

Abstract
Camptothecin‐induced locking of topoisomerase 1 on DNA generates a physical barrier to replication fork progression and creates topological stress. By allowing replisome rotation, absence of the Tof1/Csm3 complex promotes the conversion of impending topological stress to DNA catenation and causes camptothecin hypersensitivity. Through synthetic viability screening, we discovered that histone H4 K16 deacetylation drives the sensitivity of yeast cells to camptothecin and that inactivation of this pathway by mutating H4 K16 or the genes SIR1‐4 suppresses much of the hypersensitivity of tof1∆ strains towards this agent. We show that disruption of rDNA or telomeric silencing does not mediate camptothecin resistance but that disruption of Sir1‐dependent chromatin domains is sufficient to suppress camptothecin sensitivity in wild‐type and tof1∆ cells. We suggest that topoisomerase 1 inhibition in proximity of these domains causes topological stress that leads to DNA hypercatenation, especially in the absence of the Tof1/Csm3 complex. Finally, we provide evidence of the evolutionarily conservation of this mechanism.
Keywords: camptothecin, H4‐K16, SIR complex, synthetic viability, Tof1
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Introduction
Separation of the two parental DNA strands during DNA replication creates positive supercoiling ahead of the replication fork. Such over‐winding hinders replisome progression and must be removed for DNA replication to be completed. In eukaryotes, two DNA topoisomerases, Top1 and Top2, cooperate to allow DNA replication and segregation. The main DNA topoisomerase that relaxes positive supercoiling during DNA replication is considered to be Top1, a type‐IB topoisomerase, while Top2 activity seems to be concentrated behind replication forks 1, 2, 3. Despite the importance of DNA uncoiling for replication, Saccharomyces cerevisiae cells lacking Top1 can fully replicate their genome because in the absence of Top1, positive supercoils can either be relaxed directly by Top2 4, 5 or indirectly by rotation of replication forks along their axes, converting impending positive supercoiling into intertwines/catenation between the two daughter DNA strands 6. The catenation generated in this way is an obstacle to chromosome segregation and must be resolved by Top2, a type II topoisomerase, before the onset of mitosis 3, 7. In contrast to Top1, Top2 is essential in yeast cells because a certain amount of catenation is generated even in wild‐type cells, possibly because Top1 cannot relieve topological stress between replisomes converging towards replication termination zones 8. Consistent with this model, increased fork rotation has been observed when replication forks approach stable fork‐pausing structures, such as centromeres, tRNA genes, inactive replication origins 9, and potentially retrotransposon long terminal repeats (LTRs) and transcriptionally repressed chromatin 10, 11.
To reduce the requirement for decatenation, replisome rotation is normally restricted by the Tof1/Csm3 complex 9, the yeast homolog of the mammalian Timeless/Tipin complex. Tof1 and Csm3 are also crucial for proper pausing of replication forks at replication fork barriers present in the tandem arrays that form the large ribosomal DNA (rDNA) locus 12. Independently of these functions, the Tof1/Csm3 complex also interacts with Mrc1 13, which functions as an adaptor to transmit signals from the apical replication‐checkpoint kinase Mec1 to the transducer kinase Rad53 during replication stress induced by nucleotide depletion 14. The fact that tof1∆ strains, similar to mrc1∆ strains, show synergistic phenotypes in combination with loss of Rad9—the other major checkpoint adaptor protein in S. cerevisiae—suggests that the Tof1/Csm3 complex recruits Mrc1 for the purpose of Rad53 activation 12, 15. In this regard, it is noteworthy that Mrc1 also has checkpoint‐independent functions and can be recruited to replication forks independently of Tof1/Csm3 14, 16, 17.
Despite the above findings, certain results have remained unexplained, and the precise roles of the Tof1/Csm3 complex have remained elusive. For instance, tof1∆ and csm3∆ yeast strains were shown to be hypersensitive to high doses of camptothecin 18, a drug that induces DNA double‐strand DNA breaks (DSBs) during S phase by trapping Top1 in a covalent complex with DNA. These strains, however, are not hypersensitive to other agents that induce DSBs, such as ionising radiation, or to drugs such as hydroxyurea that affect S phase progression 18, suggesting that the camptothecin hypersensitivity of tof1∆ and csm3∆ strains might arise through topologically stressed DNA structures generated by Top1 inhibition rather than from DNA damage per se 19, 20.
Here, we show that histone H4 K16 deacetylation by the yeast sirtuin complex drives the sensitivity of wild‐type cells to camptothecin. Our results also show that the disruption of chromatin domains bearing deacetylated H4 K16 rescues the camptothecin hypersensitivity of tof1∆ and csm3∆ cells, suggesting that the increased sister chromatid catenation generated in the absence of these proteins promotes camptothecin toxicity. Finally, we show that the role of sirtuins in driving camptothecin sensitivity in S. cerevisiae is evolutionarily conserved in the yeast Schizosaccharomyces pombe and in human cells.
Results
To better understand the roles of the Tof1/Csm3 complex during DNA replication, we investigated the basis for the camptothecin hypersensitivity of TOF1‐ or CSM3‐deleted cells. This hypersensitivity arises from the well‐established trapping of Top1 in a covalent complex with DNA, as shown by the fact that it was rescued by TOP1 deletion (Fig 1A). Notably, mrc1∆ strains were not hypersensitive to camptothecin (Fig 1A; 18), indicating that a defect in replication‐checkpoint activation does not explain the camptothecin hypersensitivity of tof1∆ or csm3∆ strains. Moreover, this hypersensitivity does not appear to arise from issues connected to fork pausing at the replication fork barrier on rDNA, as pausing‐deficient fob1∆ strains were not hypersensitive to camptothecin, and FOB1 deletion did not alleviate the camptothecin hypersensitivity of a csm3∆ strain (Fig 1B).
Figure 1. A synthetic viability screening to identify the cause for the hypersensitivity of tof1∆ yeast cells to camptothecin.
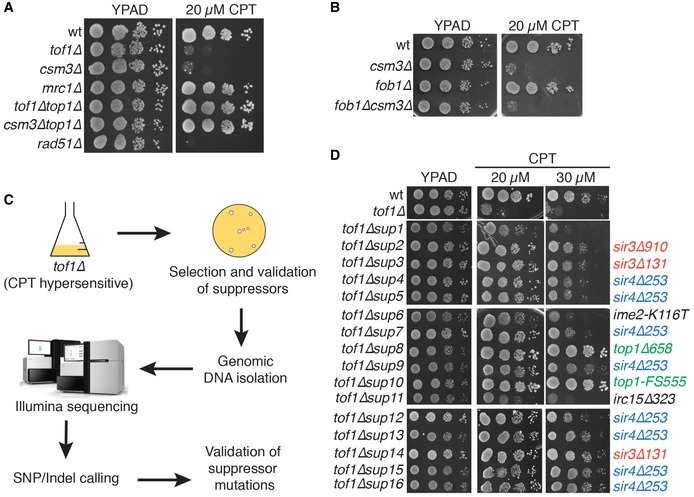
- Loss of Tof1 and Csm3 but not Mrc1 causes hypersensitivity to camptothecin in a Top1‐dependent manner.
- Loss of pausing at the replication fork barrier on rDNA does not affect camptothecin hypersensitivity.
- Outline of the procedure for a synthetic viability screen.
- Synthetic viability screening identifies sir3 and sir4 alleles as suppressors of the camptothecin hypersensitivity of tof1∆ strains.
SIR gene mutations suppress camptothecin hypersensitivity of tof1∆/csm3∆ cells
To understand the origin of the hypersensitivity of tof1∆ and csm3∆ strains to camptothecin, we carried out a synthetic viability genomic screening 21 to identify mutations capable of suppressing such hypersensitivity (Fig 1C). We plated approximately 1 × 107 cells on a YPD plate supplemented with 20 μM camptothecin (Fig EV1A), isolated sixteen resistant colonies, and verified that they indeed displayed both resistance to camptothecin and to the antibiotic G418, a readout for TOF1 deletion, which was later confirmed by whole‐genome sequencing (Figs 1D and EV1B). This validation ensured that the cells isolated did not merely survive camptothecin treatment, but carried genetic (or epigenetic) marks conferring camptothecin resistance. We then sequenced their genomic DNAs to identify candidate mutations responsible for the suppression phenotype (all the mutations identified in each strain are listed in Table EV1). Two of the sixteen strains—the most resistant ones—carried mutations that inactivated TOP1, which encodes the drug target. Three strains carried either of two nonsense mutations that inactivated SIR3, while eight of the remaining strains carried a nonsense mutation inactivating SIR4 (Fig 1D; premature stop codons are designated by a ∆ following the position of the last amino acid residue encoded by the truncated gene). Importantly, we validated these putative drivers of camptothecin resistance by directly introducing deletions of SIR3 and SIR4 in tof1∆ and csm3∆ strains and establishing that SIR3 or SIR4 inactivation suppressed camptothecin hypersensitivity (Fig 2A). In the three remaining suppressor strains—the weakest suppressors—we could not identify any mutation responsible for the suppression. In one of these, no mutations were detected, while the other two carried point mutations in IME2 (inducer of MEiosis, which is not expressed in exponentially growing cells) or IRC15. However, ensuing studies established that neither IME2 nor IRC15 deletion suppressed the camptothecin hypersensitivity of tof1∆ cells (Fig EV1C and D; the reasons for the decreased camptothecin sensitivity of these three strains therefore remain to be defined).
