

Summary
Mucopolysaccharidosis IV A, better known as Morquio-A syndrome, is a rare condition with severe skeletal and multiorgan involvement. Sometimes is not easy to differentiate from other skeletal dysplasias. Prior to definitive diagnosis, patients have been delayed or misdiagnosis due to lack of knowledge of local physicians about this disease. The aim of this study is to compare the age of onset of clinical manifestations, age of diagnosis, as seen by the parent or primary caregiver and compare this age with other population reports worldwide. Self-administered questionnaires were conducted to the primary caregiver of confirmed patients, collecting information about the onset of symptoms, age, previous diagnoses and biological variables (age, gender, sex). Data from 50 patients, 23 men and 27 women was obtained. Mean age at definitive diagnosis was 5.6 years, age at onset of signs or symptoms was 4.14 years starting with pigeon chest deformity, valgus knees at 4.5 years, stiff hands and increasing mobility of wrists to the 5.8 years, followed by limitation to lift shoulders to 7.1 years. In 78% of patients the diagnosis was by a geneticist. First and subsequent observed clinical changes were orthopedic, starting as early as 4.4 years as noted by parents. Rise of suspicious may delay 16 months' average to definitive diagnosis based on other multi-systemic findings. The most frequent specialist aid in diagnosis is a clinical geneticist followed by orthopedic surgeon. The diagnosis of Morquio-A disease in Mexico is as early as reports from other centers.
Keywords: Morquio-A syndrome, diagnosis, Mexico, delay diagnosis, height, dwarphism, skeletal dysplasia, orphan disease
1. Introduction
Mucopolysaccharidoses IV A, better known as Morquio-A syndrome, is a rare disease that represents one of the multi-organic and progressive diseases with the most severe musculoskeletal system involvement (1,2). It has a wide clinical spectrum, that may not be easy to recognize from other non-systemic skeletal dysplasias, which represents a challenge in diagnosis in young patients (3,4). This condition has progressive multisystem dysfunction and impaired functional capacity. Skeletal abnormalities may be first line clinical findings to aid in diagnosis (1,5,6).
Since Morquio-A patients have not primary neurological involvement (1,2,4,5), an early detection, as well as early treatment could prevent the severe functional impairment seen on these patients, improving outcomes and extending their autonomy (2,7–9). Generally, before definitive diagnosis, patients are delayed or misdiagnosed due the lack of knowledge about the disease (10,11).
International series describe a widespread amount of skeletal manifestations such as short trunk and neck, hip and knee dysplasia, spinal abnormalities, pigeon chest, among other less frequent characteristics (12,13). Early onset findings are kyphosis, scoliosis, growth retardation and altered gait (4,14). The vast majority of papers detail clinical features but only few mention the age of onset. Knowing average time of features may help raise clinical suspicion of the disorder at early time (5,15,16).
We made this research trying to compare age of diagnosis versus age of onset of evident clinical signs as observed from the parents of Morquio-A patients in Mexico. To describe time delay in diagnosis since the first manifestation until definite diagnosis. This work may help guide the intervention in improving suspicion and early detection of Morquio-A syndrome in our population as well as in other similar Hispanic communities.
2. Materials and Methods
During the first Mexican Morquio-A meeting, a self-administered questionnaire was given to caregivers and patients with this syndrome. All of them have had diagnosis confirmation through determination of deficient enzymatic activity in leukocytes or fibroblasts. The patients were enrolled by accepting an informed consent and agreement of confidentiality. They were all informed about the reaching of the present study.
The questionnaire consisted in open questions about biological variables (age, gender, height) as well as age at diagnosis, concomitant and familial diseases. Questions about first symptoms and clinical features of the disease were dichotomic in order to avoid to forget any major or minor joint, followed to blank spaces to write down kind of alteration and the age it was noticed. Other questions referred with time of suspicion, health care provider in charge of suspicious and age at diagnosis were open blank spaces.
All data were collect in electronic datasheets to group similar answers and findings. All answers and results are described and analyzed with measures of central tendency.
3. Results and Discussion
We received full answered questionnaires from 50 patients or first relatives, all of them had enzymatic activity confirmation of Morquio-A syndrome; 23 were of male gender (46%) and 27 females (54%). Phenotypically all correspond to non-attenuated form of the disease. Mutational analyses of all patients were not possible, so it was not included on this study.
