (1 s timepoint P < 0.01 vs. 50 and 100 ms; Sidak's post hoc test; Fig. 7 B). Of note, blockade of GABAA receptors (20 μm BIC) significantly attenuated but did not completely block the inhibitory influence of GHT pre‐stimulation, with the effect being most pronounced for short latency inhibitions (Fig. 7 B). By contrast, when we then co‐applied BIC with the GABAB receptor antagonist CGP55845 (3 μm), GHT‐driven inhibitory responses were almost completely abolished at all latencies (Fig. 7 B).
We also noted that blocking GABA receptor signalling produced a general enhancement in the magnitude of OC‐driven responses at these GHT‐responsive channels (mean ± SEM: 1.0 ± 0.1, 1.6 ± 0.2 and 2.5 ± 0.3 evoked spikes at 0 ms delay for baseline, BIC and BIC + CGP, respectively; one‐way RM ANOVA with Sidak's multiple comparison test, P < 0.001 for baseline vs. BIC and BIC vs. BIC + CGP). Similar results were also obtained for GHT‐unresponsive channels (mean ± SEM: 1.2 ± 0.1, 2.0 ± 0.2 and 2.9 ± 0.3 evoked spikes for baseline, BIC and BIC + CGP, respectively, P < 0.001 for baseline vs. BIC and BIC vs. BIC + CGP).
Collectively, our data indicate that GABA signalling is a major regulator of SCN responses to retinal input, at least a portion of which derives from the activity of afferents from the GHT. Indeed, the modulatory activity of the GHT revealed in the present study appears to arise almost entirely via GABA signalling, with GABAA receptors mediating initial components of the response and GABAB receptors driving the longer lasting elements. We subsequently confirmed this view with an additional experiment focusing on early components (< 100 ms) of the GHT‐mediated inhibition (n = 14 GHT responsive sites from three slices; Fig. 7 C). Consistent with the 20–30 ms latency reported by antidromic activation of IGL cells from the SCN (Zhang & Rusak, 1989; Blasiak & Lewandowski, 2013), we observed statistically significant reductions in OC‐evoked spiking with GHT‐prestimulation latencies as low as 25 ms (two‐way RM ANOVA with Sidak's post hoc tests). Importantly, the inhibitory effects of GHT‐stimulation at all latencies tested in the present study were almost completely blocked by BIC alone at all delays tested (Fig. 7 C).
Manipulating GHT signalling modulates specific features of SCN‐driven light responses
Collectively, our data above suggest that GABAergic cells contributing to the GHT exert very little effect on spontaneous activity across the SCN but play a more substantial role in gating SCN responses to optic input. Based on these findings, we next aimed to understand the extent to which light‐driven changes in GHT output might modulate SCN responses to visual signals. Superficially, it might be assumed that, because the route for any potential light‐driven GHT output to reach the SCN is longer than that for direct retinal input to this region, these GHT‐derived inhibitory signals would be too slow to modulate SCN visual responses. On the other hand, visual stimuli relevant to the circadian system would be expected to produce sustained firing in RGCs (Wong, 2012) which, given the very persistent nature of the GHT‐driven inhibition (Fig. 7), could quite substantially modulate on‐going SCN light responses. To determine then how/whether GHT activity contributes to SCN visual signalling, we monitored light responses directly from the SCN via multi‐electrode recordings in anaesthetized mice, before and after unilateral inhibition of spiking in the visual thalamus.
To achieve robust inhibition of GHT signalling, we performed relatively large infusions of the GABAA receptor agonist muscimol (1 mm; 2–3 μl injection at 1 μl min−1) directly into the visual thalamus at the same time as monitoring multi‐unit firing via a linear multi‐electrode array attached to the drug‐delivery cannula (Fig. 8 A and B). This manipulation rapidly abolished spontaneous and light‐driven spiking activity at recording sites located up to 750 μm dorsal to the tip of the drug delivery cannula (Fig. 8 C). Assuming an approximately spherical spread of the drug (as observed for dye used to mark the electrode track; Fig. 8 A), we inferred then that this method of drug delivery inhibits spike firing across the vast majority of the visual thalamus (including that of cells contributing to the GHT).
We next turned our attention to the impact of GHT inhibition on light‐evoked activity within the SCN. Although our in vitro data suggested that GHT‐driven signals exert a stronger inhibitory effect during the projected day, we instead chose to perform these in vivo experiments during the early‐mid projected night (ZT 12–17). Hence, across this epoch, light‐driven SCN activity is more readily quantifiable and is associated with more robust circadian phase resetting effects (Meijer et al. 1996, 1998; Brown et al. 2011; van Oosterhout et al. 2012). Despite performing these recordings at a time when the GHT exerts its most modest effects, we still observed pronounced changes in multi‐unit SCN light responses following inhibition of the ipsilateral GHT projection (Fig. 8 D and G). In particular, there were substantially enhanced responses when light pulses (405 nm, 5 s, 11.4–15.4 log effective photons/cm−2 s−1) were applied to just the contralateral or to both eyes (F test for identical irradiance response relationship pre‐ vs. post‐muscimol, P = 0.001 and P = 0.002 respectively; n = 63 light responsive sites from five mice; Fig. 8 G). This effect stemmed primarily from an increase in maximal response amplitude rather than sensitivity because, in both cases, sigmoidal fit parameters for pre‐ vs. post‐GHT inhibition data were identical when we normalized those datasets to exhibit a common maximal value (F test; P = 0.999 and P = 0.984; data not shown). We did not, however, detect any statistically significant changes in the irradiance response relationship for stimulation of the ipsilateral retina following muscimol application (F test; P = 0.118; Fig. 8 G). Taken together, these data suggest that the (inhibitory) GHT projection is primarily activated by crossed retinal inputs.
In additional sets of experiments, we evaluated the impact of inhibiting the contralateral GHT projection (Fig. 8 E and H) and assessed the possibility that our data above may have been influenced by spontaneous/circadian changes in SCN photic sensitivity over the tested recording epochs (with an identical protocol to the above but with no muscimol injection; Fig. 8 F and I). Importantly, when we did not infuse muscimol, there was no significant change in responsiveness to any of the stimuli employed over the tested recording epochs (F test; P = 0.250, P = 0.101 and P = 0.994, respectively, for changes in irradiance response relationship to contralateral, ipsilateral and binocular light steps; n = 127 light responsive sites from six mice; Fig. 8 I). Despite a trend towards increased SCN responses to stimulation of the ipsilateral retina, there was a similar lack of statistically significant differences in responsiveness following inhibition of the contralateral GHT (F test; P = 0.481, P = 0.06, P = 0.318, respectively, for contralateral, ipsilateral and binocular light steps; n = 33 light responsive sites from three mice; Fig. 8 F and H). These later data thus broadly align with findings from anatomical studies (Pickard, 1982; Mikkelsen, 1990; Moore et al. 2000) and our in vitro experiments (Fig. 6C) indicating that the contralateral GHT projection is weaker than that arising from the ipsilateral IGL/vLGN.