Figure EV1. A synthetic viability screening identifies the SIR complex as the major driver of tof1∆ camptothecin hypersensitivity.
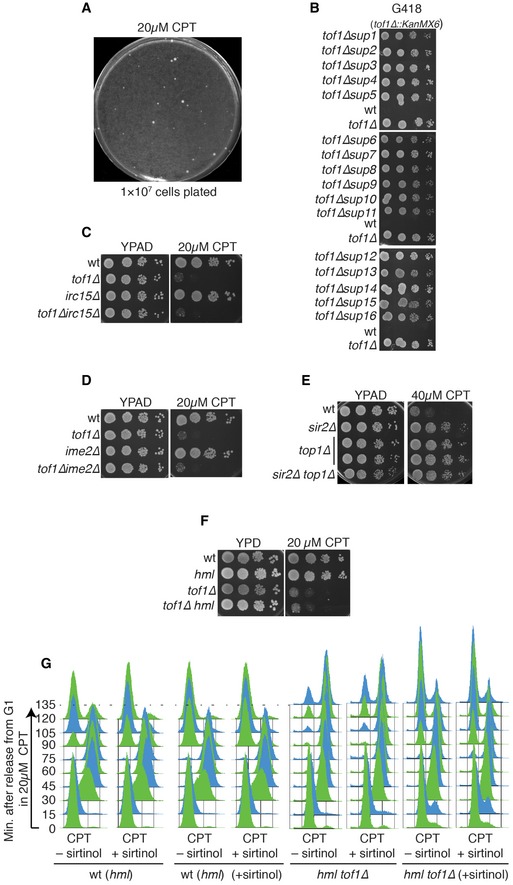
- Spontaneous suppressors of camptothecin sensitivity of tof1∆ cells.
- Suppressor strains recovered from the tof1∆ synthetic viability screen are G418 resistant, suggesting presence of the TOF1 deletion cassette.
- Deletion of IRC15 does not suppress tof1∆ camptothecin hypersensitivity.
- Deletion of IME2 does not suppress tof1∆ camptothecin hypersensitivity.
- Deletion of SIR2 and deletion of TOP1 lead to similar levels of camptothecin resistance, and combining the two mutations does not further increase camptothecin resistance.
- Deletion of HML does not alter the camptothecin hypersensitivity of tof1∆ cells.
- tof1∆ cells and congenic wild‐type cells were pre‐grown either in the absence or in the presence of sirtinol. They were subsequently synchronised in G1 and released into S phase in the presence of camptothecin, either with or without sirtinol. Cell cycle progression was monitored by FACS analysis.
Figure 2. Loss of the SIR complex suppresses camptothecin hypersensitivity of tof1∆ yeast strains.
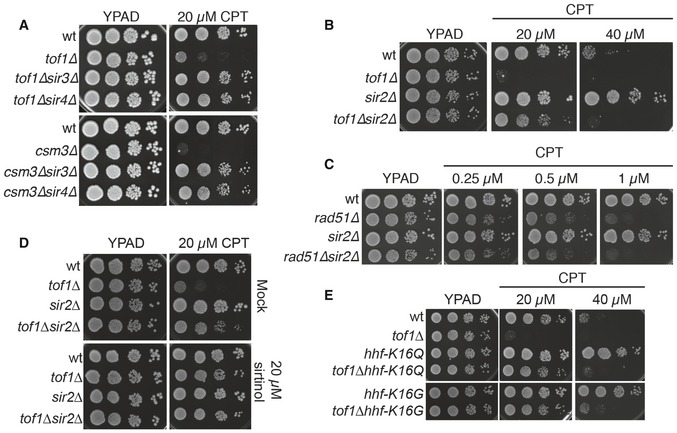
- Deletion of SIR3 or SIR4 suppresses the camptothecin hypersensitivity of tof1∆ and csm3∆ cells.
- Deletion of SIR2 also suppresses camptothecin hypersensitivity of tof1∆ cells and reduces camptothecin sensitivity of a wild‐type strain.
- Deletion of SIR2 cannot suppress camptothecin hypersensitivity of a rad51∆ strain.
- Inhibition of Sir2 deacetylase activity with sirtinol suppresses camptothecin hypersensitivity of tof1∆ cells.
- Mutations that mimic a permanently acetylated H4‐K16 (K16Q) or that remove the binding site for Sir3 (K16G) also suppress camptothecin sensitivity of wild‐type and tof1∆ strains.
Sir3 and Sir4 form a ternary protein complex with the histone deacetylase catalytic subunit Sir2 (reviewed in 22), with removal of any of the three subunits inactivating the transcriptional silencing functions of the complex 23. Significantly, we established that loss of Sir2 alleviated the camptothecin hypersensitivity of tof1∆ cells to a similar extent as conferred by Sir3 or Sir4 loss (Figs 2B and EV2B, lower panel). Furthermore, by increasing the concentration of camptothecin, we found that deletion of SIR2, SIR3, and SIR4 also promoted camptothecin resistance in a wild‐type yeast background (Figs 2B and EV2B, upper panel). Interestingly, in wild‐type cells, deletion of SIR2 suppressed camptothecin sensitivity to a similar extent as conferred by TOP1 deletion, and combining the deletions did not further increase resistance to camptothecin (Fig EV1E). By contrast, SIR2 deletion did not alleviate the strong camptothecin hypersensitivity of a rad51∆ strain, which is severely defective in repairing DSBs induced by camptothecin (Fig 2C). These data indicated that the SIR complex is a major mediator of camptothecin sensitivity, but crucially, inactivation of the SIR complex does not act as a general suppressor of camptothecin toxicity, for example by reducing Top1 activity, cell permeability to camptothecin, or DSB induction by camptothecin.
Figure EV2. Binding of the SIR complex to highly expressed genomic loci mediates camptothecin sensitivity.
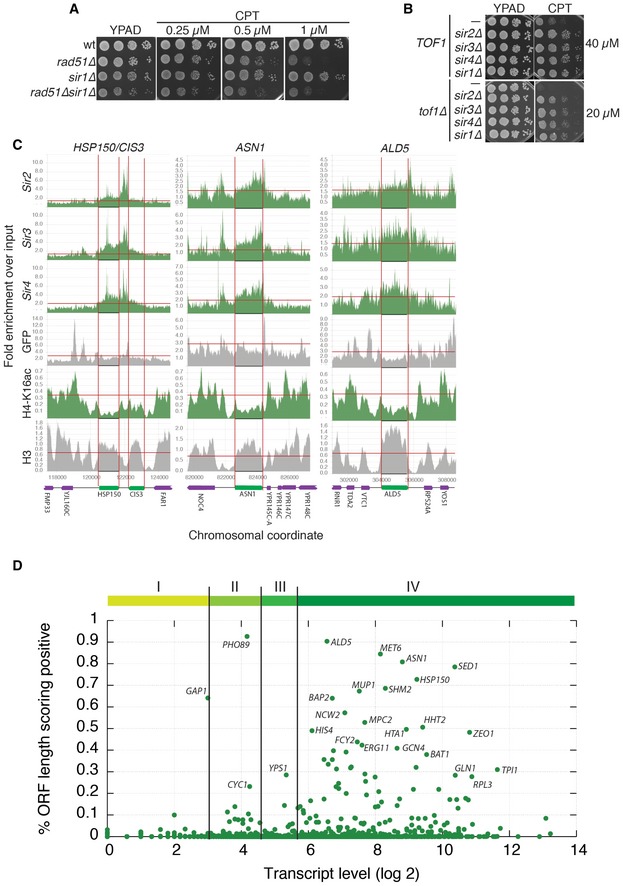
- Deletion of SIR1 does not rescue camptothecin hypersensitivity in rad51∆ cells.
