

Abstract
Brown adipose has the potential to counteract obesity, and thus, identifying signaling pathways that regulate the activity of this tissue is of great clinical interest. PRDM16 is a transcription factor that activates brown fat‐specific genes while repressing white fat and muscle‐specific genes in adipocytes. Whether PRDM16 also controls other gene programs to regulate adipocyte function was unclear. Here, we identify a novel role for PRDM16 in suppressing type I interferon (IFN)‐stimulated genes (ISGs), including Stat1, in adipocytes in vitro and in vivo. Ectopic activation of type I IFN signaling in brown adipocytes induces mitochondrial dysfunction and reduces uncoupling protein 1 (UCP1) expression. Prdm16‐deficient adipose displays an exaggerated response to type I IFN, including higher STAT1 levels and reduced mitochondrial gene expression. Mechanistically, PRDM16 represses ISGs through binding to promoter regions of these genes and blocking the activating function of IFN regulatory factor 1 (IRF1). Together, these data indicate that PRDM16 diminishes responsiveness to type I IFN in adipose cells to promote thermogenic and mitochondrial function.
Keywords: brown adipose, interferon regulatory factor, mitochondria, Prdm16, Ucp1
Subject Categories: Immunology, Metabolism
Introduction
There are three general classes of adipocytes: white, brown, and beige. White adipocytes store and release energy according to systemic demand, whereas brown and beige adipocytes burn energy to produce heat. Brown and beige adipocytes are characterized by a high density of mitochondria that contain uncoupling protein 1 (UCP1) in their inner membrane. UCP1, when activated by fatty acids, permits proton leak across the inner mitochondrial membrane (Klingenberg et al, 1999). Dissipation of the mitochondrial proton gradient by UCP1 drives the oxidation of available substrates and results in heat production. The thermogenic function of brown and beige fat defends mammals against hypothermia upon cold exposure. Additionally, brown and beige fat activity counteracts many of the harmful metabolic effects of a high‐fat diet in mice, including obesity and insulin resistance (Guerra et al, 1998; Cederberg et al, 2001; Feldmann et al, 2009; Seale et al, 2011; Auffret et al, 2012). In humans, brown adipose tissue (BAT) activity levels correlate with leanness (van Marken Lichtenbelt et al, 2009; Saito et al, 2009).
PRD1‐BF1‐RIZ1 homologous domain‐containing protein 16 (PRDM16) is a critical regulator of the brown fat‐selective gene program in brown and beige adipocytes (Seale et al, 2007, 2008, 2011; Kajimura et al, 2008; Ohno et al, 2012; Harms et al, 2015). PRDM16 increases the transcription of brown fat‐specific genes such as Ucp1 by co‐activating various transcription factors, including peroxisome proliferator‐activated receptor gamma (PPARγ), PPARγ coactivator 1‐alpha (PGC1α), and CCAAT/enhancer‐binding protein beta (CEBP‐β) (Seale et al, 2007, 2008; Kajimura et al, 2009). The co‐activator function of PRDM16 is mediated, at least in part, through recruitment of the Mediator complex to brown fat‐specific gene enhancers (Harms et al, 2015; Iida et al, 2015). PRDM16 also represses the transcription of certain white adipocyte‐specific and muscle‐specific genes in adipose cells by interacting with C‐terminal‐binding proteins (CtBPs) and euchromatic histone‐lysine N‐methyltransferase 1 (EHMT1) (Kajimura et al, 2008; Ohno et al, 2013; Harms et al, 2014). Of note, the N‐terminal PR domain of PRDM16 contains methyltransferase activity and is able to methylate histone H3K9 (Pinheiro et al, 2012; Li et al, 2015; Zhou et al, 2016).
Genetic loss‐of‐function studies in mice show that PRDM16 is required for the maintenance of BAT activity and for beige adipocyte biogenesis in white adipose tissue (WAT) (Seale et al, 2011; Cohen et al, 2014; Harms et al, 2014). PRDM16 also plays an important role in the development and function of other cell types, including hematopoietic stem cells (HSCs) and neural stem cells (NSCs) (Chuikov et al, 2010; Aguilo et al, 2011). Deletion of PRDM16 in HSCs and NSCs increases the levels of reactive oxygen species (ROS) and promotes cell death (Chuikov et al, 2010). Similarly, loss of PRDM16 in astrocytoma cells leads to mitochondrial dysfunction and apoptosis (Lei et al, 2016). In HSCs, PRDM16 induces the expression of Mitofusin 2 to promote mitochondrial function and reduce endoplasmic reticulum stress (Luchsinger et al, 2016).
In this study, we identify a previously unrecognized role for PRDM16 as a repressor of type I interferon (IFN) responses. The type I IFN pathway is best known for its critical role in antiviral defense. However, type I IFN also regulates the activity of certain stem cell populations (Essers et al, 2009; Sato et al, 2009; Yu et al, 2015). We found that PRDM16 blocked both the basal and IFNα‐induced expression of type I IFN‐stimulated genes (ISGs) in adipogenic cells. Conversely, deletion of Prdm16 from brown adipose cells and from BAT in vivo increased ISG expression. Prdm16‐deficient BAT was also hyper‐responsive to induced IFN signaling in vivo. Ectopic activation of type I IFN signaling in brown adipocytes caused profound mitochondrial dysfunction and reduced thermogenic capacity. Mechanistically, PRDM16 blocked the binding and transcriptional activating function of IFN regulatory factor 1 (IRF1) at ISG promoters. We conclude that PRDM16‐mediated ISG repression plays an important role in maintaining mitochondrial and thermogenic function in adipocytes.
Results
PRDM16 is required to repress type I IFN‐stimulated genes (ISGs) in adipocytes
PRDM16 binds and activates the transcription of many brown fat‐specific genes in adipocytes (Harms et al, 2015). To identify additional PRDM16‐regulated genes in adipocytes, we performed an unbiased analysis of gene expression in Prdm16‐deleted versus control adipocytes using cDNA microarrays. Adipogenic precursor cells were isolated from the inguinal (subcutaneous) WAT (ingWAT) of Rosa26 CreER; Prdm16 flox (R26 Cre+) mice and treated with 4‐hydroxytamoxifen (4OHT) to induce Prdm16 deletion or with vehicle (ethanol) as control. Prdm16‐knockout (KO) and control cells were then induced to undergo adipocyte differentiation in the presence or absence of the PPARγ ligand rosiglitazone (rosi), which activates mitochondrial and brown fat‐selective genes (Tai et al, 1996; Digby et al, 1998; Petrovic et al, 2008; Ohno et al, 2012). Prdm16 KO and control cells underwent efficient conversion into mature lipid droplet‐containing adipocytes that expressed equivalent levels of general adipocyte‐specific genes such as Adiponectin (AdipoQ) and Pparg2 (Fig 1A). Prdm16‐deleted adipocytes expressed drastically reduced levels of Ucp1 and other brown fat‐selective genes in response to rosi (Fig 1A), in agreement with published results (Ohno et al, 2012).
Figure 1. PRDM16 is required to repress type I IFN‐stimulated genes (ISGs) in adipocytes.
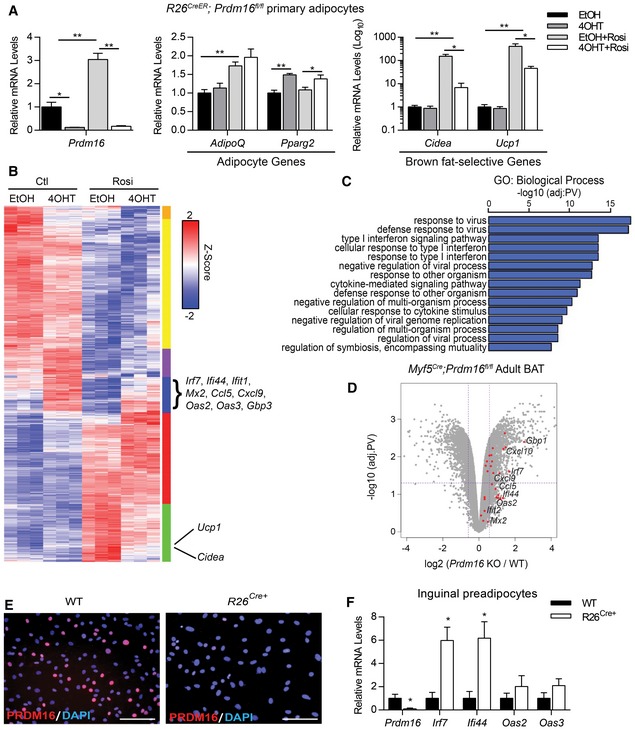
- Relative mRNA levels of Prdm16, pan‐adipogenic genes (AdipoQ, Pparg2), and brown fat‐selective genes (Cidea, Ucp1) in R26 CreER; Prdm16 fl/fl inguinal adipocytes treated with ethanol (EtOH) or 1 μM 4‐hydroxytamoxifen (4OHT) to induce knockdown of Prdm16, then differentiated +/− 1 μM rosiglitazone (rosi).
- Heat map depicting global gene expression levels in control (EtOH) and Prdm16 KO (4OHT) cells under control (Ctl) or rosi treatment.
- Gene ontology (GO) analysis of upregulated genes (blue cluster, B).
- Volcano plot depicting log‐fold change of gene expression in Prdm16 fl/fl (WT) and Myf5 Cre ; Prdm16 fl/fl (KO) adult mice. Red dots identify type I IFN‐stimulated genes (ISGs) found in the blue cluster of the heat map in (B).
- Immunofluorescence analysis of PRDM16 expression (red) and nuclei (DAPI, blue) in WT and R26 CreER; Prdm16 fl/fl (R26Cre+) primary inguinal preadipocytes treated with 4OHT. Scale bar = 100 μm.
- Relative mRNA levels of Prdm16 and ISGs in WT and R26Cre+ primary inguinal preadipocytes.
Global analyses revealed that many brown fat‐selective and mitochondrial genes were induced by rosi in a PRDM16‐dependent manner (Figs 1B and EV1A; green cluster). Gene ontology (GO) analysis of the most upregulated genes in Prdm16 KO versus control adipocytes, both with and without rosi treatment, identified the type I IFN and viral defense pathways as prominent PRDM16‐repressed pathways (Fig 1B and C; blue cluster). The majority of the genes in this group were ISGs, including Irf7, Ifi44, Mx2, Cxcl9, and Oas2. These ISGs were also greatly increased in 4OHT‐treated adipocytes from R26 Cre+ but not from Prdm16 fl/fl wild‐type mice, showing that ISG activation was not caused by 4OHT (Fig EV1B). Importantly, ISG levels were increased in Prdm16 KO BAT of adult Myf5 Cre;Prdm16 fl/fl mice compared to control WT BAT (Fig 1D). The induction of ISGs was not apparent in the BAT of young Prdm16 KO mice (Fig EV1C), which have intact thermogenic function (Harms et al, 2014). Additionally, the cold‐induced beiging of subcutaneous inguinal (ing) WAT, which occurs in a PRDM16‐dependent manner (Cohen et al, 2014), was accompanied by decreased expression levels of many ISGs, including Ifi27l2 and Ccl5 (Fig EV1D).