We also investigated whether inhibition of GHT signalling changed spontaneous activity within the SCN in any way. Although we found statistically significant increases in spontaneous firing under all conditions tested in Fig. 8 D and F (mean ± SEM: 123 ± 38 vs. 45 ± 15 and 9 ± 3% above baseline for ipsilateral, contralateral or no GHT inhibition, respectively, paired t tests, all P < 0.05), the magnitude of this effect was substantially greater following inhibition of the ipsilateral GHT (one‐way ANOVA with Dunnett's post hoc test, P < 0.001 vs. no inhibition). It is also worth noting that the average change in spontaneous population activity following inhibition of the ipsilateral GHT (6.1 ± 2.4 spikes s−1) was significantly smaller than the change in light‐evoked firing evoked by this manipulation (19.6 ± 2.4 spikes s−1 for 15.4 log photons cm−2 s−1 presented to both eyes; paired t test, P < 0.001).
Collectively, the data above suggest that, in vivo, inhibitory input from the ipsilateral GHT is active under low light conditions and is further increased by the activity of crossed retinal projections. This conclusion that GHT input is primarily stimulated by crossed retinal inputs derives from the lack of overt changes in the irradiance response relationship for SCN responses to ipsilateral retinal stimulation following thalamic inhibition (Fig. 8 G). However, because we have previously shown that many SCN neurons preferentially receive input from just one eye (Walmsley & Brown, 2015), an alternate explanation for these findings is that GHT inputs preferentially converge on SCN neurons receiving contralateral retinal input.
To evaluate this latter possibility, we next investigated whether artificially stimulating GHT activity (by optogenetic stimulation of GABAergic neurons in the IGL/vLGN region) could adjust SCN responses to contralateral and/or ipsilateral retinal stimulation. Accordingly, we implanted recording probes with attached optical fibres (‘optrodes’; for further details, see Methods) into the IGL region of GAD2‐Cre; Ai32 mice, allowing us to illuminate large portions of the IGL/vLGN and to monitor the resulting activity (Fig. 9 A and C). Using this approach, we then evaluated SCN multi‐unit responses (Fig. 9 D) to retinal stimulation (5 s steps, 15.4 log effective photons cm−2 s−1) in the absence or presence of trains of optogenetic stimulation (10 ms flashes at 5–20 Hz for 5 s).
Because there is a delay of 30–40 ms before visually evoked activity becomes first detectable within the SCN and LGN (van Diepen et al. 2013; Howarth et al. 2014; Walmsley & Brown, 2015), we reasoned that the first pulse of these stimulus trains should be sufficient to increase the inhibitory influence of the GHT prior to the arrival of any retinal input. Thus, assuming that GHT inputs do not preferentially converge on SCN neurons receiving contralateral retinal input, we expected that initial components of the SCN visual response should be inhibited regardless of which eye was stimulated. By contrast, we reasoned that the effect of continued optogenetic stimulation on SCN responses should be much greater under conditions when GHT activity is normally low (i.e. during ipsilateral visual stimulation).
Consistent with this reasoning, we found that the earliest components of the light‐evoked multi‐unit firing (0–250 ms after visual stimulation) were reliably reduced by concomitant optogenetic stimulation of the GHT regardless of which eye or stimulation frequency was used (n = 72 light responsive SCN sites from six mice; two‐way RM ANOVA with Sidak's post hoc tests; Fig. 9 E and F). These data thus confirm that the GHT is capable of modulating SCN responses to input from either retina. We also found that, although optogenetic stimulation at 5 and 10 Hz could also suppress later components (3–5 s after stimulus onset) of the SCN response to ipsilateral retinal inputs, there were no statistically significant reductions in the responses to visual stimuli that also involved the contralateral retina at any frequency tested (two‐way RM ANOVA with Sidak's post hoc tests; Fig. 9 E and G).
We interpret these latter data as reflecting the fact that the GHT is already much more active during contralateral retinal stimulation and, as such, further increasing this activity with optogenetic stimuli has negligible effects on the inhibitory signals sent to the SCN. Analysis of optogenetically‐driven changes in thalamic activity broadly supported this hypothesis (Fig. 9 H and I). Here, we specifically focused on recording sites in the IGL/vLGN region where we observed both visual and optogenetically evoked responses (n = 41 sites from six mice). Because only a subset of IGL/vLGN neurons contribute to the GHT, the population activity observed is not necessarily directly reflective of the signals being sent to the SCN. Nonetheless, although all sites exhibited very pronounced light‐induced firing during contralateral retinal stimulation, activity during stimulation of the ipsilateral retina was negligible (and, on average, slightly suppressed; Fig. 9 H and I).
Moreover, although optogenetic stimuli could significantly elevate thalamic activity spike firing during stimulation of either or both retinas (two‐way RM ANOVA with Sidak's post hoc tests; Fig. 9 H), the magnitude of the resulting changes was always substantially smaller than the nominal stimulation frequencies tested (mean ± SEM = 5.1±0.4 spikes s−1 for 20 Hz stimulation during binocular illumination; the largest change observed). We assume these observations reflect the fact that our optogenetic stimuli also substantially increase local inhibition within the thalamus, limiting the amount that we are able to increase overall firing rates. Regardless, the net result was that optogenetic stimulation produced only a marginal increase in the already high activity when paired with illumination of the contralateral retina but resulted in a qualitative change in (the much lower) thalamic activity when paired with stimulation of just the ipsilateral retina (Fig. 9 H and I).
As expected based on our in vitro data, optogenetic stimulation in the absence of concomitant retinal stimulation did not significantly alter SCN firing rates at any frequency tested (two‐way RM ANOVA with Sidak's post hoc test; all P > 0.05; not shown) despite producing significant, albeit modest changes in LGN firing activity (two‐way RM ANOVA with Sidak's post hoc test; P > 0.001 at all frequencies; maximal change in firing = 1.5 ± 0.3 spikes s−1 for 20 Hz stimulation).
In summary, the data above support our view that the primary action of the GHT is to modulate SCN responses to on‐going retinal input and establish that, although the GHT is capable of modulating SCN responses to input from either eye, the primary sensory signal driving GHT output is light detected by the contralateral retina.
Discussion
In the present study, we show that GHT neurons play an important role in gating SCN neuronal responses to optic input and actively modulate specific features of the SCN light response. Although existing data imply that GHT activity can oppose light‐induced phase resetting (Yannielli & Harrington, 2000; Lall & Biello, 2002, 2003; Yannielli et al. 2004), to our knowledge, this is the first report showing that the GHT directly modulates acute electrophysiological responses to light/retinal input. In addition, we establish a novel in vitro model for studying network processing within the circadian system. Using that model, we show that GABA released from the GHT specifically modulates SCN responses to retinal input with little effect on spontaneous activity. Moreover, we show that this modulatory action exhibits a pronounced circadian variation. Collectively, our data (Fig. 10) provide a mechanistic basis by which the GHT can shape temporal variations in the SCN response to retinal input and influence both circadian photoentrainment, as well as much more rapid light‐dependent changes in downstream physiology (e.g. hormone secretion).
Figure 10. Schematic of proposed geniculohypothalamic circuitry and its impact on SCN output.