- Deletion of SIR1, SIR2, SIR3 or SIR4 increases camptothecin resistance to a similar extent, both in a wild‐type and in a tof1∆ background.
- Analysis of ChIP‐seq data for the indicated proteins: enrichment is plotted as a function of the genomic coordinate; in green is the protein/modification tested; in grey are controls. The position of each ORF is indicated at the bottom of the panel and by two vertical red lines. The horizontal red lines indicate the thresholds used in this work to determine enrichment (Sir2/Sir3/Sir4/GFP/H3) or loss (H4‐K16) of ChIP signal.
- The majority of SIR‐positive ORFs are highly expressed genes, but high expression does not necessarily correlate with high SIR score. Vertical lines identify quartiles (I, II, III, IV) of the gene expression distribution.
SIR proteins mediate camptothecin sensitivity via histone H4‐K16 deacetylation
To assess whether loss of the deacetylase activity of the Sir complex was responsible for the suppression of tof1∆ hypersensitivity to camptothecin, we used the small‐molecule Sir2 inhibitor, sirtinol 24. This work established that addition of 20 μM sirtinol strongly suppressed the camptothecin sensitivity of a tof1∆ strain (Fig 2D). While Sir2 homologs in higher eukaryotes have been implicated in deacetylating proteins involved in DNA repair, such as PARP1, Ku70, and CtIP 25, 26, 27, the prime target for S. cerevisiae Sir2 is histone H4 lysine 16 (H4‐K16), which is found in an acetylated state through much of the transcriptionally active yeast genome. In S. cerevisiae, deacetylation of this residue by Sir2 allows binding of Sir3, thus recruiting further Sir2 that removes acetylation marks from flanking H4‐K16 residues, a process that is then propagated to produce a transcriptionally silent heterochromatic state 22. To explore whether the relevant target for Sir2 in relation to its effects on the camptothecin sensitivity of tof1∆ cells was H4‐K16, we mutated this residue to glutamine (Q), a residue that mimics a constitutively acetylated lysine and abrogates Sir3 binding 28. Strikingly, this hhf‐K16Q mutation suppressed the camptothecin hypersensitivity of a tof1∆ strain, and at higher doses also reduced the camptothecin sensitivity of a wild‐type strain (Fig 2E). Similarly, mutation of H4‐K16 to glycine (G), which prevents binding by Sir3 28, strongly counteracted the camptothecin sensitivity of both tof1∆ and wild‐type cells. Taken together, these results highlighted a correlation between chromatin association of the SIR complex and camptothecin sensitivity.
A deacetylated H4‐K16 template promotes camptothecin‐induced mitotic arrest
To further explore how TOF1 or CSM3 deletion causes camptothecin hypersensitivity, we took advantage of the fact that camptothecin treatment of synchronised wild‐type cells released from G1 into S phase leads to a prolonged G2/M cell cycle delay 29. We first assessed the effect of TOF1 and CSM3 deletion on this particular phenotype by arresting wild‐type, tof1∆ and csm3∆ cultures in G1 by alpha‐factor treatment, and then releasing them from this arrest either in the presence or in the absence of camptothecin. As expected, wild‐type cells treated in this way with camptothecin did not delay bulk DNA replication compared to strains released in the absence of camptothecin, although they did exhibit delayed exit from the subsequent mitosis (Fig 3A). Significantly, compared to wild‐type controls, cells deleted for TOF1 or CSM3 arrested for longer periods of time in G2/M following camptothecin treatment (Fig 3A bottom panels), a phenotype that correlated with persistence of the mitotic cyclin, Clb2 (Fig 3B). Nevertheless, these cells eventually re‐entered the cell cycle and continued proliferating, consistent with the fact that tof1∆ and csm3∆ strains were not killed by acute camptothecin treatment (Fig 3C; note that a repair‐defective rad51∆ strain was hypersensitive even to acute camptothecin treatment).
Figure 3. A deacetylated H4‐K16 template mediates sensitivity to camptothecin during DNA replication.
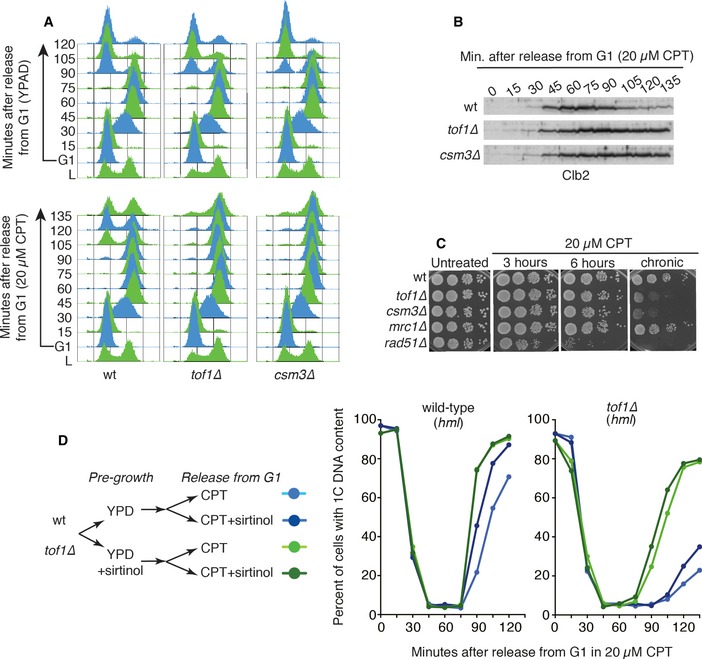
- A wild‐type yeast strain released into S phase in the presence of 20 μM camptothecin does not exhibit delayed progression through S phase, but exhibits delayed progression through the subsequent mitosis. In the absence of Tof1 or Csm3, camptothecin‐treated cells remain arrested in G2/M for longer periods of time than wild‐type cells.
- tof1∆ and csm3∆ cells released into S phase in the presence of camptothecin exhibit delayed destruction of the mitotic cyclin Clb2.
- tof1∆ and csm3∆ cells are not hypersensitive to transient camptothecin treatment.
- tof1∆ cells and congenic wild‐type cells were pre‐grown either in the absence or in the presence of sirtinol. They were subsequently synchronised in G1 and released into S phase in the presence of camptothecin, either with or without sirtinol. Cell cycle progression was monitored by FACS analysis. Quantification of G1 cells shows that sirtinol addition during camptothecin treatment does not suppress the mitotic delay of tof1∆ cells, while pre‐growth in the presence of sirtinol is sufficient to suppress the camptothecin hypersensitivity phenotype of tof1∆ cells. A representative experiment is shown.
Source data are available online for this figure.
Collectively, the data we had obtained supported a model in which the mechanism by which the SIR complex yields camptothecin sensitivity is via effects on H4‐K16 deacetylation. In this regard, we reasoned that the SIR complex might impart camptothecin sensitivity either by deacetylating newly incorporated histone H4 during DNA replication, or by it broadly promoting a condensed chromatin template that impairs DNA replication in the presence of camptothecin. If Sir2 deacetylation activity during S phase promoted camptothecin sensitivity, one would expect that addition of sirtinol after the release from G1 would circumvent the extended mitotic delay induced by camptothecin in tof1∆ cells. Conversely, if broad acetylation of the chromatin template was required to rescue the tof1∆ phenotype, sirtinol should lead to suppression of extended mitotic delay only if tof1∆ cells were pre‐grown in the presence of sirtinol. To discriminate between these two hypotheses, we grew hml and hml, tof1∆ cells either in the presence or in the absence of sirtinol, and then synchronised them in G1 by addition of alpha‐factor (Fig 3D). We used a mutant hml background because sirtinol makes wild‐type cells insensitive to alpha‐factor by derepressing the HML/R (HM) loci 24 (importantly, as shown in Fig EV1F, HML mutation did not affect camptothecin sensitivity). We then released the G1‐synchronised cells into S phase in the presence of camptothecin alone, or in the presence of camptothecin plus sirtinol. While addition of sirtinol after the G1 release was not sufficient to rescue the mitotic delay of tof1∆ cells (Figs 3D and EV1G), pre‐growing tof1∆ cells in the presence of sirtinol suppressed their mitotic delay, whether or not sirtinol was present during the subsequent camptothecin treatment. A similar effect, albeit smaller, could be observed in a wild‐type strain (Figs 3D and EV1G). Taken together, these findings supported a model in which camptothecin leads to replication‐associated problems that arise within chromatin regions containing deacetylated H4‐K16, with cells lacking Tof1 or Csm3 being particularly sensitive to such problems.
