

Summary
Cardiomyopathy associated with dystrophinopathies [Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), X-linked dilated cardiomyopathy (XL-dCM) and cardiomyopathy of Duchenne/Becker (DMD/BMD) carriers] is an increasing recognized manifestation of these neuromuscular disorders and notably contributes to their morbidity and mortality. Dystrophinopathic cardiomyopathy (DCM) is the result of the dystrophin protein deficiency at the myocardium level, parallel to the deficiency occurring at the skeletal muscle level. It begins as a “presymptomatic” stage in the first decade of life and evolves in a stepwise manner toward pictures of overt cardiomyopathy (hypertrophic stage, arrhythmogenic stage and dilated cardiomyopathy). The final stage caused by the extensive loss of cardiomyocytes results in an irreversible cardiac failure, characterized by frequent episodes of acute congestive heart failure (CHF), despite a correct pharmacological treatment. The picture of a severe dilated cardiomyopathy with intractable heart failure is typical of BMD, XL-dCM and cardiomyopathy of DMD/BMD carriers, while it is less frequently observed in patients with DMD. Heart transplantation (HT) is the only curative therapy for patients with dystrophinopathic end-stage heart failure who remain symptomatic despite an optimal medical therapy. However, no definitive figures exist in literature concerning the number of patients with DCM transplanted, and their outcome. This overview is to summarize the clinical outcomes so far published on the topic, to report the personal series of dystrophinopathic patients receiving heart transplantation and finally to provide evidence that heart transplantation is a safe and effective treatment for selected patients with end-stage DCM.
Keywords: Cardiomyopathy, Duchenne muscular dystrophy, Becker muscular dystrophy, X-linked dilated cardiomyopathy, Duchenne/Becker carrier's cardiomyopathy
1. Introduction
Dystrophinopathies are X-linked muscle disorders caused by mutations in the dystrophin gene, located at Xp21, that encodes for the sarcolemmal protein dystrophin, virtually present in all tissues, but most abundant in skeletal muscle cells and heart (1–3). Dystrophin provides the connection between a large complex of glycoproteins called the dystrophin-associated glycoprotein complex (DAG) on the muscle cell membrane and the intracellular actin filaments, that transmit forces generated by the sarcomere contraction to the extracellular matrix (4,5). Absence, reduced levels or abnormal structure of dystrophin lead to membrane fragility, making muscle fibres more prone to injury during contraction (6). As muscle disease progresses, muscle repair cannot adequately compensate for damage, leading to necrosis of skeletal and cardiac myocytes and the progressive replacement by fibro-fatty tissue (7–15). Dystrophinopathies include four different clinical presentations: Duchenne muscular dystrophy (DMD), the more severe form, Becker muscular dystrophy (BMD), the more benign form, the X-linked dilated cardiomyopathy (XL-dCM) (8) and the cardiomyopathy of DMD/BMD carriers.
Duchenne muscular dystrophy (DMD, OMIM 310200) is the most common muscle disorder in infancy, affecting 1 in 3.500 male newborns (9). It is the most severe form of dystrophinopathy, caused by the complete absence of the dystrophin protein at both skeletal and cardiac muscle level. Becker muscular dystrophy (BMD, OMIM 300376) is the milder dystrophinopathy affecting 1 in 18.450 males (9). Symptoms typically begin between ages 3 and 21, with a mean age of onset of 11 years. The disease is usually caused by “in frame” mutations able to produce a certain amount of dystrophin (16). BMD, unlike Duchenne-type muscular dystrophy, is characterized by a slow progression of muscle weakness so that in many patients the independent activity is maintained until late adulthood.
XL-dCM, first described in 1993 by Towbin (17), is a rare cardiomyopathy caused by mutations in the 5' end of the dystrophin gene that lead to the absence of the M-isoform of dystrophin only in the heart (17). The preserved skeletal muscle strength increases cardiac demands, leading to dilated cardiomyopathy (18). The clinical picture is characterized by the onset of congestive heart failure (CHF) in young men with a mean age of 15–30, with a rapid fatal evolution within 2 years from the onset, although older patients can also be affected.