A, based on the data reported in the present study, we propose that GABAergic neurons contributing to the GHT are primarily excited by contralateral retina inputs and convey the resulting inhibitory signals to SCN neurons that receive input from either (or both) eyes. An SCN‐intrinsic mechanism then adjusts SCN responses to retinal and GHT inputs according to time of day. B and C, a schematic illustrating inferred excitatory and inhibitory synaptic influences on SCN neurons (B) and the resulting spike output under varying illumination (C) for SCN cells receiving input from just the contra‐ (upper) and or ipsilateral (lower) eye, respectively. Following experimental illumination of just the contralateral retina (left), GHT input is activated and inhibitory inputs reach the SCN 50–100 ms after arrival of excitatory retinal inputs. For SCN cells that receive contralateral retinal inputs, the resulting long‐lasting inhibitory influence attenuates the excitatory retinal drive, leading to a suppression of steady state light‐driven firing rates (especially during the projected day when SCN neurons are more sensitive to GHT signals). By contrast, in the absence of any driving excitatory input (SCN cells receiving only ipsilateral input), spontaneous firing is unaffected. When only the ipsilateral retina is illuminated, the activity of the GHT is very low and therefore does not influence SCN responses (middle). Under the more commonly encountered situation in the natural world where both eyes are illuminated (right), inhibitory GHT signals provided by the GHT are able to reduce SCN responses regardless of which eye a cell receives input from.
Existing attempts to define how GHT signals act within the central clock have been hampered by the lack of an in vitro model that allows for experimental manipulation of this pathway. As such, in vitro studies have focused on pharmacological manipulation of NPY signalling as a proxy for GHT activity (Shibata & Moore, 1993; Biello et al. 1997; Gribkoff et al. 1998; Yannielli & Harrington, 2000). Although such information is useful, this approach provides at best an incomplete picture of the influence of the GHT. Hence, not all GHT cells produce NPY (Moore & Card, 1994; Morin & Blanchard, 2001), nor are the circumstances that trigger NPY release well understood. Similarly, although studies using pharmacological manipulation of GABA signalling provide data consistent with the idea that GHT‐derived GABA release may modulate circadian photic phase resetting (Gillespie et al. 1996, 1997; Novak & Albers, 2004), such approaches are unable to distinguish the specific role of the GHT vs. the many other possible sources of GABAergic modulation. Our angled slice model now provides a direct way of assessing the mechanisms by which GHT signals influence SCN activity.
In the present study, we employed two approaches to manipulate GHT signalling: electrical stimulation and targeted optogenetic activation of GAD2‐positive neurons. The former has the advantage that it should activate any cell contributing to the GHT, regardless of neurochemical phenotype. The downside of this approach, however, is that it carries the risk of indirectly triggering retinal input to the SCN as a result of antidromic activation of ipRGCs that send axon collaterals to the hypothalamus (Pickard, 1985; Treep et al. 1995; Morin et al. 2003). We consider that the modest number of excitatory SCN responses observed following electrical stimulation of the GHT was probably driven by this route. Hence, ipRGC inputs to the SCN are glutamatergic (Gompf et al. 2015), whereas almost all IGL/vLGN neurons are GABAergic (Moore & Card, 1994; Harrington, 1997; Lein et al. 2007) and the excitatory responses observed in the present study were blocked by glutamate but not GABA receptor antagonists.
As such, we used optogenetics to selectively activate GABAergic GHT projections. In line with the results obtained using electrical stimulation, changes in spontaneous SCN firing were very rarely detectable following optogenetic activation of the GABAergic GHT projection. By contrast, an influence of this pathway was readily apparent because a long‐lasting inhibition of evoked spiking (persisting for at least 1 s) at the majority of hypothalamic sites (∼60%) responding to stimulation of retinal afferents. In line with this in vitro data, optogenetic activation of the GHT in vivo could modulate visual responses in the SCN but did not result in any detectable changes in spontaneous firing. Similarly, pharmacological inhibition of the GHT in vivo produced much larger changes in light‐evoked vs. spontaneous SCN activity. Collectively, these observations indicate that our in vitro preparation preserves a substantial proportion of the GHT intact and suggest that the principle action of the GHT, at least under the conditions investigated in the present study, is to modulate SCN neuronal responses to excitatory input.
A number of features of our experimental design are worthy of further discussion. First, to experimentally demonstrate GHT‐driven modulatory effects in vitro, we stimulated the GHT before activating retinal afferents. Here, we find that GHT‐driven inhibition of OT‐evoked responses becomes detectable within at least 25 ms, consistent with what would be predicted from the latency of antidromically evoked IGL spikes following hypothalamic stimulation (Zhang & Rusak, 1989; Blasiak & Lewandowski, 2013). In the intact animal, RGC impulses will actually arrive in the SCN slightly earlier than in the IGL/vLGN (latencies for visual responses ∼30 vs. 40 ms: present study; van Diepen et al. 2013; Howarth et al. 2014; Walmsley & Brown, 2015). As such, the SCN response to a single RGC impulse should have terminated before any visually driven GHT‐dependent signal arrives. Importantly, however, visual stimuli sufficient to trigger meaningful circadian responses must necessarily generate prolonged trains of RGC impulses. Indeed, even after many hours of continuous light exposure, ipRGCs continue to fire at ∼10 spikes s−1 (Wong, 2012). Accordingly, during physiologically relevant trains of retinal input to the SCN, the GHT‐driven inhibition that we demonstrate in the present study is easily sufficiently long‐lasting (hundreds of milliseconds) to modulate SCN responses to all but the very earliest components of the SCN response to a visual stimulus (illustrated in schematic form in Fig. 10).
Second, most of the analysis presented here derives from multi‐unit recordings. It is formally possible that, by combining signals from a heterogeneous population neurons, only some of which receive GHT input, this use of multi‐unit data leads us to underestimate the impact of GHT signals on the recipient neurons. Analysis of single cell responses form a subset of our recordings indicates that such a possibility probably does not result in a substantial underestimation of the impact of GHT input. Hence, there were very few instances where we could observe an effect of GHT stimulation at the multi‐unit but not single unit level and, among single cells where we detected a significant influence of GHT‐input, the magnitude of the resulting responses was very similar to those reported for multi‐unit data.
By contrast, perhaps a more significant factor that could result in our in vitro data under‐representing the magnitude of GHT‐driven inhibitory input to SCN neurons is that we focused on investigating the effects of single‐pulse GHT stimulation. It is formally possible that GHT‐driven signals (arising via GABA and/or NPY release) could summate during high‐frequency stimulation to produce a greater inhibition. This might explain then why we find that in vivo inhibition of GHT activity (discussed in more detail below) produces a greater increase in visual response magnitude (∼100%) than would be predicted from our in vitro data at an equivalent circadian phase (∼25%). Alternatively/additionally, it is worth noting that, because the thalamic input to the SCN runs via the OT, the electrical stimulation used in our in vitro studies to activate retinal afferents will probably also directly stimulate GHT terminals, potentially occluding the effect of prior optical stimulation of the GHT.