Multiple HM‐like chromatin regions govern camptothecin sensitivity
The yeast genome contains three well‐studied heterochromatic regions that are transcriptionally silenced by SIR proteins: the rDNA array, subtelomeric regions and the cryptic mating‐type loci (Fig 4A–C). To establish whether loss of rDNA silencing mediated the suppression of tof1∆ camptothecin hypersensitivity upon SIR protein loss, we used a strain carrying a deletion of the entire rDNA locus complemented by a multi‐copy plasmid containing the rDNA repeat unit 30. We found that deletion of the rDNA locus did not reduce the hypersensitivity of tof1∆ cells to camptothecin (Fig 4A), indicating that this genomic region is not the prime target of the SIR complex that mediates camptothecin toxicity in tof1∆ cells. This notion was also supported by the fact that, while we observed suppression of camptothecin sensitivity with sir2∆, sir3∆, or sir4∆, silencing of the rDNA locus only requires Sir2, with SIR4 deletion actually increasing rDNA silencing by delocalising Sir2 from telomeres 31.
Figure 4. Disruption of Sir1‐dependent silencing suppresses camptothecin hypersensitivity of tof1∆ cells.
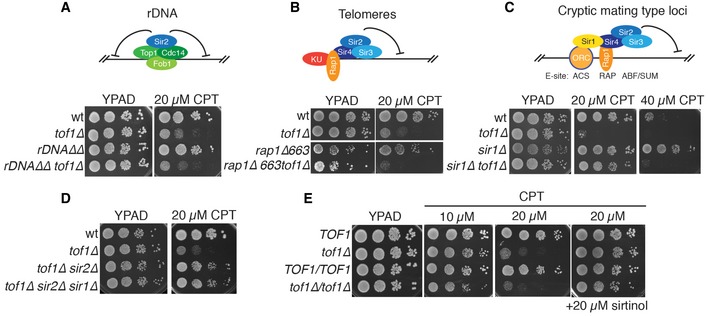
- Disruption of the rDNA locus is not sufficient to suppress camptothecin hypersensitivity of tof1∆ cells.
- A mutation in RAP1 that disrupts telomeric silencing does not suppress camptothecin hypersensitivity of tof1∆ cells.
- Deletion of SIR1 suppresses camptothecin sensitivity in wild‐type and tof1∆ cells.
- SIR1 deletion is epistatic with SIR2 deletion with respect to suppression of camptothecin hypersensitivity of tof1∆ strains.
- Homozygous tof1∆/tof1∆ diploid cells are as sensitive to camptothecin as tof1∆ haploids, and their hypersensitivity can be alleviated by sirtinol.
To determine if loss of subtelomeric silencing could rescue the camptothecin hypersensitivity of tof1∆ cells, we employed a strain carrying a C‐terminal truncation of Rap1 (rap1∆663), the so‐called rap1‐17 allele. This mutation completely disrupts transcriptional silencing at telomeres (telomere position effect) and partially affects silencing of the cryptic mating‐type locus HML 32. While strains carrying the rap1∆663 allele grew slower than wild‐type strains, presumably due to the role of Rap1 in regulating transcription of genes involved in ribosome formation and glycolysis 33, 34, they did not display altered sensitivity to camptothecin (Fig 4B). The rap1∆663 mutation also failed to suppress the camptothecin hypersensitivity of tof1∆ cells (Fig 4B), indicating that loss of telomere position effect does not promote survival in the presence of this drug.
At the cryptic mating‐type loci HML and HMR, silencing is established by replication origin recognition complex (ORC)‐mediated recruitment of Sir1, which then attracts the SIR complex via an interaction with Sir4 35, 36. Sir4 binding is also stabilised by an interaction with Rap1, which binds to its DNA consensus sequence located next to the ORC binding site ACS (ARS consensus sequence, Fig 4C). For these reasons, deletion of SIR1 results in partial loss of silencing at the cryptic mating‐type loci, but does not affect telomeric or rDNA silencing 37. Strikingly, we found that SIR1 deletion strongly alleviated the camptothecin hypersensitivity of a tof1∆ strain (Fig 4C). Importantly, SIR1 deletion did not further improve the survival of tof1∆sir2∆ strains, suggesting that Sir1 mediates camptothecin sensitivity entirely via its connection to the Sir2/3/4 complex (Fig 4D). Furthermore, similar to what we had observed for SIR2, SIR3, or SIR4 deletion, disruption of SIR1 also decreased the sensitivity of a wild‐type strain to high levels of camptothecin but it did not rescue the camptothecin hypersensitivity of a rad51∆ strain (Figs 4C and EV2A and B). These data were thus consistent with our conclusions that camptothecin sensitivity is not mainly generated via the rDNA or telomeric loci. Moreover, they indicated that the features of the chromatin template that are toxic to tof1∆ and wild‐type cells in the presence of camptothecin are generated in a Sir1‐dependent manner.
Various SIR‐bound genomic regions mediate camptothecin sensitivity
Our findings suggested that loss of the SIR complex might promote camptothecin resistance via effects on the HM loci. To test whether this might be connected to changes on the HM chromatin template itself or associated expression of genetic information from the normally silenced HML locus, we analysed the sensitivity of diploid tof1∆/tof1∆ cells that simultaneously express the genetic information encoded by HMR and HML. If transcription of genetic information from the HML locus reduced the camptothecin hypersensitivity of MATa tof1∆ strains, one would expect a homozygous tof1∆ diploid strain to be less camptothecin sensitive than the corresponding haploid strain; however, this was not the case (Fig 4E). Moreover, the camptothecin hypersensitivity of diploid tof1∆/tof1∆ cells was also rescued by sirtinol, clearly establishing that chromatin alterations, rather than expression of HM genetic information, are responsible for suppression of camptothecin hypersensitivity (Fig 4E).
While the above findings suggested that the chromatin status of the HM locus governs camptothecin sensitivity, when we deleted the HML and HMR loci, we were surprised to observe that this did not rescue the camptothecin hypersensitivity of tof1∆ cells (Fig 5A). This observation therefore strongly suggested the existence of other genomic loci targeted by Sir1‐4 as governing camptothecin sensitivity. To attempt to identify such loci, we analysed chromatin immunoprecipitation‐sequencing (ChIP‐seq) data for Sir2, Sir3, Sir4, GFP, acetylated histone H4‐K16, and histone H3 38, 39. In these datasets, we searched for genomic regions displaying higher association with Sir2, Sir3, and Sir4 compared to neighbouring regions. From the ensuing list, we then removed regions displaying increased GFP binding to exclude ChIP bias towards highly expressed genes 39. We also removed regions where we did not observe decreased histone H4‐K16 acetylation (the consequence SIR complex binding) compared to neighbouring regions, as well as regions also displaying reduced histone H3 ChIP signals suggesting depletion of nucleosomes. Genomic regions identified in this manner localised to confirmed open reading frames (ORFs; Figs 5B and EV2C).
Figure 5. Disruption of ORC1‐mediated binding of the SIR complex to highly transcribed genes suppresses camptothecin hypersensitivity of tof1∆ cells.
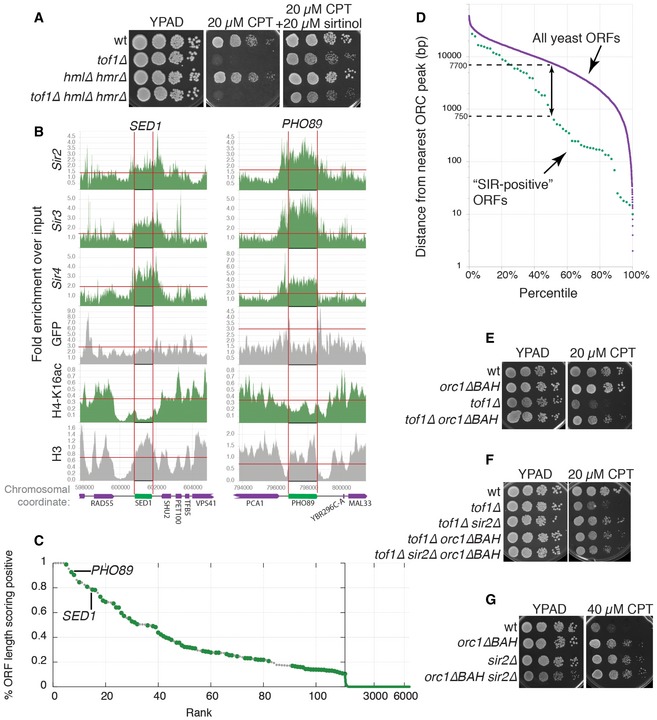
- Deletion of HML and HMR does not suppress camptothecin hypersensitivity of tof1∆ strains.