The majority of DMD and BMD female carriers are asymptomatic; however, few of them may show muscle symptoms (8,16) such as mild muscle weakness and elevated serum CK, while a larger part present cardiomyopathy (20–25). The onset of symptoms in DMD/BMD carriers may have several causes, the most frequent of them represented by a skewed X-chromosome inactivation (XCI) (26–28).
The above mentioned clinical presentations arise from different pathogenic conditions with consequent variable degrees of skeletal muscle and cardiac involvement. Cardiac death usually occurs from ventricular dysfunction and dilated cardiomyopathy, that represents the end-stage of dystrophinopathic cardiomyopathy (DCM) (10–11,29–30). DCM is constantly progressive and evolves in a stage-wise manner, passing from a presymptomatic condition to dilated cardiomyopathy through a number of pathophysiologically distinct stages (10,11) (Figure 1).
Figure 1.
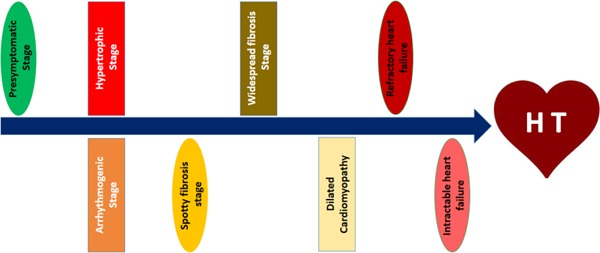
Flow-chart showing the evolution of dystrophinopathic cardiomyopathy.
In brief, the presymptomatic stage is followed by a stage in which the myocardial fibrosis is limited to few foci (spotty or focal fibrosis), often inducing a compensatory hypertrophy of the surrounding areas still expressing dystrophin, with consequent regional heterogeneity of repolarization that can favor the onset of arrhythmias. Later, the confluence of these areas determines a widespread diffuse fibrosis leading to dilated cardiomyopathy, a stage characterized by ventricle enlargement, tinning of ventricular walls, and reduced left ejection fraction. Dilated cardiomyopathy in turn evolves toward the stage of heart failure (HF). The first episodes of HF are usually responsive to pharmacological treatment; subsequently, HF evolves towards the stage of intractable HF not amenable to adequate drug treatment. Therefore heart failure represents an important contributor to mortality in these patients (9,10) for which the heart transplantation (HT) remains the ultimate intervention to preserve life. However, despite the high incidence of end-stage DCM in patients with dystrophinopathies, there is a reluctance to perform heart transplantation in these patients due to the paucity of donors and the concerns that the accompanying myopathy will limit the benefits obtained through the HT.
Furthermore no definitive figures exist in literature about the number of patients with DCM transplanted and their outcome, nor clear indications in the current guidelines considering heart transplantation as an option for dystrophinopathic patients with end-stage heart failure.
This overview aims to i) report the so far published frequencies of heart transplantation in patients with DCM and related clinical outcomes; ii) report the personal series of dystrophinopathic patients who received HT and iii) provide evidence that HT is a safe and effective treatment for selected patients with end-stage DCM.
2. Heart transplantation in patients with dystro-phinopathic cardiomyopathy
2.1. Review of the literature
Inherited myopathies in patients with secondary end-stage cardiomyopathies have always been considered a relative contraindication for cardiac transplantation. High operative risk related to muscle impairment and potential graft involvement secondary to the underlying myopathy have been the two main reasons implicated in the poor prognosis. For these reasons, HT has not been considered an appropriate treatment option for DMD patients with drug-resistant dilated cardiomyopathy, because of pulmonary function and skeletal muscles impairment (31). The literature contains only two isolated reports of successful heart transplantation in these patients (31,32). Cripe et al. (31) reported the case of a 14-year-old DMD patient with severe dilated cardiomyopathy, but preserved pulmonary function who underwent a successful cardiac transplantation surviving four years later. Rees et al. (32) described HT in 3 patients with DMD with a mean duration of follow-up of 40 months. All the patients tolerated immunosuppression, had no complications in post-operative intubation and were able to be rehabilitated, suggesting that cardiac transplantation can be successfully performed in DMD patients presenting a severe cardiomyopathy, preserved pulmonary function, and a discrete muscle function. Recently, Amodeo et al. (33) explored at the Bambino Gesù Children's Hospital in Rome, the use of left ventricular assist devices (LVAD-Jarvik 2000) as destination therapy in 7 DMD patients, as an alternative treatment for selected end-stage heart failure in DMD. All patients survived to hospital discharge and resumed previous activities; however data about their long-term outcomes remain limited.
