

Abstract
SMAD4 is a common intracellular effector for TGF‐β family cytokines, but the mechanism by which its activity is dynamically regulated is unclear. We demonstrated that ubiquitin‐specific protease (USP) 4 strongly induces activin/BMP signaling by removing the inhibitory monoubiquitination from SMAD4. This modification was triggered by the recruitment of the E3 ligase, SMURF2, to SMAD4 following ligand‐induced regulatory (R)‐SMAD–SMAD4 complex formation. Whereas the interaction of the negative regulator c‐SKI inhibits SMAD4 monoubiquitination, the ligand stimulates the recruitment of SMURF2 to the c‐SKI‐SMAD2 complex and triggers c‐SKI ubiquitination and degradation. Thus, SMURF2 has a role in termination and initiation of TGF‐β family signaling. An increase in monoubiquitinated SMAD4 in USP4‐depleted mouse embryonic stem cells (mESCs) decreased both the BMP‐ and activin‐induced changes in the embryonic stem cell fate. USP4 sustained SMAD4 activity during activin‐ and BMP‐mediated morphogenic events in early zebrafish embryos. Moreover, zebrafish depleted of USP4 exhibited defective cell migration and slower coordinated cell movement known as epiboly, both of which could be rescued by SMAD4. Therefore, USP4 is a critical determinant of SMAD4 activity.
Keywords: embryonic stem cells, SMAD4, TGF‐β, USP4, zebrafish
Subject Categories: Development & Differentiation, Signal Transduction
Introduction
Transforming growth factor β (TGF‐β) family members, which include the TGF‐βs, activins, and bone morphogenetic proteins (BMPs), play pivotal roles in controlling stem cell fate, embryogenesis and the maintenance of tissue homeostasis. Dysregulation of TGF‐β family activity causes developmental defects and diseases (Massagué, 2008; Ikushima & Miyazono, 2010). TGF‐β family ligands bind and activate specific heteromeric type I and type II serine–threonine kinase receptor complexes. The TGF‐β/activin type I receptors, also termed activin receptor‐like kinase (ALK) 5 and 4, mediate carboxy terminal phosphorylation of receptor‐regulated (R)‐SMADs, that is, SMAD2 and SMAD3, whereas BMP type I receptors (i.e. ALK1/2/3/6) phosphorylate R‐SMAD1, SMAD5, and SMAD8. Activated R‐SMADs hetero‐oligomerize with the common mediator (Co)‐SMAD4, translocate into the nucleus, and regulate the expression of target genes (Massagué, 2008; Kang et al, 2009; Moustakas & Heldin, 2009; Ikushima & Miyazono, 2010). SMAD3 and SMAD4 directly bind to a 5′CAGACA3′ DNA sequence, called the SMAD‐binding element (SBE) (ten Dijke & Hill, 2004).
The TGF‐β family receptors and downstream SMAD proteins are subject to complex regulation by reversible interactions with other proteins, changes in subcellular localization and post‐translational modifications. Emerging studies have revealed key roles for ubiquitin addition and removal from TGF‐β family components, processes that are mediated by specific E3 ubiquitin ligases and deubiquitinating enzymes (DUBs), respectively (Lonn et al, 2009; De Boeck & ten Dijke, 2012). The E3 ligases SMURF1/2 play prominent roles in the poly‐ubiquitination and degradation of TGF‐β family signaling components. SMURFs have also been shown to indirectly ubiquitinate proteins by forming complexes with R‐SMADs or I‐SMADs that contain proline‐rich PPXY motifs that are recognized by a tryptophan‐rich WW domain in SMURFs (Bonni et al, 2001; Morén et al, 2005). Ubiquitin‐specific proteases (USP)4, 11, and 15 act as DUBs for TGF‐β type I receptor and/or R‐SMADs (Inui et al, 2011; Al‐Salihi et al, 2012; Eichhorn et al, 2012; Zhang et al, 2012). SMAD4 is monoubiquitinated at Lysine 519, which inhibits the formation of a complex with activated, phosphorylated SMAD2 (Dupont et al, 2009). The E3 ligase TRIM33/Ectodermin, which contains a PHD domain and a bromodomain, has been implicated in mediating the inhibitory monoubiquitination of SMAD4 in a histone‐dependent manner (Agricola et al, 2011). Other reports, however, have concluded that TRIM33/Ectodermin interacts with activated SMAD2/3 and functions to control erythroid differentiation independently of SMAD4 (He et al, 2006). Moreover, in mammalian embryonic cells, TRIM33/Ectodermin was found to be required for SMAD2/3‐SMAD4 binding and activation of poised promoters (Xi et al, 2011).
c‐SKI negatively regulates TGF‐β family signaling by interacting with R‐SMADs and SMAD4 and recruiting transcriptional suppressors, such as histone deacetylates and protein arginine methyltransferases. The resulting complexes compete with the co‐activator p300/CBP (Akiyoshi et al, 1999; Luo et al, 1999; Sun et al, 1999). c‐SKI inactivates R‐SMAD–SMAD4 complex formation (Wu et al, 2002) and can inhibit monoubiquitination of SMAD4 (Wang et al, 2008). Moreover, by binding SMADs and co‐repressors, c‐SKI can maintain TGF‐β‐responsive promoters in a repressed state (Luo et al, 1999; Suzuki et al, 2004; Tabata et al, 2009). Interestingly, TGF‐β stimulation rapidly induces c‐SKI degradation (Sun et al, 1999) and the E3 ligase RNF111 has been implicated in this response (Le Scolan et al, 2008).
In this study, we demonstrated that USP4 acts as a potent activator of both activin and BMP signaling by directly de‐ubiquitinating the monoubiquitinated SMAD4 that is formed by R‐SMAD‐SMURF2‐induced monoubiquitination. USP4 ensures that the levels of SMAD hetero‐oligomers are sustained and the SMAD4 DNA binding affinity is enhanced. It also enables an efficient re‐use of SMAD4. Moreover, we have provided evidence that SMURF2 is a mediator of the TGF‐β/SMAD‐induced c‐SKI degradation that permits SMAD signaling to initiate. Our results show that USP4 enhances SMAD4‐mediated activin and BMP pathways in mouse embryonic stem cells (mESCs) and is required for early zebrafish embryogenesis. Because SMAD4 dysfunction is involved in cancer and other diseases, our findings could have relevance for understanding and treating these conditions.
Results
USP4 is required for SMAD signaling
To search for DUBs critical to activin/SMAD signaling, we performed a gain‐of‐function screen by overexpressing 74 DUB cDNAs (Sowa et al, 2009), and measuring their effects on a SMAD2‐SMAD4‐dependent transcriptional luciferase reporter (Labbé et al, 1998). Several DUBs, including USP4, potently activated the activin A (hereafter activin)‐induced signal (Figs 1A and EV1A and B). Of note, USP9X was previously reported to activate TGF‐β/nodal signaling (Dupont et al, 2009); however, when it was tested separately (because it was not included in the DUB library), USP9X had no significant effect on the activity of the reporter (Fig EV1C). Wild‐type USP4 (USP4‐WT) potentiated the activin‐SMAD signaling in HepG2 cells. By contrast, a mutant harboring a point mutation in the active site cysteine (USP4 C311S, hereafter called USP4‐CS) was unable to stimulate the reporter (Fig 1B), showing that the catalytic activity was essential. Knockdown assays demonstrated that USP4 was required for optimal induction of the activin‐SMAD‐mediated transcriptional response (Figs 1C and EV1D). Ectopic expression of USP4‐WT, but not USP4‐CS, increased the magnitude and the duration of the activin‐induced SMAD2‐SMAD4 complex formation, but not the levels of SMAD2 phosphorylation in HepG2 cells (Fig 1D). Consistent with these data, depletion of endogenous USP4 with two independent shRNAs had the opposite effect in HepG2 cells (Fig 1E). Moreover, the activin‐induced SMAD2‐SMAD4 complex formation was attenuated in USP4‐deficient mouse embryonic fibroblasts (USP4 −/− MEFs) (Fig 1F). The impaired expression of the activin/SMAD target genes in USP4 −/− MEFs was rescued by ectopic expression of USP4‐WT, but not the USP4‐CS mutant (Fig EV2A).
Figure 1. USP4 is required for activin‐ and BMP‐induced SMAD signaling.
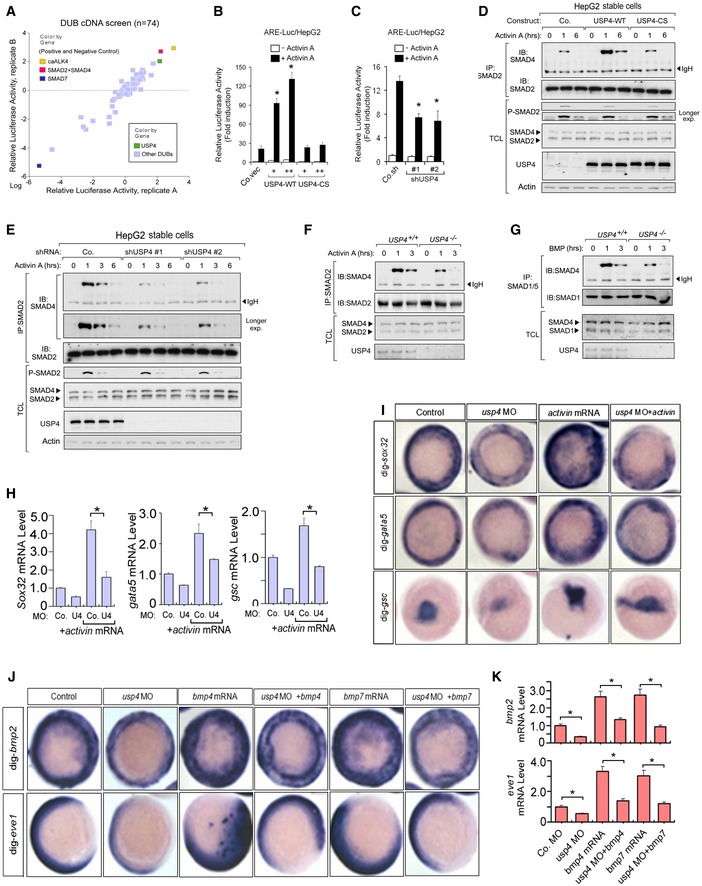
-
ADUB cDNA screening data from HEK293T cells. USP4, which strongly activates the TGF‐β‐induced SMAD2‐SMAD4‐dependent ARE‐Luc transcriptional reporter, is indicated (with green square). Testing refers to the analysis of individual cDNAs. Transfection with constitutively active (ca)ALK4 or SMAD2 together with SMAD4 was included as controls that potently activate the response (indicated with yellow and red squares, respectively). The inhibitory SMAD7 was used as control that potently inhibits the response (blue square); the x‐ and y‐axes are the relative luciferase activity in two replicates, P < 0.01.
-
B, CEffect of USP4 wild‐type (USP4‐WT) and the USP4 mutant that lacked de‐ubiquitinating enzyme activity (USP4‐CS) (B) or knockdown (shUSP4 #1 and #2) (C). The ARE‐Luc transcriptional response induced by activin A (50 ng/ml) in HepG2 cells. The data are presented as means ± SD. Co.vec, empty vector; Co.sh, non‐targeting shRNA. * indicates statistical significance (P < 0.05). Experiments were performed in triplicate.
-
D, EImmunoprecipitation (IP) and immunoblot (IB) analyses of SMAD2‐SMAD4 complex formation and SMAD2 phosphorylation in HepG2 cells following stable overexpression of USP4‐WT or USP4‐CS (D) or stable knockdown of USP4 with shRNA (shUSP4 #1 and #2) (E), after stimulation with activin A (50 ng/ml) for the indicated times. IB for actin was included as loading control.
-
F, GIP and IB analyses of SMAD2‐SMAD4 (F) or SMAD1/5‐SMAD4 (G) formation in wild‐type (USP4 +/+) and USP4 knockout (USP4 −/−) MEFs after stimulation with activin A (50 ng/ml) or BMP2 50 ng/ml) for the indicated times.
-
HRelative expression levels of zebrafish sox32, gata5, and goosecoid (gsc) in embryos, as indicated by quantitative real‐time PCR (qRT–PCR). The quantified mRNA levels were normalized to β‐actin and presented relative to control embryos. * indicated the statistical significance (P < 0.05). Experiments were performed on at least three independent injected embryos.