- Analysis of ChIP‐seq data for the proteins indicated on the y‐axes. Enrichments are plotted as a function of the genomic coordinate; in green is the protein/modification tested; in grey are controls. The ORFs indicated at the top of each graph are identified by two vertical red lines. The horizontal red lines indicate the thresholds used in this work to determine enrichment (Sir2/Sir3/Sir4/GFP/H3) or loss (H4‐K16) of ChIP signal.
- Identification of regions bound by the SIR complex: for each ORF in the genome, a “SIR score” was calculated as the fraction of the ORF for which both increased Sir2, Sir3, Sir4, and decreased H4‐K16ac were observed. ORFs were sorted based of their “SIR score”. Subtelomeric ORFs and ORFs proximal to HML and HMR are shown with small grey dots, while remaining ORFs are shown with large green dots.
- SIR‐positive ORFs are on average located closer to sites of ORC binding than ORFs in general. All yeast ORFs are shown in purple as a function of their distance from the nearest site of ORC binding. Non‐telomeric and non‐HM SIR‐positive ORFs (SIR score > 0.2) are shown in green.
- Deletion of the BAH domain of ORC1 partially rescues camptothecin hypersensitivity of tof1∆ cells.
- In a tof1∆orc1∆BAH background, deletion of SIR2 does not further increase camptothecin resistance.
- SIR2 deletion and orc1∆BAH mutation are epistatic with regard to camptothecin resistance.
We then defined a “SIR‐binding score” (the fraction of nucleotides for which the above conditions held) for every ORF in the yeast genome. While the majority of all ORFs essentially had a null SIR score (indicative of no enrichment of SIR complex binding), we found that 82 of them showed an enrichment of Sir2/3/4 and concomitant loss of H4‐K16 acetylation along more than 20% of their sequence (Table EV4). Of these 82 ORFs, 28 were localised in subtelomeric regions or in regions proximal to the HM loci (Fig 5C, small grey dots), while the remaining 54 hits were positioned along chromosome lengths (Fig 5C, green dots). Although the majority of the identified ORFs are expressed at high levels during exponential growth, high expression was not sufficient for a high SIR score (Fig EV2D based on data from 40). Taken together, these findings highlighted how, in addition to functioning at its well‐defined target loci, the SIR complex may also act at a variety of loci scattered throughout the genome, and suggested that these loci might also promote camptothecin toxicity in wild‐type and tof1∆ cells.
Recruitment of Sir1 at HM loci requires its interaction with the bromo‐adjacent domain (BAH) region of Orc1 36, 41, 42. We therefore assessed whether any of the loci we identified above were also positioned in proximity to a site bound by ORC. Thus, we calculated the distance between the centre of each ORF and the nearest ORC binding site 43. This revealed that ~50% of SIR‐enriched ORFs were located less than ~750 bp from a site of ORC binding (Fig 5D), a distance considerably shorter than the median value of 7.7 kbp for all yeast ORFs. We therefore reasoned that, if ORC has a functional role in recruiting the SIR complex to these genomic loci, it should be possible to suppress the camptothecin hypersensitivity of tof1∆ cells by preventing ORC‐mediated recruitment of Sir1. In line with this hypothesis—and in contrast to what we had observed upon deleting the HM locus—deleting the BAH domain of Orc1 markedly suppressed the camptothecin hypersensitivity of tof1∆ cells (Fig 5E; effects of ORC1 deletion could not be studied because it is an essential gene). In agreement with the proposed role of Orc1 BAH domain in recruiting the SIR complex, deletion of SIR2 did not further enhance camptothecin resistance of orc1∆BAH cells, either in a wild‐type or a tof1∆ background (Fig 5F and G). These findings thus suggested that the chromatin substrates that become toxic to tof1∆ cells exposed to camptothecin are at least partially formed in an ORC‐dependent manner.
Evidence for an evolutionarily conserved connection between sirtuin function and camptothecin sensitivity
Consistent with a model in which SIR proteins might promote camptothecin sensitivity in other organisms, we found that sir2∆ S. pombe strains were more resistant to camptothecin than control sir2+ strains (Fig 6A). Moreover, in line with our findings in budding yeast, addition of sirtinol to the growth medium reduced the camptothecin sensitivity of wild‐type S. pombe cells (Fig 6B).
Figure 6. The role of sirtuins in driving camptothecin sensitivity appears to be evolutionarily conserved.
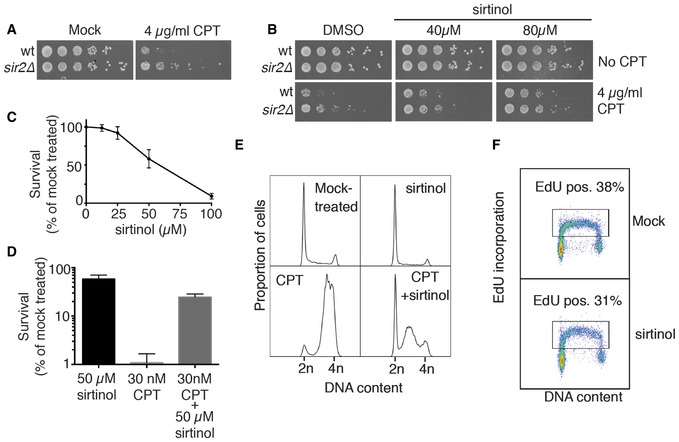
- Schizosaccharomyces pombe sir2∆ cells are more resistant to camptothecin than congenic wild‐type controls.
- Sirtinol alleviates camptothecin sensitivity of wild‐type S. pombe cells.
- Sirtinol is cytotoxic for non‐transformed human cells. Clonogenic capacity of cells was measured after 48‐h treatment with indicated doses of sirtinol. Average and standard deviation (n = 4) are shown for each point.
- Sirtinol rescues camptothecin‐mediated lethality in human cells. Cells were pre‐treated with 50 μM sirtinol for 24 h and then incubated for another 24 h in the presence of 50 μM sirtinol, 30 nM camptothecin or a combination of both drugs. Average and standard deviation (n = 4) are shown for each point.
- 48‐h treatment with sirtinol does not affect the cell cycle distribution, but partially alleviates the G2/M arrest caused by camptothecin. A representative experiment is shown.
- Sirtinol does not inhibit DNA replication in hTERT RPE‐1 cells. A representative experiment is shown.
Next, we tested whether sirtinol affected the camptothecin sensitivity of hTERT‐immortalised, non‐transformed human RPE‐1 cells. In line with previous studies 44, 45, we found that sirtinol induced a dose‐dependent killing of human cells (Fig 6C). However, when the cells were pre‐incubated with sirtinol for 24 h prior to camptothecin addition, a protective effect on the survival to camptothecin was observed (Fig 6D). As expected, flow cytometry analysis based on DNA content showed that camptothecin induced a replication‐dependent cell cycle arrest, with the majority of cells in G2/M after 24 h of camptothecin treatment. By contrast, camptothecin‐induced G2/M accumulation was much less pronounced when cells had been pre‐treated with sirtinol (Fig 6E). Importantly, when we used EdU pulse‐labelling to quantify DNA replication, we found that neither the proportion of EdU‐incorporating cells nor the average intensity of EdU per cell was significantly affected by sirtinol (Fig 6F), indicating that replication was not inhibited by the sirtinol concentration used in our experimental setting. Collectively, these results supported a model in which cell killing induced by camptothecin is to a large degree mediated by the action of sirtuins, via a mechanism that is conserved from yeast to human cells.