Likewise, few cases of HT in patients with Becker muscular dystrophy have been so far reported in literature (34–39), though heart failure is the most frequent cause of death in these patients. In fact patients with BMD seem to pay the advantage of a prolonged ambulation with a higher occurrence of heart involvement and dilated cardiomyopathy, that represents the major cause of their morbidity and mortality (19,40–43). Some dystrophin mutations have been correlated to an increased incidence of severe cardiomyopathy (3) in these patients in which the presence of cardiomyopathy dramatically reduces the life expectancy, otherwise only slightly limited compared to normal subjects (44). Wu et al. (34) reported the results of a retrospective review of the Cardiac Transplant Research Database, a multi-institutional registry of 29 transplant centres in the United States, in the period 1990–2005. In their review - the largest study describing the long-term outcomes of cardiac transplantation in patients with BMD with a follow-up period extending more than 10 years - the post-transplant cardiac outcomes of 15 BMD patients were compared with those of 275 non-muscular dystrophy patients with non-ischemic cardiomyopathy, matched for age, body mass index, gender, and race. The survival rate in BMD patients was similar to that of controls at 1 year (89% vs. 91%; p = 0.5) and 5 years (83% vs. 78%; p = 0.5). The differences in rates of cumulative infection, rejection, or allograft vasculopathy between the 2 groups were not significant (p = 0.5 for all comparisons).
Ruiz-Cano et al. (35) described a Spanish single-centre experience with HT in 3 BMD patients with a mean follow-up duration of 57.4 months. Both intra-operative and post-operative course of these individuals did not show higher complication rates than other patients. All recipients experienced successful rehabilitation; no evidence of graft dysfunction was detected during the follow-up. All patients were alive at the time of the study, and in good performance. The AA concluded that HT for patients with BMD end-stage cardiomyopathy is not associated with a poorer prognosis when there is a mild degree of muscular impairment.
Casazza et al. (36), reported the case of a successful cardiac transplantation because of a severe and rapidly progressive dilated cardiomyopathy complicated by terminal heart failure in a BMD patient with a mild muscle impairment. Unfortunately, no data about the long-term prognosis of this patient have been reported.
Patanè et al. (37) described a 27-year-old BMD patient with end-stage dilated cardiomyopathy and CHF who was fairly well, one year after a successful transplantation. Melacini et al. (38) reported the case of a 24-years-old BMD patient, with end-stage dilated cardiomyopathy and moderate myopathy, who successfully underwent heart transplantation. The patient received triple immunosuppressive therapy consisting of azathioprine, cyclosporineA (CyA) and prednisone in the post-operative period.
Steger et al. (39) extrapolated 4 patients (1.19 %) affected by end-stage cardiomyopathy related to inherited myopathies from a large cohort of 335 patients undergoing HT at the Innsbruck Medical University, between January 1994 and December 2011. Three of them had BMD, and 1- female- was affected by limb-girdle muscular dystrophy; the mean age of patients was 38.5 years (range 16–56) and the post-operative follow-up period was on average 68.5 months (range 16–139). All patients had an uneventful immediate post-operative course. One of them (BMD) died 16 months after the HT because of pulmonary embolism and right heart failure, and one 11 years after HT because of myocardial and cerebral infarction following tricuspid valve replacement. The remaining 2 patients were in good general condition without progression of the muscle disease, for the entire period of the study. The AA concluded that HT may increase the quality of life and life expectancy in selected patients suffering from end-stage heart failure due to inherited benign muscular dystrophies. These data seem to disclaim the long-held belief that patients with muscular dystrophy and advanced stages of cardiomyopathy may have worse outcomes after HT compared with patients without it.
To date there is only one report on HT in patients with XL-dCM (45). Out of 4 patients (mean age 24, 4 years; range 16–31) who were heart transplanted in the period August 1989 – January 2000, only one died suddenly at 66 months of follow-up. At the time of heart transplantation all patients were in NYHA functional class IV. After 44 months of FU on average (range 22–66) all the patients were fine and in NYHA functional class I, stressing the concept that HT should be the first choice option for patients with XL-dCM and preserved muscle function, for whom myocardial end-stage disease and serious rhythm disturbances are severely disabling and life-threatening (46).