-
I, JInhibitory action of usp4 depletion on activin‐ and bmp4‐induced phenotype. Embryos were fixed and hybridized with probes for sox32, gata5 and goosecoid (gsc) (I) and bmp2 and eve1 (J). See also the overview images of the embryos in the Fig EV2F and G.
-
KThe relative expression levels of zebrafish bmp2 and eve1 as indicated by qRT–PCR. The quantified mRNA levels were normalized to β‐actin are presented relative to control embryos. * indicates statistical significance (P < 0.05). Experiments were performed on at least three independent injected embryos.
Figure EV1. DUB cDNA screening identified USP4 as the most potent activator of activin signaling.
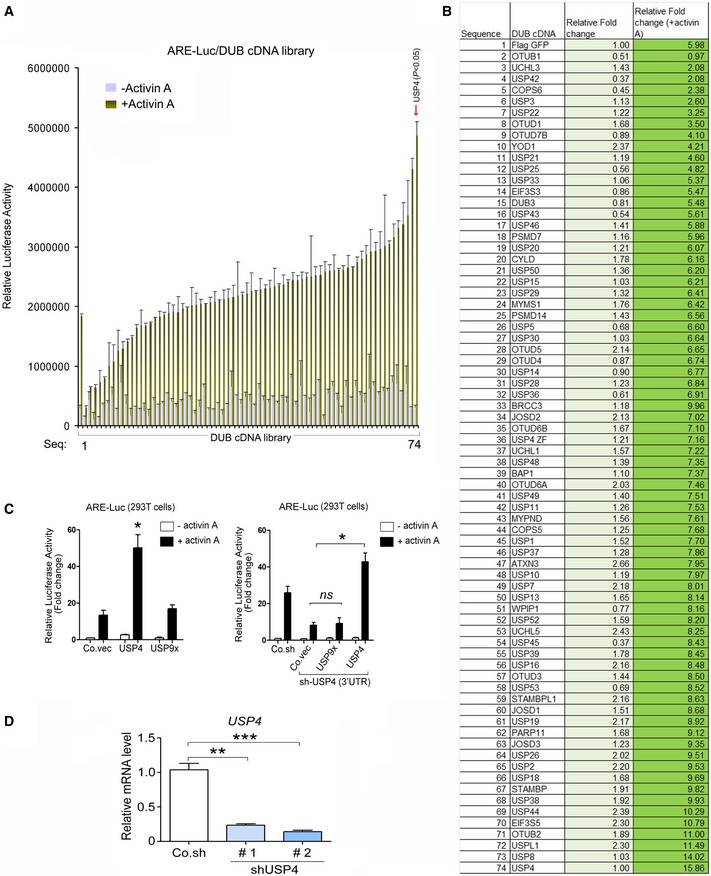
- This graph depicts the activin (5 ng/ml)‐induced ARE‐Luc transcriptional reporter activity compared to control cells and the effects of a representative set of DUB cDNAs, n = 3. The top activator USP4 is indicated with an arrow. The data are presented as means ± SD.
- List of the results of screen performed in Appendix Fig S1A.
- Left panel: effect of USP4 and USP9x on the ARE‐Luc transcriptional response induced by activin A (50 ng/ml) in HepG2 cells. Right panel (mentioned in the discussion): USP4, but not the USP9x rescued the defective ARE‐Luc response caused by sh‐USP4 that target USP4 3'UTR. The data are presented as means ± SD. Co.vec, empty vector; Co.sh, non‐targeting shRNA. * indicates statistical significance (P < 0.05). Two‐tailed, unpaired t‐test. Experiments were performed in triplicate.
- Q‐PCR analysis of USP4 mRNA level in HepG2 cells infected with lentivirus encoding control (Co.sh) or USP4 shRNA (#1). Experiments were performed in triplicate. Two‐tailed, unpaired t‐test. **P < 0.01 and ***P < 0.001. The data are presented as means ± SD.
Figure EV2. USP4 is required for BMP‐SMAD signaling.

-
AqRT–PCR analysis of activin target genes SMAD7, PAI‐1, P21, and P15 in USP4 +/+ and USP4 −/− MEFs in the absence or presence of ectopically expressed USP4‐WT or USP4‐CS and treated with activin A (50 ng/ml) for 1 h. The values and error bars represent the mean ± SD of triplicate assays and at least two independent experiments.
-
B, CThe effect of USP4 wild‐type (USP4‐WT) and the USP4 mutant in which the de‐ubiquitinating enzyme activity is inactive (USP4‐CS) (B) or USP4 knockdown (shUSP4 #1 and #2) (C) on the BRE‐Luc transcriptional response induced by BMP6 (25 ng/ml) in HepG2 cells. These data are presented as the means ± SD (n = 3). Co.vec, empty vector; Co.sh, non‐targeting shRNA.
-
D, EImmunoprecipitation (IP) and immunoblotting (IB) analyses of SMAD1/5‐SMAD4 complex formation and SMAD1/5 phosphorylation in HepG2 cells that stably overexpressed USP4‐WT or USP4‐CS (D) or in which stable knockdown of USP4 by shRNA (shUSP4 #1 and #2) (E) was achieved and after stimulation with BMP6 (25 ng/ml) for the indicated times. IB for actin on TCL is included as loading control.
-
F, GEffect of morpholino‐mediated depletion of usp4 on activin‐ and bmp4‐induced phenotype as measured by expression of gata5 and sox32 (F) and eve1 and bmp2 (G). These figures relate to Fig 1I and J.
-
HThe relative expression levels of zebrafish vox and vent as determined by qRT–PCR. The quantified mRNA levels were normalized to β‐actin mRNA level and are presented relative to control embryos. * indicates statistical significance (P < 0.05), and ** indicates statistical significance (P < 0.005). Two‐tailed, unpaired t‐test. These data are presented as the means ± SD (n = 3).
Source data are available online for this figure.
Subsequently, we investigated the role of USP4 in BMP/SMAD signaling. USP4‐WT, but not USP4‐CS, promoted the BMP/SMAD‐mediated transcriptional response, and USP4 depletion reduced BMP‐induced reporter activation (Fig EV2B and C). The BMP‐induced SMAD complex (SMAD1/5‐SMAD4) was stabilized by USP4‐WT, whereas BMP‐induced SMAD1 phosphorylation remained unchanged (Fig EV2D). USP4 depletion impaired the formation of the BMP‐induced SMAD complex (SMAD1/5‐SMAD4), but had no effect on SMAD1 phosphorylation (Fig EV2E). Compared to the wild‐type MEFs, the formation of the BMP‐SMAD (SMAD1)‐SMAD4 complex was reduced in USP4 −/− MEFs (Fig 1G). We further examined USP4 function in zebrafish embryos. Consistent with the results obtained with cultured cells, quantitative real‐time PCR (qRT–PCR) analysis and in situ hybridization showed that usp4‐depleted zebrafish embryos express only low amounts of the activin signaling targets sox32, gata5, and gsc (Figs 1H and I, and EV2F and G). In addition, usp4 depletion was associated with a severe defect in the nodal‐related squint (sqt)‐initiated responses (Fig EV3). These results are in line with our previous observations that ectopic expression of usp4 mRNA in zebrafish strongly promotes the expression of the Nodal target genes goosecoid (gsc) and notail (ntl) (Zhang et al, 2012). Similarly, induction of the bmp2 and eve1 mesodermal markers by ectopic expression of bmp4 and bmp7 was impaired in usp4 morphants (Fig 1J and K). Expression of target BMP genes such as vox and vent was stimulated by usp4 mRNA ectopic expression and impaired by MO‐mediated usp4 depletion (Fig EV2H). Taken together, these results indicated that USP4 is a critical activator of activin‐ and BMP‐SMAD signaling in mammalian cells and zebrafish embryos.
Figure EV3. Genetic interaction between zebrafish usp4 and sqt .
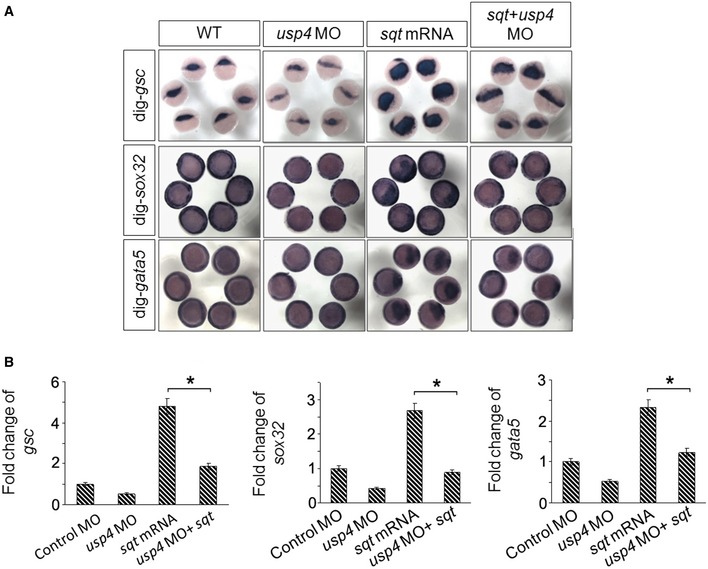
- The depletion of usp4 partially inhibited the sqt‐induced phenotype. Single‐cell embryos were injected with control MO, usp4 MO, sqt mRNA, or their combination. Embryos at the shield stage or 70% epiboly stage were fixed and hybridized with probes for markers of the endoderm and mesoderm including sox32, gata5, and goosecoid (gsc). Each group of embryos is oriented differently: animal pole view for sox32, and gata5 panels; organizer view for the gsc panel.
- The relative expression levels of zebrafish goosecoid (gsc), sox32, and gata5 were monitored by quantitative real‐time PCR (qRT–PCR). The quantified mRNA levels were normalized to β‐actin and are presented relative to the control embryos. * indicates statistical significance (P < 0.05). Two‐tailed, unpaired t‐test. Experiments were performed on at least three independent injected embryos. These data are presented as the means ± SD.
USP4 interacts with SMAD4 and targets monoubiquitinated SMAD4 for deubiquitination in vitro
The fact that USP4 did not affect activin/BMP‐induced SMAD phosphorylation but did promote maintenance of the SMAD hetero‐oligomer complex indicated that USP4 acts downstream of the activin/BMP type I receptor. Indeed, unlike its activity toward the TGF‐β type I receptor (Zhang et al, 2012), USP4 did not act as a DUB for the activin/BMP type I receptors. We therefore tested whether USP4 targets the Co‐SMAD or the I‐SMAD. Overexpressed USP4 did bind to SMAD4, but not to SMAD7 (Fig 2A). Moreover, USP4 co‐localized with SMAD4 in the cytoplasm (Appendix Fig S1) and recombinant USP4 associated with SMAD4 in vitro using a glutathione S‐transferase (GST) pull‐down assay (Fig 2B). Next, we investigated whether USP4 affects SMAD4 ubiquitination. Figure 2C shows that a SMAD4‐USP4‐WT complex was not ubiquitinated, whereas SMAD4 bound to the USP4‐CS mutant was highly ubiquitinated (of which the main fraction was monoubiquitinated). Purified (GST)‐USP4 WT but not GST–USP4‐CS removed the monoubiquitin from SMAD4 (Fig 2D). We therefore tested whether USP4 specifically acts as a DUB for the monoubiquitinated SMAD4 by performing in vitro de‐ubiquitination assays. We purified active USP4 and determined its de‐ubiquitinating activity by measuring its ability to cleave a ubiquitin‐AMC substrate (Fig 2E). As shown in Fig 2F, two forms of Flag‐SMAD4 were purified from transfected HEK293T cells: unmodified SMAD4 and a higher molecular weight form, which corresponded (by size) to monoubiquitinated SMAD4. This modified SMAD4 was rapidly converted to free SMAD4 when it was incubated with increasing doses of USP4 consistent with the idea that USP4 cleaves the monoubiquitin from SMAD4 in vitro. To further test this idea, we purified ubiquitinated SMAD4 (mainly monoubiquitinated SMAD4) and showed that it was cleaved upon incubation with USP4 (Fig 2G). This occurred rapidly; 50% of the monoubiquitinated SMAD4 was cleaved within 20 min of the addition of USP4 (Fig 2H). Taken together, our results demonstrate that monoubiquitinated SMAD4 is a substrate of USP4.
Figure 2. USP4 interacts with SMAD4 and de‐ubiquitinates mono‐ubiquitinated SMAD4 in vitro .
