

Abstract
Mitochondrial fusion and fission events, collectively known as mitochondrial dynamics, act as quality control mechanisms to ensure mitochondrial function and fine‐tune cellular bioenergetics. Defective mitofusin 2 (Mfn2) expression and enhanced mitochondrial fission in skeletal muscle are hallmarks of insulin‐resistant states. Interestingly, Mfn2 is highly expressed in brown adipose tissue (BAT), yet its role remains unexplored. Using adipose‐specific Mfn2 knockout (Mfn2‐adKO) mice, we demonstrate that Mfn2, but not Mfn1, deficiency in BAT leads to a profound BAT dysfunction, associated with impaired respiratory capacity and a blunted response to adrenergic stimuli. Importantly, Mfn2 directly interacts with perilipin 1, facilitating the interaction between the mitochondria and the lipid droplet in response to adrenergic stimulation. Surprisingly, Mfn2‐adKO mice were protected from high‐fat diet‐induced insulin resistance and hepatic steatosis. Altogether, these results demonstrate that Mfn2 is a mediator of mitochondria to lipid droplet interactions, influencing lipolytic processes and whole‐body energy homeostasis.
Keywords: brown adipose tissue, insulin resistance, lipid droplet, mitochondrial dynamics, mitofusin 2
Subject Categories: Membrane & Intracellular Transport, Metabolism
Introduction
Mitochondria occupy a central stage in cellular energy metabolism. Besides energy production, they play a major role in many cellular functions including apoptosis, cell proliferation, steroidogenesis, cellular signaling, and lipid metabolism. Mitochondria are dynamic and undergo architectural rearrangements through continuous fusion and fission cycles depending on environmental cues such as the nutritional status (for review, Zorzano et al, 2009). In mammals, mitochondrial dynamics are controlled by a few GTPase enzymes; namely, mitofusins 1 and 2 (Mfn1 and Mfn2) control outer mitochondrial membrane fusion, while optic atrophy 1 (OPA1) governs inner mitochondrial membrane fusion. Conversely, mitochondrial fission is controlled mainly by the dynamin‐related protein‐1 (Drp1) (Zorzano et al, 2009).
Mitochondrial dynamics are altered in insulin‐resistant states in humans and rodents. Indeed, in skeletal muscle from obese and type 2 diabetic (T2D) patients the mitochondrial network is characterized by increased fission levels and reduced mitochondrial size (Kelley et al, 2002; Bach et al, 2003; Toledo et al, 2006), which is linked to a decrease in Mfn2 expression (Bach et al, 2003, 2005; Hernandez‐Alvarez et al, 2010). In mice, the whole‐body Mfn2 deficiency is lethal at mid‐gestation due to impairment of placental function (Chen et al, 2003). However, transgenic mice with tissue‐specific ablations of Mfn2 have unveiled the multiple metabolic roles of Mfn2 (Sebastian et al, 2012; Schneeberger et al, 2013). The liver‐specific deletion of Mfn2 led to systemic metabolic abnormalities, such glucose intolerance and enhanced hepatic gluconeogenesis (Sebastian et al, 2012). In brain, POMC neuron‐specific deletion of Mfn2 induced hyperphagia and obesity while reducing energy expenditure (Schneeberger et al, 2013). Many of these effects have been attributed to the ability of Mfn2 to mediate not only mitochondria–mitochondria contacts, but also to influence the interaction of mitochondria with other cellular membrane organelles. For example, Mfn2 has a critical role in the maintenance of mitochondria–endoplasmic reticulum (ER) interactions (de Brito & Scorrano, 2008; Cosson et al, 2012; Filadi et al, 2015), and ablation of Mfn2 triggered ER stress in virtually all models tested (Sebastian et al, 2012; Schneeberger et al, 2013).