Very few cardiac transplantation case reports have been so far reported in cardiological manifesting DMD carriers (47,48) for whom once again the only hope for survival remains heart transplantation. Melacini et al. (47) reported the first case of successful HT in a symptomatic DMD carrier, with severe dilated cardiomyopathy, showing a deletion of exons 50–52. Davies et al. (48) reported the case of a 25-year-old DMD carrier who during the third trimester of her pregnancy developed a severe cardiac failure and performed successful heart transplantation, after 311 days of mechanical circulatory support.
2.2. Personal series
Among 55 patients affected by dystrophin related end-stage cardiomyopathy followed in our division in the last 15 years, 4 of them (7.2%) underwent heart transplantation. Three of them had Becker's muscular dystrophy and the fourth was affected by XL-dCM. The clinical characteristics of the patients are summarised in Table 1.
Table 1. Patients' clinical characteristics.
| Patient | Diagnosis | Dystrophin Gene Defect | Gender | Age at diagnosis (years) | CK (U/L) pre HT | Age at HT (years) | NYHA class at HT | Follow-up (months) |
|---|---|---|---|---|---|---|---|---|
| 1 | BMD | del ex. 45–49 | M | 15 | 1152 | 34 | IV | 153 |
| 2 | BMD | del ex. 45–49 | M | 17 | 1263 | 33 | IV | 121 |
| 3 | BMD | del ex 3–4 | M | 29 | 856 | 27 | IV | 174 |
| 4 | XL-dCM | del promoter | M | 22 | 320 | 27 | IV | 131 |
| Mean ± SD | 20 ± 6.2 | 897.8 ± 421.7 | 30.2 ± 3.8 | IV | 144.7 ± 23.6 |
BMD: Becker's muscular dystrophy; CK: creatinkinase; HT: heart transplantation; M: male; XL-dCM: X-linked dilated cardiomyopathy.
After transplantation, all patients received triple drug immunosuppressive therapy with cyclosporine, everolimus and steroids. High-dose steroid therapy was withdrawn within 12 months following HT in all patients.
Becker Cases: Cases 1 and 2 were diagnosed as BMD at the age of 15 and 17 years respectively, when they complained of mild muscle weakness, associated with increased serum creatin-kinase (CK) levels (mean values 6.3 times the maximum normal values). Molecular investigation confirmed the diagnosis, showing a deletion of exons 45–49 in dystrophin gene in both cases.
At the age of 31 years, both developed dilated cardiomyopathy that required orthotopic HT in the following few years, performed at the age of 34 years in case 1, and at the age of 33 years in case 2 (Figure 2A).
Figure 2.
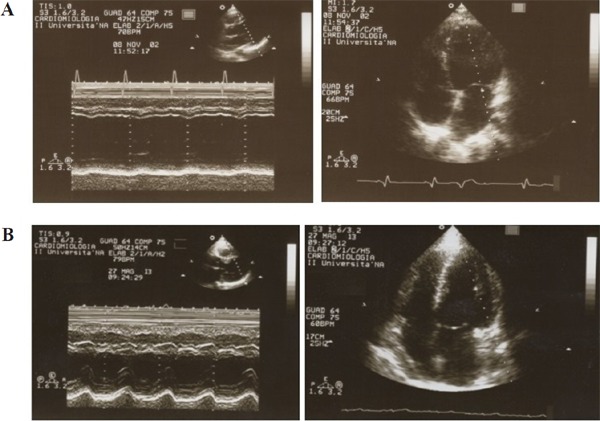
(A) Echocardiography (M-mode scan and apical view of the left ventricle) in the BMD patient (case 2). To be noted: the enlargement of the left atrium (systolic dimension = 48 mm) and the marked left ventricular dilatation (end diastolic diameter = 72 mm); the hypokinesis of the interventricular septum and the virtually absent motion of the left ventricular posterior wall. (B) Echocardiography in the same patient 10 years after heart transplantation. The M-mode scan and the apical view of the left ventricle show normal chamber diameters and ventricular systolic function.