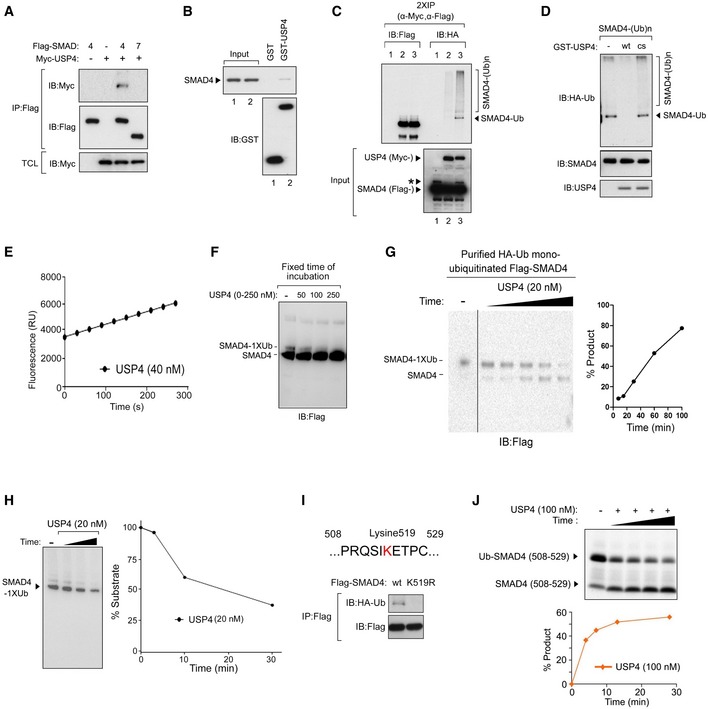
- Immunoblot (IB) analysis of whole‐cell lysate (TCL) and immunoprecipitates derived from HEK293T cells transfected with Myc‐USP4 and Flag‐SMAD4 or SMAD7.
- IB analysis of purified SMAD4 bound to GST–USP4 in vitro.
- IB analysis of whole‐cell lysate and immunoprecipitates derived from HEK293T cells transfected with Flag‐SMAD4, Myc‐USP4 WT, or USP4 C311S and immunoprecipitated with anti‐Myc and subsequently anti‐Flag antibodies. 1, Flag‐SMAD4; 2, Flag‐SMAD4+Myc‐USP4 WT; 3, Flag‐SMAD4+Myc‐USP4‐CS. Position of mono‐ and polyubiquitinated SMAD4 on the IB is indicated.
- The HA‐ubiquitinated SMAD4 substrate (purified using denaturing conditions) was incubated with purified recombinant USP4 WT or USP4 CS at 37°C for 1 h. The reaction was terminated by the addition of SDS sample buffer followed by a 2‐min heat denaturation at 95°C. The reaction products were evaluated using IB analysis.
- Analysis of the kinetic hydrolysis of the ubiquitin‐AMC substrate by USP4. Fluorescence excitation wavelength, 380 nm; emission wavelength, 460 nm.
- IB analysis of purified Flag‐SMAD4 protein incubated with increasing concentrations of recombinant USP4 for 30 min. The mono‐ubiquitinated SMAD4 was removed by USP4, while the amount of free SMAD4 remains constant.
- The effect of an optimal concentration (20 nM) of recombinant USP4 on the cleavage of monoubiquitinated SMAD4 shown by anti‐Flag Western blotting. Right panel shows quantified percentage of free SMAD4. The line indicates where the gel has been cut; all samples were run on the same gel. See also the source data.
- The kinetic effect of an optimal concentration (20 nM) of recombinant USP4 on the cleavage of monoubiquitinated SMAD4. Right panel shows quantified percentage of substrate.
- Lysine 519 of SMAD4 is monoubiquitinated. IB analysis of whole‐cell lysate and immunoprecipitates derived from HEK293T cells that stably expressed HA‐Ub and were transfected with Flag SMAD4 or Flag‐SMAD4 K519R.
- Deubiquitination of TAMRA‐labeled SMAD4 508–529 peptide covalently ubiquitinated on K519, incubated with 100 nM USP4. Samples taken from the reaction at various time points were loaded on SDS–PAGE, visualized (upper panel), and quantified (lower panel) using the TAMRA (tetramethylrhodamine) fluorescence.
Source data are available online for this figure.
Consistent with a previous report (Dupont et al, 2009), site‐specific mutation of Lysine 519 to arginine abolished the SMAD4 monoubiquitination (Fig 2I). To address whether USP4 specifically targets SMAD4 Lysine 519 monoubiquitination, we covalently linked a 21‐amino acid SMAD4 peptide containing Lysine 519 (SMAD4 amino acids 508–529) to a ubiquitin molecule in vitro and incubated this substrate with 100 nM of active USP4. The level of the ubiquitinated SMAD4 peptide decreased when it was incubated with USP4 and there was a concomitant increase in free SMAD4 peptide (Fig 2J). These data suggest that the ubiquitin molecule covalently linked to SMAD4 is cleaved by USP4 and that a minimal SMAD4 region that includes the putative monoubiquitination site is sufficient for USP4‐mediated de‐ubiquitination (Fig 2J).
USP4 removes SMAD4 monoubiquitination in vivo
Next, we investigated the physical and functional interactions between USP4 and SMAD4 in cells. Interestingly, co‐expression of SMAD4 and USP4 (WT or mutant versions) revealed that whereas monoubiquitinated SMAD4 efficiently interacted with USP4, the SMAD4‐K519R mutant (lacking the putative monoubiquitination site) failed to bind to USP4. Consistent with this finding, an artificial monoubiquitinated SMAD4 peptide mimic, that is, the C‐terminal ubiquitin‐fused SMAD4, had enhanced affinity for USP4 (Fig 3A). Thus, USP4 preferentially interacts with monoubiquitinated SMAD4, and the two proteins appear to dissociate following its deubiquitination.
Figure 3. USP4 removes monoubiquitin from SMAD4 in vivo .
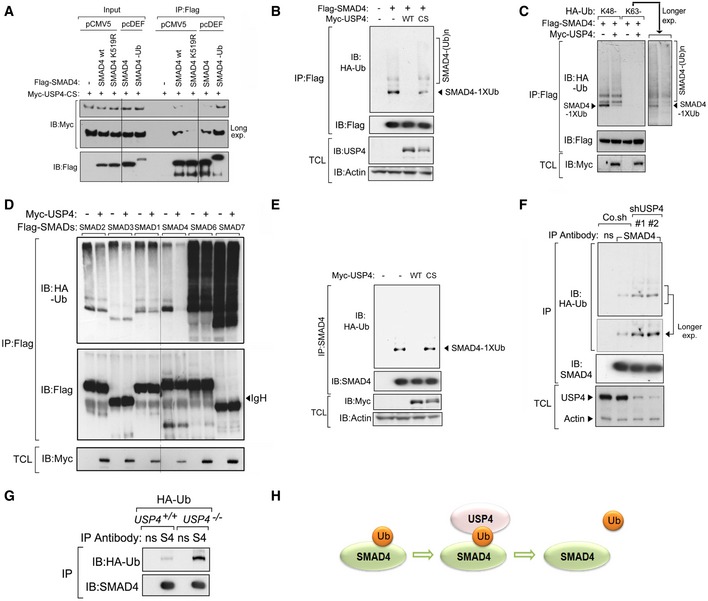
- Cells were transfected with Myc‐USP4‐CS as well as Flag‐SMAD4 wt, SMAD4 K519R or SMAD4‐Ub fusion. The black lines indicate where the Western blot image was cut, see also source data.
- Cells that stably expressed HA‐Ub were transfected with Flag‐SMAD4, Myc‐USP4 WT, or Myc‐USP4‐CS.
- Cells were transfected with Flag‐SMAD4, Myc‐USP4‐WT, and Lysine 48 mutated (K48‐) or Lysine 63 mutated (K63‐) Ub.
- Cells that stably expressed HA‐Ub were transfected with Flag‐SMADs and Myc‐USP4. The black line indicates where the Western blot image was cut, see also source data.
- Cells that stably expressed HA‐Ub were transfected with Myc‐USP4‐WT or Myc USP4‐CS.
- Cells that stably expressed HA‐Ub were transfected with control non‐targeting shRNA (Co.sh) or USP4 shRNA (#1 or #2).
- IB analysis of whole‐cell lysate and immunoprecipitates derived from wild‐type (USP4 +/+) and USP4‐deficient (USP4 −/−) MEFs that had been infected with a lentivirus that expressed HA‐Ub.
- Schematic that depicts USP4 removing SMAD4 monoubiquitination.
To analyze whether USP4 acts as a DUB for monoubiquitinated SMAD4 in vivo, we transfected HEK293T cells with expression plasmids encoding the HA‐tagged wild‐type ubiquitin or mutants (K48‐ and K63‐, that form only Lys‐48 or only Lys‐63 polyubiquitin chains, respectively). The Flag‐tagged SMAD4 proteins were then affinity‐purified, and their ubiquitination status was visualized by immunoblotting for HA‐ubiquitin. Both mono‐ and poly‐ubiquitination appeared as modifications of the Flag‐tagged SMAD4 (Fig 3B). Both the K48‐ and K63‐ Ub mutants were found to be conjugated to SMAD4, indicating that multiple types of polyubiquitination can occur on SMAD4 (Fig 3C). Ectopic expression of wild‐type USP4 but not the catalytically inactive USP4‐CS mutant inhibited SMAD4 ubiquitination (Fig 3B). Importantly, when we used ubiquitin mutants that form only Lys‐48 or only Lys‐63 polyubiquitin chains, USP4 only inhibited the conjugation of monoubiquitin to SMAD4 in a deubiquitinating activity‐dependent manner (Fig 3C). USP4 appeared to act specifically on SMAD4, because it did not affect the ubiquitination of other SMAD molecules (Fig 3D). Endogenous SMAD4 was mainly modified by monoubiquitination, and only trace amounts of polyubiquitination was observed (Fig 3E). Ectopically expressed USP4‐WT, but not the USP4‐CS mutant, abolished endogenous SMAD4 monoubiquitination (Fig 3E). Importantly, loss of endogenous USP4 enhanced the monoubiquitination of endogenous SMAD4 (Fig 3F), indicating that USP4 is a critical regulator of SMAD4 in cells. Consistently, enhanced SMAD4 monoubiquitination was observed in USP4‐deficient MEF cells compared with control cells (Fig 3G). Taken together, we have shown that USP4 targets monoubiquitinated SMAD4 for deubiquitination (Fig 3H).
R‐SMAD/SMURF2 competes with USP4 for interaction with SMAD4 and enables SMAD4 monoubiquitination upon TGF‐β stimulation
We found that SMAD4 monoubiquitination interferes with its ability to form heteromeric complexes with R‐SMADs evidenced by the finding that SMAD1 and SMAD2 associated more efficiently with free SMAD4 compared to monoubiquitinated SMAD4 (Figs EV4A and 4A). Activated SMAD complexes recognize SMAD‐binding elements (SBEs) and we found that free SMAD4, but not the monoubiquitinated SMAD4, was captured by an SBE oligonucleotide (Fig 4B) suggesting that monoubiquitination blocks SMAD4 binding to DNA. Moreover, ectopically expressed free SMAD4, but not the SMAD4‐ubiquitin fusion protein, enhanced the association of SMAD2/SMAD3 to DNA (Fig 4C).
Figure EV4. Regulation of SMAD4 monoubiquitination.
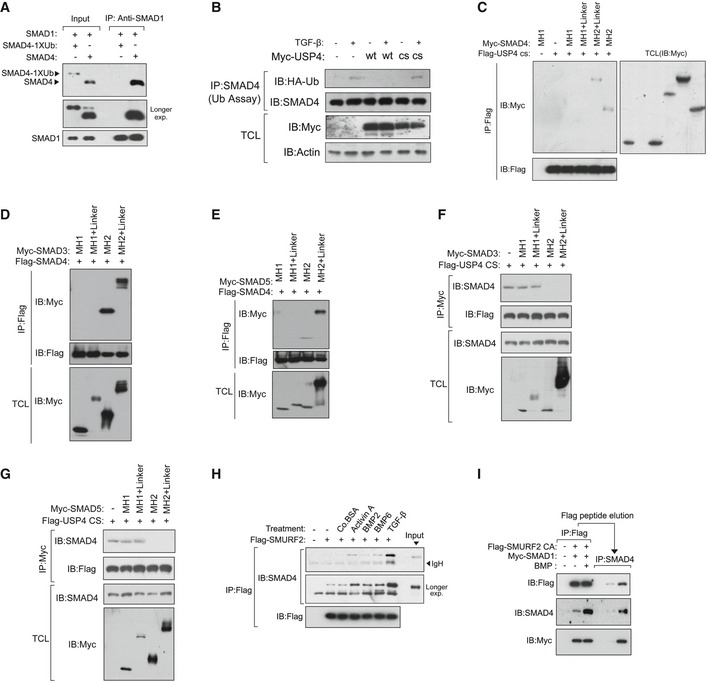
-
AR‐SMAD preferred to associate with free SMAD4 rather than monoubiquitinated SMAD4. IB analysis of the input and immunoprecipitates derived from the incubation of purified SMAD1 with SMAD4 or SMAD4 ‐1xUb.