Figure EV1. PRDM16 is required for repression of interferon‐stimulated genes.
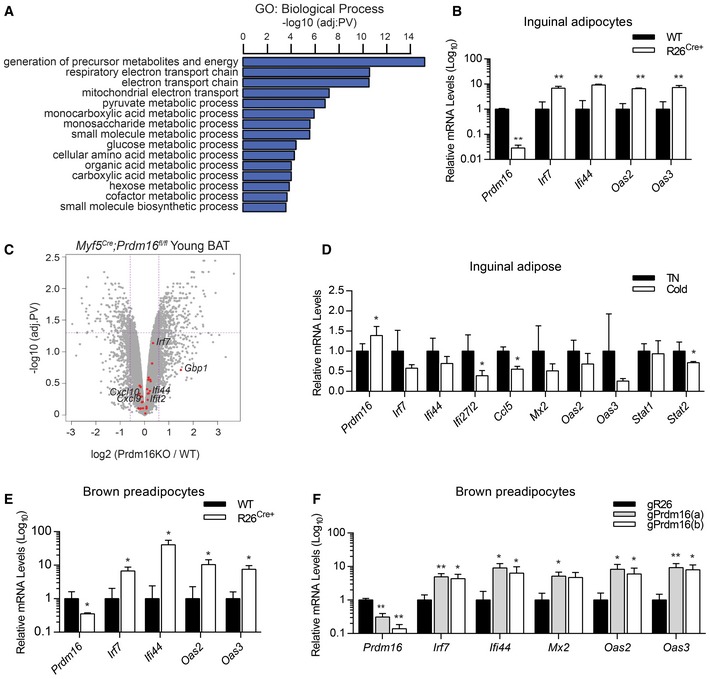
- Gene ontology (GO) of downregulated genes in Prdm16 KO cells (green cluster Fig 1B).
- Relative mRNA levels of Prdm16 and ISGs in Prdm16 fl/fl (WT) and R26 CreER; Prdm16 fl/fl (R26 Cre+) inguinal adipocytes treated with 1 μM 4‐hydroxytamoxifen (4OHT).
- Volcano plot comparing gene expression between young Prdm16 fl/fl (WT) and Prdm16 KO BAT. Red dots indicate type I ISGs found in the blue cluster of Fig 1B heat map.
- Relative mRNA levels of Prdm16 and ISGs in inguinal adipose from wild‐type mice incubated in TN (n = 5) or cold (n = 5).
- Relative mRNA levels of Prdm16 and ISGs in WT (n = 7) and R26 Cre+ (n = 13) brown preadipose cells treated with 4OHT.
- Relative mRNA of Prdm16 and ISGs in brown adipocyte precursor cells transduced with CRISPR lentiviral vectors expressing Cas9 and guide RNA sequences for Rosa26 (gR26) or Prdm16 (gPrdm16a, gPrdm16b) (n = 3).
PRDM16 expression increases during the course of adipocyte differentiation (Seale et al, 2007, 2011), and thus, the role of PRDM16 in regulating genes at the preadipocyte stage is largely unknown. We reliably detected nuclear PRDM16 protein in precursor cells isolated from the ingWAT of WT mice, while the addition of 4OHT eliminated PRDM16 protein signal only from R26Cre+‐derived cells (Fig 1E). As observed in mature adipocytes, Prdm16 deletion in ingWAT‐derived (Fig 1F) and BAT‐derived (Fig EV1E) precursor cells led to increased expression of many ISGs. Similarly, CRISPR/Cas9‐mediated reduction in PRDM16 (PRDM16‐CRISPR) expression in brown adipocyte precursors increased ISG expression (Fig EV1F). Together, these results establish a requirement for PRDM16 in repressing a broad set of type I ISGs in adipocytes and adipocyte precursor cells both in vitro and in vivo.
PRDM16 blocks type I IFN responses downstream of IFNAR receptor
To determine whether ectopic PRDM16 expression is sufficient to repress ISGs, we transduced Prdm16 KO brown adipocyte precursors with either control or PRDM16‐expressing retroviral vectors. PRDM16 decreased both the mRNA and protein levels of signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2) (Fig 2A and B), which are transcription factors that mediate many effects of type I IFN (Leung et al, 1995; Bromberg et al, 1996; Horvath et al, 1996; Meraz et al, 1996; Park et al, 2000). The reduced protein levels of STAT1 and STAT2 corresponded with reduced levels of the phosphorylated (active) forms of these factors (Fig 2B). PRDM16 also strongly blocked the expression of many other ISGs, including a 10‐ to 20‐fold reduction in the mRNA levels of Irf7, Ifi44, Oas2, and Oas3 (Fig 2A). PRDM16 did not reduce the levels of STAT3, another transcription factor involved in the IFN cascade (Fig 2B).
Figure 2. PRDM16 blocks type I IFN signaling downstream of IFNAR receptor.
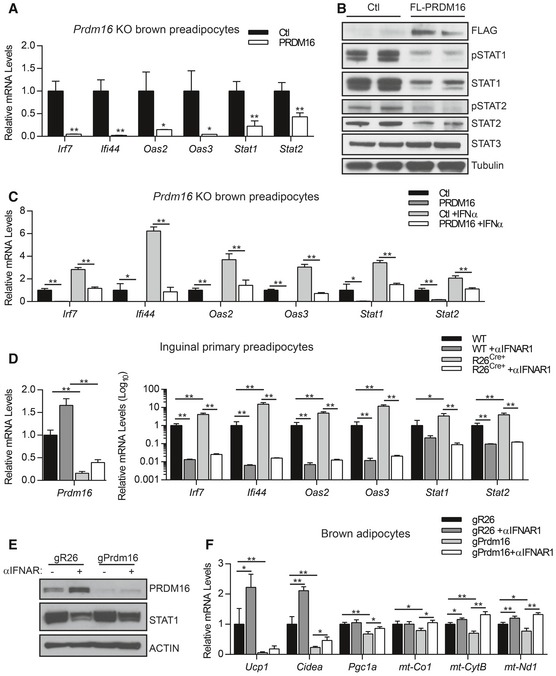
- Relative mRNA levels of IFN‐stimulated genes (ISGs) in Prdm16 KO brown adipocyte precursors infected with control (Ctl) or PRDM16 retrovirus. Data represent (n = 3) mean ± standard deviation and are consistent with results from duplicate independent experiments. *P ≤ 0.05, **P ≤ 0.01 (Student's t‐test).
- Western blot analysis of FLAG, phosphorylated STAT1 (pSTAT1), STAT1, phosphorylated STAT2 (pSTAT2), STAT3, and tubulin (loading control) protein in Prdm16 KO precursors infected with control (Ctl) or FLAG‐PRDM16 retrovirus.
- Relative mRNA levels of ISGs in control (Ctl) and PRDM16‐expressing preadipocytes +/− recombinant mouse IFNα. Data represent (n = 3) mean ± standard deviation and are consistent with results from duplicate independent experiments. *P ≤ 0.05, **P ≤ 0.01 (Student's t‐test).
- Relative mRNA levels of ISGs in WT and R26Cre+ inguinal preadipocytes treated with 4OHT and vehicle or anti‐IFNAR1 neutralizing antibody (αIFNAR1) for 4 days. Data represent (n = 3) mean ± standard deviation and are consistent with results from duplicate independent experiments. *P ≤ 0.05, **P ≤ 0.01 (paired two‐way ANOVA).
- Western blot analysis of PRDM16, STAT1, and actin (loading control) protein in brown adipocytes expressing gR26 (control) and gPrdm16 CRISPR/Cas9 constructs and treated +/− αIFNAR1 throughout differentiation.
- Relative mRNA levels of brown‐selective (Ucp1, Cidea, Pgc1a) and mitochondrial (mt‐Co1, mt‐CytB, mt‐Nd1) genes in brown adipocytes expressing gR26 and gPrdm16 CRISPR/Cas9 constructs +/− αIFNAR1 throughout differentiation. Data represent (n = 3) mean ± standard deviation and are consistent with results from duplicate independent experiments. *P ≤ 0.05, **P ≤ 0.01 (paired two‐way ANOVA).
Source data are available online for this figure.
We next examined whether PRDM16 regulates transcriptional responses to exogenous type I IFN. Ectopic PRDM16 expression reduced basal ISG levels and blunted IFNα‐induced ISG expression, including Stat1 and Stat2 (Fig 2C). Conversely, ISGs were induced to higher levels in Prdm16 KO cells than in control cells upon treatment with varying doses of IFNα (Fig EV2A), indicating that Prdm16 deficiency sensitizes cells to IFNα.
Figure EV2. PRDM16 blocks type I IFN signaling downstream of IFNAR receptor.
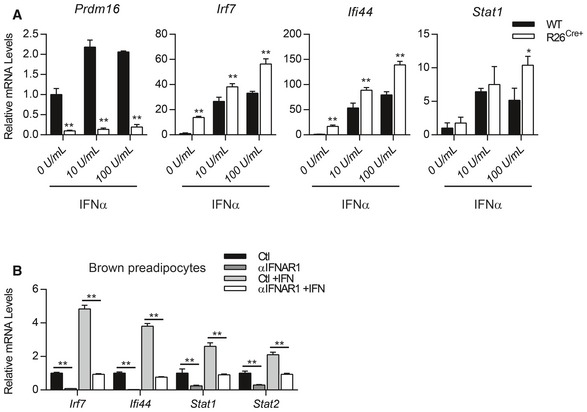
- Relative mRNA levels of Prdm16, Irf7, Ifi44, and Stat1 in WT and R26 Cre+ inguinal precursors treated with increasing doses of recombinant mouse IFNα.
- Relative mRNA levels of ISGs in brown preadipocytes treated with vehicle, anti‐IFNAR (αIFNAR) neutralizing antibody, mouse IFNα, or a combination of αIFNAR and IFNα.