At the time of transplant pre-evaluation, both patients had very mild muscular impairment and no respiratory involvement. The intra-operative and early post-operative courses did not reveal any complications; mechanical ventilation was withdrawn within 10 hours following surgery. Both patients had a successful functional rehabilitation reaching a good performance status. Case 1, nine months following HT, experienced mild signs of laboratory rejection, promptly resolved by the immunosuppressive therapy optimization. Case 2 showed, in the last 2 years, a slight progressive worsening of kidney function related to immunosuppressive therapy (serum creatinine value of 2.6 mg/dL; CrCl 40.96 mL/min; stage III of renal failure), which required cyclosporine dose reduction. To date, more than 10 years following HT, no evidence of graft dysfunction was detected (Figure 2B), while a very slow progression of the pre-existing muscular impairment was observed. Both patients continue to have a good quality of life.
Case 3 was initially diagnosed as idiopathic dilated cardiomyopathy, complicated by terminal heart failure. He required 2 years later an orthotopic HT. The early post-surgical period was uneventful. Two years after HT, the patient was addressed to our division for muscle weakness at lower limbs and increased serum CK values. Based on clinical evaluation and molecular investigation (presence of a deletion of exons 3 and 4 in dystrophin gene), a diagnosis of BMD was made. Seven years after HT, the patient showed clinical and echocardiographic evidence of acute graft dysfunction, and a reduction (< 35%) of left ventricular ejection fraction (LVEF). The replacement of everolimus with mycophenolate mofetil produced a progressive improvement of patient's condition and of echocardiographic parameters (LVEF: 50%). Twelve years later, he developed an acute complete (3rd degree) atrio-ventricular block requiring a pacemaker implantation; after two years a new heart transplant was indicated, for evidence of clinical and echocardiographic graft rejection (LVEF 25%). However, the transplant was not performed as the patient died suddenly during sleep, before performing it.
Case 4: The patient was addressed to our department for signs and symptoms of low cardiac output, history of reduced exercise tolerance and exertional dyspnea. At the first examination, no muscle and respiratory weakness were noted, while CK values were increased. The patient underwent both muscle and endomyocardial biopsies, who showed the absence of the dystrophin staining with the antibody anti-NH2 terminal domain only at the cardiac level, not in the muscle. The dystrophin staining was instead normal at both cardiac and muscle level with the antibodies anti-ROD and anti-COOH domains. A diagnosis of X-linked dilated cardiomyopathy was made and the patient treated accordingly.
During the following years an intractable congestive heart failure required HT, performed at the age of 27 years. The early post-operative course was uncomplicated; mechanical ventilation was withdrawn within the following 24 hours. The patient returned to work and till now he continues to have a good exercise tolerance and quality of life.
3. Discussion
The long-term clinical outcomes of cardiac transplantation in patients with dystrophinopathic cardiomyopathy properly selected, seem to be similar to that of a matched cohort of patients undergoing HT for idiopathic dilated cardiomyopathy, by analysing the few evidence available in literature.
However, special care and consideration are necessary during the peri-operative and post-operative period to avoid life-threatening complications and the progression of the primary disease. Among them, an intensive neuromyological observation is necessary in order to i) adequately adapt the dosage of immunosuppressants to avoid the onset of a secondary myopathy and the occurrence of rhabdomyolysis due to the toxic affect of cyclosporine and ii) to limit the post-operative complications and the progression of the primary muscle disease. In fact the combined use of cyclosporine and lipid-lowering agent, such as statins and gemfibrozil, could be toxic for muscle cells (49).
Also in our experience, HT remains the treatment of choice for patients with advanced DCM and poor muscle impairment. No other treatment option such as medical therapy or electrical and/or mechanical devices, can compete with HT long-term results, particularly when compared with the natural course of end-stage heart failure. Even the fear of a possible heart rejection is limited - at least for patients suffering from BMD, XL-dCM and relative carriers - because the presence of some dystrophin makes it very unlikely the production of antibodies against the dystrophin-deleted-region. As a consequence, a more favourable prognosis may be expected in these subjects.
Given for that a special attention should be given to the extent of skeletal myopathy, respiratory muscle involvement and survival in these patients, however it's time that cardiac surgeons overcome the reluctance to heart transplant patients with dystrophinopathic cardiomyopathy, due to the supposed reduced life expectancy. The long-term prognosis in these patients in fact is closely related to the possibility to be transplanted.