Mfn2 expression is ubiquitous in mammalian tissues, albeit it finds its highest expression levels in tissues enriched in mitochondria, such as heart and the brown adipose tissue (BAT) (Bach et al, 2003). Recently, the BAT has gained significant attraction after its identification and localization in humans (for review, see Peirce et al, 2014). Both white adipose tissue (WAT) and BAT are characterized by a high lipid storage capacity in intracellular lipid droplets (LDs), but they differ in their physiological functions. While WAT is the primary site for lipid storage, BAT is specialized for non‐shivering thermogenic functions by increasing energy expenditure through uncoupled respiration (Nedergaard et al, 2007). Activation of the BAT has been shown to enhance energy expenditure and glucose tolerance in mice and humans (Peirce et al, 2014). Upon cold exposure, β3‐adrenergic signaling ignites the lipolysis process in the BAT, where triglycerides from the LDs are hydrolyzed into glycerol and fatty acids (FAs). These FAs then enter the mitochondria for lipid oxidation aimed to feed the electron transport chain and heat production. Decades ago, several studies already documented the close apposition of LDs with mitochondria in the BAT, adipocytes, and other tissues (Cushman, 1970; Blanchette‐Mackie & Scow, 1983). This close connectivity would ensure the efficient transfer of FAs for beta‐oxidation without moving long distance in the cytosol. Interestingly, the juxtaposition of both organelles is regulated, and enhanced upon increased energy demand, such as exercise in skeletal muscle (Tarnopolsky et al, 2007) and cold exposure in the BAT (Yu et al, 2015). However, the molecular mechanisms controlling the interaction between these two organelles remain largely unknown.
In the present study, we aimed to understand the role of Mfn2 in BAT function. For this, we generated adipose‐specific Mfn2 knockout (Mfn2‐adKO) mice. We provide evidence that Mfn2 deletion in BAT dampens thermogenic function by blunting lipolysis and respiratory capacity. This leads to an increased fat accumulation in the BAT of Mfn2‐adKO mice, even on a low‐fat diet (LFD). We further uncover that Mfn2 directly interacts with perilipin 1, facilitating this way mitochondria–LDs contacts and the coupling of triglyceride hydrolysis with FA oxidation (FAO) after adrenergic stimulation.
Results
Mfn2‐adKO mice display thermogenic dysfunction
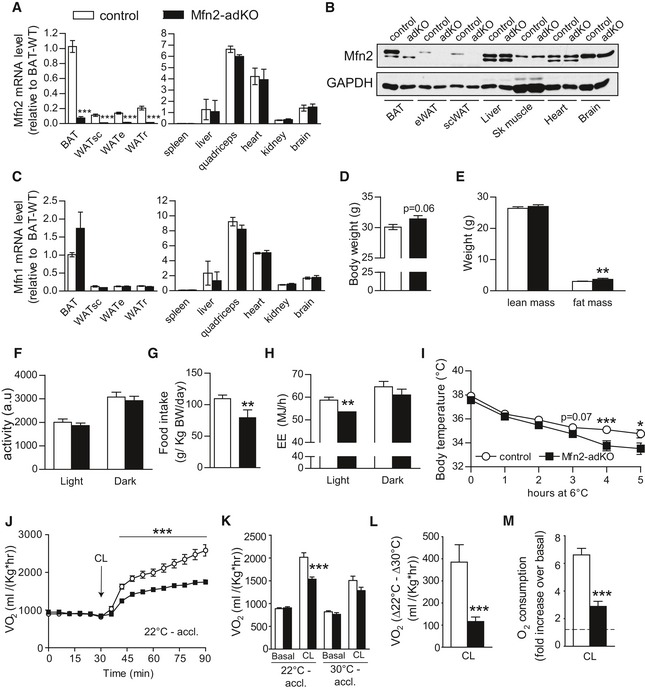
We first generated adipose‐specific Mfn2 knockout mice (Mfn2‐adKO) by crossing Mfn2loxP/loxP mice, backcrossed to a C57BL/6 background, with mice expressing the Cre recombinase under the adiponectin promoter (Eguchi et al, 2011). This led to the specific disruption of Mfn2 mRNA and protein expression in brown and white adipose tissues (BAT and WAT, respectively), but not in any other tissue examined (Fig 1A and B). Importantly, while weakly expressed in all WAT depots tested, BAT was one of the highest Mfn2 expressing tissues among the panel analyzed (Fig 1B). Importantly, no compensation by Mfn1 was observed in Mfn2 defective tissues (Figs 1C and EV1).