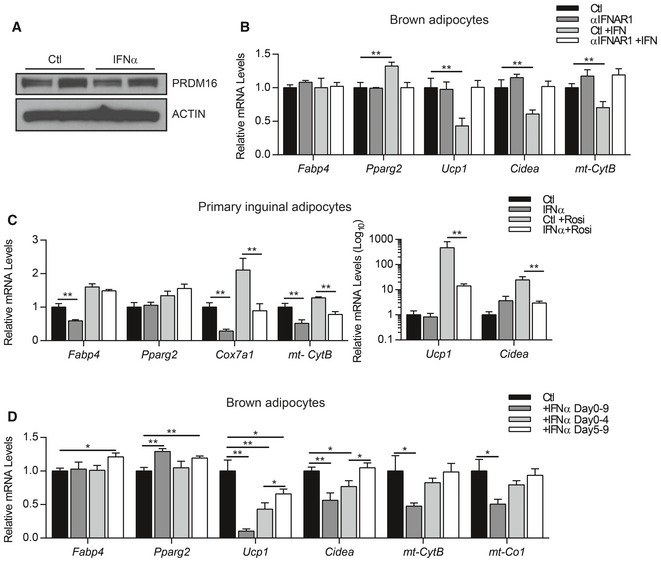
Type I IFNs bind to the interferon alpha and beta receptor (IFNAR) on the cell surface which activates a downstream signaling cascade. This response can be efficiently and specifically blocked in brown preadipocytes through treatment with an IFNAR‐neutralizing antibody (αIFNAR) (Fig EV2B). Notably, αIFNAR1 treatment eliminated ISG expression in both WT and Prdm16 KO cells, indicating that receptor signaling is active under basal conditions and that ISG induction due to loss of PRDM16 requires IFNAR function (Fig 2D).
To determine whether basal IFN signaling influences brown adipogenesis, we differentiated control and PRDM16‐depleted brown preadipose cells with or without αIFNAR1. PRDM16 expression was efficiently reduced using CRISPR/Cas9 technology (Fig 2E), resulting in a corresponding decrease in the levels of brown fat‐specific (Ucp1, Cidea, Pgc1a) and mitochondrial (mt‐Co1, mt‐CytB, mt‐Nd1) genes in differentiated brown adipocytes (Fig 2F). Anti‐IFNAR treatment rescued the expression of mitochondrial genes but not Ucp1 in Prdm16‐deleted adipocytes (Fig 2F). Altogether, these results demonstrate that PRDM16 acts downstream of the type I IFN receptor to repress transcriptional responses to type I IFN and safeguard mitochondrial gene expression in adipocytes.
Activation of type I IFN signaling disrupts mitochondrial structure and function in adipocytes
The above studies suggested that IFN activation may have an inhibitory effect on brown fat cell differentiation and/or function. To evaluate this, we treated brown adipocyte precursor cells with recombinant IFNα or vehicle control and induced adipocyte differentiation. We used a dose of IFNα that increased STAT1 mRNA and protein levels and elevated ISG levels to a similar extent as that observed in Prdm16 KO cells (Fig 3A and C). A previous study reported that IFNα inhibits adipogenesis of 3T3‐L1 cells (Lee et al, 2016). By contrast, we found that IFNα‐treated and control‐treated cells differentiated into Oil‐Red‐O‐stained mature adipocytes with equivalent efficiency and expressed similar levels of the general adipocyte marker genes Fabp4 and Pparg2 (Fig 3B). Strikingly however, IFNα‐treated adipocytes expressed drastically lower levels of UCP1 at the mRNA and protein level (Fig 3C and D) with no change in PRDM16 protein levels (Fig EV3A). IFNα treatment also decreased the expression of the brown fat marker gene Cidea and several mitochondrial genes, including Cox7a1 and mitochondrial‐encoded genes mt‐Cytb and mt‐Co1 (Fig 3D). Pre‐treatment of cells with αIFNAR prevented the decrease in Ucp1 and mt‐CytB expression in mature adipocytes (Fig EV3B), confirming that the inhibitory effect of IFNα on brown fat and mitochondrial programing was due to elevated canonical IFN signaling. IFNα treatment similarly inhibited the beige fat program in inguinal adipocytes, including reducing the basal levels of mitochondrial genes and repressing (by ~50‐fold) the rosi‐stimulated expression of Ucp1 (Fig EV3C).
Figure 3. Type I IFN disrupts mitochondrial structure and function in adipocytes.
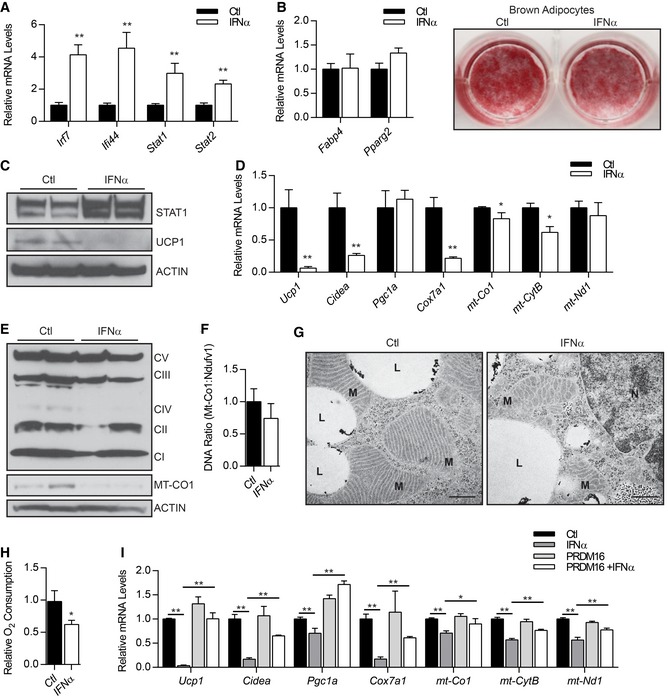
- Relative expression levels of ISGs. Data represent (n = 3) mean ± standard deviation. **P ≤ 0.01 (Student's t‐test).
- Oil Red O staining of lipid droplets and relative mRNA levels of pan‐adipogenic genes (Fabp4, Pparg2). Data represent (n = 3) mean ± standard deviation.
- Western blot analysis of STAT1, UCP1, and actin (loading control) protein levels.
- Relative mRNA levels of brown fat‐selective (Ucp1, Cidea, Pgc1a) and mitochondrial (Cox7a1, mt‐Co1, mt‐Cytb, mt‐Nd1) genes. Data represent (n = 3) mean ± standard deviation and are consistent with duplicate independent experiments. *P ≤ 0.05, **P ≤ 0.01 (Student's t‐test).
- Western blot analysis of mitochondrial complex proteins and actin (loading control).
- Relative ratio of mitochondrial DNA (mt‐Co1) to nuclear DNA (Ndufv1) (n = 6). Data are presented as mean ± standard deviation.
- Transmission electron micrograph of representative brown adipocytes showing mitochondria (M), lipid droplets (L), and nuclei (N). Scale bar = 500 nm.
- Relative oxygen consumption rates of adipocytes (n = 6). Data are presented as mean ± standard deviation. *P ≤ 0.05 (Student's t‐test).
- Relative mRNA levels of brown‐selective genes (Ucp1, Cidea, Pgc1a) and mitochondrial genes (mt‐Co1, mt‐CytB, mt‐Nd1) in brown adipocytes infected with control (Ctl) or PRDM16 retrovirus +/− mouse IFNα. Data represent (n = 3) mean ± standard deviation. *P ≤ 0.05, **P ≤ 0.01 (paired two‐way ANOVA).
Figure EV3. Type I IFN disrupts mitochondrial structure and function in adipocytes.
-
A, BWestern blot analysis of PRDM16 and actin protein (A) and relative mRNA levels of pan‐adipogenic genes (Fabp4, Pparg2) and brown‐selective genes (Ucp1, Cidea) (B) in brown adipocytes treated with vehicle, anti‐IFNAR (αIFNAR) neutralizing antibody, mouse IFNα, or αIFNAR + IFNα.
-
CRelative mRNA levels of general adipocyte markers (Fabp4, Pparg2), mitochondrial genes (Cox7a1, mt‐Cytb), and brown fat‐selective genes (Ucp1, Cidea) in primary inguinal adipocytes treated with IFNα or vehicle (Ctl) +/− 1 μM rosiglitazone (Rosi).
-
DRelative mRNA levels of general adipocyte markers (Fabp4, Pparg2), brown fat‐selective genes (Ucp1, Cidea), and mitochondrial genes (mt‐Cytb, mt‐Co1) in brown adipocytes treated with vehicle (Ctl) or mouse IFNα for varying periods during differentiation.
We found that IFN treatment early in brown fat differentiation (day 0–4) led to a permanent reduction in the expression of brown fat and mitochondrial genes in mature adipocytes (5 days later); this included a ~60% reduction in Ucp1 levels (Fig EV3D). By contrast, IFNα treatment during later stagers (days 5–9) had less of an impact on the brown fat gene program, including a ~35% reduction in Ucp1 and no significant change in Cidea expression. Overall, these results show that activation of the type I IFN system in brown preadipocytes and during early stages of differentiation specifically impairs the activation of brown fat‐specific genes.
IFNα reduced the expression of specific mitochondrial proteins in brown fat cells, including MT‐CO1, a subunit of complex IV, without affecting the levels of other mitochondrial components (Fig 3E). Control and IFNα‐treated cells had comparable amounts of mitochondria DNA (Fig 3F), suggesting that IFNα treatment does not reduce mitochondrial biogenesis per se. However, transmission electron microscopic analyses revealed that IFNα treatment had profound effects on mitochondrial morphology. The mitochondria in control brown adipocytes contained dense and well‐organized cristae whereas the mitochondria in IFNα‐treated adipocytes had severely disorganized cristae with a highly reticular morphology (Fig 3G). Consistent with these morphological effects on mitochondria, IFNα‐treated adipocytes displayed a 40% reduction in oxygen consumption as compared to control adipocyte cultures (Fig 3H).
To determine whether increased PRDM16 expression can protect brown fat cells against the inhibitory effects of exogenous IFNα, we transduced brown preadipocytes with control or PRDM16‐expressing retroviral vectors and induced the cells to differentiate in the presence of IFNα or vehicle control. Remarkably, PRDM16 expression completely rescued Ucp1 expression in IFNα‐treated adipocytes. PRDM16 also mitigated the inhibitory effects of IFNα on the expression of Cidea and mitochondrial genes (Cox7a1, mt‐Cytb) (Fig 3I). Taken together, these results demonstrate that type I IFN signaling suppresses mitochondrial function and decreases the thermogenic capacity of brown and beige adipocytes and this effect can be blocked by elevating PRDM16 levels.