In line with the idea that electrical stimulation of the OC does itself trigger some baseline level of GHT activity, we and others (Moldavan & Allen, 2013) find that GABA receptor blockade does indeed result in a general increase in the magnitude of OC‐evoked responses. On the other hand, the GHT is far from the only source of GABA release in the SCN. Hence, facilitatory effects of GABA receptor blockade on OC‐evoked responses do not necessarily confirm an endogenous GHT‐contribution of this kind. In this regard, it is noteworthy that the reductions in early visual responses observed following optical activation of the GHT in vivo (where we expect one optogenetically‐driven GHT impulse to reach the SCN before any light‐driven GHT signals do) are broadly similar in magnitude to those we report in vitro at an equivalent circadian phase.
On balance then, although our present study leaves these latter issues unresolved to some extent, the inhibitory actions of GHT‐stimulation that we report in vitro should perhaps be considered as a conservative estimate of the extent to which these inputs regulate SCN activity. Despite this caveat, it is abundantly clear from both our in vitro and in vivo data that GHT‐derived signals exert a much more robust effect on SCN responses to retinal input than on spontaneous firing. Such an arrangement may, in principle arise simply because the low spontaneous activity of SCN neurons ensures that the functional impact of GHT‐dependent inhibitory signals is hard to detect in the absence of a driving input. We consider it most improbable that this constitutes the primary explanation for the apparent lack of effect of GHT‐stimulation on spontaneous firing. Indeed, the design of these experiments (where we evaluated responses to hundreds of stimulus repeats) was specifically tailored to enable us to detect inhibitory responses even when spontaneous activity was relatively low.
An alternative explanation then is that GHT inputs might be arranged such that they can specifically gate SCN responses to retinal signals without exerting major effects on spontaneous firing. In this regard, NPY cells reportedly form axodendritic contacts with SCN neurons (Pelletier, 1990). Moreover, it has previously been demonstrated that retinal and GABAergic terminals (potentially including those originating from the GHT) are typically found in very close proximity within the SCN, often converging on the same postsynaptic dendrite (Castel et al. 1993). Because spontaneous SCN firing is primarily generated within the soma, such an arrangement should indeed allow GABA released by this population of GHT cells to preferentially gate responses to excitatory input. In addition, the presence of GABAB receptors almost exclusively located on (nearby) retinal terminals leaves open the possibility that GABA spillover might also presynaptically modulate retinal input (Jiang et al. 1995; Moldavan et al. 2006; Belenky et al. 2008). In any of the above situations, however, the net effect is that the functional impacts of GHT signals are most clearly evident in the face of on‐going retinal transmission.
It is worth noting that pharmacological activation of NPY receptors is able to suppress spontaneous firing in the SCN (Gribkoff et al. 1998). By contrast, we found that inhibitory responses to optogenetic activation of the GHT could be almost entirely blocked by GABA receptor antagonists. It appears then that, under our experimental conditions, NPY released from the GHT does not exert a major effect. In principle, it is formally possible that the approaches we used preferentially activate non‐NPY cells. This is probably not the case, however, because almost all GABAergic neurons produce GAD2 (Zhao et al. 2013), the expression of which is itself very reliably reported by the GAD2‐Cre line (Taniguchi et al. 2011). Certainly, our data indicate very robust ChR2/EYFP expression in the IGL/vLGN region. Instead then, we suggest that specific patterns of presynaptic terminal activity may be required to evoke endogenous NPY release, with the short bursts of firing evoked by our test stimuli primarily acting via triggering GABA release.
Significantly, we identify a pronounced rhythm in the functional impact of this GHT‐derived GABA release, with two‐fold greater inhibition of OC‐evoked spiking during the mid‐late projected day vs. the early projected night. As discussed below, these observations provide a potential mechanism by which signals from the GHT could contribute to shaping the characteristic temporal gating of the SCN light response/clock resetting (Meijer et al. 1996, 1998; Brown et al. 2011; van Oosterhout et al. 2012). The effect that we observe in vitro is not, however, associated with any appreciable change in responses recorded directly from the IGL/vLGN region. Accordingly, we interpret these data as reflecting a circadian variation in the impact of GHT‐derived GABA release at the level of the SCN neurons and/or retinal terminals. In line with this latter view, the results of a previous study indicate that GABAB receptor‐mediated presynaptic inhibition of retinohypothalamic transmission is enhanced during the projected day (Moldavan & Allen, 2013). We should note, however, that it is formally possible that GHT‐derived GABA release might exhibit circadian variation even though IGL/vLGN firing does not itself change over the circadian day.
In interpreting the functional significance of the in vitro data discussed above, a key area to consider is the nature of the sensory signals that actually activate GHT transmission. As a result of the challenging nature of directly identifying GHT cells in electrophysiological recordings, existing attempts to address this deficit have not provided a clear answer (Harrington & Rusak, 1989; Zhang & Rusak, 1989; Thankachan & Rusak, 2005; Blasiak & Lewandowski, 2013). Although we do not directly investigate this in the present study, our in vivo experiments employing thalamic inhibition clearly indicate that, during visual stimulation, activity of the ipsilateral GHT reduces SCN population responses specifically to stimuli that target the contralateral retina.
As a result of the extensive interconnections among the SCN, IGL/vLGN and pretectum (Morin & Allen, 2006), thalamic inhibition could influence SCN light responses via various routes. Nonetheless, the simplest interpretation of our data is that the inhibitory GABAergic neurons that form the GHT themselves primarily receive ON‐excitatory input from crossed retinal projections. Consistent with this view, we rule out one important alternative explanation for the above data (i.e. that the GHT preferentially impinges on SCN neurons receiving contralateral retinal input; Walmsley & Brown, 2015). Hence, we show that optogenetic activation of the GHT in vivo is capable of reducing early components (first 250 ms) of the SCN response to both ipsilateral and contralateral input. We also show that maintained optogenetic stimulation has negligible effects on later components of the SCN response to contralateral retinal stimulation, as expected if endogenous activity of the GHT is already high. By contrast, maintained optogenetic stimulation at 5 and 10 Hz produced continued inhibition of responses driven by the ipsilateral retina, consistent with a much lower level of endogenous GHT activity under these conditions.
Regarding interpretation of this latter element of our data, we should note that, at a higher optogenetic stimulation frequency (20 Hz), this inhibition of on‐going ipsilateral retinal‐driven activity disappeared. This is despite the fact that our analysis of population activity in the IGL/vLGN region during the same recordings revealed significantly increased spiking activity (of a broadly similar magnitude to that observed at lower frequencies). Because only a subset of neurons in the IGL/vLGN contribute to the GHT, we assume then that the thalamic data recorded in the present study is not entirely representative of the activity of GHT neurons. Alternatively, it is formally possible that the specific spiking pattern of GHT afferents is also an important determinant of the overall impact on the SCN. In either case, our general conclusion that GHT signalling is primarily activated by the illumination of the contralateral retina is certainly supported by our observations of light evoked activity in the IGL/vLGN (i.e. present study and previous large scale recordings of sensory responses in this region: Howarth et al. 2014), which indicates that excitatory responses to stimulation of the ipsilateral retina are rare (present in only ∼25% of cells).