Figure 1. Energy homeostasis in Mfn2‐adKO mice.
-
A–CMfn2 gene expression (A) and protein levels (B), as well as Mfn1 gene expression (C), were measured in tissues from 20‐week‐old male control and Mfn2‐adKO mice.
-
D, EThree‐month‐old male control and Mfn2‐adKO mice were used to analyze body weight (D) and composition (E) through EchoMRI.
-
F–HDaily activity (F), food intake (G), and energy expenditure (EE) (H) were measured during indirect calorimetry tests using a comprehensive laboratory animal monitoring system (CLAMS).
-
IThermogenic capacity was evaluated by placing 16‐week‐old male control and Mfn2‐adKO mice at 6°C for 5 h.
-
JNon‐shivering thermogenesis in mice kept at regular housing temperature (˜22°C) was evaluated by measuring baseline and CL (1 mg/kg)‐induced O2 consumption in anesthetized mice at 30°C (n = 8 mice per genotype).
-
K, LImmediately after the experiment in (J), the mice were housed at thermoneutrality (˜30°C) for 4 weeks. Then, non‐shivering thermogenesis was measured as in (J). Total O2 consumption (K) and the difference between CL‐induced increases in O2 consumption at 22°C and 30°C (L) are shown.
-
MBaseline and CL (1 μM)‐induced O2 consumption rates were evaluated in isolated mature adipocytes using an O2K Oxygraph. Then, the relative effect of CL was calculated for each preparation (n = 11 per genotype).
Figure EV1. Brown adipose tissue function is not affected in Mfn1‐adKO mice.
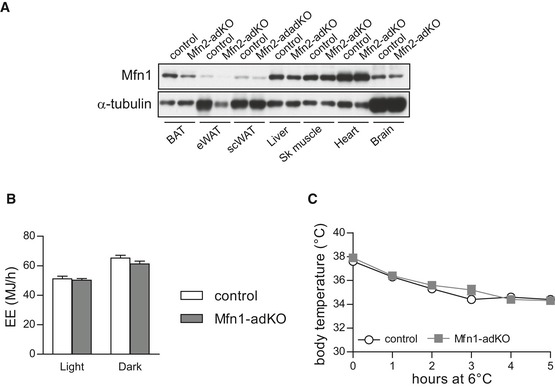
- Mfn1 protein levels were measured in tissues from 20‐week‐old male control and Mfn2‐adKO mice.
- Three‐month‐old control and Mfn1‐adKO mice were fed ad libitum with a low‐fat diet, and energy expenditure (EE) was measured during indirect calorimetry tests using a comprehensive laboratory animal monitoring system (CLAMS).
- A cold resistance test was used to evaluate thermogenic activity in control and Mfn1‐adKO mice.
On a LFD, Mfn2‐adKO mice showed a tendency toward a higher body weight due to a slightly higher fat mass content (Fig 1D and E). Whereas subcutaneous and interscapular WAT depots were slightly bigger in Mfn2‐adKO mice compared to control mice, other fat depots were not affected (data not shown). These differences were not due to changes in daily physical activity, as the activity of Mfn2‐adKO mice did not differ from its control littermates (Fig 1F). Similarly, the higher fat mass could not be explained by enhanced food intake, as, actually, food intake was decreased in Mfn2‐adKO mice compared to control mice (Fig 1G). Most likely, the slight differences in fat mass stem from the marked decreased energy expenditure observed during the light phase in Mfn2‐adKO mice (Fig 1H). Given the role of BAT thermogenesis in whole‐body energy expenditure, we speculated that BAT thermogenic function might be affected by Mfn2 deficiency. To test this hypothesis, we first subjected mice to a 5‐h cold exposure. While no differences in core body temperature were observed at baseline, Mfn2‐adKO mice displayed a sharper decrease in body temperature during the test compared to control mice (Fig 1I). To further examine BAT thermogenic function in a more direct fashion, we next assessed non‐shivering thermogenesis by measuring whole‐body O2 consumption after β3‐adrenergic stimulation in anesthetized mice. While Mfn2‐adKO and control littermates showed similar O2 consumption rates at baseline, the increase in O2 consumption triggered by the specific β3‐adrenergic receptor agonist CL326,243 (CL) was largely blunted in Mfn2‐adKO mice (Fig 1J), further supporting deficient BAT thermogenesis. When a similar experiment was performed in mouse that were not housed at regular housing temperature (~22°C), but acclimated for a month at thermoneutrality (~30°C) in order to blunt BAT activity, there was no difference in O2 consumption either in the basal state or after CL injection between control and Mfn2‐adKO mice (Fig 1K). The differential increase in CL‐stimulated O2 consumption at 22°C and thermoneutrality testifies for adaptive non‐shivering thermogenesis capacity by the BAT (Cannon & Nedergaard, 2011), and was severely impaired in Mfn2‐adKO mice (Fig 1L). Similar conclusions were reached when respirometry experiments were performed ex vivo in isolated mature brown adipocytes, as the response to CL was impaired by more than 60% in brown adipocytes from Mfn2‐adKO mice (Fig 1M). All these tests demonstrate beyond doubt that Mfn2‐adKO mice display a dramatic impairment in adaptive BAT thermogenic function.