PRDM16 opposes type I IFN signaling in vivo
An important question is whether PRDM16 regulates the response of BAT to type I IFN in vivo. To address this question, we treated 6‐ to 7‐week‐old BAT‐selective Prdm16 KO (KO) (Myf5 Cre; Prdm16 flox) and littermate control mice with either vehicle or recombinant IFNα over a 2‐week period. At this young age, ISGs are expressed at similar levels in control and KO BAT (Fig EV1C). However, the IFNα treatment of mice induced STAT1 protein to much higher levels in KO BAT than in WT BAT (Fig 4A). Furthermore, the mRNA levels of Stat1 and several other ISGs were increased by IFNα treatment only in KO BAT (Figs 4B and EV4A). ISGs were expressed at comparable levels in the ingWAT of WT and BAT‐selective Prdm16 KO mice (Fig EV4C).
Figure 4. PRDM16 opposes type I IFN signaling in vivo .
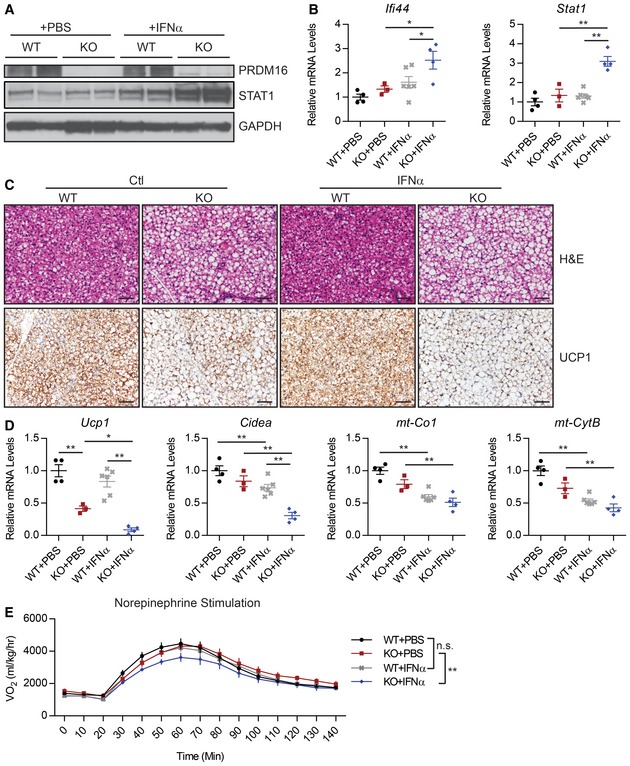
-
A–DPrdm16 fl/fl (WT) and Myf5 Cre ; Prdm16 fl/fl (KO) mice treated with IFNα or phosphate‐buffered saline (PBS) for 2 weeks prior to analysis of brown adipose tissue (BAT). Experimental groups: WT+PBS (n = 4), KO+PBS (n = 3), WT+IFN (n = 6), KO+IFN (n = 4). (A) Western blot analysis of PRDM16, STAT1, and GAPDH (loading control) protein levels. (B) qPCR analysis of Ifi44 and Stat1 mRNA levels. Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01 (paired two‐way ANOVA). (C) Hematoxylin and eosin (H&E) and anti‐UCP1 immunohistochemical staining. Scale bar = 50 μm. (D) Relative mRNA levels of brown fat‐specific genes (Ucp1, Cidea) and mitochondrial genes (mt‐Co1, mt‐CytB). Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01 (paired two‐way ANOVA).
-
EVolume of O2 (VO2) consumed before and after norepinephrine injection. Experimental groups: WT+PBS (n = 9), KO+PBS (n = 6), WT+IFN (n = 6), KO+IFN (n = 7). Data are presented as mean ± SEM. **P ≤ 0.01 (paired two‐way ANOVA).
Source data are available online for this figure.
Figure EV4. PRDM16 opposes type I IFN signaling in vivo .
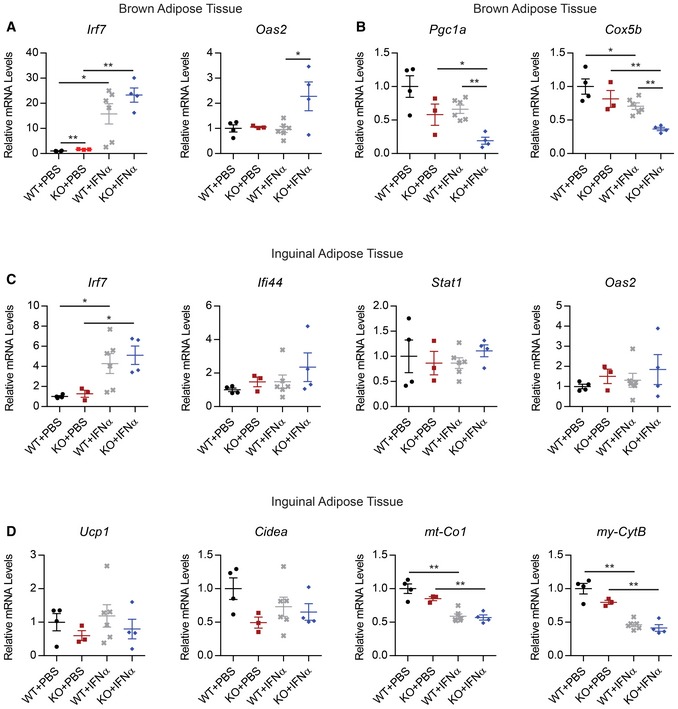
-
A, BRelative mRNA levels of ISGs (A) and mitochondrial genes (B) in brown adipose of Prdm16 fl/fl (WT) and Myf5 Cre ; Prdm16 fl/fl (KO) mice treated with IFNα or phosphate‐buffered saline (PBS) for 2 weeks.
-
C, DRelative mRNA levels of ISGs (C), as well as brown fat‐selective genes (Ucp1, Cidea) and mitochondrial genes (mt‐Co1, mt‐CytB) (D) in inguinal tissue from the same experimental mice in (A, B).
IFNα treatment had little impact on the morphology of BAT from control mice. Under basal conditions, the KO BAT had larger lipid droplets and reduced Ucp1 gene levels (Fig 4C and D). The loss of brown fat character in KO BAT was exacerbated by IFNα treatment. This included diminished expression of UCP1 and brown fat‐selective genes in BAT from IFNα‐treated relative to control‐treated KO animals (Figs 4C and D, and EV4B). Hematoxylin and eosin (H&E) staining of BAT sections revealed that there was greater lipid accumulation in the BAT of IFNα‐treated KO mice relative to that in saline‐treated KO mice (Fig 4C). In the ingWAT, where basal PRDM16 expression is low, the IFN treatment caused equivalent reduction in mitochondrial‐encoded genes (mt‐Co1, mt‐Cytb) in both WT and KO mice.
We studied the effect of IFNα treatment on the thermogenic capacity of WT and BAT‐selective Prdm16 KO mice by measuring oxygen consumption (respiration) using metabolic cages. To specifically evaluate BAT activity, we monitored respiration in anesthetized mice before and after stimulation with norepinephrine (NE), the physiological inducer of brown fat thermogenesis. Interestingly, there was no significant difference in NE‐stimulated respiration between WT (control) mice treated with vehicle or IFNα. However, KO mice treated with IFNα displayed a significant reduction in NE‐induced respiration compared to vehicle‐treated KO mice (P = 0.002) (Fig 3E). These results suggest that the PRDM16‐mediated suppression of type I IFN responses is required for preserving BAT function.
PRDM16 represses ISG transcription through direct binding at promoter/enhancers
PRDM16 is a transcriptional factor that binds at brown fat‐selective gene enhancers to activate gene transcription. Chromatin immunoprecipitation combined with high‐throughput sequencing (ChIP‐seq) in brown preadipocytes showed that PRDM16 also binds at or near the promoter regions of many ISGs, including Ifi44 and Oas3 (Fig 5A). These PRDM16‐bound regions had lower levels of the activating histone mark H3K27 acetylation in PRDM16‐expressing cells, suggesting that they are functional sites (Fig 5A). ChIP‐qPCR experiments confirmed that PRDM16 binds proximal to many of the ISGs that are reduced in expression by PRDM16 (Fig 5B).
Figure 5. PRDM16 represses ISGs through direct binding at gene promoters.
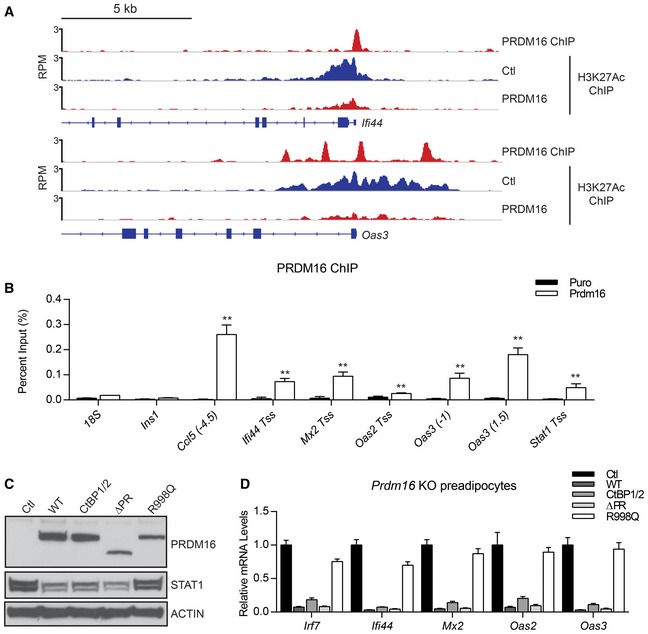
- ChIP‐seq stack‐height profiles in reads per million (RPM) for PRDM16 and H3K27 acetylation (Ac) at Ifi44 and Oas3 in Prdm16 KO brown adipocyte precursors that express PRDM16 or a control (Ctl) retrovirus.
- ChIP‐qPCR analysis of PRDM16 binding at ISG promoters/enhancers in control (Ctl) or PRDM16‐expressing brown preadipose cells. Ins1 and 18S were used as non‐specific binding site controls. Data represent (n = 3) mean ± standard deviation and are consistent with results from duplicate independent experiments. **P ≤ 0.01 (Student's t‐test).
- Western blot analysis of STAT1 and PRDM16 protein levels in Prdm16 KO brown preadipose cells transduced with retroviral vectors that express different forms of PRDM16: wild‐type (WT), CtBP‐binding mutant (CtBP1/2), PR‐domain deletion mutant (∆PR), DNA‐binding mutant (R998Q), or empty vector (Ctl). Loading control, actin.