3.1. Height and weight
Mean age of the patients in present study was 15.3 years (range 3.1 to 41 years). The mean height of patients studied is 99 cm (ranging from 60 to 140 cm; n = 47), and mean weight of 20.4 kg (10 to 49 kg; n = 48).
Excluding the younger patients, the final height of adult Morquio-A patients is 87 cm (94 to 125 cm) and final adult weight of 24.5 kg (10 to 49 kg) with a body mass index average of 22.08 (range 10.0 to 34.02 kg/m2). This data is only from patients older than 18 years and excluding both the tallest and shortest patients (n = 15) (Figure 1).
Figure 1.
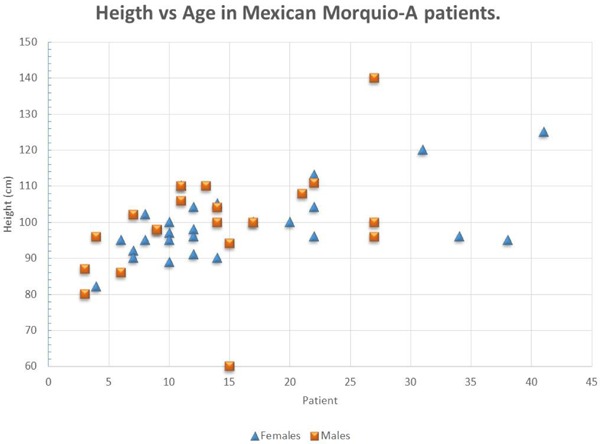
Height according age in Morquio-A patients. Notice all of them are between 90 and 110 cm. Most patients reach final height during second decade of life.
3.2. Definitive diagnosis
Mean age at definite diagnosis was 5.6 years, ranging from 1 month to 15 years in all patients in this cohort (n = 50). Eighteen patients had a positive familiar history of Morquio-A syndrome (9 siblings and 9 cousins or other relatives). In patients with brothers affected with Morquio-A, the diagnosis was made at mean 5.5 years old (from one month to 15 years); and in patients with relatives other than sibs, diagnosis was made at mean 5.8 years (from 1 year to 15 years). In the remain (n = 32) patients without family history, diagnosis was made between 18 months and 14 years (mean 4.6 years).
For earliest diagnosis, two patients were diagnosed before one year of life: at age of one and nine months, respectively. Both had one sibling affected as a source of clinical suspicion.
Ten patients had different previous misdiagnoses before Morquio-A confirmation: among them skeletal dysplasia (n = 4), achondroplasia (n = 2), Ehlers-Danlos syndrome (n = 1), Sotos syndrome (n = 1), scoliosis (n = 1) and chronic bronchitis (n = 1). Eleven patients did not respond (22%) and more than half of the patients did not have a previous presumptive diagnosis (58%, n = 29) (Figure 2).
Figure 2.
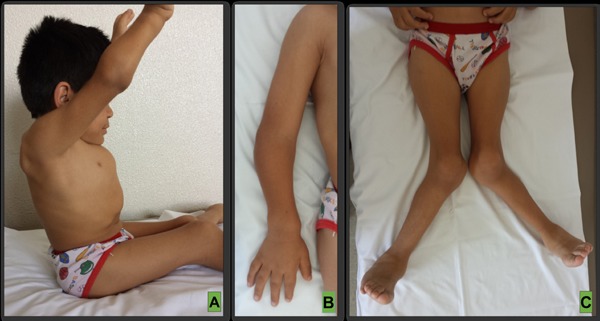
Clinical features of Morquio-A patients. (A), Progressive inability to raise arms upperhead and full extend elbows, pigeon chest and relative short neck; (B), Wrist, elbow and hand deformations; (C), Severe genu valgus. Personal archive files. Patient and guardian consent was obtained prior attachment of present clinical images.