Discussion
By using a synthetic viability screening approach 21, we identified the SIR3 and SIR4 genes as major mediators of the sensitivity of both wild‐type and tof1∆ cells to camptothecin. We subsequently established that SIR2 and SIR1 also function in a similar way (these genes were likely not found in our initial screen because of the relatively small number of suppressor strains analysed). We established that, rather than by reducing camptothecin action, deletion of these SIR genes removes a factor that hinders cell proliferation in the presence of camptothecin in wild‐type cells and that is particularly toxic to cells lacking the Tof1‐Csm3 replication‐pausing complex. Camptothecin promotes the accumulation of positive supercoiling during DNA replication by locking topoisomerase 1 on DNA in a non‐functional state 19, 20. Since Tof1 and Csm3 function to restrict replisome rotation during DNA replication 9, and since an important factor driving fork rotation is positive supercoiling 19, we hypothesise that an excess of camptothecin‐induced positive supercoiling is the factor that is alleviated by deletion of SIR genes.
While camptothecin is a Top1 inhibitor, lack of Top1 activity is not sufficient to explain the hypersensitivity of tof1∆ and csm3∆ cells, as deletion of TOP1 is not toxic to tof1∆ cells. Instead, camptothecin‐mediated locking of Top1 on DNA could be the source of topological stress, either directly by creating topologically closed domains, or indirectly by preventing Top2 action—similar to what is thought to happen when a catalytically inactive Top2 that still retains an ability to bind DNA is expressed 46.
Lack of Sir2, Sir3, or Sir4 leads to loss of histone H4 lysine 16 (H4‐K16) deacetylation and subsequent impairment in heterochromatin formation. We have observed that inhibition of Sir2 deacetylase activity or mutation of H4‐K16 to glutamine (a residue that mimics an acetylated lysine) also increases the camptothecin resistance of both wild‐type and tof1∆ cells. Importantly, we have established that the cell cycle delay observed in camptothecin‐treated tof1∆ cells can only be suppressed if Sir2 activity is inhibited prior to camptothecin treatment, suggesting that it is the state of the chromatin template itself that becomes toxic to cells when replicated in the presence of camptothecin.
Yeast genomes contain three well‐characterised regions of transcriptionally silenced chromatin: the ribosomal DNA, subtelomeric regions and the cryptic mating‐type loci HML and HMR; and of these, only the cryptic mating‐type loci require Sir1 for their silencing 37. The fact that SIR1 deletion also suppresses the camptothecin sensitivity of tof1∆ cells initially suggested to us that HML and HMR represent the chromatin templates that are toxic to tof1∆ cells in the presence of camptothecin. However, we did not observe a reduction in tof1∆ sensitivity to camptothecin by deleting HML and HMR, meaning that these two genomic loci alone are not responsible for the strong camptothecin sensitivity phenotype displayed by tof1∆ cells.
Analysis of publicly available ChIP‐seq data allowed us to identify various genomic loci that exhibit enhanced localisation of Sir2, Sir3 and Sir4 as well as H4‐K16 under‐acetylation. Notably, we found that these genomic loci colocalise with confirmed ORFs and are located closer to sites of ORC binding than the average yeast ORF. Indeed, we found that many of these sites colocalise with genomic loci that were previously shown to bind ORC despite not having replication origin activity 43. Importantly, we note that some of the SIR‐enriched loci also colocalise with sites of replication fork pausing and sites enriched in binding of Rrm3, a DNA helicase that relieves replication fork pauses 47, 48, suggesting that SIR‐enriched loci are inherently difficult to replicate even in the absence of camptothecin. The fact that these ORFs are amongst the most highly expressed yeast genes and yet exhibit enhanced recruitment of the SIR silencing complex and markers of histone H4 deacetylation is enigmatic. One possibility is that strong transcription could prevent heterochromatin formation despite the presence of the SIR complex. Indeed, it has been shown that promoter strength affects the efficiency of silencing 49. In this regard, the sensitivity of yeast cells to camptothecin might stem from DNA catenation that is generated when replication forks approach barriers created by the Sir2/3/4 complex, a phenotype that would be exacerbated by the absence of the Tof1/Csm3 complex. We note that increased catenation would likely require time to be resolved, thereby potentially accounting for the M/G1 delay observed following camptothecin treatment in wild‐type cells and more strongly in tof1∆ cells. Another possibility is that SIR‐mediated genomic loci could be particularly prone to replication‐induced topological stress and would therefore be more frequently targeted by Top1 and more susceptible to camptothecin‐induced DNA damage. In this context, replisome instability caused by TOF1 deletion would increase the chance of fork breakdown or failure to rescue fork reversal events.
We have also provided evidence that the role of sirtuins in driving camptothecin sensitivity is evolutionarily conserved from yeast to humans. In the case of S. pombe, either loss or inhibition of Sir2, the fission yeast SIR2 ortholog, results in camptothecin resistance. As in S. cerevisiae, fission yeast Sir2 is involved in the heterochromatin assembly within the mating‐type locus, subtelomeric regions and centromeric DNA 50, 51 by deacetylating histone H3‐K9 and histone H4‐K16. Furthermore, we have found that sirtinol protects non‐transformed human RPE‐1 cells from killing by camptothecin via a mechanism that does not appear to reflect effects on DNA replication per se. As in yeast, human SIRT1 and SIRT2 deacetylate H3‐K9 and H4‐K16, among other substrates, and promote heterochromatin formation and gene silencing 52, 53. This suggests that the role of sirtuins in camptothecin‐mediated lethality in human cells may be similar to that in yeast, although alternative mechanisms cannot be ruled out due to the considerable number of sirtuin‐dependent pathways documented in mammalian systems.
Inhibition of topoisomerase 1 is a widely used therapeutic strategy to selectively kill proliferating cancer cells, with camptothecin analogues being part of the standard of care provided by many cancer clinics worldwide. Various mechanisms of camptothecin resistance have been observed, ranging from overexpression of drug‐efflux transporters, which actively reduce intracellular drug concentration 54, to specific Top1 mutations that prevent its interaction with camptothecin 55, 56. On the other hand, several sirtuin inhibitors have been shown to exhibit cytotoxic activity against various cancer cell lines (reviewed in 57) and are currently being assessed for their potential clinical applicability 58, 59. Using budding yeast as a model system, we have found that inhibition of histone H4‐K16 deacetylation by inactivation of the SIR protein complex represents an additional mechanism of camptothecin resistance and that this mechanism is likely conserved in fission yeast and in human cells. Further studies will be required to determine the precise mechanism‐of‐action of sirtinol in both transformed and non‐transformed human cells and whether sirtuins play a role in the emergence of resistance to camptothecin analogues in cancers.
Materials and Methods
Yeast strains and plasmids
Yeast strains used for this work are haploid derivatives of W303 unless otherwise indicated and are listed in Table EV1. All deletions were introduced by one‐step gene disruption/tagging 60. Strains carrying histone H4 mutations were obtained by plasmid shuffling, transforming the strain JHY6 (hht1‐hhf1∆::KanMX6 hht2‐hhf2∆::HPH) with plasmids obtained by site‐directed mutagenesis of plasmid pMR206 (HHT2‐HHF2; TRP1). Isolation of suppressor strains was carried out as previously described 21. The number of colonies sequenced was determined by reason of economics. S. pombe strains used were 49 (h+ ade6‐M210 leu1‐32 ura4‐D18) and 34 (h+ sir2::kanMX6 ade6‐M216 leu1‐32 ura4‐D18).
Whole‐genome paired‐end DNA sequencing and data analysis
Whole‐genome paired‐end DNA sequencing and data analysis were performed as previously described 21. All raw sequencing data are available from the European Nucleotide Archive (ENA) under the accession codes detailed in Table EV2. SNPs and indels were identified by using the SAMtools (v0.1.19) mpileup function, which finds putative variants and indels from alignments and assigns likelihoods, and BCFtools that performs the variant calling 61. The following parameters were used: for SAMtools (v0.1.19) mpileup “‐EDS ‐C50 ‐m2 ‐F0.0005 ‐d 10000” and for BCFtools (v0.1.19) view “‐p 0.99 –vcgN”. Functional consequences of the variants were produced by using the Ensembl VEP 62.
Drug sensitivity assays
Overnight‐grown saturated cultures of the indicated strains were serially diluted (10 fold) in water; 10‐μl drops of each dilution were deposited on each plate. Images were scanned 2–3 days after plating and growth at 30°C. Each experiment was repeated at least twice (n ≥ 2).