To certify to which extent this phenotype was linked to mitochondrial fission, we generated Mfn1‐adKO mice. Mfn1 is an essential protein for mitochondria fusion events. Accordingly, decreased Mfn1 function has been shown to trigger a profound shift in the fusion/fission balance, toward a dramatically fragmented mitochondrial network in most, if not all, cells and tissues tested to date (Chen et al, 2003; Park et al, 2008; Papanicolaou et al, 2012; Dietrich et al, 2013; Kulkarni et al, 2016). Further, While Mfn1 mRNA levels did not compensate for Mfn2 loss, we observed that Mfn1 protein slightly decreased in the BAT and WAT form Mfn2‐adKO mice (Fig EV1A). However, Mfn1‐adKO mice do not display differences in energy expenditure or in response to cold exposure (Fig EV1B and C). This certifies that the alterations observed in Mfn2‐adKO mice are not simply due to enhanced mitochondrial fission and prompted us to further explore the role of Mfn2 in adipose tissues.
Mfn2‐adKO BAT displays impaired lipolytic and mitochondrial functions
The interscapular fat depot from Mfn2‐adKO mice had a whiter appearance compared to control mice, suggesting higher fat accumulation in the BAT of these mice (Fig 2A). H&E stainings revealed an increase of the lipid droplets (LD) size in BAT from Mfn2‐adKO mice (Fig 2B). This led us to explore whether this abnormal lipid was due to alterations in either lipogenic or lipolytic processes. When lipogenic markers were analyzed (Fig 2C), no changes or even some decreases in their mRNA levels were observed, as in the case of the peroxisome proliferator‐activated receptor γ (PPARγ), a key transcriptional regulator of lipogenesis, or the acetyl‐CoA carboxylase 2 (ACC2) enzyme, one of the two enzymes catalyzing the carboxylation of acetyl‐CoA to malonyl‐CoA, the main building block for de novo fatty acid synthesis. We additionally evaluated global ACC phosphorylation, a critical negative determinant of ACC activity. As suggested by the mRNA data, ACC protein levels were lower in Mfn2‐adKO mice and ACC phosphorylation was increased by 35% (Fig 2D), indicative of a downregulation of its lipogenic activity. As a whole, these results argue that changes in lipogenesis are not at the root of the higher lipid accumulation in the BAT of Mfn2‐adKO mice. Therefore, we next evaluated the lipolytic capacity of Mfn2 defective BAT. Strikingly, both basal and stimulated lipolysis rates were decreased by almost 80% in Mfn2‐adKO mice compared to control mice (Fig 2E). In further support, FAO‐related genes expression was decreased in Mfn2‐adKO BAT compared to control mice (Fig 2F).
Figure 2. Impaired brown adipose tissue function in Mfn2‐adKO mice.