Definitive diagnosis of Morquio-A was achieved by a genetic specialist in 78% of patients (n = 39), 8% by an orthopedic surgeon (n = 4), by the neurologist (n = 1) and by endocrinologist (n = 1). Five patients (10%) did not remember the doctor who gave them the accurate diagnosis. Most of the patients (28%, n = 14) were not referred for assessment by any specialist, 32% (n = 16) did not answer and 40% (n = 20) had a referral for specialized assessment from pediatrics, physical therapist or family doctor. We did not have the mutational analysis of these patients, so clinico-mutational correlation was not possible to achieve.
3.3. Clinical manifestations
First clinical syndromic manifestation observed by parents and patients was progressive deformity in the chest (pigeon chest) starting from 8 months to 9 years (mean of 4.14 years; n = 34), followed by valgus knees between 1.6 and 13 years (mean 4.5 years; n = 37). Progressive deformity, weakness or functional limitation in wrists ranged from 1 to 13 years (average 5.8 years; n = 29), limitation or progressive inability to raise overhead shoulders was noticed since the 2 until 20 years (mean 7.1 years; n = 16).
Before a known accurate diagnosis, seven patients had surgical procedure history (14%), three for umbilical hernia repair, two for neurosurgical stabilization in cervical spine, one for ear nose and throat and one for orthopedic knee procedure. Other 19 patients had surgery after the Morquio-A diagnosis. The remaining 24 patients (48%) had no history of surgery.
Since this syndrome has not any primary neurological involvement (17), the earlier abnormal signs in our population were seen around 4 years old and described as pigeon chest or thoracic deformities (including triangular cifosis, widened chest and thoracic asymmetry), however this is not corresponding with Bhattacharya, who describes this first manifestation evident as early as 6 to 12 months (11).
The second relevant finding to make parents search for medical advice was mayor joint involvement as seen in knee valgus and limitation to lift shoulders upper head and finally, small joint affection with wrist deformity and functional worsen hand function towards 5 or 6 years.
All patients in this population belong to the so-called severe phenotype, with all similar physical features that make an early diagnosis possible. unlike patients with the attenuated phenotype, in whom the diagnosis is more difficult and thus delayed.
None of the parents mentioned as an early sign the growth delay, although this is showed in all patients since the 4 years old (18), but as shown on the record, this was the parent or patient perspective, not the medical course, and all patients showed short stature at mature age.
The most evident clinical features of this syndrome are orthopedic type (14), nonetheless in only one patient the diagnosis was suspected due a non-skeletal feature (chronic bronchitis).
Diagnosis was made almost one year earlier on patients without family history of the disease, compared to those with positive familiar history (4.6 years vs. 5.5 years, respectively). We don't have a clear explanation for this, but the hypothesis is that similar phenotype observed among family members is responsible for not raising suspect of the syndrome in affected individuals.
These early signs found in our population, are considered the most common in all population reports, regardless of the type of mutation of the patient, and this is consistent with world literature (19). All these Hispanic patients share ethnic characteristics which may be regionally oriented than other populations around the globe. Is the need to early diagnose these patients in order to avoid clinical deterioration and worst surgery outcomes (20).
In conclusion, according to our results, the main suspected data for this disease are of the orthopedic type, drawing attention before the first four years, with thoracic deformity and knee valgus, followed for limitation to raise the arms above the shoulder level. Any of these alterations will merit assessment by the physician, who needs to have the suspected diagnosis or reference to the specialist to assist in the earliest detection. We consider that diagnosis of Morquio-A syndrome in Mexico is as early in childhood as in other world centers. Further studies are needed to determine the most frequent errors as well as delays in diagnosis, which help diagnose patients and prevent complications arising from delayed treatment.
Acknowledgements
Authors wish to thank the nonprofit foundation “Pide un Deseo Mexico, IAP” and “MPS JaJax” for all the facilities provided to the patients in assist into this meeting, as well as always make efforts to promote the fight and desire to better live every day.