Analysis of yeast cell cycle progression and Western blotting
Exponentially growing cultures (30°C) were synchronised in G1 by addition of 5 μg/ml alpha‐factor for 2 h. G1‐synchronised cultures were then transferred to fresh YPD and released into S phase in the presence or in the absence of camptothecin and/or sirtinol; 45 min after the release, 20 μg/ml alpha‐factor was added to allow quantification of G1 cells by preventing re‐entry into the cell cycle. To detect Clb2, trichloroacetic acid protein extracts were separated on 10% polyacrylamide gels and Clb2 detection was carried out using anti‐Clb2 antibodies (Santa Cruz sc9071).
Analysis of ChIP‐seq data
Reads were aligned using BWA‐MEM, and duplicates marked with Picard. For each genomic coordinate, coverage was calculated using samtools and bedtools (samtools view ‐q10 ‐b $filename|genomeCoverageBed ‐d ‐ibam stdin ‐g) and normalised using the genomewide median of each sample. For each coordinate, the enrichment (E) was calculated as the ratio of the normalised coverages of IP and input samples. Every genomic position showing E sir2 > 1.75 and E sir3 > 1.5 and E sir4 > 2 and E GFP < 3 and E H4‐K16ac < 0.3 and E H3 > 0.75 was exported to a bed file. These values were determined empirically, and small adjustments did not substantially alter the final results. For every ORF, the total number (T) of positions (nucleotides) for which the above conditions held was calculated by querying the bed file. The final SIR score was obtained by dividing this number (T) by the length of the ORF.
Human cell culture
hTERT RPE‐1 cells were cultured in Dulbecco's modified Eagle's (DME)/F12 1:1 medium (Sigma‐Aldrich) supplemented with 10% foetal bovine serum (BioSera), 2 mM L‐glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin (Sigma‐Aldrich) and buffered with 0.2% Na(CO3)2.
Clonogenic survival assays
Cells were treated for 48 h with 50 μM sirtinol (Tocris), with camptothecin 30 nM for 24 h or pre‐treated with sirtinol for 24 h and then incubated with camptothecin and sirtinol for another 24 h as indicated. Cells were then washed three times with PBS and left to form colonies for 7–14 days. Colonies were stained with 0.1% (w/v) crystal violet in 20% (v/v) ethanol for counting. Results were normalised to plating efficiencies of untreated cells.
Human flow cytometry assays
Flow cytometry assays were performed as described in 63. A 1‐h pulse of 10 μM EdU was performed after 48 h of the indicated treatment. After fixation and permeabilisation, Alexa Fluor 488 azide (Invitrogen) was used for the click reaction to detect the incorporated EdU. Finally, cells were resuspended in FACS buffer with DAPI and analysed in a BD LSRFortessa™ cell analyser.
Data availability: referenced data
ChIP‐seq data were downloaded from the Sequence Read Archive (NCBI) using accession numbers specified in Table EV3 and originated from the following publications: (i) Thurtle and Rine 38; (ii) Teytelman et al 39.
Author contributions
The initial project was conceived by FP, VC and SPJ. Screening for suppressors and DNA extractions were carried out by FP and NJG. Analysis of whole‐genome sequencing data was carried out by MH and FP. Subsequent in vivo experiments and analyses of ChIP‐seq data were designed and carried out by FP; experiments in S. pombe and human cells were carried out by IS. The manuscript was largely written by FP, IS and SPJ, with contributions made by all the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Source Data for Figure 3
Acknowledgements
We thank Jasper Rine, Catherine A. Fox, Takehiko Kobayashi, and Tony Kouzarides for the gift of strains and plasmids, all the members of the SPJ laboratory for helpful discussions, and Helena Santos‐Rosa for advice with designing histone point mutations. Research in the Jackson laboratory is funded by Cancer Research UK [C6/A11224, C6/A18796, C6946/A14492] and the Wellcome Trust [WT092096]; FP was supported by the Associazione Italiana per la Ricerca sul Cancro [10932] and the European Molecular Biology Organization [ALTF1287‐2011]; IS, MH, and NJG were supported by the Wellcome Trust [101126/Z/13/Z (Strategic Award—COMSIG)] and [098051 to MH];VC is supported by the Armenise‐Harvard Foundation and the Fondazione Telethon.
EMBO Reports (2017) 18: 1000–1012
Contributor Information
Fabio Puddu, Email: f.puddu@gurdon.cam.ac.uk.
Stephen P Jackson, Email: s.jackson@gurdon.cam.ac.uk.
References
- 1. Schvartzman JB, Stasiak A (2004) A topological view of the replicon. EMBO Rep 5: 256–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Postow L, Crisona NJ, Peter BJ, Hardy CD, Cozzarelli NR (2001) Topological challenges to DNA replication: conformations at the fork. Proc Natl Acad Sci USA 98: 8219–8226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lucas I, Germe T, Chevrier‐Miller M, Hyrien O (2001) Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J 20: 6509–6519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim RA, Wang JC (1989) Function of DNA topoisomerases as replication swivels in Saccharomyces cerevisiae . J Mol Biol 208: 257–267 [DOI] [PubMed] [Google Scholar]
- 5. French SL, Sikes ML, Hontz RD, Osheim YN, Lambert TE, El Hage A, Smith MM, Tollervey D, Smith JS, Beyer AL (2011) Distinguishing the roles of topoisomerases I and II in relief of transcription‐induced torsional stress in yeast rRNA genes. Mol Cell Biol 31: 482–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sundin O, Varshavsky A (1980) Terminal stages of SV40 DNA replication proceed via multiply intertwined catenated dimers. Cell 21: 103–114 [DOI] [PubMed] [Google Scholar]
- 7. Baxter J, Sen N, Martínez VL, De Carandini MEM, Schvartzman JB, Diffley JFX, Aragón L (2011) Positive supercoiling of mitotic DNA drives decatenation by topoisomerase II in eukaryotes. Science 331: 1328–1332 [DOI] [PubMed] [Google Scholar]
- 8. Fachinetti D, Bermejo R, Cocito A, Minardi S, Katou Y, Kanoh Y, Shirahige K, Azvolinsky A, Zakian VA, Foiani M (2010) Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol Cell 39: 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schalbetter SA, Mansoubi S, Chambers AL, Downs JA, Baxter J (2015) Fork rotation and DNA precatenation are restricted during DNA replication to prevent chromosomal instability. Proc Natl Acad Sci USA 112: E4565–E4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA (2003) The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein‐DNA complexes. Mol Cell 12: 1525–1536 [DOI] [PubMed] [Google Scholar]
- 11. Zaratiegui M, Vaughn MW, Irvine DV, Goto D, Watt S, Bähler J, Arcangioli B, Martienssen RA (2011) CENP‐B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature 469: 112–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tourrière H, Versini G, Cordón‐Preciado V, Alabert C, Pasero P (2005) Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol Cell 19: 699–706 [DOI] [PubMed] [Google Scholar]
- 13. Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S‐phase checkpoint proteins Tof1 and Mrc1 form a stable replication‐pausing complex. Nature 424: 1078–1083 [DOI] [PubMed] [Google Scholar]
- 14. Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JFX, Carr AM, Elledge SJ (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 3: 958–965 [DOI] [PubMed] [Google Scholar]
- 15. Foss EJ (2001) Tof1p regulates DNA damage responses during S phase in Saccharomyces cerevisiae . Genetics 157: 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Osborn AJ, Elledge SJ (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 17: 1755–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bando M, Katou Y, Komata M, Tanaka H, Itoh T, Sutani T, Shirahige K (2009) Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J Biol Chem 284: 34355–34365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Redon C, Pilch DR, Bonner WM (2006) Genetic analysis of Saccharomyces cerevisiae H2A serine 129 mutant suggests a functional relationship between H2A and the sister‐chromatid cohesion partners Csm3‐Tof1 for the repair of topoisomerase I‐induced DNA damage. Genetics 172: 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M (2012) Topoisomerase I poisoning results in PARP‐mediated replication fork reversal. Nat Struct Mol Biol 19: 417–423 [DOI] [PubMed] [Google Scholar]
- 20. Koster DA, Palle K, Bot ESM, Bjornsti M‐A, Dekker NH (2007) Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 448: 213–217 [DOI] [PubMed] [Google Scholar]
- 21. Puddu F, Oelschlaegel T, Guerini I, Niu H, Ochoa‐Montaño B, Viré E, Sung P, Keane TM, Adams DJ, Keane TM et al (2015) Synthetic viability genomic screening defines Sae2 function in DNA repair. EMBO J 34: 1509–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kueng S, Oppikofer M, Gasser SM (2013) SIR proteins and the assembly of silent chromatin in budding yeast. Annu Rev Genet 47: 275–306 [DOI] [PubMed] [Google Scholar]