- Relative mRNA levels of ISGs in cells from (C). Data represent (n = 3) mean ± standard deviation.
Source data are available online for this figure.
PRDM16 has several domains, including an N‐terminal PR domain with methyltransferase function, two zinc‐finger clusters (ZF1 and ZF2) that can bind to DNA, and a transcriptional repressor domain that interacts with C‐terminal binding proteins (CtBPs) (Nishikata et al, 2003; Ishibashi & Seale, 2015). To investigate which, if any, of these PRDM16 domains/activities are critical for ISG repression, we expressed mutant forms of PRDM16 in Prdm16 KO brown preadipose cells. PRDM16 mutants that lack CtBP‐binding or methyltransferase activity (e.g. PR‐domain mutant) repressed the expression of STAT1 and other ISGs to a similar degree as wild‐type PRDM16 (Fig 5C and D). However, a DNA‐binding mutant form of PRDM16 harboring a point mutation in the second zinc‐finger cluster (R998Q) had almost no capacity to repress ISGs (Fig 5C and D), though it activates brown fat genes and represses white fat genes normally (Fig EV5A and B, and Seale et al, 2007). These results suggest that DNA‐binding is critical for PRDM16‐mediated suppression of ISGs, but not for activation of brown fat‐selective genes.
Figure EV5. PRDM16 represses ISGs through direct binding at gene promoters.
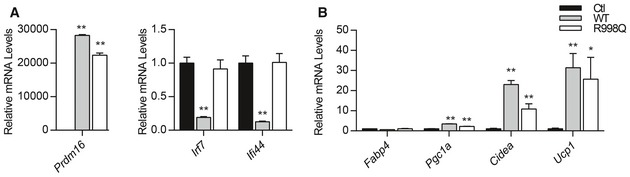
-
A, BRelative mRNA levels of Prdm16 and ISGs (A) and relative mRNA levels of adipogenic (Fabp4) and brown fat‐selective genes (Pgc1a, Cidea, Ucp1) in Prdm16 KO brown adipocytes cells transduced with retroviral vectors expressing wild‐type (WT) or DNA‐binding mutant (R998Q) PRDM16, or empty vector (Ctl).
PRDM16 blocks the activation of ISGs by IRF1
We identified an overlapping IFN‐stimulated response element (ISRE) and IRF‐binding element (IRF‐E) at many of PRDM16 binding sites at ISG promoters, including at the Ifi44 promoter (Fig EV6A). IFN regulatory factors (IRFs) are critical transcriptional effectors of the IFN signaling circuitry (Fujita et al, 1989; Harada et al, 1990; Kimura et al, 1994; Schafer et al, 1998; Sato et al, 2000). Moreover, various IRFs have been shown to regulate adipocyte differentiation and function (Eguchi et al, 2008, 2011; Kong et al, 2014; Kumari et al, 2016). Among the IRF family members, we found that Irf1 and Irf7 were particularly highly expressed in brown preadipocytes (Fig EV6B). IRF1 was a prime candidate for further study because it is known to activate a similar ISG signature as IFNα, including STAT1 (Xu et al, 2016). IRF1 expression levels are relatively constant throughout the process of adipocyte differentiation (Fig EV6C). To test whether IRF1 was required for ISG induction in Prdm16‐deficient brown adipocytes, we used lentiviral delivery of short‐hairpin RNAs (shRNAs) to knock down Irf1 expression. Two shRNA sequences were effective in reducing IRF1 protein levels and resulted in 50–70% reductions in the expression of many ISGs, including Irf7, Ifi44, and Stat1 (Fig 6A and B). The shRNA‐mediated reduction in ISG expression was reversed by co‐expression of the shRNA‐resistant human form of IRF1 (Fig EV6D). CRISPR/Cas9‐mediated reduction in IRF1 in Prdm16‐deficient brown adipocytes also decreased ISG expression (Fig EV6F). In mature brown adipocytes with endogenous levels of Prdm16 expression, knockdown of Irf1 did not affect ISG or brown fat gene expression (Fig EV6E), whereas ectopic IRF1 expression in fibroblasts increased ISG levels (Fig 6C and D).
Figure EV6. PRDM16 blocks IRF1 function at IRF‐E elements.
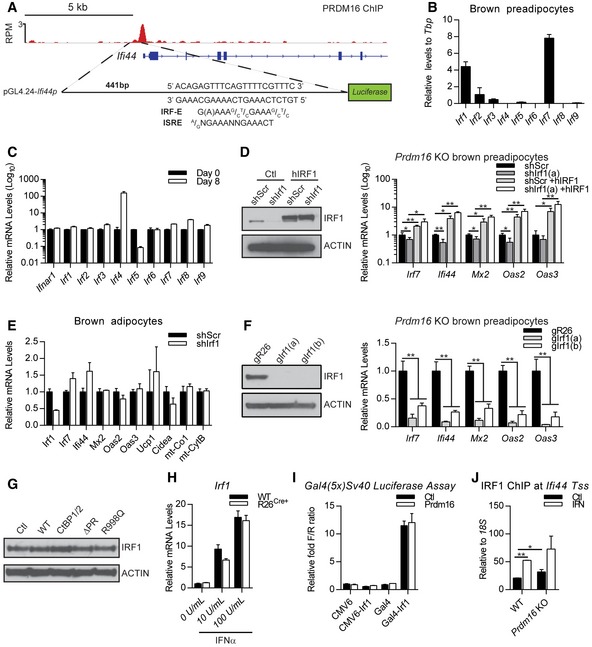
- Schematic showing the ChIP‐seq track of PRDM16 binding at Ifi44 promoter and the identified IFN‐stimulated response element (ISRE)/IRF‐binding element (IRF‐E) that was inserted into the luciferase reporter plasmid (pGL4.24‐Ifi44p).
- Relative mRNA levels of IRF genes in brown preadipose cells.
- Relative mRNA levels of Ifnar1 and Irfs in brown preadipocytes (D0) and mature brown adipocytes (D8).
- Western blot analysis of IRF1 and actin protein levels and relative mRNA levels of ISGs in Prdm16 KO brown adipocytes cells transduced with lentiviral short‐hairpin RNA directed against Irf1 (shIrf1) or a scrambled control (shScr) and either retroviral expression vectors expressing human IRF1 (hIRF1) or puromycin control (Ctl).
- Relative mRNA levels of Irf1 and ISGs and of brown fat‐selective genes (Ucp1, Cidea) and mitochondrial genes (mt‐Co1, mt‐CytB) in brown adipocytes transduced with lentiviral short‐hairpin RNA directed against Irf1 (shIrf1) or a scrambled control (shScr).
- Western blot analysis of IRF1 and actin protein levels and relative mRNA levels of ISGs in Prdm16 KO brown adipocytes cells transduced with CRISPR lentiviral vectors expressing Cas9 and guide RNA sequences for Rosa26 (gR26) or Irf1 (gIrf1a, gIrf1b).
- Western blot analysis of IRF1 and actin (loading control) protein levels in cells from Fig 5C.
- Relative mRNA levels of Irf1 in Prdm16 fl/fl (WT) and R26 CreER; Prdm16 fl/fl (R26 Cre+) inguinal adipocytes treated 1 μM 4OHT and increasing doses of IFNα.
- Transcriptional activity of a Gal4 UAS‐driven luciferase gene in response to expression of GAL4 DNA‐binding domain alone (Gal4), IRF1, or GAL4‐IRF1+/− PRDM16.
- ChIP‐qPCR showing IRF1 binding at Ifi44 transcriptional start site (Tss) in WT and Prdm16 KO cells +/− IFNα.
Figure 6. PRDM16 blocks the activation of ISGs by IRF1.
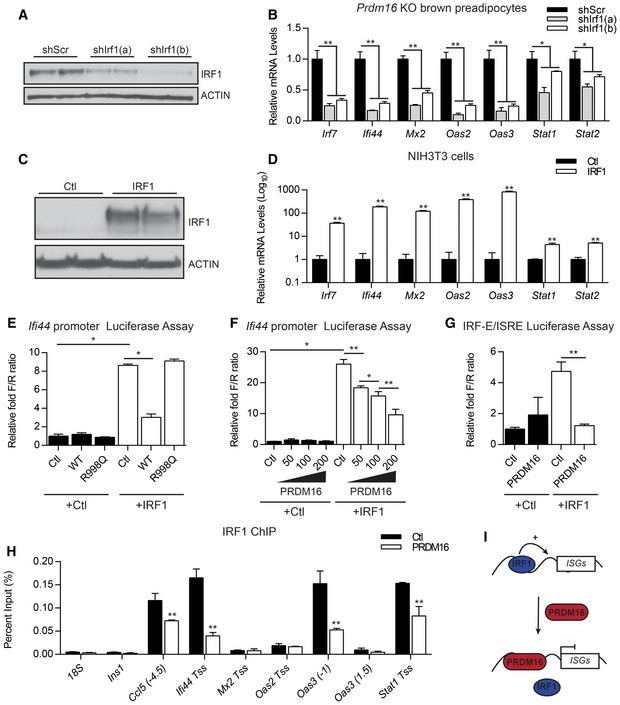
-
A, BWestern blot analysis of IRF1 and actin (loading control) protein levels (A) and relative mRNA levels of ISGs (B) in Prdm16 KO brown preadipose cells transduced with lentiviral short‐hairpin RNA directed against Irf1 (shIrf1a, shIrf1b) or a scrambled control (shScr).
-
C, DWestern blot analysis of IRF1 and actin (loading control) protein levels (C) and relative mRNA levels of ISGs (D) in cells from in NIH3T3 cells transfected with CMV6 (Ctl) or CMV6‐IRF1.
-
ETranscriptional activity of the Ifi44 promoter in NIH3T3 cells upon expression of IRF1 and wild‐type (WT) or DNA‐binding mutant (R998Q) forms of PRDM16.
-
FTranscriptional activity of the Ifi44 promoter in response to IRF1 expression and increasing amounts of PRDM16 expression.
-
GTranscriptional activity of the IFN regulatory factor‐binding element (IRF‐E)/IFN‐stimulated response element (ISRE) in Ifi44 in response to IRF1 and/or PRDM16.
-
HChIP‐qPCR analysis of IRF1 binding at ISGs in brown preadipose cells transduced with PRDM16 or control (Ctl) retrovirus. Ins1 and 18S were used as non‐specific binding site controls.
-
IProposed model for PRDM16‐action at ISG promoter regions.