References
- 1. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003; 2:731-740. [DOI] [PubMed] [Google Scholar]
- 2. Sadoulet-Puccio HM, Kunkel LM. Dystrophin and its isoforms. Brain Pathol. 1996; 6:25-35. [DOI] [PubMed] [Google Scholar]
- 3. Nigro G, Politano L, Nigro V, Petretta VR, Comi LI. Mutation of dystrophin gene and cardiomyopathy. Neuromuscul Disord. 1994; 4:371-379. [DOI] [PubMed] [Google Scholar]
- 4. Rafael JA, Cox GA, Corrado K, Jung D, Campbell KP, Chamberlain JS. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol. 1996; 134:93-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Corrado K, Rafael JA, Mills PL, Cole NM, Faulkner JA, Wang K, Chamberlain JS. Transgenic mdx mice expressing dystrophin with a deletion in the actin-binding domain display a “mild Becker” phenotype. J Cell Biol. 1996; 134:873-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993; 90:3710-3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wallace GQ, McNally EM. Mechanisms of muscle degeneration, regeneration and repair in the muscular dystrophies. Annu Rev Physiol. 2009; 71:37-57. [DOI] [PubMed] [Google Scholar]
- 8. Obler D, Wu BL, Lip V, Estrella E, Keck S, Haggan C, Semigran M, Smoot LB. Familial dilated cardiomyopathy secondary to dystrophin splice site mutation. J Card Fail. 2010; 16:194-199. [DOI] [PubMed] [Google Scholar]
- 9. Emery AE. Population frequencies of inherited neuromuscular diseases. A world survey. Neuromuscul Disord. 1991; 1:19-29. [DOI] [PubMed] [Google Scholar]
- 10. Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990; 26:271-277. [DOI] [PubMed] [Google Scholar]
- 11. Politano L, Nigro G. Managing dystrophinopathic cardiomyopathy. Expert Opin Orphan Drugs. 2016; 4:1159-1178. [Google Scholar]
- 12. Passamano L, Taglia A, Palladino A, Viggiano E, D'Ambrosio P, Scutifero M, Rosaria Cecio M, Torre V, DE Luca F, Picillo E, Paciello O, Piluso G, Nigro G, Politano L. Improvement of survival in Duchenne muscular dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012; 31:121-125. [PMC free article] [PubMed] [Google Scholar]
- 13. Politano L, Nigro G. Treatment of dystrophinopathic cardiomyopathy: Review of the literature and personal results. Acta Myol. 2012; 31:24-30. [PMC free article] [PubMed] [Google Scholar]
- 14. Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EO, Towbin JA. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005; 112:2799-2804. [DOI] [PubMed] [Google Scholar]
- 15. Nigro G, Comi LI, Politano L, Nigro V. Dilated cardiomyopathy of muscular dystrophy: A multifaceted approach to management. Semin Neurol. 1995; 15:90-92. [DOI] [PubMed] [Google Scholar]
- 16. Cohn RD, Campbell KP. Molecular pathways for dilated cardiomyopathy. In: Molecular basis of cardiovascular disease: A companion to Braunwald′s heart disease (Chien K. editor). 2nd ed. Saunders Philadelphia, USA, 2004; pp. 306-310. [Google Scholar]
- 17. Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, McCabe ER, Swift M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993; 87:1854-1865. [DOI] [PubMed] [Google Scholar]
- 18. Spurney CF. Cardiomyopathy of Duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve. 2011; 44:8-19. [DOI] [PubMed] [Google Scholar]
- 19. Kaspar RW, Allen HD, Montanaro F. Current under-standing and management of dilated cardiomyopathy in Duchenne and Becker muscular dystrophy. J Am Acad Nurse Pract. 2009; 21:241-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nigro G, Di Somma S, Comi LI, Politano L, Papparella S, Restucci B, Petretta VR, Giugliano MA, Carotenuto A, Limongelli FM, DE Divitiis O. Structural basis of cardiomyopathy in Duchenne/Becker carriers. Endomyocardial biopsy evaluation. Ann N Y Acad Sci. 1995; 752:108-110. [DOI] [PubMed] [Google Scholar]