-
BUSP4 prevented the monoubiquitination of SMAD4 upon stimulation of cells with TGF‐β. IB analysis of total cell lysates (TCL) and immunoprecipitates derived from HA‐Ub expressing HEK293T cells that were transfected with Myc‐USP4 wild‐type (WT) or Myc‐USP4 C311S mutant (CS),with or without TGF‐β treatment (5 ng/ml) for 1 h.
-
CUSP4 bound to the SMAD4 MH2 domain. HEK293T cells were transfected with Myc‐tagged SMAD4 deletions and Flag‐tagged USP4 CS. The cells were harvested for immunoprecipitation (IP) with anti‐Flag affinity resin and subjected to IB analysis with indicated antibodies (left panel). TCL were analyzed by IB using myc antibodies (right panel).
-
D, EThe SMAD4 bound to the MH2 domain of SMAD3 or SMAD5. HEK293T cells were transfected with Myc‐tagged SMAD3 deletions (D) or SMAD5 deletion (E) and Flag‐tagged SMAD4. The cells were harvested for IP with anti‐Flag affinity resin and IB with the indicated antibodies (upper two panels). TCL were analyzed by IB using myc antibodies (bottom panel).
-
F, GThe MH2 domain of SMAD3 or SMAD5 displaced USP4 from SMAD4. IB analysis of total cell lysate (TCL) and immunoprecipitates derived from HEK293T cells that were transected with Flag‐USP4 CS and Myc‐SMAD3 (F) or Myc‐SMAD5 (G) deletions as indicated.
-
HIB analysis of immunoprecipitates derived from Flag‐SMURF2‐transfected HEK293T cells treated with or without activin A (50 ng/ml), BMP2 (50 ng/ml), or TGF‐β (5 ng/ml) for 1 h.
-
IBMP6 promoted the formation of a trimeric complex of SMURF2, SMAD1, and SMAD4. Flag‐SMURF2 CA and Myc‐SMAD1 were co‐expressed in HEK293T cells. The cells were treated with BMP6 (50 ng/ml) for 1 h and then harvested for immunoprecipitation with anti‐Flag beads and elution with Flag‐peptide. The second IP with anti‐SMAD4 antibody was performed, and the resulting samples were subjected to IB with anti‐Flag, anti‐SMAD4, or anti‐Myc antibodies.
Source data are available online for this figure.
Figure 4. The R‐SMAD‐SMURF2 complex mediates the ligand‐induced SMAD4 monoubiquitination and termination of the SMAD signaling.
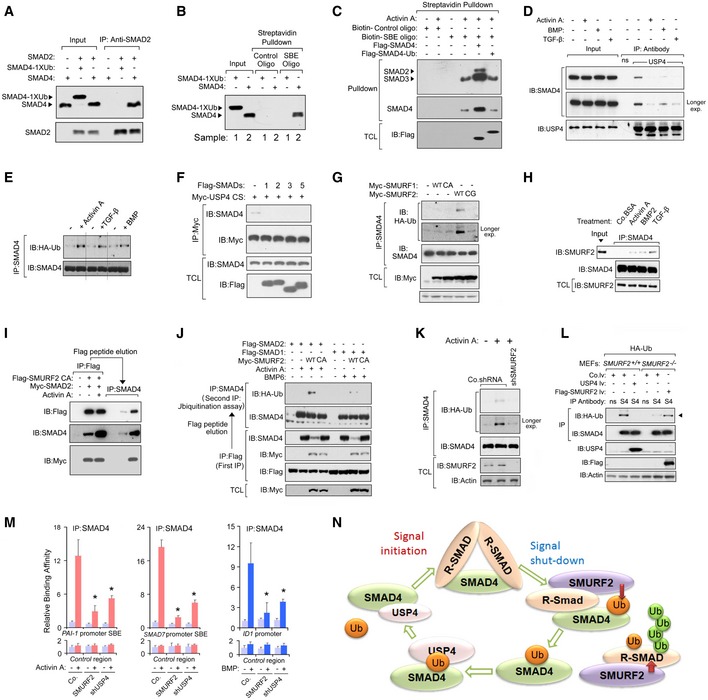
- R‐SMAD preferentially associates with free SMAD4. Immunoblot (IB) analysis of input and anti‐SMAD2 immunoprecipitates derived from in vitro incubation of purified SMAD2 protein together with SMAD4 or SMAD4‐1xUb.
- IB analysis of purified SMAD4 and SMAD4‐1xUb of the input and the streptavidin SMAD‐binding element (SBE) or the control oligonucleotide pull‐down.
- IB analysis of the total cell lysate (TCL) and streptavidin SBE pull‐down derived from HEK293T cells that had been transfected with Flag‐SMAD4 or Flag‐SMAD4‐1xUb. The cells were treated with activin A (50 ng/ml) for 1 h.
- TGF‐β family ligands cause the disassociation of SMAD4 and USP4. IB analysis of whole‐cell lysates (input) and immunoprecipitates derived from HEK293T cells that were treated with activin A (50 ng/ml), BMP (50 ng/ml), or TGF‐β (5 ng/ml) for 1 h.
- Treatment with TGF‐β (5 ng/ml)), activin A (50 ng/ml), and BMP2 (50 ng/ml) promotes SMAD4 monoubiquitination. IP with SMAD4 antibody followed with IB analysis of HEK293T cells that stably express HA‐Ub. The black lines indicate where the Western blot image was cut, see also source data.
- R‐SMAD displaces USP4 from SMAD4. IB analysis of total cell lysate (TCL) and immunoprecipitates derived from HEK293T that had been transected with Myc‐USP4 C311S and Flag‐SMADs (SMAD1, ‐2, ‐3, or 5).
- SMURF2 promotes SMAD4 monoubiquitination. HEK293T cells that stably expressed HA‐Ub were transfected with Myc‐tagged SMURF1 (wt/CA) or SMURF2 (WT/CG). The cells were harvested for the SMAD4 ubiquitination assay (IP for SMAD4 followed by IB for HA‐Ub).
- The interaction between endogenous forms of SMAD4 and SMURF2 was promoted by stimulation with ligand (activin A (50 ng/ml), BMP2 (50 ng/ml), or TGF‐β (5 ng/ml) or BSA control for 1 h in HeLa Cells.
- Activin A promotes the formation of the trimeric complex of SMURF2, SMAD2, and SMAD4. Flag‐SMURF2 CA and Myc‐SMAD2 were co‐expressed in HEK293T cells. Cells were harvested for immunoprecipitation, and Flag‐peptide elution was conducted. A second IP with anti‐SMAD4 antibody was performed, and the immunoprecipitates were immunoblotted with anti‐Flag, anti‐SMAD4, and anti‐Myc as indicated.
- SMAD4 was monoubiquitinated in the SMAD4‐R/SMAD/SMURF2 complex following stimulation with activin A (50 ng/ml) or BMP6 (50 ng/ml). HEK293T cells were transfected with the indicated plasmids, and 36 h later, the cells were stimulated with the ligands for 1 h and then harvested for 2 sequential IPs; the first IP used Flag beads, followed by Flag‐peptide elution. The eluates were then denatured for the second IP with an anti‐HA antibody.
- Endogenous SMURF2 is required for efficient monoubiquitination of SMAD4. MDA‐MB‐231 cells that were infected with lentiviruses that encoded control (Co.shRNA) or SMURF2 (shSMURF2) shRNA were harvested for the SMAD4 ubiquitination assay.
- Wild‐type SMURF2 +/+ and SMURF2‐deficient (SMURF2 −/−) MEFs that expressed HA‐Ub were infected with lentivirus that encoded USP4 or SMURF2 and harvested for the SMAD4 ubiquitination assay (IP for SMAD4 followed by IB for HA‐Ub). Position of the monoubiquitinated SMAD4 is indicated with an arrowhead.
- MDA‐MB 231 cells that expressed the Control shRNA (Co.), Flag‐SMURF2 (SMURF2), or USP4 shRNA (shUSP4) were treated with or without activin A (50 ng/ml) or BMP (50 ng/ml). The cells were then harvested for ChIP using an anti‐SMAD4 antibody. The SMAD‐responsive region (PAI‐1/SMAD7/ID1 promoter) and an unresponsive control region of the same gene (control region) were analyzed by qRT–PCR. The data are presented as mean ± SD of triplicate assays and represent at least two independent experiments. * indicates statistical significance (P < 0.05). Two‐tailed, unpaired t‐test.
- Schematic that depicts the dynamic control of monoubiquitination of SMAD4 mediated by the enzymatic activities of the R‐SMAD‐SMURF2 complex and USP4.
Source data are available online for this figure.
We observed that challenging cells with TGF‐β family members reduced the interaction between USP4 and SMAD4 (Fig 4D). Accordingly, sharply increased SMAD4 monoubiquitination occurred upon ligand stimulation (Fig 4E). In addition, USP4‐WT, but not the USP4‐CS mutant, reduced ligand‐induced SMAD4 monoubiquitination (Fig EV4B). These results suggest that the inhibitory SMAD4 monoubiquitination might provide a negative feedback signal for TGF‐β family/SMAD signaling. Next, we mapped the USP4‐binding domain in the SMAD4 protein and found that USP4 bound to the SMAD4 MH2 domain (Fig EV4C), the same region that is responsible for its binding to R‐SMAD following C‐terminal phosphorylation by the type I receptor (Fig EV4D and E). We hypothesized that the activated R‐SMAD might compete with USP4 for SMAD4 binding. Indeed, ectopically expressed R‐SMADs (shown as SMAD1/2/3/5) disrupted the USP4‐SMAD4 association (Fig 4F). Moreover, overexpression of the R‐SMAD MH2 domain (shown as SMAD3 and SMAD5 MH2 domain) alone was sufficient to cause the observed disruption (Fig EV4F and G). These results suggest that ligand‐induced R‐SMAD–SMAD4 complex formation is necessary for SMAD4 monoubiquitination.
Next, we screened the ability of different E3 ligases to stimulate SMAD4 monoubiquitination. We found that SMURF2 was the most active ligase. SMURF2 (but not SMURF1) promoted endogenous SMAD4 monoubiquitination in an E3 ligase activity‐dependent manner (Fig 4G). Of note, we found that TRIM33/Ectodermin did not affect SMAD4 monoubiquitination when co‐transfected with SMAD4. SMURF2 interacts with R‐SMADs via a PY motif that is absent in SMAD4. SMURF2 might thus be recruited to SMAD4 via R‐SMADs upon ligand stimulation. Consistent with this notion (and with the results shown in Fig 4E), when cells were stimulated with TGF‐β, endogenous SMAD4 was recruited to both ectopically expressed and endogenous SMURF2 (Figs 4H and EV4H). In addition, SMAD4‐SMAD1/SMAD2–SMURF2 formed a trimeric complex (Figs 4I and EV4I). To further test whether R‐SMAD might mediate SMAD4 monoubiquitination by SMURF2, we isolated endogenous SMAD4 from the ligand (activin or BMP6)‐induced SMAD4‐SMAD2–SMURF2 complex and determined its level of ubiquitination. Monoubiquitination of SMAD4 was triggered by SMURF2 when it was part of a R‐SMAD‐containing complex (Fig 4J). Furthermore, we depleted endogenous SMURF2 and observed decreased SMAD4 monoubiquitination (Fig 4K). Consistent with this, the SMAD4 monoubiquitination level was lower in SMURF2‐deficient (SMURF2 −/−) MEFs. In SMURF2 +/+ and SMURF2 −/− MEFs, the monoubiquitination of SMAD4 was reduced or increased by the lentivirus‐encoded expression of USP4 or SMURF2, respectively (Fig 4L). We next determined whether R‐SMAD–SMAD4 complex binding to DNA requires USP4‐mediated removal of the monoubiquitin from SMAD4. For this, we used chromatin immunoprecipitation (ChIP) to measure the recruitment of the SMAD4 to specific SMAD target genes (PAI1, SMAD7, and ID1) in MDA‐MB‐231 cells. Our results show that activin‐ or BMP‐dependent SMAD association with chromatin was lost upon USP4 depletion or SMURF2 overexpression (Fig 4M). Taken together, our results indicate a dynamic interplay between USP4 and SMURF2. The deubiquitination of monoubiquitinated SMAD4 by USP4 allows signal initiation, and subsequent ligand‐induced R‐SMAD–SMAD4 complex formation is required for SMURF2‐mediated inhibitory monoubiquitination of SMAD4 (Fig 4N).