References
- 1. Harmatz P, Mengel KE, Giugliani R, et al. The Morquio-A: Clinical Assessment Program: Baseline Results Illustrating Progressive, Multisystemic Clinical Impairments in Morquio-A Subjects. Mol Genet Metab. 2013; 109:54-61. [DOI] [PubMed] [Google Scholar]
- 2. Hendriksz CJ, Berger KI, Giugliani R, Harmatz P, Kampmann C, Mackenzie WG, Raiman J, Villarreal MS, Savarirayan R. International Guidelines for the Management and Treatment of Morquio-A Syndrome. Am J Med Genet A. 2015; 167:11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rush ET. Atypical Presentation of Mucopolysaccharidosis Type IVA. Mol Genet Metab Rep. 2016; 8:8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wood TC, Harvey K, Beck M, et al. Diagnosing Mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013; 36:293-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hendriksz CJ, Harmatz P, Beck M, Jones S, Wood T, Lachman R, Gravance CG, Orii T, Tomatsu S. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013; 110:54-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clarke LA, Harmatz P, Fong EW. Implementing Evidence-Driven Individualized Treatment Plans within Morquio A Syndrome. Mol Genet Metab. 2016; 117:217. [DOI] [PubMed] [Google Scholar]
- 7. Regier DS, Tanpaiboon P. Role of Elosulfase Alfa in Mucopolysaccharidosis IV A. Appl Clin Genet. 2016; 9:67-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lavery C, Hendriksz C. Mortality in Patients with Morquio Syndrome A. JIMD Reports. 2015; 15:59-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tomatsu S, Sawamoto K, Alméciga-Díaz CJ, et al. Impact of Enzyme Replacement Therapy and Hematopoietic Stem Cell Transplantation in Patients with Morquio A Syndrome. Drug Des Devel Ther. 2015; 9:1937-1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Solanki GA, Martin KW, Theroux MC, Lampe C, White KK, Shediac R, Lampe CG, Beck M, Mackenzie WG, Hendriksz CJ, Harmatz PR. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): Presentation, diagnosis and management. J Inher Metab Dis. 2013; 36:339-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhattacharya K, Balasubramaniam S, Choy YS, et al. Overcoming the Barriers to Diagnosis of Morquio A Syndrome. Orphanet J Rare Dis. 2014; 9:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yabe H, Tanaka A, Chinen Y, Kato S, Sawamoto K, Yasuda E, Shintaku H, Suzuki Y, Orii T, Tomatsu S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol Genet Metab. 2016; 117:84-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z, Zhang W, Wang Y, Meng Y, Su L, Shi H, Huang S. Mucopolysaccharidosis IVA Mutations in Chinese Patients: 16 Novel Mutations. J Hum. Genet. 2010; 55:534-540. [DOI] [PubMed] [Google Scholar]
- 14. Yasuda E, Fushimi K, Suzuki Y, et al. Pathogenesis of Morquio A syndrome: An autopsied case reveals systemic storage disorder. Mol Genet Metab. 2013; 109:301-311. [DOI] [PubMed] [Google Scholar]
- 15. Suarez-Guerrero JL, Gómez HP, Arias FJ, Contreras-García GA. Mucopolysaccharidosis: Clinical features, diagnosis and management. Rev chil Pediatr. 2015; 87:295-304. [DOI] [PubMed] [Google Scholar]
- 16. Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: Clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007; 30:165-174. [DOI] [PubMed] [Google Scholar]
- 17. Davison JE, Kearney S, Horton J, Foster K, Peet AC, Hendriksz CJ. Intellectual and neurological functioning in Morquio syndrome (MPS IV A) J Inherit Metab Dis. 2013; 36:323-328. [DOI] [PubMed] [Google Scholar]
- 18. Montaño AM, Tomatsu S, Brusius A, Smith M, Orii T. Growth charts for patients affected with Morquio A disease. Am J Med Genet A. 2008;146A:1286-1295. [DOI] [PubMed] [Google Scholar]
- 19. Lachman RS, Burton BK, Clarke LA, Hoffinger S, Ikegawa S, Jin DK, Kano H, Kim OH, Lampe C, Mendelsohn NJ, Shediac R, Tanpaiboon P, White KK. Mucopolysaccharidosis IVA (Morquio A syndrome) and VI (Maroteaux-Lamy syndrome): Under-recognized and challenging to diagnose. Skeletal Radiol. 2014; 43:359-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Colmenares-Bonilla D, Vasconcelos-Martinez M, Guerra-Jasso J, Ocampo-Perez L. Guided growth may not be the best option for knee valgus deformity in adolescent patients with Morquio-A. Musculoskelet Surg. 2016; DOI: 10.1007/s12306-016-0441-0 [DOI] [PubMed] [Google Scholar]