- 23. Rine J, Herskowitz I (1987) Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae . Genetics 116: 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL (2001) Identification of a class of small molecule inhibitors of the sirtuin family of NAD‐dependent deacetylases by phenotypic screening. J Biol Chem 276: 38837–38843 [DOI] [PubMed] [Google Scholar]
- 25. Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP (2009) SIRT1 promotes cell survival under stress by deacetylation‐dependent deactivation of poly(ADP‐ribose) polymerase 1. Mol Cell Biol 29: 4116–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeong J, Juhn K, Lee H, Kim S‐H, Min B‐H, Lee K‐M, Cho M‐H, Park G‐H, Lee K‐H (2007) SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med 39: 8–13 [DOI] [PubMed] [Google Scholar]
- 27. Kaidi A, Weinert BT, Choudhary C, Jackson SP (2010) Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329: 1348–1353 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28. Onishi M, Liou G‐G, Buchberger JR, Walz T, Moazed D (2007) Role of the conserved Sir3‐BAH domain in nucleosome binding and silent chromatin assembly. Mol Cell 28: 1015–1028 [DOI] [PubMed] [Google Scholar]
- 29. Redon C, Pilch DR, Rogakou EP, Orr AH, Lowndes NF, Bonner WM (2003) Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint‐blind DNA damage. EMBO Rep 4: 678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wai HH, Vu L, Oakes M, Nomura M (2000) Complete deletion of yeast chromosomal rDNA repeats and integration of a new rDNA repeat: use of rDNA deletion strains for functional analysis of rDNA promoter elements in vivo. Nucleic Acids Res 28: 3524–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith JS, Brachmann CB, Pillus L, Boeke JD (1998) Distribution of a limited Sir2 protein pool regulates the strength of yeast rDNA silencing and is modulated by Sir4p. Genetics 149: 1205–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kyrion G, Liu K, Liu C, Lustig AJ (1993) RAP1 and telomere structure regulate telomere position effects in Saccharomyces cerevisiae . Genes Dev 7: 1146–1159 [DOI] [PubMed] [Google Scholar]
- 33. Vignais ML, Woudt LP, Wassenaar GM, Mager WH, Sentenac A, Planta RJ (1987) Specific binding of TUF factor to upstream activation sites of yeast ribosomal protein genes. EMBO J 6: 1451–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lieb JD, Liu X, Botstein D, Brown PO (2001) Promoter‐specific binding of Rap1 revealed by genome‐wide maps of protein‐DNA association. Nat Genet 28: 327–334 [DOI] [PubMed] [Google Scholar]
- 35. Triolo T, Sternglanz R (1996) Role of interactions between the origin recognition complex and SIR1 in transcriptional silencing. Nature 381: 251–253 [DOI] [PubMed] [Google Scholar]
- 36. Gardner KA, Rine J, Fox CA (1999) A region of the Sir1 protein dedicated to recognition of a silencer and required for interaction with the Orc1 protein in Saccharomyces cerevisiae . Genetics 151: 31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith JS, Boeke JD (1997) An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev 11: 241–254 [DOI] [PubMed] [Google Scholar]
- 38. Thurtle DM, Rine J (2014) The molecular topography of silenced chromatin in Saccharomyces cerevisiae . Genes Dev 28: 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Teytelman L, Thurtle DM, Rine J, van Oudenaarden A (2013) Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci USA 110: 18602–18607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hou Z, Bernstein DA, Fox CA, Keck JL (2005) Structural basis of the Sir1‐origin recognition complex interaction in transcriptional silencing. Proc Natl Acad Sci USA 102: 8489–8494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Z, Hayashi MK, Merkel O, Stillman B, Xu R‐M (2002) Structure and function of the BAH‐containing domain of Orc1p in epigenetic silencing. EMBO J 21: 4600–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shor E, Warren CL, Tietjen J, Hou Z, Müller U, Alborelli I, Gohard FH, Yemm AI, Borisov L, Broach JR et al (2009) The origin recognition complex interacts with a subset of metabolic genes tightly linked to origins of replication. PLoS Genet 5: e1000755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peck B, Chen C‐Y, Ho K‐K, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao C‐D, Lam EW‐F (2010) SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther 9: 844–855 [DOI] [PubMed] [Google Scholar]
- 45. Wang J, Kim TH, Ahn MY, Lee J, Jung JH, Choi WS, Lee BM, Yoon KS, Yoon S, Kim HS (2012) Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF‐7 human breast cancer cells. Int J Oncol 41: 1101–1109 [DOI] [PubMed] [Google Scholar]
- 46. Baxter J, Diffley JFX (2008) Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell 30: 790–802 [DOI] [PubMed] [Google Scholar]
- 47. Azvolinsky A, Giresi PG, Lieb JD, Zakian VA (2009) Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae . Mol Cell 34: 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mohanty BK, Bairwa NK, Bastia D (2006) The Tof1p‐Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae . Proc Natl Acad Sci USA 103: 897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ren J, Wang C‐L, Sternglanz R (2010) Promoter strength influences the S phase requirement for establishment of silencing at the Saccharomyces cerevisiae silent mating type Loci. Genetics 186: 551–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Freeman‐Cook LL, Gómez EB, Spedale EJ, Marlett J, Forsburg SL, Pillus L, Laurenson P (2005) Conserved locus‐specific silencing functions of Schizosaccharomyces pombe sir2 + . Genetics 169: 1243–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shankaranarayana GD, Motamedi MR, Moazed D, Grewal SIS (2003) Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr Biol 13: 1240–1246 [DOI] [PubMed] [Google Scholar]
- 52. Vaquero A, Scher M, Lee D, Erdjument‐Bromage H, Tempst P, Reinberg D (2004) Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 16: 93–105 [DOI] [PubMed] [Google Scholar]
- 53. Vaquero A, Scher MB, Lee DH, Sutton A, Cheng H‐L, Alt FW, Serrano L, Sternglanz R, Reinberg D (2006) SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev 20: 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maliepaard M, van Gastelen MA, de Jong LA, Pluim D, van Waardenburg RC, Ruevekamp‐Helmers MC, Floot BG, Schellens JH (1999) Overexpression of the BCRP/MXR/ABCP gene in a topotecan‐selected ovarian tumor cell line. Cancer Res 59: 4559–4563 [PubMed] [Google Scholar]
- 55. Jensen NF, Agama K, Roy A, Smith DH, Pfister TD, Rømer MU, Zhang H‐L, Doroshow JH, Knudsen BR, Stenvang J et al (2016) Characterization of DNA topoisomerase I in three SN‐38 resistant human colon cancer cell lines reveals a new pair of resistance‐associated mutations. J Exp Clin Cancer Res 35: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chrencik JE, Staker BL, Burgin AB, Pourquier P, Pommier Y, Stewart L, Redinbo MR (2004) Mechanisms of camptothecin resistance by human topoisomerase I mutations. J Mol Biol 339: 773–784 [DOI] [PubMed] [Google Scholar]
- 57. Bruzzone S, Parenti MD, Grozio A, Ballestrero A, Bauer I, Del Rio A, Nencioni A (2013) Rejuvenating sirtuins: the rise of a new family of cancer drug targets. Curr Pharm Des 19: 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seifert T, Malo M, Kokkola T, Engen K, Fridén‐Saxin M, Wallén EAA, Lahtela‐Kakkonen M, Jarho EM, Luthman K (2014) Chroman‐4‐one‐ and chromone‐based sirtuin 2 inhibitors with antiproliferative properties in cancer cells. J Med Chem 57: 9870–9888 [DOI] [PubMed] [Google Scholar]
- 59. Rotili D, Tarantino D, Carafa V, Paolini C, Schemies J, Jung M, Botta G, Di Maro S, Novellino E, Steinkühler C et al (2012) Benzodeazaoxaflavins as sirtuin inhibitors with antiproliferative properties in cancer stem cells. J Med Chem 55: 8193–8197 [DOI] [PubMed] [Google Scholar]
- 60. Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- 61. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F (2010) Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics 26: 2069–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Forment JV, Jackson SP (2015) A flow cytometry‐based method to simplify the analysis and quantification of protein association to chromatin in mammalian cells. Nat Protoc 10: 1297–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Source Data for Figure 3
Data Availability Statement
ChIP‐seq data were downloaded from the Sequence Read Archive (NCBI) using accession numbers specified in Table EV3 and originated from the following publications: (i) Thurtle and Rine 38; (ii) Teytelman et al 39.