SMURF2‐induced degradation of c‐SKI
To gain further insight into the dynamics of SMAD4 monoubiquitination, we compared this with other SMAD signaling events. TGF‐β stimulated transient SMAD4 monoubiquitination peaked at 1 h, and this broadly mirrored the kinetics of SMAD2 phosphorylation and SMAD2‐SMAD4 complex formation (Fig 5A). We found that the detectable SMAD4 monoubiquitination occurred only after TGF‐β‐dependent degradation of c‐SKI (Fig 5A), which suggests that c‐SKI may antagonize SMAD4 monoubiquitination. Consistent with a previous report (Wang et al, 2008), ectopic co‐expression of c‐SKI abolished SMAD4 monoubiquitination (but not SMAD4 polyubiquitination) (Fig 5B). Moreover, c‐SKI blocked TGF‐β‐dependent endogenous SMAD4 monoubiquitination (Fig 5C).
Figure 5. c‐SKI inhibits SMAD4 monoubiquitination and SMURF2 Mediates Rapid Degradation of c‐SKI.
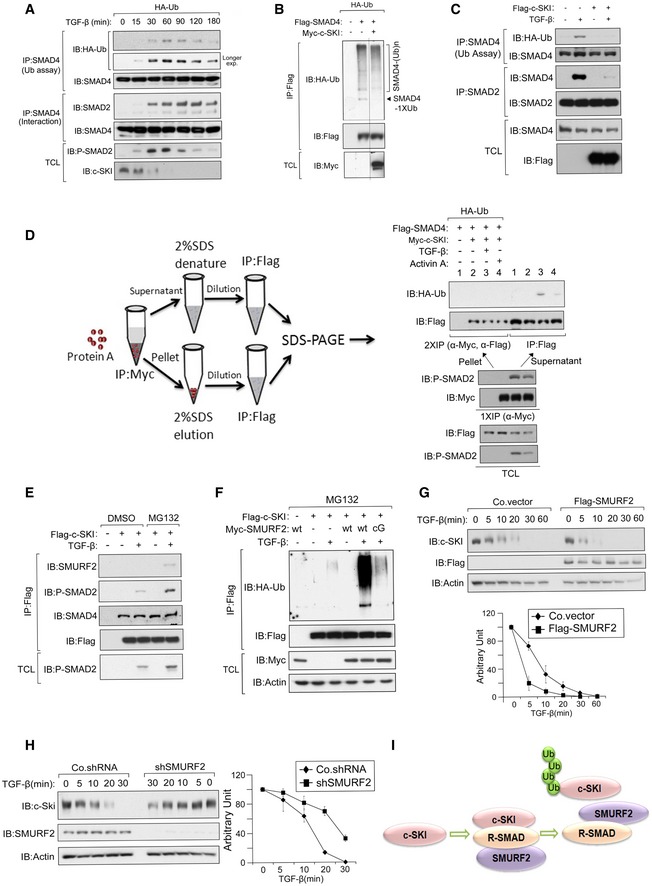
-
AHA‐Ub‐expressing MDA‐MB‐231 cells that had been treated with TGF‐β (5 ng/ml) as indicated were harvested for immunoblot analysis (IB) and the SMAD4 ubiquitination assay.
-
BSKI abolishes the monoubiquitination of ectopically expressed SMAD4. HA‐Ub‐expressing HEK293T cells that had been transfected with Flag‐SMAD4 and Myc‐c‐SKI were harvested for IB and the Flag‐SMAD4 ubiquitination assay. The black line indicates where the Western blot image was cut; the samples were run on the same gel.
-
Cc‐SKI abolishes the monoubiquitination of endogenous SMAD4. HEK293T cells with or without Flag‐c‐SKI expression were treated with TGF‐β (5 ng/ml) for 1 h and then harvested for the SMAD4 ubiquitination assay.
-
DThe c‐SKI‐associated SMAD4 is not monoubiquitinated following ligand stimulation. HA‐Ub‐expressing HEK293T cells that had been transfected with Flag‐SMAD4 and Myc‐c‐SKI were treated with 5 ng/ml TGF‐β or 50 ng/ml activin A for 1 h, and then, the cells were harvested for anti‐Myc immunoprecipitation (IP). Next, the precipitates were denatured with 2% SDS and diluted for the anti‐Flag ubiquitination assay. The cell lysates after the anti‐Myc IP were harvested and split to provide a sample for the co‐IP assay to detect the phosphorylated SMAD2 (p‐SMAD2), and a denatured sample for the SMAD4 ubiquitination assay.
-
ESMURF2 is recruited to c‐SKI in a ligand‐dependent manner. Flag‐c‐SKI‐transfected HEK293T cells treated with or without TGF‐β (5 ng/ml) for 1 h and harvested for IP and IB. Cells were treated with 5 μM MG132 or vehicle control DMSO for 5 h before harvest.
-
FSMURF2 stimulated the c‐SKI ubiquitination in a ligand‐dependent manner. Flag‐c‐SKI and Myc‐SMURF2‐WT/CG‐transfected HEK293T cells were treated with or without TGF‐β (5 ng/ml) for 1 h and harvested for the anti‐Flag ubiquitination assay. The cells were treated with 5 μM MG132 or the vehicle (DMSO) for 5 h before harvest.
-
G, Hc‐SKI stability is prolonged upon depletion of SMURF2. Upper panel: IB of MDA‐MB‐231 cells infected with lentivirus that encoded the Control vector (Co. vector), Flag‐SMURF2 (G), or Control shRNA (Co. shRNA), or SMURF2 shRNA (shSMURF2) (H) and treated with cycloheximide (CHX) (20 μg/ml) for the indicated times. Lower panel: quantification of band intensities from the upper panel. The band intensities were normalized to the t = 0 controls. The results are presented as he mean ± SD of three independent sets of experiments.
-
ISchematic that depicts the SMURF2‐mediated degradation of c‐SKI.
Source data are available online for this figure.
To detect c‐SKI‐associated SMAD4 ubiquitination, we eluted the SMAD4 from the c‐SKI immunoprecipitates, disrupted the protein complexes with SDS‐containing buffer, and re‐precipitated the c‐SKI‐bound SMAD4. The c‐SKI‐associated SMAD4 was not monoubiquitinated, whereas the free SMAD4 was monoubiquitinated (Fig 5D). We also identified an interaction between phosphorylated SMAD2 (p‐SMAD2) and c‐SKI following ligand stimulation. This suggests that activated SMAD2 binds transiently to c‐SKI and might mediate rapid degradation of c‐SKI (Fig 5D). Additionally, following TGF‐β stimulation, SMURF2 (and an increased level of P‐SMAD2) was found in a c‐SKI complex only after MG132 treatment (Fig 5E). This finding suggest that SMAD2–SMURF2 associated with c‐SKI and that c‐SKI might be targeted for degradation in this complex. Consistent with these data, SMURF2 induced c‐SKI ubiquitination only when the TGF‐β pathway was activated (Fig 5F and Appendix Fig S2). The TGF‐β‐induced c‐SKI instability was augmented by ectopic expression of SMURF2 and decreased by SMURF2 depletion (Fig 5G and H). Taken together, these results argue that ligand‐induced recruitment of SMURF2 to the c‐SKI‐R‐SMAD complex triggers c‐SKI ubiquitination and degradation (Fig 5I).
USP4 sustains activin/BMP signaling in mESCs
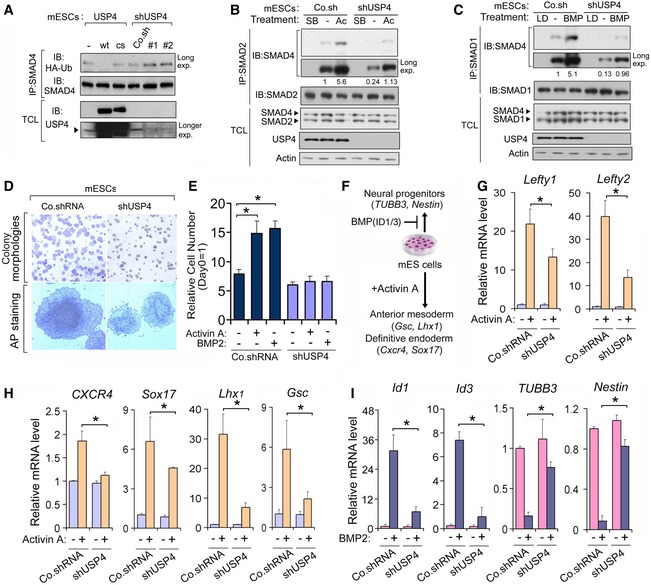
Activin and BMP signals suppress differentiation and sustain embryonic stem cell self‐renewal (Ying et al, 2003a,b; Ogawa et al, 2007). We found that USP4 also controlled SMAD4 monoubiquitination in mESCs (Fig 6A). mESCs in which USP4 had been depleted were less sensitive to activin than wild‐type mESCs, as shown by a reduced formation of SMAD2‐SMAD4 complexes (Fig 6B). Similarly, knockdown of USP4 strongly inhibited BMP‐induced SMAD1‐SMAD4 complex formation (Fig 6C). These results show that USP4 depletion provokes a defective activin/BMP response. In addition, feeder‐free cultures of USP4‐depleted mESCs in response to activin displayed much smaller colony sizes (Fig 6D) consistent with an impaired mESC capacity for self‐renewal and propagation, by comparison with wild‐type mESCs (Fig 6E). Similar results were observed when cells were treated with BMP (Fig 6E).
Figure 6. USP4 Sustains the Activin/BMP Signals in mESCs.
-
AImmunoblot (IB) analysis of total cell lysate (TCL) and immunoprecipitates derived from mouse embryonic stem cells (mESCs) that stably overexpressed USP4 WT/CS or those in which USP4 was knocked down with shRNA (shUSP4 #1 and #2).
-
B, CImmunoprecipitation (IP) and IB analyses of the SMAD2‐SMAD4 (B) or SMAD1‐SMAD4 (C) complex formation in mESCs following stable knockdown of USP4 and treatment with or without ALK4 kinase inhibitor SB431542 (SB) or activin A (50 ng/ml) (B) or BMP type I receptor kinase inhibitor LDN‐193189 (LD), or BMP2 (50 ng/ml) (C) for 1 h.
-
DUpper panel: morphological analysis of colonies of mESCs that had been infected with control shRNA (Co.shRNA) or shUSP4. Lower panel: alkaline phosphatase (AP) staining of random single mESC clones from the upper panel.
-
ERelative numbers of control and USP4‐depleted mESCs of day 5 versus day 0 after treatment with activin A (50 ng/ml) or BMP2 (50 ng/ml). Each bar represents the mean ± SD (n = 3).
-
FSchematic of mESC differentiation upon addition of BMP2/4 (upper part) or activin A (lower part).
-
GControl shRNA transfected (Co.shRNA) mESCs and those in which USP4 was stably depleted (shUSP4) were allowed to differentiate for 4 days on collagen IV‐coated plates in the absence or presence of activin A. The expression levels of two activin‐induced target genes, Lefty1 and Lefty2, were assessed by qRT–PCR. The values and error bars represent the mean ± SD of triplicate assays and at least two independent experiments.
-
HThe differentiation of activin A‐induced anterior mesoderm and definitive endoderm was inhibited in USP4‐depleted mESCs. The expression of the anterior mesoderm marker genes Goosecoid (Gsc) and LIM/homeobox protein 1 (Lhx1), and definitive endoderm markers Cxcr4 and Sox17 were measured using qRT–PCR. The values and error bars represent the mean ± SD of triplicate assays and at least two independent experiments.