- 21. Politano L, Nigro V, Nigro G, Petretta VR, Passamano L, Papparella S, Di Somma S, Comi LI. Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA. 1996; 275:1335-1338. [PubMed] [Google Scholar]
- 22. Comi LI, Nigro G, Politano L, Petretta VR. The cardiomyopathy of Duchenne/Becker consultands. Int J Cardiol. 1992; 34:297-305. [DOI] [PubMed] [Google Scholar]
- 23. Hoogerwaard EM, van der Wouw PA, Wilde AA, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, van Essen AJ, Leschot NJ, de Visser M. Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 1999; 9:347-351. [DOI] [PubMed] [Google Scholar]
- 24. Kamakura K, Kawai M, Arahata K, Koizumi H, Watanabe K, Sugita H. A manifesting carrier of Duchenne muscular dystrophy with severe myocardial symptoms. J Neurol. 1990; 237:483-485. [DOI] [PubMed] [Google Scholar]
- 25. Mirabella M, Servidei S, Manfredi G, Ricci E, Frustaci A, Bertini E, Rana M, Tonali P. Cardiomyopathy may be the only clinical manifestation in female carriers of Duchenne muscular dystrophy. Neurology. 1993; 43:2342-2345. [DOI] [PubMed] [Google Scholar]
- 26. Viggiano E, Picillo E, Cirillo A, Politano L. Comparison of X-chromosome inactivation in Duchenne muscle/myocardium-manifesting carriers, non-manifesting carriers and related daughters. Clin Genet. 2013; 84:265-270. [DOI] [PubMed] [Google Scholar]
- 27. Viggiano E, Ergoli M, Picillo E, Politano L. Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. Hum Genet. 2016; 135:685-698. [DOI] [PubMed] [Google Scholar]
- 28. Viggiano E, Picillo E, Ergoli M, Cirillo A, Del Gaudio S, Politano L. Skewed X-chromosome inactivation plays a crucial role in the onset of symptoms in carriers of Becker muscular dystrophy. J Gene Med. 2017; 10.1002/jgm.2952. [DOI] [PubMed] [Google Scholar]
- 29. Finsterer J, Stöllberger C. The heart in human dystrophinopathies. Cardiology. 2003; 99:1-19. [DOI] [PubMed] [Google Scholar]
- 30. Michele DE, Campbell KP. Cardiomyopathy in muscular dystrophies. In: Molecular mechanisms of cardiac hypertrophy and failure (Walsh RA. editor). Taylor & Francis, London, UK, 2005; pp.541-567. [Google Scholar]
- 31. Cripe L, Kinnett K, Uzark K, Eghtesady P, Wong B, Spicer R. P1.14 Cardiac transplantation in Duchenne muscular dystrophy: A case report. Neuromuscul Disord. 2011; 21:645 Doi: 10.1016/j.nmd.2011.06.774 [DOI] [Google Scholar]
- 32. Rees W, Schüler S, Hummel M, Hetzer R. Heart transplantation in patients with muscular dystrophy associated with end-stage cardiomyopathy. J Heart Lung Transplant. 1993; 12:804-807. [PubMed] [Google Scholar]
- 33. Amodeo A, Adorisio R. Left ventricular assist device in Duchenne cardiomyopathy: Can we change the natural history of cardiac disease? Int J Cardiol. 2012; 29:161:e43. [DOI] [PubMed] [Google Scholar]
- 34. Wu RS, Gupta S, Brown RN, Yancy CW, Wald JW, Kaiser P, Kirklin NM, Patel PC, Markham DW, Drazner MH, Garry DJ, Mammen PP. Clinical outcomes after cardiac transplantation in muscular dystrophy patients. J Heart Lung Transplant. 2010; 29:432-438. [DOI] [PubMed] [Google Scholar]
- 35. Ruiz-Cano MJ, Delgado JF, Jiménez C, Jiménez S, Cea-Calvo L, Sánchez V, Escribano P, Gómez MA, Gil-Fraguas L, Sáenz de la Calzada C. Successful heart transplantation in patients with inherited myopathies associated with end-stage cardiomyopathy. Transplant Proc. 2003; 35:1513-1515. [DOI] [PubMed] [Google Scholar]