Of note, the enhancement of SCN visual responses that we observed following thalamic inhibition showed no signs of diminishing across the duration of the visual stimulations tested in the present study. Again, this is consistent with both the long‐lasting effects of GHT activity that we observe in vitro and the sustained visual responses driven by ipRGCs that provide a major source of input to the IGL/vLGN (Brown et al. 2010; Wong, 2012). As such, it appears that the GHT modulates the irradiance coding properties of SCN neurons. Based on the relationship between SCN firing and irradiance, the magnitude of this effect is large for relatively bright stimuli (>1014 photons cm−2 s−1; equivalent to those encountered around sunrise/sunset). Indeed, for contralateral light steps, we found that ∼10‐fold lower light levels were required to produce SCN population firing responses equivalent to those attained before inhibition of the GHT.
Most significantly, the in vivo recordings discussed above occurred across a time epoch when the GHT was expected to exert its weakest effects. Indeed, assuming a two‐fold greater effect of the GHT input during the day (as predicted by our in vitro data), it appears that input from the GHT could be sufficient to account for a substantial component of the circadian modulation in light‐driven SCN activity observed previously (three‐ to four‐fold variation in magnitude at submaximal intensity; Meijer et al. 1998; Brown et al. 2011; van Oosterhout et al. 2012). An intriguing question then is to what extent does the GHT helps to define the phase response curve for behavioural photoentrainment?
Certainly, based on data from IGL‐lesioned hamsters, the GHT is not an absolute requirement for photoentrainment under most circumstances (Harrington & Rusak, 1986; Pickard et al. 1987; Johnson et al. 1989; Maywood et al. 1997; Edelstein & Amir, 1999a,b; Morin & Pace, 2002; Muscat & Morin, 2006; Evans et al. 2012). Nonetheless, we consider it noteworthy that, in mice, behavioural phase resetting responses to light are largest during the first half of the projected night (Daan & Pittendrigh, 1976), a portion of the day when the inhibitory effects of GHT‐activation are at their lowest ebb. Insofar as we have previously shown that the magnitude of light‐evoked SCN firing is predictive of mouse circadian phase resetting responses (Brown et al. 2011), our data are thus consistent with the possibility that GHT inputs contribute to reducing circadian responses to light around dawn and across the projected day.
In summary, our data provide new insight into the contributions of the GHT to visual processing within the SCN. We show that the GHT actively modulates SCN responses to retinal input and demonstrate a circadian variation in the magnitude of this effect that is consistent with an important role in the temporal gating of photic responses, a fundamental property of the circadian system. We also establish a new model system for studying geniculohypothalamic communication, which we expect to have widespread utility for many further investigations of how network interactions across the extended circadian system contribute to daily rhythms in physiology and behaviour.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
TMB, LH and LW designed the experiments. LH, LW, AP, MH and TMB collected and analysed the data. TMB and LH wrote the manuscript. All authors approved the final version of the manuscript, agree to be accountable for all aspects of the work and all persons designated as authors qualify for authorship. All experiments were performed at the University of Manchester, UK.
Funding
This work was funded by a Biotechnology and Biological Sciences Research Council (BBSRC, UK) David Philips Fellowship to TMB (BB/I017836/1). LW was funded by a BBSRC doctoral training grant and strategic skills award.
References
- Abrahamson EE & Moore RY (2001). Suprachiasmatic nucleus in the mouse: retinal innervation, intrinsic organization and efferent projections. Brain Res 916, 172–191. [DOI] [PubMed] [Google Scholar]
- Belenky MA, Yarom Y & Pickard GE (2008). Heterogeneous expression of gamma‐aminobutyric acid and gamma‐aminobutyric acid‐associated receptors and transporters in the rat suprachiasmatic nucleus. J Comp Neurol 506, 708–732. [DOI] [PubMed] [Google Scholar]
- Biello SM, Golombek DA & Harrington ME (1997). Neuropeptide Y and glutamate block each other's phase shifts in the suprachiasmatic nucleus in vitro. Neuroscience 77, 1049–1057. [DOI] [PubMed] [Google Scholar]
- Blasiak T & Lewandowski MH (2013). Differential firing pattern and response to lighting conditions of rat intergeniculate leaflet neurons projecting to suprachiasmatic nucleus or contralateral intergeniculate leaflet. Neuroscience 228, 315–324. [DOI] [PubMed] [Google Scholar]
- Brown TM, Banks JR & Piggins HD (2006). A novel suction electrode recording technique for monitoring circadian rhythms in single and multiunit discharge from brain slices. J Neurosci Methods 156, 173–181. [DOI] [PubMed] [Google Scholar]
- Brown TM, Gias C, Hatori M, Keding SR, Semo M, Coffey PJ, Gigg J, Piggins HD, Panda S & Lucas RJ (2010). Melanopsin contributions to irradiance coding in the thalamo‐cortical visual system. PLoS Biol 8, e1000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TM, McLachlan E & Piggins HD (2008). Angiotensin II regulates the activity of mouse suprachiasmatic nuclei neurons. Neuroscience 154, 839–847. [DOI] [PubMed] [Google Scholar]
- Brown TM & Piggins HD (2007). Electrophysiology of the suprachiasmatic circadian clock. Prog Neurobiol 82, 229–255. [DOI] [PubMed] [Google Scholar]
- Brown TM & Piggins HD (2009). Spatiotemporal heterogeneity in the electrical activity of suprachiasmatic nuclei neurons and their response to photoperiod. J Biol Rhythms 24, 44–54. [DOI] [PubMed] [Google Scholar]
- Brown TM, Wynne J, Piggins HD & Lucas RJ (2011). Multiple hypothalamic cell populations encoding distinct visual information. J Physiol 589, 1173–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel M, Belenky M, Cohen S, Ottersen OP & Storm‐Mathisen J (1993). Glutamate‐like immunoreactivity in retinal terminals of the mouse suprachiasmatic nucleus. Eur J Neurosci 5, 368–381. [DOI] [PubMed] [Google Scholar]
- Chen SK, Badea TC & Hattar S (2011). Photoentrainment and pupillary light reflex are mediated by distinct populations of ipRGCs. Nature 476, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Lee CJ, Schroeder A, Kim YS, Jung SH, Kim JS, dY Kim, Son EJ, Han HC, Hong SK, Colwell CS & Kim YI (2008). Excitatory actions of GABA in the suprachiasmatic nucleus. J Neurosci 28, 5450–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler DJ, Haraura M, Reed HE, Shen S, Sheward WJ, Morrison CF, Marston HM, Harmar AJ & Piggins HD (2003). The mouse VPAC2 receptor confers suprachiasmatic nuclei cellular rhythmicity and responsiveness to vasoactive intestinal polypeptide in vitro. Eur J Neurosci 17, 197–204. [DOI] [PubMed] [Google Scholar]