-
IControl transfected shRNA (Co.shRNA) mESCs expressing control and those in which USP4 was stably depleted (shUSP4) were cultured in N2B27‐supplemented serum‐free media in the presence or absence of BMP2 and harvested 5 days later. The expression levels of the direct BMP target genes Id1 and Id3, as well as the neural differentiation markers β‐III tubulin (Tubb3) and Nestin, were measured using qRT–PCR. The value for the non‐BMP2 treated cells was set as “1”. The values and error bars represent the mean ± SD of triplicate assays and at least two independent experiments.
We also investigated the possible role of USP4 in ESC differentiation (Fig 6F). USP4‐depleted cells exhibited a reduced activin‐induced upregulation of the SMAD2/3 target genes Lefty1 and Lefty2 (Fig 6G). In mitogen‐poor medium, the activin‐induced mESC differentiation into anterior mesoderm (via induction of Goosecoid (Gsc) and LIM homeobox (Lhx)1) and definitive endoderm (via induction of C‐X‐C motif receptor 4 (Cxcr4) and sex determining region Y‐box (Sox)17) were suppressed upon loss of USP4 expression (Fig 6H). Induction of Id1/Id3 transcripts by BMP inhibits mESC differentiation into neural progenitor cells. USP4 depletion impaired the Id1/Id3 gene response, which led to inefficient neuronal differentiation, as indicated by the expression of maker genes such as β‐III tubulin (TUBB3) and Nestin (Fig 6I). Together, these data identify USP4 as a downstream effector of activin/BMP signaling in mESCs.
USP4 controls activin/BMP‐induced signaling and cell movements in zebrafish embryonic development
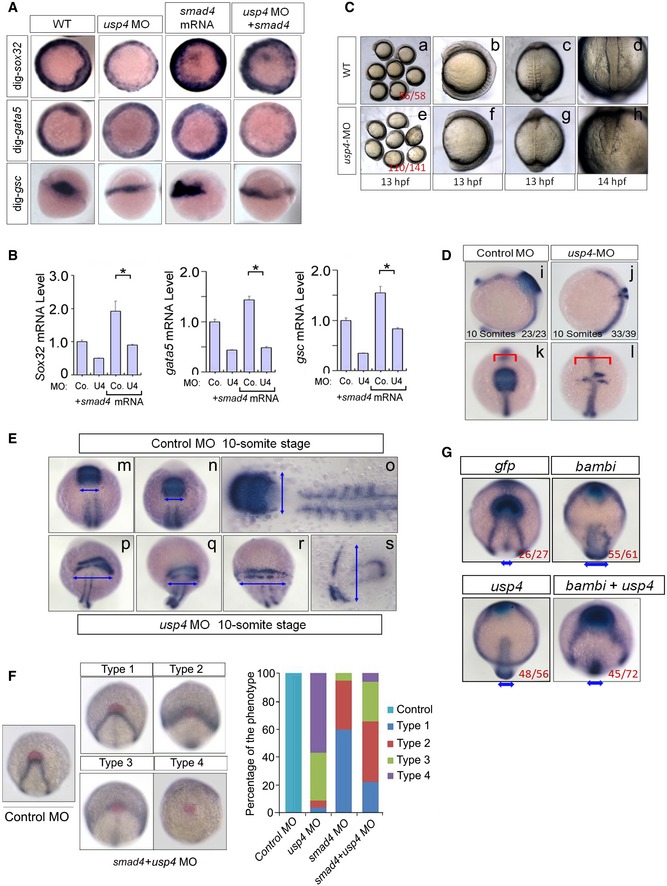
The zebrafish genome encodes more than 90 putative DUBs, and USP4 has been identified as one of the 57 DUB genes that are required for zebrafish early development (Tse et al, 2009). To investigate the significance of the interplay between USP4‐SMAD4 in vivo, we analyzed the development of zebrafish embryos with altered expression levels of SMAD4 and USP4 that are required for zebrafish early development (Tse et al, 2009). Injection of smad4 mRNA moderately upregulated the mesodermal and endodermal markers, and this effect was reversed by the depletion of usp4 (Figs 7A and B, and EV5A). This indicated that USP4 was required for SMAD4‐mediated transcription in vivo.
Figure 7. USP4 controls activin/BMP signaling and affects cell movements during Zebrafish embryonic development.
-
AThe depletion of usp4 moderately inhibits the smad4‐induced phenotype. Single‐cell embryos were injected with control morpholino (MO), usp4 MO, smad4 mRNA or their combination. Embryos at the shield stage or 70% epiboly stage were fixed and hybridized with probes for endoderm and mesoderm markers including sox32, gata5, and goosecoid (gsc). The groups of embryos in each panel are oriented differently: animal pole view for sox32 and gata5; organizer view for the gsc panel. See Fig EV5A for overview on the zebrafish embryos.
-
BSingle‐cell embryos were injected by control MO, usp4 MO, smad4 mRNA, or their combination. The relative expression levels of zebrafish sox32, gata5, and goosecoid (gsc) monitored by real‐time PCR (qPCR). The quantified mRNA levels were normalized to β‐actin and presented relative to the control embryos. The significance levels are indicated by *P < 0.05. Experiments were performed on at least three independent injected embryos. Two‐tailed, unpaired t‐test. The data are presented as means ± SD.
-
C–EEffects of usp4 knockdown on embryonic cell movements. The injection of usp4 morpholino led to impaired CE movement. Single‐cell embryos were injected with usp4 MO or control MO at the indicated doses. The embryos in panel C‐(c) and panel C‐(g) are shown as dorsal views with animal pole to the top. Embryos in panel C‐(d) and panel C‐(h) are shown as animal pole views. The somite stage is indicated in each subfigure. (D) Zebrafish usp4 morphants demonstrated defects in cell movement and differentiation of cell fate. The embryos were fixed at the ten‐somite stage and simultaneously hybridized to probes for the hind brain marker krox20 and middle line marker shha. The red line shows the krox20 expression domain. The embryo in panels i and j is shown in the lateral view, and embryo in panels k and l is shown in the animal pole view. (E) Zebrafish usp4 morphants demonstrated defects in cell movement and differentiation of cell fate. The embryos were fixed at the ten‐somite stage and simultaneously hybridized to probes for the hind brain marker krox20 and the somites marker myod. The double arrowhead shows the krox20‐expressing domain. Panels m, n, o refer to control MO, 10‐somite stage; panels p, q, r, and s refer to usp4 MO1, 10‐somite stage. Panels o and s display the flattened images of the control embryo and the usp4 morphant, respectively.
-
FThe rescue effects of ectopic smad4 mRNA expression in the usp4 morphants. The left panels show four distinct types of expression patterns indicated by the hybridization to the hgg1 (red), dlx3 (black), and ntl (black) probes. The right panel is a bar graph that shows the ratios of various embryonic patterns following injection with usp4 MO, smad4 mRNA, and their combination.
-
GZebrafish bambi mRNA injection rescues the CE defect that was observed in the usp4 morphants. After bambi or usp4 mRNA injection, the embryos were fixed at the two‐somite stage and simultaneously hybridized to hgg1 (blue), dlx3 (black), and ntl (black) probes. Each embryo is shown in an anterodorsal views to demonstrate the relative positions of the hgg1 and dlx3 expression domains. In the bottom of each panel, the double arrow heads show the width of the ntl expression domain.
Figure EV5. Effect of usp4 morphants on embryonic cell movements.
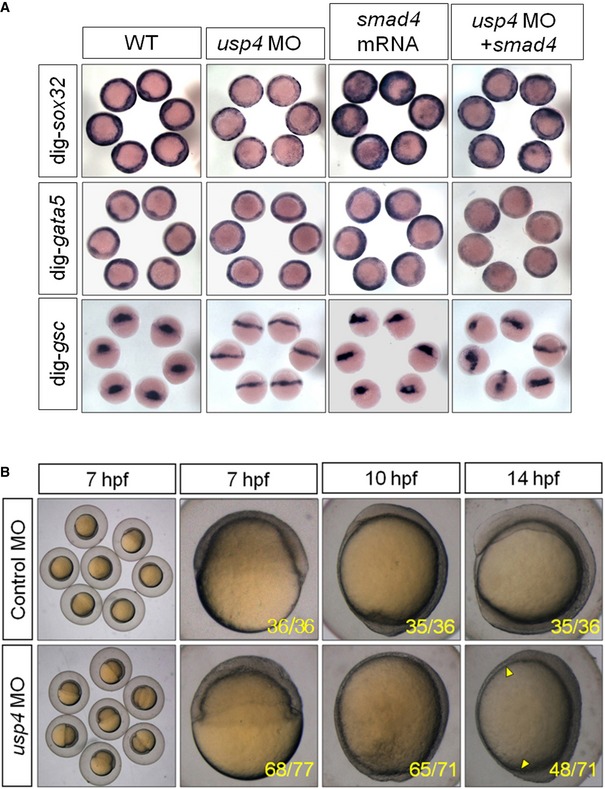
- Related to Fig 7A, overview of zebrafish embryos. The usp4 depletion partially inhibits smad4‐induced phenotype. Single‐cell embryos were injected with control morpholino (MO), usp4 MO, smad4 mRNA, or their combination (as indicated).
- Injection of usp4 morpholino led to impaired epiboly and convergent extension (CE) movement. Single‐cell embryos were injected with usp4 MO or control morpholino (control MO). The stages of the embryos are shown, and the number of described embryos and total number are listed in the bottom.
Compared to wild‐type or control MO (cMO)‐injected embryos, embryos injected with usp4 MO exhibited defective migration and slower epiboly (Fig EV5B). Whereas wild‐type or control MO‐injected embryos developed to the bud stage [10 h post‐fertilization (hpf)], the usp4 morphants developed to ~90% epiboly stage. At the nine‐somite stage (~13 hpf), embryos injected with 4 ng of usp4 MO had a wider embryonic axis and incomplete somites (Figs 7D and EV5), which suggests that the usp4 depletion in zebrafish embryos could impair the proper formation of convergent extension (CE).
To validate the effect of ectopic usp4 expression on CE, we examined the expression of a set of marker genes that have been used to indicate CE movement during gastrulation (Topczewski et al, 2001; Marlow et al, 2002). In wild‐type embryos at the two‐somite stage, the neural plate (the boundaries of which are marked by dlx3 expression), becomes narrow, and the polster (which is derived from the anterior hypoblast cells and expresses hgg1 (ctslb—Zebrafish Information Network)), is positioned in front of the anterior edge of the neural plate. In contrast, embryos injected with usp4 mRNA demonstrated a much broader neural plate, at the same stage, and the polster was located within or fell behind the anterior edge of the neural plate (Figs 7E and F, and EV5). Furthermore, ectopic expression of smad4 in zebrafish induced slower movement of the neural plate and essentially rescued the serious CE defects observed in the usp4 morphants (Fig 7F).
Taken together, these observations indicated that USP4 functions upstream of SMAD4 that is a shared mediator of activin and BMP signaling, to control cell movements and the early development of zebrafish embryos. BAMBI is a pseudo‐type I receptor that antagonizes both activin and BMP signaling (Onichtchouk et al, 1999). It was apparent that the injection of bambi mRNA resulted in a separate, but wider, ntl expression domain, or unmerged stripes. The co‐injection of both usp4 or bambi mRNA rescued their respective CE defects at the same stage (Fig 7G) and lends support to the hypothesis that USP4 and BAMBI have opposing effects on SMAD4 signaling during zebrafish embryogenesis.
Discussion
USP4 is a critical positive regulator of TGF‐β family signaling
We have identified USP4 as an essential determinant of the intensity and the duration of both activin and BMP signaling. Our data reveal that USP4 controls the output of activin/BMP‐induced signaling downstream of the receptor‐activated R‐SMAD C‐terminal phosphorylation: USP4 depletion reduced activin‐ and BMP‐induced SMAD complex formation and target gene expression, whereas ectopic expression of USP4 increased these biological responses. Thus, USP4 amplifies SMAD signaling and the resulting biological effects of SMAD signaling. Furthermore, the activin and BMP‐induced responses in mESCs and zebrafish development also depended critically on USP4. We found that in the activin/BMP pathways, USP4 specifically targeted monoubiquitinated SMAD4 for de‐ubiquitination and transcriptional activation.