- 36. Casazza F, Brambilla G, Salvato A, Morandi L, Gronda E, Bonacina E. Cardiac transplantation in Becker muscular dystrophy. J Neurol. 1988; 235:496-498. [DOI] [PubMed] [Google Scholar]
- 37. Patanè F, Zingarelli E, Attisani M, Sansone F. Successful heart transplantation in Becker's muscular dystrophy. Eur J Cardiothorac Surg. 2006; 29:250. [DOI] [PubMed] [Google Scholar]
- 38. Melacini P, Gambino A, Caforio A, Barchitta A, Valente ML, Angelini A, Fanin M, Thiene G, Angelini C, Casarotto D, Danieli GA, Dalla-Volta S. Heart transplantation in patients with inherited myopathies associated with end-stage cardiomyopathy: Molecular and biochemical defects on cardiac and skeletal muscle. Transplant Proc. 2001; 33:1596-1599. [DOI] [PubMed] [Google Scholar]
- 39. Steger CM, Höfer D, Antretter H. Cardiac manifestation in muscular dystrophies leading to heart transplantation. European Surgery. 2013; 45:245-250. [Google Scholar]
- 40. Yilmaz A, Gdynia HJ, Baccouche H, Mahrholdt H, Meinhardt G, Basso C, Thiene G, Sperfeld AD, Ludolph AC, Sechtem U. Cardiac involvement in patients with Becker muscular dystrophy: New diagnostic and pathophysiological insights by a CMR approach. J Cardiovasc Magn Reson. 2008; 10:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nigro G, Comi LI, Politano L, et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle Nerve. 1995; 18:283-291. [DOI] [PubMed] [Google Scholar]
- 42. Politano L, Palladino A, Nigro G, Scutifero M, Cozza V. Usefulness of heart rate variability as a predictor of sudden cardiac death in muscular dystrophies. Acta Myol. 2008;27:114-122. [PMC free article] [PubMed] [Google Scholar]
- 43. Ducceschi V, Nigro G, Sarubbi B, Comi LI, Politano L, Petretta VR, Nardi S, Briglia N, Santangelo L, Nigro G, Iacono A. Autonomic nervous system imbalance and left ventricular systolic dysfunction as potential candidates for arrhythmogenesis in Becker muscular dystrophy. Int J Cardiol. 1997. May 23;59:275-279. [DOI] [PubMed] [Google Scholar]
- 44. Fayssoil A, Abasse S. Cardiac resynchronization therapy in Becker muscular dystrophy: For which patients? Hellenic J Cardiol. 2010; 51:377-378. [PubMed] [Google Scholar]
- 45. Grande AM, Rinaldi M, Pasquino S, D'Armini AM, Viganò M. Heart transplantation in X-linked dilated cardiomyopathy. Ital Heart J. 2002; 3:476-478. [PubMed] [Google Scholar]
- 46. Diegoli M, Grasso M, Favalli V, Serio A, Gambarin FI, Klersy C, Pasotti M, Agozzino E, Scelsi L, Ferlini A, Febo O, Piccolo G, Tavazzi L, Narula J, Arbustini E. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J Am Coll Cardiol. 2011; 58:925-934. [DOI] [PubMed] [Google Scholar]
- 47. Melacini P, Fanin M, Angelini A, Pegoraro E, Livi U, Danieli GA, Hoffman EP, Thiene G, Dalla Volta S, Angelini C. Cardiac transplantation in a Duchenne muscular dystrophy carrier. Neuromuscul Disord. 1998; 8:585-590. [DOI] [PubMed] [Google Scholar]
- 48. Davies JE, Winokur TS, Aaron MF, Benza RL, Foley BA, Holman WL. Cardiomyopathy in a carrier of Duchenne's muscular dystrophy. J Heart Lung Transplant. 2001; 20:781-784. [DOI] [PubMed] [Google Scholar]
- 49. Ketelsen UP, Trenk D, Eschenbruch EM, Tollenaere PJ. Myopathy/rhabdomyolisis in patients after heart transplantation by presurgical treatment with lipid-lowering drugs? Interaction of cyclosporine and HMG-CoA reductase inhibitor therapy? Neuromuscul Disord. 1997; 7:446.9132144 [Google Scholar]