- Daan, S & Pittendrigh C (1976). A Functional Analysis of Circadian Pacemakers in Nocturnal Rodents: II. The Variability of Phase Response Curves. J Comp Physiol 106, 253–266. [Google Scholar]
- Edelstein K & Amir S (1999a). The intergeniculate leaflet does not mediate the disruptive effects of constant light on circadian rhythms in the rat. Neuroscience 90, 1093–1101. [DOI] [PubMed] [Google Scholar]
- Edelstein K & Amir S (1999b). The role of the intergeniculate leaflet in entrainment of circadian rhythms to a skeleton photoperiod. J Neurosci 19, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JA, Carter SN, Freeman DA & Gorman MR (2012). Dim nighttime illumination alters photoperiodic responses of hamsters through the intergeniculate leaflet and other photic pathways. Neuroscience 202, 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farajnia S, van Westering TL, Meijer JH & Michel S (2014). Seasonal induction of GABAergic excitation in the central mammalian clock. Proc Natl Acad Sci USA 111, 9627–9632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie CF, Huhman KL, Babagbemi TO & Albers HE (1996). Bicuculline increases and muscimol reduces the phase‐delaying effects of light and VIP/PHI/GRP in the suprachiasmatic region. J Biol Rhythms 11, 137–144. [DOI] [PubMed] [Google Scholar]
- Gillespie CF, Mintz EM, Marvel CL, Huhman KL & Albers HE (1997). GABA(A) and GABA(B) agonists and antagonists alter the phase‐shifting effects of light when microinjected into the suprachiasmatic region. Brain Res 759, 181–189. [DOI] [PubMed] [Google Scholar]
- Gompf HS, Fuller PM, Hattar S, Saper CB & Lu J (2015). Impaired circadian photosensitivity in mice lacking glutamate transmission from retinal melanopsin cells. J Biol Rhythms 30, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González JA & Dyball RE (2006). Pinealectomy reduces optic nerve but not intergeniculate leaflet input to the suprachiasmatic nucleus at night. J Neuroendocrinol 18, 146–153. [DOI] [PubMed] [Google Scholar]
- Govardovskii VI, Fyhrquist N, Reuter T, Kuzmin DG & Donner K (2000). In search of the visual pigment template. Vis Neurosci 17, 509–528. [DOI] [PubMed] [Google Scholar]
- Green DJ & Gillette R (1982). Circadian rhythm of firing rate recorded from single cells in the rat suprachiasmatic brain slice. Brain Res 245, 198–200. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Pieschl RL, Wisialowski TA, van den Pol AN & Yocca FD (1998). Phase shifting of circadian rhythms and depression of neuronal activity in the rat suprachiasmatic nucleus by neuropeptide Y: mediation by different receptor subtypes. J Neurosci 18, 3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groos G & Hendriks J (1982). Circadian rhythms in electrical discharge of rat suprachiasmatic neurones recorded in vitro. Neurosci Lett 34, 283–288. [DOI] [PubMed] [Google Scholar]
- Güler AD, Altimus CM, Ecker JL & Hattar S (2007). Multiple photoreceptors contribute to nonimage‐forming visual functions predominantly through melanopsin‐containing retinal ganglion cells. Cold Spring Harb Symp Quant Biol 72, 509–515. [DOI] [PubMed] [Google Scholar]
- Harrington ME ( 1997). The ventral lateral geniculate nucleus and the intergeniculate leaflet: interrelated structures in the visual and circadian systems. Neurosci Biobehav Rev 21, 705–727. [DOI] [PubMed] [Google Scholar]
- Harrington ME & Rusak B (1986). Lesions of the thalamic intergeniculate leaflet alter hamster circadian rhythms. J Biol Rhythms 1, 309–325. [DOI] [PubMed] [Google Scholar]
- Harrington ME & Rusak B (1989). Photic responses of geniculo‐hypothalamic tract neurons in the Syrian hamster. Vis Neurosci 2, 367–375. [DOI] [PubMed] [Google Scholar]
- Hattar S, Kumar M, Park A, Tong P, Tung J, Yau KW & Berson DM (2006). Central projections of melanopsin‐expressing retinal ganglion cells in the mouse. J Comp Neurol 497, 326–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL (1998). An alternate pathway for visual signal integration into the hypothalamo‐pituitary axis: retinorecipient intergeniculate neurons project to various regions of the hypothalamus and innervate neuroendocrine cells including those producing dopamine. J Neurosci 18, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth M, Walmsley L & Brown TM (2014). Binocular integration in the mouse lateral geniculate nuclei. Curr Biol 24, 1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhman KL & Albers HE (1994). Neuropeptide Y microinjected into the suprachiasmatic region phase shifts circadian rhythms in constant darkness. Peptides 15, 1475–1478. [DOI] [PubMed] [Google Scholar]
- Huhman KL, Gillespie CF, Marvel CL & Albers HE (1996). Neuropeptide Y phase shifts circadian rhythms in vivo via a Y2 receptor. Neuroreport 7, 1249–1252. [DOI] [PubMed] [Google Scholar]
- Huhman KL, Marvel CL, Gillespie CF, Mintz EM & Albers HE (1997). Tetrodotoxin blocks NPY‐induced but not muscimol‐induced phase advances of wheel‐running activity in Syrian hamsters. Brain Res 772, 176–180. [DOI] [PubMed] [Google Scholar]
- Irwin RP & Allen CN (2009). GABAergic signalling induces divergent neuronal Ca2+ responses in the suprachiasmatic nucleus network. Eur J Neurosci 30, 1462–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itri JN, Michel S, Vansteensel MJ, Meijer JH & Colwell CS (2005). Fast delayed rectifier potassium current is required for circadian neural activity. Nat Neurosci 8, 650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs GH & Williams GA (2007). Contributions of the mouse UV photopigment to the ERG and to vision. Doc Ophthalmol 115, 137–144. [DOI] [PubMed] [Google Scholar]
- Janik D & Mrosovsky N (1994). Intergeniculate leaflet lesions and behaviourally‐induced shifts of circadian rhythms. Brain Res 651, 174–182. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Allen CN & North RA (1995). Presynaptic inhibition by baclofen of retinohypothalamic excitatory synaptic transmission in rat suprachiasmatic nucleus. Neuroscience 64, 813–819. [DOI] [PubMed] [Google Scholar]
- Johnson RF, Moore RY & Morin LP (1989). Lateral geniculate lesions alter circadian activity rhythms in the hamster. Brain Res Bull 22, 411–422. [DOI] [PubMed] [Google Scholar]
- Johnson RF, Smale L, Moore RY & Morin LP (1988). Lateral geniculate lesions block circadian phase‐shift responses to a benzodiazepine. Proc Natl Acad Sci USA 85, 5301–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsbeek A, Teclemariam‐Mesbah R & Pévet P (1993). Efferent projections of the suprachiasmatic nucleus in the golden hamster (Mesocricetus auratus). J Comp Neurol 332, 293–314. [DOI] [PubMed] [Google Scholar]