We then explored whether PRDM16 functionally interacts with IRF1 to regulate ISG expression. We used the proximal Ifi44 promoter, containing an identified PRDM16‐binding site (Fig EV6A), to drive expression of a Luciferase reporter. IRF1 robustly activated the Ifi44 promoter, and this induction was very effectively blocked by co‐expression of WT but not the R998Q mutant form of PRDM16 (Fig 6E). We were unable to detect any evidence of a physical interaction between IRF1 and PRDM16 using a variety of approaches, and PRDM16 expression did not change IRF1 levels (Fig EV6G and H). Furthermore, PRDM16 did not repress the activating function of a GAL4 DBD (DNA‐binding domain)‐IRF1 fusion protein on a Gal4 DBD‐driven reporter (Fig EV6I). These results suggest that PRDM16 does not repress IRF1 function through physical binding and that the repressive effect of PRDM16 requires the IRF‐binding site and/or other nearby promoter elements.
PRDM16 suppressed IRF1‐mediated gene activation in a dose‐dependent manner (Fig 6F), suggesting that PRDM16 may compete with IRF1 for binding to the Ifi44 promoter. Consistent with this, the isolated IRF‐E/ISRE site alone was sufficient to confer responsiveness to both IRF1 and PRDM16 in transcription assays (Fig 6G). Additionally, we found that PRDM16 expression decreased IRF1 binding at several native ISG promoters using ChIP‐qPCR (Fig 6H). Finally, in WT brown preadipocytes with endogenous PRDM16 levels, recombinant IFNα increased IRF1 binding to various ISGs, while Prdm16 KO cells displayed higher IRF1 binding (Fig EV6J). Together, these results suggest that PRDM16 represses ISG target genes by binding to IRF elements and blocking access to the transcriptional activator, IRF1 (Fig 6I).
Discussion
PRDM16 is a critical transcriptional regulator of brown and beige adipocyte identity. PRDM16 co‐regulates other DNA‐binding factors to promote the transcription of brown fat genes in adipocytes (Seale et al, 2007, 2008; Kajimura et al, 2009). We show here that, in addition to its direct transcriptional activating effect on brown fat‐specific genes, PRDM16 reinforces and maintains brown fat identity by suppressing the type I IFN response. PRDM16 blocks the activation of IFN‐induced genes by competing with IRF1 for binding to IRF‐E binding motifs. While the interaction between PRDM16 and IRF1 plays an important role in regulating the IFN response in adipogenic cells, whether PRDM16 also functionally interacts with other IRFs remains to be determined. Notably, PRDM1, a related family member, also binds to the IRF1‐binding element to repress activation of the IFN pathway in other cell types (Kuo & Calame, 2004; Doody et al, 2007; Mould et al, 2015), suggesting that PRDM16 and PRDM1 may have overlapping roles in regulating the type I IFN pathway.
Type I IFNs are best known for their role in mounting antiviral responses. Virus‐infected cells secrete and respond to type I IFNs, including IFNα and IFNβ. These cytokines establish an antiviral state through multiple mechanisms, including the production of antimicrobial proteins that act directly on viruses and modulation of the adaptive immune response (Honda et al, 2005). The importance of this pathway is underscored by the finding that mice lacking the type I IFN receptor (IFNAR) rapidly succumb to viral infections (Hwang et al, 1995). Similarly, humans with mutations in STAT1, a key effector of the IFN response, die from viral infection (Dupuis et al, 2003; Chapgier et al, 2006). The suppressive effect of PRDM16 on IFN responses may be important for preserving the thermogenic function of BAT in virus‐infected animals. This may be especially important in small animals such as newborns to survive cold exposure while dealing with viral infection. PRDM16 may also be needed to protect BAT activity during viral infection in order to support hyperthermia (fever).
Low levels of type I IFN, particularly IFNβ, are also present in many cells/tissues in the absence of infection (Tovey et al, 1987; Hamilton et al, 1996; Hida et al, 2000; Hata et al, 2001; Abt et al, 2012). Constitutive type I IFN expression is believed to be a priming mechanism for rapid induction of the pathway upon viral infection (Vogel & Fertsch, 1984; Hata et al, 2001; Abt et al, 2012; Ganal et al, 2012; Kawashima et al, 2013). Similarly, increased type I IFN signaling can increase cellular responsiveness to other cytokines, such as IFNγ by increasing the levels of common signaling intermediates like STAT1 (Hamilton et al, 1996; Gough et al, 2010). Interestingly, the type I IFN pathway is required to achieve the proper balance of proliferation and maturation of hematopoietic stem cells (HSCs) (Essers et al, 2009; Kim et al, 2016). In this context, elevated IFN signaling leads to stem cell exhaustion (Essers et al, 2009; Sato et al, 2009), highlighting the importance of a tightly regulated IFN system. Notably, PRDM16 is also a critical regulator of HSCs (Chuikov et al, 2010), suggesting that regulation of IFN signaling may be a key function of PRDM16 in this compartment. Along these lines, it will now be important to determine whether the PRDM16 and IFN regulate the proliferation and/or maintenance of brown adipose precursors.
A prominent effect of IFN activation in brown adipocytes is reduced mitochondrial function and abnormal mitochondrial morphology (Fig 3). This result agrees with previous studies showing that IFNα inhibits the expression of mitochondrial‐encoded genes in lymphoid cells (Shan et al, 1990; Lewis et al, 1996). We found that IFNα activation leads to a loss of cristae structure and a reduction in specific mitochondrial proteins like MT‐CO1 in brown adipocytes. The mechanism(s) by which IFN activation reduces mitochondrial function is unclear. One possibility is that IFN‐activated STAT1/2 directly represses the transcription of Ucp1 and mitochondrial‐encoded genes. However, type I IFN induces a large number of downstream ISGs, any of which could have as yet undetermined roles in regulating mitochondrial function and cellular metabolism. Importantly, blocking JAK–STAT signaling in human adipocytes decreases IFN signaling and induces brown fatlike characteristics (Moisan et al, 2015), suggesting a potentially important role for this pathway in human metabolism.
In summary, PRDM16 regulates the brown fat gene program through multiple mechanisms, including via direct actions at brown fat gene enhancers and indirectly by suppressing the type I IFN‐driven gene program. Our study also suggests potentially important links between innate immune and metabolic pathways in adipocytes that warrant further investigation and predicts a potential role for IFN signaling in metabolic regulation. In support of this, genetic manipulations that influence the type I IFN pathway in mice have revealed significant metabolic phenotypes. For example, IRF3 knockout mice have increased energy expenditure due to the browning of the inguinal adipose (Kumari et al, 2016). Moreover, both IRF7 and IRF3 knockout mice are protected from diet‐induced obesity and have improved insulin sensitivity (Wang et al, 2013; Kumari et al, 2016). Additional focus on the role of type I IFN in adipocytes may reveal new approaches to preserve and/or increase brown fat activity for the treatment of obesity and metabolic disease.
Materials and Methods
Animals
All animal experiments were approved by the University of Pennsylvania's Institutional Animal Care and Use Committee. Rosa26 CreER, Prdm16 flox mice were maintained on a mixed 129Sv/C57Black6 genetic background (Harms et al, 2014). Myf5 Cre; Prdm16 flox mice were backcrossed into the C57Black6 background for 10 generations (Harms et al, 2014). Male mice were used for all experiments. Mice (6–7 weeks old) were injected with 25,000 U of recombinant mouse interferon alpha A (PBL Assay Science) or an equal volume of phosphate‐buffered saline (PBS) as control six times over 2 weeks. For NE injections, mice were first placed in CLAMS metabolic chambers at 33°C, then sedated with 75 mg/kg Nembutal, followed 20 min later by injection with 1 mg/kg NE (Sigma A9512‐1G). Data were collected until mice recovered from barbiturate sedation.
Cell culture
Primary inguinal preadipocytes were isolated from Prdm16 flox and Rosa26 CreER, Prdm16 flox mice as previously described (Rajakumari et al, 2013). Recombination in Rosa26 CreER, Prdm16 flox adipocytes was induced by treating cells with 1 μM of 4‐hydroxy‐tamoxifen (Sigma) for 3 days in growth phase. Cells were differentiated with medium containing 10% FBS, 0.5 μM isobutylmethylxanthine, 125 nM indomethacin, 1 μM dexamethasone, 20 nM insulin, and 1 nM T3 without or with 1 μM rosiglitazone. To block type I IFN signaling, cells were treated with 1 μg/ml anti‐IFNAR1 antibody MAR1‐5A3 (Leinco Technologies, Inc.) during growth for 4 days. Immortalized brown and primary ingWAT adipocytes were treated with vehicle or 1,000 U/ml recombinant mouse interferon alpha A (PBL Assay Science) throughout differentiation to determine effects of IFN on differentiation. For CRISPR/Cas9‐mediated gene editing, guide RNA sequences against mouse Prdm16 were cloned into LentiCRISPR (Shalem et al, 2014), a gift from Feng Zhang (Addgene, 49535). A guide targeted at the mouse Rosa26 locus was used as a negative control. gRNA‐Prdm16(A): 5′ CGGCGTGCATCCGCTTGTGC 3′; gRNA‐Prdm16(B): 5′ CCAACCTGTGCCGGCACAAG 3′; gRNA‐R26: 5′ AAGATGGGCGGGAGTCTTCT 3′; gRNA‐Irf1(A): 5′ AGCACGCTGCTAAGCACGGC 3′; gRNA‐Irf1(B): 5′ GCACGCTGCTAAGCACGGCT 3′. Short‐hairpin RNA (shRNA) constructs were generated by the High‐Throughput Screening Core (University of Pennsylvania). shIrf1(a): 5′ AGATGGACATTATACCAGATA 3′; shIrf1(b): 5′ CTCTTCTGTCTATGGAGACTT 3′. Oil Red O staining and retrovirus production were performed as described previously (Seale et al, 2007).
Real‐time qPCR
Total RNA was extracted by TRIzol (Invitrogen) followed by purification using PureLink RNA columns (Invitrogen). Isolated mRNA was reverse transcribed using the High‐Capacity cDNA Synthesis kit (Applied Biosystems) and used in real‐time qPCRs with SYBR Green master mix (Applied Biosystems) on a 7900 HT (Applied Biosystems). TATA‐binding protein (Tbp) was used as an internal normalization control.
Microarray data
Microarray services were provided by the UPENN Molecular Profiling Facility, including quality control tests of the total RNA samples by Agilent Bioanalyzer and Nanodrop spectrophotometry. All protocols were conducted as described in the Ambion Expression Manual and the Affymetrix GeneChip Expression Analysis Technical Manual. In microarray data of control (EtOH) and Prdm16 KO (4OHT) cells under control (Ctl) or rosiglitazone (rosi) (GSE86018), differentially expressed genes were selected for clustering analysis by fold change > 1.5 and adjusted P‐value < 0.05. Hierarchical clustering was performed using (1−Spearman correlation coefficient) as a distance measure for genes and samples. Gene ontology analysis was conducted using Enrichr (Chen et al, 2013; Kuleshov et al, 2016), and top enriched biological process terms were presented. For Prdm16 KO BAT gene expression data, we used previously published microarray data (Harms et al, 2014, 2015).