USP4 was previously shown to deubiquitinate the TGF‐β type I receptor (Zhang et al, 2012). It may thus activate TGF‐β signaling by both stabilizing the TGF‐β type I receptor and by inhibiting SMAD4 monoubiquitination (and thereby promoting its association with target promoters). We were unable to show that USP9X affects SMAD4 monoubiquitination or activin/SMAD signaling as previously reported (Dupont et al, 2009). The two proteins do not appear to have a redundant role; the inhibitory effect on activin signaling by the shRNA‐mediated knockdown of USP4 (through targeting the 3′ untranslated sequence) was rescued by ectopic expression of USP4 but not by USP9X (Fig EV1C). A negative result does not exclude a role for USP9X in removing monoubiquitin from SMAD4. It may indicate that the cellular context is important for the effects of USP9X on the deubiquitination of SMAD4. Notably, in a recent study, the phosphorylation of SMAD4 Thr277 induced by free fatty acids was found to promote USP9X‐SMAD4 interaction in breast cancer cells (Wu et al, 2017).
Monoubiquitinated SMAD4 is a substrate of USP4
The present study provided several lines of evidence to support the conclusion that monoubiquitinated SMAD4 is a specific substrate of USP4. USP4 interacted with SMAD4 and was preferentially associated with monoubiquitinated SMAD4. In vitro, purified monoubiquitinated SMAD4 interacted with and was rapidly cleaved by active USP4. USP4 efficiently removed monoubiquitin from endogenous SMAD4 monoubiquitination, enhanced the binding of SMAD4 to its consensus DNA binding site and increased SMAD complex formation, which resulted in SMAD signal activation. In addition, monoubiquitinated SMAD4 accumulated in USP4‐depleted or ‐deficient cells, and correspondingly suppressed the expression of SMAD4 target genes. R‐SMAD and USP4 were found to compete for a common SMAD4 binding domain. In this view, upon ligand stimulation, R‐SMAD may compete with USP4 for binding to SMAD4, and allow SMAD signaling to initiate but at the same time expose SMAD4 for monoubiquitination. The levels of SMAD4 driven gene expression would then be determined by a balance between SMAD4 binding to DNA and SMAD4 monoubiquitination.
We did detect polyubiquitinated forms of SMAD4 when it was ectopically expressed, and these forms could be removed by active USP4. However, we did not detect any change in the stability of the endogenous SMAD4 and only monoubiquitinated SMAD4 was clearly altered by changes in the levels of USP4. These findings suggest that in vivo, USP4 only acts on the pool of the monoubiquitinated SMAD4.
Dynamic control of SMAD4 function by SMURF2
SMAD4 monoubiquitination is clearly induced upon signal activation via R‐SMAD‐mediated recruitment of SMURF2. At the same time, monoubiquitinated SMAD4 antagonizes SMAD complex formation and removes the SMAD complex from DNA. This suggests that SMAD4 monoubiquitination by SMURF2 controls the duration of signaling. Moreover, the USP4‐mediated reversal of this modification is required to initiate a new round of SMAD4‐dependent transcription and/or efficient re‐use of SMAD4. Apparently, in TGF‐β/SMAD signaling, this balanced, dynamic recycling of SMAD4 requires rapid degradation of the negative regulator c‐SKI, which we found was achieved through SMAD2–SMURF2‐induced c‐SKI polyubiquitination. This mechanism is reminiscent of the RNF111‐induced degradation of SKI/SKIL which also depended on an interaction with activated SMAD2/3 (Levy et al, 2007; Le Scolan et al, 2008). Taken together, our results suggest that SMURF2 has a dual function in TGF‐β family/SMAD: It promotes signal initiation by inducing instability of c‐SKI (which suppresses SMAD signaling and maintains the repression of TGF‐β family target genes), and it terminates signal transduction by mediating inhibitory SMAD4 monoubiquitination.
Previously, TRIM33/Ectodermin was identified as a SMAD4 ubiquitin ligase (Dupont et al, 2005). Our inability to detect a role for TRIM33/Ectodermin in SMAD ubiquitination might be due to our different experimental settings. Our initial screen to identify the E3 ligase mediating SMAD4 monoubiquitination was performed in the absence of ligand stimulation, and under these conditions, TRIM33/Ecodermin was ineffective as a monoubiquitin E3 ligase. TGF‐β/SMAD2 activation has been shown to potentiate the ability of TRIM33/Ectodermin to induce SMAD4 monoubiquitination (Dupont et al, 2009). The absence of ligand addition and cotransfection with SMAD2 in our experimental setup may have been the reason why we did not detect the SMAD4 monoubiquitination activity of TRIM33/Ectodermin. TRIM33/Ectodermin and SMURF2 belong to structurally different classes of E3 ligases (RING versus HECT domain containing proteins, respectively). Moreover, TRIM33/Ectodermin has a bromodomain and contact of this domain with histones is needed for its E3 ubiquitin ligase activity (Agricola et al, 2011). Therefore, TRIM33 and SMURF2 are likely to act in different subcellular compartments and be subject to distinct regulatory mechanisms. Of note, the function of TRIM33/Ectodermin in the ubiquitination of SMAD4 appears not to be conserved in Drosophila. Instead of Bonus, which is the closest Drosophila homologue to TRIM33, a SMURF2‐like HECT domain E3 ligase (NEDDL) has been suggested to mediate the ubiquitination of the Drosophila homologue of SMAD4, that is, MEDEA (Wisotzkey et al, 2014).
Function of USP4 in mESCs and zebrafish embryos
Both activin and BMP signaling control mESC self‐renewal and cell fate determination. Comparison between wild‐type and USP4‐depleted mESCs revealed a prominent role for USP4 in controlling activin‐ and BMP‐mediated responses. Monoubiquitinated SMAD4 accumulated in USP4‐depleted stem cells resulting in reduced stem cell proliferation and inhibition of the activin‐induced formation of the anterior mesoderm and definitive endoderm, as well as BMP‐mediated repression of neural induction. Morpholino silencing of zebrafish usp4 reduced SMAD4‐mediated activin/BMP signaling, which suggested that the USP4‐mediated release of the inhibitory effect of SMAD4 monoubiquitination is evolutionally conserved. In addition, zebrafish embryos that were depleted of usp4 exhibited defective cell migration and slower epiboly, which suggested that CE was impaired during gastrulation. Importantly, this CE defect could be rescued by ectopic expression of SMAD4. Moreover, the similar gastrulation defect that was induced by ectopic expression of BAMBI, a BMP and activin inhibitor, was partly rescued by co‐expression of USP4. Thus, USP4 sustained the activin/BMP signal in vivo and is a critical determinant of SMAD4 activity.
In conclusion, our findings have demonstrated that activin/BMP‐dependent monoubiquitination of SMAD4 is meditated by the relative activities of the E3 ubiquitin ligase SMURF2 and the deubiquitinase USP4, which determine both the duration and the intensity of the activated signaling pathway.
Materials and Methods
Cell culture
MEFs from wild‐type and USP4−/− mice were kindly provided by Xiongbin Lu (University of Texas MD Anderson Cancer Center, Houston, TX, USA) (Zhang et al, 2011). MEFs from wild‐type and SMURF2−/− mice were kindly provided by Ying E. Zhang (National Cancer Institute, Bethesda, MD, USA) (Tang et al, 2011). HEK293T cells, HeLa cervical cancer cells, MDA‐MB‐231 breast cancer cells, and MEFs were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS; Hyclone) and 100 U/ml penicillin/streptomycin. mESCs were cultured on gelatin‐coated dishes in DMEM supplemented with 10% FBS, 1 mM sodium pyruvate (Invitrogen), 1% non‐essential amino acids (Invitrogen), 2 mM l‐glutamine (Invitrogen), 0.1 mM β‐mercaptoethanol (Sigma), and 1,000 U/ml LIF (Millipore).
cDNA screen
HEK293T cells that had been stably transfected with SMAD2‐SMAD4‐dependent ARE‐Luc transcriptional reporter were used as a readout system to identify regulators of activin signaling. We first stimulated the cells with a suboptimal dose (5 ng/ml activin) that produced ~30% of the maximum stimulation level (50 ng/ml activin) to permit identification of both activators and inhibitors. Seventy‐four human DUB cDNAs were re‐screened in triplicate. Similar expression of each DUB was confirmed by Western blotting analysis.
Protein purification
GST–USP4 wt and mutant expression constructs were generated by subcloning into pGEX‐4T1 vectors. GST–USP4 plasmids were used to transform the Escherichia coli strain BL21 or Rosetta, respectively. The cultures were grown overnight at 37°C. The next day, the cultures were diluted 1:50 in fresh LB medium and grown at 37°C to an optical density of 0.6 at 600 nm. The cells were then induced overnight at 24°C in the presence of 0.5 mM isopropyl‐β‐D‐thiogalactopyranoside (IPTG), 20 mM HEPES at pH 7.5, 1 mM MgCl2, and 0.05% glucose. For GST–USP4 purification, the washed pellets were resuspended in lysis buffer (PBS, 0.5 M NaCl, complete protease inhibitors (Roche), 1 mM phenylmethyl sulphonyl fluoride, and 1% Triton X‐100). After sonication and a freeze–thaw step, the supernatants of the cell lysates were incubated with glutathione‐Sepharose (GE Healthcare). The beads were washed twice with lysis buffer and three times with 50 mM Tris–HCl (pH 7.5) and 0.5 M NaCl. Purified proteins were eluted in 50 mM Tris–HCl (pH 7.5), 0.5 M NaCl and 20 mM glutathione. For the purification of USP4 protein, the GST was removed biotin‐tagged thrombin. For the purification of SMAD2 protein from mammalian cells, a Flag‐SMAD2 expression plasmid was transfected in HEK293T cells and SMAD2 protein was immunoprecipitated overnight from the cell lysate with α‐Flag‐M2 resin (Sigma), followed by elution with Flag peptide (Sigma, 1 mg/ml in 50 mM HEPES (pH 7.5), 100 mM NaCl, 0.1% NP40, 5% glycerol).
Ubiquitination assay
The cells were washed with PBS and lysed in two pellet volumes of RIPA buffer (20 mM NAP pH 7.4, 150 mM NaCl, 1% Triton, 0.5% sodium‐deoxycholate, and 1% SDS) supplemented with protease inhibitors and 10 mM N‐ethylmaleimide (NEM). Lysates were sonicated, boiled at 95°C for 5 min, and diluted with RIPA buffer containing 0.1% SDS, then centrifuged at 4°C (16 × 103 g for 15 min). The supernatant was incubated with the specific antibody and protein A‐sepharose for 3 h at 4°C. After extensive washing, the bound proteins were eluted with 2× SDS sample buffer and separated by SDS–PAGE, which was followed by immunoblotting analysis.
Chromatin immunoprecipitation
ChIP was performed as previously described (Zhang et al, 2012). Detailed information is provided in the Appendix.
Transcription reporter assay
Transcriptional luciferase reporter assays were performed as described before (Zhou et al, 2014). Detailed information is provided in the Appendix Supplementary Methods. For the ARE‐luc reporter, FAST2 was co‐transfected to enable SMAD2 transcriptional response (Labbé et al, 1998).
In vitro de‐ubiquitination of SMAD4
Recombinant USP4 used in Fig 2E–H and J was purified as described in (Clerici et al, 2014). Ub‐AMC assay was performed as described (Clerici et al, 2014). For the assay shown in Fig 2G and H, HA‐ubiquitinated Flag‐SMAD4 substrates were prepared by transiently co‐transfecting HEK293T cells with Flag‐SMAD4 and HA‐ubiquitin. Thirty‐six hours later, the cells were harvested and SMAD4 was purified on Flag beads and eluted with Flag peptide (Fig 2F). Pure monoubiquitinated SMAD4 was obtained subjecting the Flag eluate to HA purification. For the assay shown in Fig 2D, HA‐ubiquitinated Flag‐SMAD4 substrates were prepared by transiently co‐transfecting HEK293T cells with Flag‐SMAD4 and HA‐ubiquitin. Thirty‐six hours later, the cells were then treated with MG132 (5 μM) for 4 h, and the HA‐ubiquitinated Flag‐SMAD4 was purified using denaturing conditions and then incubated with purified USP4 protein as previously described (Zhang et al, 2012). The reactions were quenched with SDS sample buffer followed by heat denaturation (2 min at 95°C). The reaction products were detected using Western blotting analysis.
Zebrafish embryo assays
Zebrafish (Danio rerio), AB strain, were maintained at 28.5°C under a 14:10 light:dark cycle. Fish staging and embryo production were conducted as previously described (Cao et al, 2004; Zhang et al, 2004). The embryo stages are expressed in hours post‐fertilization (hpf) at standard temperature. Detailed information is provided in the Appendix. The depletion of usp4 was performed by morpholinos that selectively inhibit splicing (MO2) or translation (MO1). The MO usp4‐induced morphological changes were rescued (in part) by the ectopic usp4 mRNA expression (Appendix Fig S3). Zebrafish embryos injected with p53 MO at 4 ng do not show a phenotype and were included as negative control.