- Kubota A, Inouye ST & Kawamura H (1981). Reversal of multiunit activity within and outside the suprachiasmatic nucleus in the rat. Neurosci Lett 27, 303–308. [DOI] [PubMed] [Google Scholar]
- Lall GS & Biello SM (2002). Attenuation of phase shifts to light by activity or neuropeptide Y: a time course study. Brain Res 957, 109–116. [DOI] [PubMed] [Google Scholar]
- Lall GS & Biello SM (2003). Attenuation of circadian light induced phase advances and delays by neuropeptide Y and a neuropeptide Y Y1/Y5 receptor agonist. Neuroscience 119, 611–618. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA & Jones AR (2007). Genome‐wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Long MA, Jutras MJ, Connors BW & Burwell RD (2005). Electrical synapses coordinate activity in the suprachiasmatic nucleus. Nat Neurosci 8, 61–66. [DOI] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AJ, Gu X, Zanella S, Kidney J, Gu H, Mao Y, Hooks BM, Boyden ES, Buzsáki G, Ramirez JM, Jones AR, Svoboda K, Han X, Turner EE & Zeng H (2012). A toolbox of Cre‐dependent optogenetic transgenic mice for light‐induced activation and silencing. Nat Neurosci 15, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant EG, Watson NV & Mistlberger RE (1997). Both neuropeptide Y and serotonin are necessary for entrainment of circadian rhythms in mice by daily treadmill running schedules. J Neurosci 17, 7974–7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, Smith E, Hall SJ & Hastings MH (1997). A thalamic contribution to arousal‐induced, non‐photic entrainment of the circadian clock of the Syrian hamster. Eur J Neurosci 9, 1739–1747. [DOI] [PubMed] [Google Scholar]
- Meijer JH, Watanabe K, Détàri L & Schaap J (1996). Circadian rhythm in light response in suprachiasmatic nucleus neurons of freely moving rats. Brain Res 741, 352–355. [DOI] [PubMed] [Google Scholar]
- Meijer JH, Watanabe K, Schaap J, Albus H & Détári L (1998). Light responsiveness of the suprachiasmatic nucleus: long‐term multiunit and single‐unit recordings in freely moving rats. J Neurosci 18, 9078–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen JD (1990). Projections from the lateral geniculate nucleus to the hypothalamus of the Mongolian gerbil (Meriones unguiculatus): an anterograde and retrograde tracing study. J Comp Neurol 299, 493–508. [DOI] [PubMed] [Google Scholar]
- Mikkelsen JD (1992). The organization of the crossed geniculogeniculate pathway of the rat: a Phaseolus vulgaris‐leucoagglutinin study. Neuroscience 48, 953–962. [DOI] [PubMed] [Google Scholar]
- Moldavan MG & Allen CN (2013). GABAB receptor‐mediated frequency‐dependent and circadian changes in synaptic plasticity modulate retinal input to the suprachiasmatic nucleus. J Physiol 591, 2475–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldavan MG, Irwin RP & Allen CN (2006). Presynaptic GABA(B) receptors regulate retinohypothalamic tract synaptic transmission by inhibiting voltage‐gated Ca2+ channels. J Neurophysiol 95, 3727–3741. [DOI] [PubMed] [Google Scholar]
- Moore RY & Card JP (1994). Intergeniculate leaflet: an anatomically and functionally distinct subdivision of the lateral geniculate complex. J Comp Neurol 344, 403–430. [DOI] [PubMed] [Google Scholar]
- Moore RY, Weis R & Moga MM (2000). Efferent projections of the intergeniculate leaflet and the ventral lateral geniculate nucleus in the rat. J Comp Neurol 420, 398–418. [DOI] [PubMed] [Google Scholar]
- Morin LP & Allen CN (2006). The circadian visual system, 2005. Brain Res Rev 51, 1–60. [DOI] [PubMed] [Google Scholar]
- Morin LP & Blanchard J (1995). Organization of the hamster intergeniculate leaflet: NPY and ENK projections to the suprachiasmatic nucleus, intergeniculate leaflet and posterior limitans nucleus. Vis Neurosci 12, 57–67. [DOI] [PubMed] [Google Scholar]
- Morin LP & Blanchard JH (2001). Neuromodulator content of hamster intergeniculate leaflet neurons and their projection to the suprachiasmatic nucleus or visual midbrain. J Comp Neurol 437, 79–90. [DOI] [PubMed] [Google Scholar]
- Morin LP, Blanchard JH & Provencio I (2003). Retinal ganglion cell projections to the hamster suprachiasmatic nucleus, intergeniculate leaflet, and visual midbrain: bifurcation and melanopsin immunoreactivity. J Comp Neurol 465, 401–416. [DOI] [PubMed] [Google Scholar]
- Morin LP, Goodless‐Sanchez N, Smale L & Moore RY (1994). Projections of the suprachiasmatic nuclei, subparaventricular zone and retrochiasmatic area in the golden hamster. Neuroscience 61, 391–410. [DOI] [PubMed] [Google Scholar]
- Morin LP & Pace L (2002). The intergeniculate leaflet, but not the visual midbrain, mediates hamster circadian rhythm response to constant light. J Biol Rhythms 17, 217–226. [DOI] [PubMed] [Google Scholar]
- Mrugala M, Zlomanczuk P, Jagota A & Schwartz WJ (2000). Rhythmic multiunit neural activity in slices of hamster suprachiasmatic nucleus reflect prior photoperiod. Am J Physiol Regul Integr Comp Physiol 278, R987–R994. [DOI] [PubMed] [Google Scholar]
- Muscat L & Morin LP (2006). Intergeniculate leaflet: contributions to photic and non‐photic responsiveness of the hamster circadian system. Neuroscience 140, 305–320. [DOI] [PubMed] [Google Scholar]
- Nakamura W, Yamazaki S, Nakamura TJ, Shirakawa T, Block GD & Takumi T (2008). In vivo monitoring of circadian timing in freely moving mice. Curr Biol 18, 381–385. [DOI] [PubMed] [Google Scholar]
- Novak CM & Albers HE (2004). Circadian phase alteration by GABA and light differs in diurnal and nocturnal rodents during the day. Behav Neurosci 118, 498–504. [DOI] [PubMed] [Google Scholar]
- Novak CM, Ehlen JC, Huhman KL & Albers HE (2004). GABA(B) receptor activation in the suprachiasmatic nucleus of diurnal and nocturnal rodents. Brain Res Bull 63, 531–535. [DOI] [PubMed] [Google Scholar]
- Nygård M, Hill RH, Wikström MA & Kristensson K (2005). Age‐related changes in electrophysiological properties of the mouse suprachiasmatic nucleus in vitro. Brain Res Bull 65, 149–154. [DOI] [PubMed] [Google Scholar]
- Paxinos G & Franklin K (2001). The Mouse Brain in Stereotaxic Coordinates, 2nd edn Academic Press, San Diego, CA. [Google Scholar]
- Pelletier G (1990). Ultrastructural localization of neuropeptide Y in the hypothalamus. Ann NY Acad Sci 611, 232–246. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, de Jeu MT, Bos NP, Schaap J & Geurtsen AM (2002). Diurnal modulation of pacemaker potentials and calcium current in the mammalian circadian clock. Nature 416, 286–290. [DOI] [PubMed] [Google Scholar]
- Pickard GE (1982). The afferent connections of the suprachiasmatic nucleus of the golden hamster with emphasis on the retinohypothalamic projection. J Comp Neurol 211, 65–83. [DOI] [PubMed] [Google Scholar]