Cell immunostaining
Briefly, cells were fixed with 4% (wt/vol) paraformaldehyde (PFA) for 10 min, permeabilized with 0.5% Triton X‐100 for 15 min, and then blocked in 4% goat serum for 30 min. Cells were then incubated with primary antibody anti‐Prdm16 1:200 (Seale et al, 2007), followed by secondary antibody Alexa Fluor 647 donkey anti‐rabbit IgG 1:500 (Invitrogen), and DAPI (Invitrogen) for nuclear staining.
Western blot
Protein extracts were prepared as previously described (Rajakumari et al, 2013). Proteins were separated in 4–12% Bis–Tris NuPAGE gels (Invitrogen) and transferred to PVDF membranes. For Western blot, antibodies used were as follows: anti‐PRDM16 (Seale et al, 2007), anti‐FLAG (Sigma, F1804), anti‐pSTAT1 (Santa Cruz, sc7988), anti‐STAT1 (Santa Cruz, sc‐346), anti‐pSTAT2 (Millipore, 07‐224), anti‐STAT2 (Cell Signaling Technology, 4597S), anti‐STAT3 (Cell Signaling Technology, 9139S), anti‐tubulin (Sigma, T6199), anti‐UCP1 (R&D Systems, MAB6158), anti‐actin (Millipore), total OXPHOS antibody cocktail (Abcam, ab110413), anti‐MT‐CO1 (Abcam, ab14705), and anti‐IRF1 (Cell Signaling Technology, 8478S).
Chromatin immunoprecipitation
Immortalized brown preadipocytes infected with MSCV‐Puromycin or MSCV–Prdm16 were grown to confluency and fixed in 1% formaldehyde for 15 min, then quenched with 125 mM glycine for 5 min. ChIP was performed as described previously (Harms et al, 2015). Chromatin was probed with 1 μg of the following antibodies: anti‐PRDM16 (Harms et al, 2014) or anti‐histone H3K27Ac (Abcam, ab4729). Bound fragments were eluted at 65°C overnight in 20 mM Tris pH 8, 1 mM EDTA, and 1% SDS and subsequently treated with RNaseA and proteinase K before undergoing column purification (Qiagen, 28104). Target enrichment was calculated as percent input. ChIP‐seq reads for Prdm16 and H3K27Ac (GSE86017) were aligned to mouse genome, mm9, and further processed for peak‐calling and genome browser track creation as previously described (Harms et al, 2015).
Tissue O2 consumption
Differentiated brown adipocytes were trypsinized, pelleted, and resuspended in a buffer comprised of 2% BSA, 1.1 mM sodium pyruvate, and 25 mM glucose in PBS. Samples were placed in an MT200A Respirometer Cell (Strathkelvin), and oxygen consumption was measured for ~5 min. Oxygen consumption was normalized to total cell number.
Histology
For immunohistochemistry, BAT was fixed in 4% PFA overnight, dehydrated, and embedded in paraffin for sectioning. Sections were stained with hematoxylin and eosin or probed with antibodies for UCP1 (R&D Systems). For transmission electron microscopy, adipocytes were fixed with 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) overnight at 4°C, and then post‐fixed with 2.0% osmium tetroxide for 1 h at room temperature. Thin sections were stained with uranyl acetate and lead citrate and examined with a JEOL 1010 electron microscope.
Transcription assays
The Ifi44 promoter/luciferase reporter plasmid (pGL4‐Ifi44p) was constructed by PCR cloning of genomic sequence from C57Bl/6 DNA corresponding to 441 bp of Ifi44 proximal promoter and 76‐bp 5′ UTR into the XhoI and NcoI sites of pGL4.24, replacing the existing minimal promoter (Promega). pGL4‐Ifi44p‐ISRE (IRF‐E/ISRE) was built by inserting a 55‐bp linker centered at the Ifi44 transcriptional start site (and ISRE) into the KpnI and XhoI sites of pGL4.24, retaining the minimal promoter. CMX‐Gal4(DBD)‐hIRF1 was cloned by PCR amplifying human IRF1 from CMV6‐hIRF1, with BamHI and NotI sites appended for insertion into CMX‐Gal4(DBD). pRL‐CMV was used for internal normalization of the dual luciferase assays. The CMX‐Gal4(DBD) (containing 447 bp of the Gal4 DNA‐binding domain), Gal4(5x)SV40‐Luc, and pRL‐CMV plasmids were provided by Mitch Lazar (University of Pennsylvania). CMX‐hIRF1 was provided by Kathleen Sullivan (Children's Hospital of Pennsylvania). Reporter and expression plasmids were co‐transfected into NIH3T3 cells (ATCC) using Lipofectamine 2000 (Invitrogen; 11668019). At 48 h post‐transfection, cells were harvested into passive lysis buffer for dual luciferase assays (Promega; E1960) using a Synergy HT plate reader (BioTek).
Statistical analysis
Energy expenditure data were analyzed in R using a paired three‐way ANOVA over all time points after NE injection with significance level, α = 0.05. Subsequent paired two‐way ANOVAs for treatment effects over all time points were performed in individual genotype arms if interaction terms were significant at α = 0.05.
Author contributions
MK, JI, and MJH performed the experiments shown here. H‐WL and K‐JW analyzed microarray and ChIP‐Seq data. RRS provided advice on experimental setup and project development. MK and PS conceived the experiments and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We would like to thank the Histology and Gene Expression Core of the Penn Cardiovascular Institute for immunohistochemistry; the Electron Microscopy Core for processing and imaging; the UPENN Molecular Profiling Facility for microarray services; and the Functional Genomics Core of the Penn Diabetes and Endocrinology Center (DK19525) for ChIP sequencing. This work was funded by NIH/NIDDK grants R01DK103008 and R01DK107589 (P. Seale).
The EMBO Journal (2017) 36: 1528–1542
References
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, Wherry EJ, Artis D (2012) Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37: 158–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, Lemischka IR, Zhou BY, Snoeck HW (2011) Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood 117: 5057–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffret J, Viengchareun S, Carre N, Denis RG, Magnan C, Marie PY, Muscat A, Feve B, Lombes M, Binart N (2012) Beige differentiation of adipose depots in mice lacking prolactin receptor protects against high‐fat‐diet‐induced obesity. FASEB J 26: 3728–3737 [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Horvath CM, Wen Z, Schreiber RD, Darnell JE Jr (1996) Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc Natl Acad Sci USA 93: 7673–7678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederberg A, Gronning LM, Ahren B, Tasken K, Carlsson P, Enerback S (2001) FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet‐induced insulin resistance. Cell 106: 563–573 [DOI] [PubMed] [Google Scholar]
- Chapgier A, Wynn RF, Jouanguy E, Filipe‐Santos O, Zhang S, Feinberg J, Hawkins K, Casanova JL, Arkwright PD (2006) Human complete Stat‐1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo . J Immunol 176: 5078–5083 [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov S, Levi BP, Smith ML, Morrison SJ (2010) Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol 12: 999–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, Wu J, Gunawardana SC, Banks AS, Camporez JP, Jurczak MJ, Kajimura S, Piston DW, Mathis D, Cinti S, Shulman GI et al (2014) Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156: 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digby JE, Montague CT, Sewter CP, Sanders L, Wilkison WO, O'Rahilly S, Prins JB (1998) Thiazolidinedione exposure increases the expression of uncoupling protein 1 in cultured human preadipocytes. Diabetes 47: 138–141 [DOI] [PubMed] [Google Scholar]
- Doody GM, Stephenson S, McManamy C, Tooze RM (2007) PRDM1/BLIMP‐1 modulates IFN‐gamma‐dependent control of the MHC class I antigen‐processing and peptide‐loading pathway. J Immunol 179: 7614–7623 [DOI] [PubMed] [Google Scholar]
- Dupuis S, Jouanguy E, Al‐Hajjar S, Fieschi C, Al‐Mohsen IZ, Al‐Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, Al Ghonaium A, Tufenkeji H, Frayha H, Al‐Gazlan S, Al‐Rayes H, Schreiber RD, Gresser I, Casanova JL (2003) Impaired response to interferon‐alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 33: 388–391 [DOI] [PubMed] [Google Scholar]
- Eguchi J, Yan QW, Schones DE, Kamal M, Hsu CH, Zhang MQ, Crawford GE, Rosen ED (2008) Interferon regulatory factors are transcriptional regulators of adipogenesis. Cell Metab 7: 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos‐Flier E, Rosen ED (2011) Transcriptional control of adipose lipid handling by IRF4. Cell Metab 13: 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco‐Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A (2009) IFNalpha activates dormant haematopoietic stem cells in vivo . Nature 458: 904–908 [DOI] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J (2009) UCP1 ablation induces obesity and abolishes diet‐induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab 9: 203–209 [DOI] [PubMed] [Google Scholar]
- Fujita T, Kimura Y, Miyamoto M, Barsoumian EL, Taniguchi T (1989) Induction of endogenous IFN‐alpha and IFN‐beta genes by a regulatory transcription factor, IRF‐1. Nature 337: 270–272 [DOI] [PubMed] [Google Scholar]
- Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, Diefenbach A (2012) Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37: 171–186 [DOI] [PubMed] [Google Scholar]
- Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson AP, Trapani JA, Levy DE, Hertzog PJ, Clarke CJ, Johnstone RW (2010) Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol 8: e1000361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP (1998) Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest 102: 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA, Whitty GA, Kola I, Hertzog PJ (1996) Endogenous IFN‐alpha beta suppresses colony‐stimulating factor (CSF)‐1‐stimulated macrophage DNA synthesis and mediates inhibitory effects of lipopolysaccharide and TNF‐alpha. J Immunol 156: 2553–2557 [PubMed] [Google Scholar]
- Harada H, Willison K, Sakakibara J, Miyamoto M, Fujita T, Taniguchi T (1990) Absence of the type I IFN system in EC cells: transcriptional activator (IRF‐1) and repressor (IRF‐2) genes are developmentally regulated. Cell 63: 303–312 [DOI] [PubMed] [Google Scholar]
- Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, Sato T, Kurokawa M, Won KJ, Seale P (2014) Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab 19: 593–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms MJ, Lim HW, Ho Y, Shapira SN, Ishibashi J, Rajakumari S, Steger DJ, Lazar MA, Won KJ, Seale P (2015) PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes Dev 29: 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata N, Sato M, Takaoka A, Asagiri M, Tanaka N, Taniguchi T (2001) Constitutive IFN‐alpha/beta signal for efficient IFN‐alpha/beta gene induction by virus. Biochem Biophys Res Commun 285: 518–525 [DOI] [PubMed] [Google Scholar]
- Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T, Sato T, Hirose S, Shirai T, Taki S, Taniguchi T (2000) CD8(+) T cell‐mediated skin disease in mice lacking IRF‐2, the transcriptional attenuator of interferon‐alpha/beta signaling. Immunity 13: 643–655 [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Takaoka A, Taniguchi T (2005) Regulation of the type I IFN induction: a current view. Int Immunol 17: 1367–1378 [DOI] [PubMed] [Google Scholar]
- Horvath CM, Stark GR, Kerr IM, Darnell JE Jr (1996) Interactions between STAT and non‐STAT proteins in the interferon‐stimulated gene factor 3 transcription complex. Mol Cell Biol 16: 6957–6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I (1995) A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci USA 92: 11284–11288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida S, Chen W, Nakadai T, Ohkuma Y, Roeder RG (2015) PRDM16 enhances nuclear receptor‐dependent transcription of the brown fat‐specific Ucp1 gene through interactions with Mediator subunit MED1. Genes Dev 29: 308–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi J, Seale P (2015) Functions of Prdm16 in thermogenic fat cells. Temperature 2: 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Tomaru T, Erdjument‐Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM (2008) Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev 22: 1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, Spiegelman BM (2009) Initiation of myoblast to brown fat switch by a PRDM16‐C/EBP‐beta transcriptional complex. Nature 460: 1154–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Kosaka A, Yan H, Guo Z, Uchiyama R, Fukui R, Kaneko D, Kumagai Y, You DJ, Carreras J, Uematsu S, Jang MH, Takeuchi O, Kaisho T, Akira S, Miyake K, Tsutsui H, Saito T, Nishimura I, Tsuji NM (2013) Double‐stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon‐beta. Immunity 38: 1187–1197 [DOI] [PubMed] [Google Scholar]
- Kim PG, Canver MC, Rhee C, Ross SJ, Harriss JV, Tu HC, Orkin SH, Tucker HO, Daley GQ (2016) Interferon‐alpha signaling promotes embryonic HSC maturation. Blood 128: 204–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, Tanaka N, Kamijo R, Vilcek J, Mak TW, Taniguchi T (1994) Involvement of the IRF‐1 transcription factor in antiviral responses to interferons. Science 264: 1921–1924 [DOI] [PubMed] [Google Scholar]
- Klingenberg M, Echtay KS, Bienengraeber M, Winkler E, Huang SG (1999) Structure‐function relationship in UCP1. Int J Obes Relat Metab Disord 23(Suppl. 6): S24–S29 [DOI] [PubMed] [Google Scholar]
- Kong X, Banks A, Liu T, Kazak L, Rao RR, Cohen P, Wang X, Yu S, Lo JC, Tseng YH, Cypess AM, Xue R, Kleiner S, Kang S, Spiegelman BM, Rosen ED (2014) IRF4 is a key thermogenic transcriptional partner of PGC‐1alpha. Cell 158: 69–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, Tenen D, Roh HC, Kong X, Kazak L, Ahmad R, Rosen ED (2016) IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest 126: 2839–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo TC, Calame KL (2004) B lymphocyte‐induced maturation protein (Blimp)‐1, IFN regulatory factor (IRF)‐1, and IRF‐2 can bind to the same regulatory sites. J Immunol 173: 5556–5563 [DOI] [PubMed] [Google Scholar]
- Lee K, Um SH, Rhee DK, Pyo S (2016) Interferon‐alpha inhibits adipogenesis via regulation of JAK/STAT1 signaling. Biochim Biophys Acta 1860: 2416–2427 [DOI] [PubMed] [Google Scholar]
- Lei Q, Liu X, Fu H, Sun Y, Wang L, Xu G, Wang W, Yu Z, Liu C, Li P, Feng J, Li G, Wu M (2016) miR‐101 reverses hypomethylation of the PRDM16 promoter to disrupt mitochondrial function in astrocytoma cells. Oncotarget 7: 5007–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung S, Qureshi SA, Kerr IM, Darnell JE Jr, Stark GR (1995) Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol 15: 1312–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Huq A, Najarro P (1996) Inhibition of mitochondrial function by interferon. J Biol Chem 271: 13184–13190 [DOI] [PubMed] [Google Scholar]
- Li X, Wang J, Jiang Z, Guo F, Soloway PD, Zhao R (2015) Role of PRDM16 and its PR domain in the epigenetic regulation of myogenic and adipogenic genes during transdifferentiation of C2C12 cells. Gene 570: 191–198 [DOI] [PubMed] [Google Scholar]
- Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW (2016) Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529: 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ (2009) Cold‐activated brown adipose tissue in healthy men. N Engl J Med 360: 1500–1508 [DOI] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver‐Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD (1996) Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK‐STAT signaling pathway. Cell 84: 431–442 [DOI] [PubMed] [Google Scholar]
- Moisan A, Lee YK, Zhang JD, Hudak CS, Meyer CA, Prummer M, Zoffmann S, Truong HH, Ebeling M, Kiialainen A, Gerard R, Xia F, Schinzel RT, Amrein KE, Cowan CA (2015) White‐to‐brown metabolic conversion of human adipocytes by JAK inhibition. Nat Cell Biol 17: 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould AW, Morgan MA, Nelson AC, Bikoff EK, Robertson EJ (2015) Blimp1/Prdm1 functions in opposition to Irf1 to maintain neonatal tolerance during postnatal intestinal maturation. PLoS Genet 11: e1005375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikata I, Sasaki H, Iga M, Tateno Y, Imayoshi S, Asou N, Nakamura T, Morishita K (2003) A novel EVI1 gene family, MEL1, lacking a PR domain (MEL1S) is expressed mainly in t(1;3)(p36;q21)‐positive AML and blocks G‐CSF‐induced myeloid differentiation. Blood 102: 3323–3332 [DOI] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Spiegelman BM, Kajimura S (2012) PPARgamma agonists induce a white‐to‐brown fat conversion through stabilization of PRDM16 protein. Cell Metab 15: 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Ohyama K, Sharp LZ, Kajimura S (2013) EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 504: 163–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Li S, Cha E, Schindler C (2000) Immune response in Stat2 knockout mice. Immunity 13: 795–804 [DOI] [PubMed] [Google Scholar]
- Petrovic N, Shabalina IG, Timmons JA, Cannon B, Nedergaard J (2008) Thermogenically competent nonadrenergic recruitment in brown preadipocytes by a PPARgamma agonist. Am J Physiol Endocrinol Metab 295: E287–E296 [DOI] [PubMed] [Google Scholar]
- Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, Mittler G, Genoud C, Goyama S, Kurokawa M, Son J, Reinberg D, Lachner M, Jenuwein T (2012) Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 150: 948–960 [DOI] [PubMed] [Google Scholar]
- Rajakumari S, Wu J, Ishibashi J, Lim HW, Giang AH, Won KJ, Reed RR, Seale P (2013) EBF2 determines and maintains brown adipocyte identity. Cell Metab 17: 562–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Okamatsu‐Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio‐Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K, Kawai Y, Tsujisaki M (2009) High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58: 1526–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T (2000) Distinct and essential roles of transcription factors IRF‐3 and IRF‐7 in response to viruses for IFN‐alpha/beta gene induction. Immunity 13: 539–548 [DOI] [PubMed] [Google Scholar]
- Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T (2009) Interferon regulatory factor‐2 protects quiescent hematopoietic stem cells from type I interferon‐dependent exhaustion. Nat Med 15: 696–700 [DOI] [PubMed] [Google Scholar]
- Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM (1998) Regulation of type I interferon gene expression by interferon regulatory factor‐3. J Biol Chem 273: 2714–2720 [DOI] [PubMed] [Google Scholar]
- Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, Tavernier G, Langin D, Spiegelman BM (2007) Transcriptional control of brown fat determination by PRDM16. Cell Metab 6: 38–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument‐Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM (2008) PRDM16 controls a brown fat/skeletal muscle switch. Nature 454: 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman BM (2011) Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest 121: 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Vazquez E, Lewis JA (1990) Interferon selectively inhibits the expression of mitochondrial genes: a novel pathway for interferon‐mediated responses. EMBO J 9: 4307–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai TA, Jennermann C, Brown KK, Oliver BB, MacGinnitie MA, Wilkison WO, Brown HR, Lehmann JM, Kliewer SA, Morris DC, Graves RA (1996) Activation of the nuclear receptor peroxisome proliferator‐activated receptor gamma promotes brown adipocyte differentiation. J Biol Chem 271: 29909–29914 [DOI] [PubMed] [Google Scholar]
- Tovey MG, Streuli M, Gresser I, Gugenheim J, Blanchard B, Guymarho J, Vignaux F, Gigou M (1987) Interferon messenger RNA is produced constitutively in the organs of normal individuals. Proc Natl Acad Sci USA 84: 5038–5042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel SN, Fertsch D (1984) Endogenous interferon production by endotoxin‐responsive macrophages provides an autostimulatory differentiation signal. Infect Immun 45: 417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XA, Zhang R, Zhang S, Deng S, Jiang D, Zhong J, Yang L, Wang T, Hong S, Guo S, She ZG, Zhang XD, Li H (2013) Interferon regulatory factor 7 deficiency prevents diet‐induced obesity and insulin resistance. Am J Physiol Endocrinol Metab 305: E485–E495 [DOI] [PubMed] [Google Scholar]
- Xu L, Zhou X, Wang W, Wang Y, Yin Y, van der Laan LJ, Sprengers D, Metselaar HJ, Peppelenbosch MP, Pan Q (2016) IFN regulatory factor 1 restricts hepatitis E virus replication by activating STAT1 to induce antiviral IFN‐stimulated genes. FASEB J 30: 3352–3367 [DOI] [PubMed] [Google Scholar]
- Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, Guha M, Li N, Chen Q, Yang T, Lengner CJ, Greenberg RA, Johnson FB, Fuchs SY (2015) DNA‐damage‐induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep 11: 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Wang J, Lee SY, Xiong J, Bhanu N, Guo Q, Ma P, Sun Y, Rao RC, Garcia BA, Hess JL, Dou Y (2016) PRDM16 suppresses MLL1r leukemia via intrinsic histone methyltransferase activity. Mol Cell 62: 222–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6