For additional information regarding materials and methods, see the Appendix Supplementary Methods.
Statistical analysis
All Q‐PCR and transcriptional reporter experiments have been repeated at least three times, and representative experiments are shown. Western blot, immunoprecipitation, and ubiquitination assays were performed at least twice. The statistical analyses were performed using a two‐tailed, unpaired t‐test. P < 0.05 was considered statistically significant. The details are provided in the Appendix.
Author contributions
FZ, FX, LZ, KJ, ZZ, and MvD performed biochemical and cell biological experiments. RG and HH designed, performed, and interpreted the zebrafish experiments. MC performed ubiquitination studies with purified components, and TKS supervised these experiments. LZ and PtD supervised, coordinated, and drafted the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Appendix and Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank David Baker for valuable comments, Midory Thorikay for technical assistance and all members of our laboratories for stimulating discussions. We are grateful to Martijn Rabelink for the shRNA constructs. We thank Marene Landström, Xiongbin Lu, Ken Iwata, Stefano Piccolo, Jeffrey L. Wrana, Ying Zhang, and Hongrui Wang for reagents. This work was supported by the Special program from Ministry of Science and Technology of China (Grant No. 2016YFA0502500), National Natural Science Foundation of China (Grant No. 31471315, 31671457, 31571460, K124924615), Zhejiang Provincial Natural Science Foundation of China (Grant No. R14C070002), Jiangsu Provincial Natural Science Foundation of China (Grant No. BK20150354); Natural Science Foundation Project of Chongqing (cstc2012jjjq10001); Cancer Genomics Centre Netherlands.
The EMBO Journal (2017) 36: 1623–1639
Contributor Information
Long Zhang, Email: l_zhang@zju.edu.cn.
Peter ten Dijke, Email: p.ten_dijke@lumc.nl.
References
- Agricola E, Randall RA, Gaarenstroom T, Dupont S, Hill CS (2011) Recruitment of TIF1γ to chromatin via its PHD finger‐bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol Cell 43: 85–96 [DOI] [PubMed] [Google Scholar]
- Akiyoshi S, Inoue H, Hanai J, Kusanagi K, Nemoto N, Miyazono K, Kawabata M (1999) c‐Ski acts as a transcriptional co‐repressor in transforming growth factor‐β signaling through interaction with Smads. J Biol Chem 274: 35269–35277 [DOI] [PubMed] [Google Scholar]
- Al‐Salihi MA, Herhaus L, Macartney T, Sapkota GP (2012) USP11 augments TGFβ signalling by deubiquitylating ALK5. Open Biol 2: 120063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo K, Wrana JL (2001) TGF‐β induces assembly of a Smad2‐Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol 3: 587–595 [DOI] [PubMed] [Google Scholar]
- Cao Y, Zhao J, Sun Z, Zhao Z, Postlethwait J, Meng A (2004) fgf17b, a novel member of Fgf family, helps patterning zebrafish embryos. Dev Biol 271: 130–143 [DOI] [PubMed] [Google Scholar]
- Clerici M, Luna‐Vargas MP, Faesen AC, Sixma TK (2014) The DUSP‐Ubl domain of USP4 enhances its catalytic efficiency by promoting ubiquitin exchange. Nat Commun 5: 5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boeck M, ten Dijke P (2012) Key role for ubiquitin protein modification in TGFβ signal transduction. Ups J Med Sci 117: 153–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke P, Hill CS (2004) New insights into TGF‐β‐Smad signalling. Trends Biochem Sci 29: 265–273 [DOI] [PubMed] [Google Scholar]
- Dupont S, Zacchigna L, Cordenonsi M, Soligo S, Adorno M, Rugge M, Piccolo S (2005) Germ‐layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 121: 87–99 [DOI] [PubMed] [Google Scholar]
- Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L, Inui M, Moro S, Modena N, Argenton F, Newfeld SJ, Piccolo S (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 136: 123–135 [DOI] [PubMed] [Google Scholar]
- Eichhorn PJ, Rodon L, Gonzalez‐Junca A, Dirac A, Gili M, Martinez‐Saez E, Aura C, Barba I, Peg V, Prat A, Cuartas I, Jimenez J, Garcia‐Dorado D, Sahuquillo J, Bernards R, Baselga J, Seoane J (2012) USP15 stabilizes TGF‐β receptor I and promotes oncogenesis through the activation of TGF‐β signaling in glioblastoma. Nat Med 18: 429–435 [DOI] [PubMed] [Google Scholar]
- He W, Dorn DC, Erdjument‐Bromage H, Tempst P, Moore MA, Massague J (2006) Hematopoiesis controlled by distinct TIF1γ and Smad4 branches of the TGFβ pathway. Cell 125: 929–941 [DOI] [PubMed] [Google Scholar]
- Ikushima H, Miyazono K (2010) TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer 10: 415–424 [DOI] [PubMed] [Google Scholar]
- Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S, Enzo E, Moro S, Polo S, Dupont S, Cordenonsi M, Piccolo S (2011) USP15 is a deubiquitylating enzyme for receptor‐activated SMADs. Nat Cell Biol 13: 1368–1375 [DOI] [PubMed] [Google Scholar]
- Kang JS, Liu C, Derynck R (2009) New regulatory mechanisms of TGF‐β receptor function. Trends Cell Biol 19: 385–394 [DOI] [PubMed] [Google Scholar]
- Labbé E, Silvestri C, Hoodless PA, Wrana JL, Attisano L (1998) Smad2 and Smad3 positively and negatively regulate TGFβ‐dependent transcription through the forkhead DNA‐binding protein FAST2. Mol Cell 2: 109–120 [DOI] [PubMed] [Google Scholar]
- Le Scolan E, Zhu Q, Wang L, Bandyopadhyay A, Javelaud D, Mauviel A, Sun L, Luo K (2008) Transforming growth factor‐β suppresses the ability of Ski to inhibit tumor metastasis by inducing its degradation. Cancer Res 68: 3277–3285 [DOI] [PubMed] [Google Scholar]
- Levy L, Howell M, Das D, Harkin S, Episkopou V, Hill CS (2007) Arkadia activates Smad3/Smad4‐dependent transcription by triggering signal‐induced SnoN degradation. Mol Cell Biol 27: 6068–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonn P, Moren A, Raja E, Dahl M, Moustakas A (2009) Regulating the stability of TGFβ receptors and Smads. Cell Res 19: 21–35 [DOI] [PubMed] [Google Scholar]
- Luo K, Stroschein SL, Wang W, Chen D, Martens E, Zhou S, Zhou Q (1999) The Ski oncoprotein interacts with the Smad proteins to repress TGFβ signaling. Genes Dev 13: 2196–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow F, Topczewski J, Sepich D, Solnica‐Krezel L (2002) Zebrafish Rho kinase 2 acts downstream of Wnt11 to mediate cell polarity and effective convergence and extension movements. Curr Biol 12: 876–884 [DOI] [PubMed] [Google Scholar]
- Massagué J (2008) TGFβ in cancer. Cell 134: 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morén A, Imamura T, Miyazono K, Heldin CH, Moustakas A (2005) Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J Biol Chem 280: 22115–22123 [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH (2009) The regulation of TGFβ signal transduction. Development 136: 3699–3714 [DOI] [PubMed] [Google Scholar]
- Ogawa K, Saito A, Matsui H, Suzuki H, Ohtsuka S, Shimosato D, Morishita Y, Watabe T, Niwa H, Miyazono K (2007) Activin‐Nodal signaling is involved in propagation of mouse embryonic stem cells. J Cell Sci 120: 55–65 [DOI] [PubMed] [Google Scholar]
- Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massagué J, Niehrs C (1999) Silencing of TGF‐β signalling by the pseudoreceptor BAMBI. Nature 401: 480–485 [DOI] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138: 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Liu X, Ng‐Eaton E, Lodish HF, Weinberg RA (1999) SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor β signaling. Proc Natl Acad Sci USA 96: 12442–12447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Yagi K, Kondo M, Kato M, Miyazono K, Miyazawa K (2004) c‐Ski inhibits the TGF‐β signaling pathway through stabilization of inactive Smad complexes on Smad‐binding elements. Oncogene 23: 5068–5076 [DOI] [PubMed] [Google Scholar]
- Tabata T, Kokura K, ten Dijke P, Ishii S (2009) Ski co‐repressor complexes maintain the basal repressed state of the TGF‐β target gene, SMAD7, via HDAC3 and PRMT5. Genes Cells 14: 17–28 [DOI] [PubMed] [Google Scholar]
- Tang LY, Yamashita M, Coussens NP, Tang Y, Wang X, Li C, Deng CX, Cheng SY, Zhang YE (2011) Ablation of Smurf2 reveals an inhibition in TGF‐β signalling through multiple mono‐ubiquitination of Smad3. EMBO J 30: 4777–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topczewski J, Sepich DS, Myers DC, Walker C, Amores A, Lele Z, Hammerschmidt M, Postlethwait J, Solnica‐Krezel L (2001) The zebrafish glypican knypek controls cell polarity during gastrulation movements of convergent extension. Dev Cell 1: 251–264 [DOI] [PubMed] [Google Scholar]
- Tse WK, Eisenhaber B, Ho SH, Ng Q, Eisenhaber F, Jiang YJ (2009) Genome‐wide loss‐of‐function analysis of deubiquitylating enzymes for zebrafish development. BMC Genom 10: 637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Suzuki H, Kato M (2008) Roles of mono‐ubiquitinated Smad4 in the formation of Smad transcriptional complexes. Biochem Biophys Res Commun 376: 288–292 [DOI] [PubMed] [Google Scholar]
- Wisotzkey RG, Quijano JC, Stinchfield MJ, Newfeld SJ (2014) New gene evolution in the bonus‐TIF1‐γ/TRIM33 family impacted the architecture of the vertebrate dorsal‐ventral patterning network. Mol Biol Evol 31: 2309–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Krawitz AR, Chai J, Li W, Zhang F, Luo K, Shi Y (2002) Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski‐mediated repression of TGF‐β signaling. Cell 111: 357–367 [DOI] [PubMed] [Google Scholar]
- Wu Y, Yu X, Yi X, Wu K, Dwabe S, Atefi M, Elshimali Y, Kemp KT II, Bhat K, Haro J, Sarkissyan M, Vadgama JV (2017) Aberrant phosphorylation of SMAD4 Thr277‐mediated USP9x‐SMAD4 interaction by free fatty acids promotes breast cancer metastasis. Cancer Res 77: 1383–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow‐Tsang LF, Liu JX, Kim H, Barlas A, Manova‐Todorova K, Kaartinen V, Studer L, Mark W, Patel DJ, Massagué J (2011) A poised chromatin platform for TGF‐β access to master regulators. Cell 147: 1511–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Chambers I, Smith A (2003a) BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self‐renewal in collaboration with STAT3. Cell 115: 281–292 [DOI] [PubMed] [Google Scholar]
- Ying QL, Stavridis M, Griffiths D, Li M, Smith A (2003b) Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol 21: 183–186 [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhou H, Su Y, Sun Z, Zhang H, Zhang L, Zhang Y, Ning Y, Chen YG, Meng A (2004) Zebrafish Dpr2 inhibits mesoderm induction by promoting degradation of nodal receptors. Science 306: 114–117 [DOI] [PubMed] [Google Scholar]
- Zhang X, Berger FG, Yang J, Lu X (2011) USP4 inhibits p53 through deubiquitinating and stabilizing ARF‐BP1. EMBO J 30: 2177–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhou F, Drabsch Y, Gao R, Snaar‐Jagalska BE, Mickanin C, Huang H, Sheppard KA, Porter JA, Lu CX, ten Dijke P (2012) USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF‐β type I receptor. Nat Cell Biol 14: 717–726 [DOI] [PubMed] [Google Scholar]
- Zhou F, Drabsch Y, Dekker TJ, de Vinuesa AG, Li Y, Hawinkels LJ, Sheppard KA, Goumans MJ, Luwor RB, de Vries CJ, Mesker WE, Tollenaar RA, Devilee P, Lu CX, Zhu H, Zhang L, ten Dijke P (2014) Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF‐β signalling. Nat Commun 5: 3388 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Appendix and Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6