- Pickard GE (1985). Bifurcating axons of retinal ganglion cells terminate in the hypothalamic suprachiasmatic nucleus and the intergeniculate leaflet of the thalamus. Neurosci Lett 55, 211–217. [DOI] [PubMed] [Google Scholar]
- Pickard GE, Ralph MR & Menaker M (1987). The intergeniculate leaflet partially mediates effects of light on circadian rhythms. J Biol Rhythms 2, 35–56. [DOI] [PubMed] [Google Scholar]
- Reinhard K, Tikidji‐Hamburyan A, Seitter H, Idrees S, Mutter M, Benkner B & Münch TA (2014). Step‐by‐step instructions for retina recordings with perforated multi electrode arrays. PLoS ONE 9, e106148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig JA, Granados‐Fuentes D & Aguilar‐Roblero R (1997). Neuronal subpopulations in the suprachiasmatic nuclei based on their response to retinal and intergeniculate leaflet stimulation. Neuroreport 8, 885–889. [DOI] [PubMed] [Google Scholar]
- Rusak B, Meijer JH & Harrington ME (1989). Hamster circadian rhythms are phase‐shifted by electrical stimulation of the geniculo‐hypothalamic tract. Brain Res 493, 283–291. [DOI] [PubMed] [Google Scholar]
- Sakhi K, Wegner S, Belle MD, Howarth M, Delagrange P, Brown TM & Piggins HD (2014). Intrinsic and extrinsic cues regulate the daily profile of mouse lateral habenula neuronal activity. J Physiol 592, 5025–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaap J, Albus H, VanderLeest HT, Eilers PH, Détári L & Meijer JH (2003). Heterogeneity of rhythmic suprachiasmatic nucleus neurons: Implications for circadian waveform and photoperiodic encoding. Proc Natl Acad Sci USA 100, 15994–15999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaap J, Bos NP, de Jeu MT, Geurtsen AM, Meijer JH & Pennartz CM (1999). Neurons of the rat suprachiasmatic nucleus show a circadian rhythm in membrane properties that is lost during prolonged whole‐cell recording. Brain Res 815, 154–166. [DOI] [PubMed] [Google Scholar]
- Shibata S & Moore RY (1993). Neuropeptide Y and optic chiasm stimulation affect suprachiasmatic nucleus circadian function in vitro. Brain Res 615, 95–100. [DOI] [PubMed] [Google Scholar]
- Shibata S, Oomura Y, Kita H & Hattori K (1982). Circadian rhythmic changes of neuronal activity in the suprachiasmatic nucleus of the rat hypothalamic slice. Brain Res 247, 154–158. [DOI] [PubMed] [Google Scholar]
- Smith RD, Inouye S & Turek FW (1989). Central administration of muscimol phase‐shifts the mammalian circadian clock. J Comp Physiol A 164, 805–814. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Kvitsani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB & Huang ZJ (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcheng TK & Gillette MU (1996). A novel carbon fibre bundle microelectrode and modified brain slice chamber for recording long‐term multiunit activity from brain slices. J Neurosci Methods 69, 163–169. [DOI] [PubMed] [Google Scholar]
- Thankachan S & Rusak B (2005). Juxtacellular recording/labelling analysis of physiological and anatomical characteristics of rat intergeniculate leaflet neurons. J Neurosci 25, 9195–9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tousson E & Meissl H (2004). Suprachiasmatic nuclei grafts restore the circadian rhythm in the paraventricular nucleus of the hypothalamus. J Neurosci 24, 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treep JA, Abe H, Rusak B & Goguen DM (1995). Two distinct retinal projections to the hamster suprachiasmatic nucleus. J Biol Rhythms 10, 299–307. [DOI] [PubMed] [Google Scholar]
- van Diepen HC, Ramkisoensing A, Peirson SN, Foster RG & Meijer JH (2013). Irradiance encoding in the suprachiasmatic nuclei by rod and cone photoreceptors. FASEB J 27, 4204–4212. [DOI] [PubMed] [Google Scholar]
- van Oosterhout F, Fisher SP, van Diepen HC, Watson TS, Houben T, VanderLeest HT, Thompson S, Peirson SN, Foster RG & Meijer JH (2012). Ultraviolet light provides a major input to non‐image‐forming light detection in mice. Curr Biol 22, 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLeest HT, Houben T, Michel S, Deboer T, Albus H, Vansteensel MJ, Block GD & Meijer JH (2007). Seasonal encoding by the circadian pacemaker of the SCN. Curr Biol 17, 468–473. [DOI] [PubMed] [Google Scholar]
- vanderLeest HT, Vansteensel MJ, Duindam H, Michel S & Meijer JH (2009). Phase of the electrical activity rhythm in the SCN in vitro not influenced by preparation time. Chronobiol Int 26, 1075–1089. [DOI] [PubMed] [Google Scholar]
- Wagner S, Castel M, Gainer H & Yarom Y (1997). GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature 387, 598–603. [DOI] [PubMed] [Google Scholar]
- Walmsley L & Brown TM (2015). Eye‐specific visual processing in the mouse suprachiasmatic nuclei. J Physiol 593, 1731–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley L, Hanna L, Mouland J, Martial F, West A, Smedley AR, Bechtold DA, Webb AR, Lucas RJ & Brown TM (2015). Colour as a signal for entraining the mammalian circadian clock. PLoS Biol 13, e1002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AG, Swanson LW & Sanchez‐Watts G (1987). Efferent projections of the suprachiasmatic nucleus: I. Studies using anterograde transport of Phaseolus vulgaris leucoagglutinin in the rat. J Comp Neurol 258, 204–229. [DOI] [PubMed] [Google Scholar]
- Wickland C & Turek FW (1994). Lesions of the thalamic intergeniculate leaflet block activity‐induced phase shifts in the circadian activity rhythm of the golden hamster. Brain Res 660, 293–300. [DOI] [PubMed] [Google Scholar]
- Wong KY ( 2012). A retinal ganglion cell that can signal irradiance continuously for 10 hours. J Neurosci 32, 11478–11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Kerbeshian MC, Hocker CG, Block GD & Menaker M (1998). Rhythmic properties of the hamster suprachiasmatic nucleus in vivo. J Neurosci 18, 10709–10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannielli PC, Brewer JM & Harrington ME (2004). Blockade of the NPY Y5 receptor potentiates circadian responses to light: complementary in vivo and in vitro studies. Eur J Neurosci 19, 891–897. [DOI] [PubMed] [Google Scholar]
- Yannielli PC & Harrington ME (2000). Neuropeptide Y applied in vitro can block the phase shifts induced by light in vivo. Neuroreport 11, 1587–1591. [PubMed] [Google Scholar]
- Zhang DX & Rusak B (1989). Photic sensitivity of geniculate neurons that project to the suprachiasmatic nuclei or the contralateral geniculate. Brain Res 504, 161–164. [DOI] [PubMed] [Google Scholar]
- Zhao C, Eisinger B & Gammie SC (2013). Characterization of GABAergic neurons in the mouse lateral septum: a double fluorescence in situ hybridization and immunohistochemical study using tyramide signal amplification. PLoS ONE 8, e73750. [DOI] [PMC free article] [PubMed] [Google Scholar]