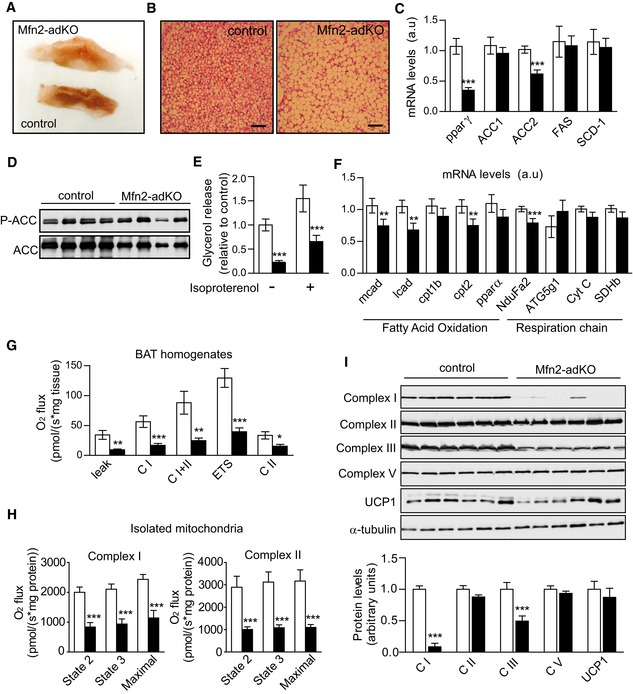
- Brown adipose tissue (BAT) pictures from male control and Mfn2‐adKO mice.
- Hematoxylin/eosin staining of BAT (scale bar, 100 μm).
- Total mRNA was extracted from BAT and used for qPCR analysis.
- Western blots were performed to evaluate the ACC phosphorylation in BAT.
- Lipolysis was evaluated by measuring glycerol release in isolated BAT from mice after 5‐h treatment with vehicle or isoproterenol (1 μM).
- Fatty acid oxidation and respiratory chain‐related gene expression in BAT was analyzed by qPCR.
- Respirometry analyses of uncoupled respiration (leak), Complex I respiration (CI), Complex I + Complex II respiration (CI + CII), maximal electron transfer system (ETS) capacity, and maximal Complex II driven respiration (ETS CII) in BAT.
- Respirometry analyses were performed in isolated mitochondria from BAT of male (n = 3 per genotype) and female (n = 3 per genotype) control and Mfn2‐adKO mice. On the left, malate (2 mM) and glutamate (10 mM) were used to stimulate Complex I and State 2, State 3 and maximal respiration were evaluated. On the right, succinate (10 mM) and rotenone (0.5 μM) were used to evaluate Complex II State 2, State 3, and maximal respiration.
- Mitochondrial proteins levels in total homogenates from BAT. Quantifications are shown below the images.
Defects in FAO are often closely linked to changes in mitochondrial function. Indeed, Mfn2 ablation in other tissues has previously been linked to mitochondrial dysfunction (Sebastian et al, 2012; Schneeberger et al, 2013). Therefore, we next assessed mitochondrial function in BAT from control and Mfn2‐adKO mice through high‐resolution respirometry analyses. Uncoupled (leak) and coupled respiration through either Complex I (CI) or CI + Complex II (CI + CII) was highly decreased in total BAT homogenates from Mfn2‐adKO mice compared to control mice (Fig 2G). Similarly, maximal electron transport system (ETS) capacity of the respiratory chain was dramatically blunted, either though CI + CII or CII alone (Fig 2G). Similar outcomes were obtained when respirometry assays were performed on isolated mitochondria. State 2, State 3, and maximal respiratory capacity through Complex I, triggered by malate and glutamate, or Complex II, triggered by succinate, were decreased in isolated mitochondria from the BAT of Mfn2‐adKO mice (Fig 2H). The decrease in uncoupled respiration was not linked to lower UCP1 levels (Fig 2I). Rather, it was consequent to an astonishing ~65% and ~40% decrease in CI and Complex III (CIII) protein levels, respectively (Fig 2I). This phenomenon was specific to these two complexes, as the protein levels of subunits from other complexes were not affected by Mfn2 deletion (Fig 2I). While a decrease in the mRNA levels of a Complex I subunit (NduFa2) was also observed (Fig 2F), it was much less marked than the deficiencies observed at the protein level, suggesting that post‐transcriptional events are at the root of the decline in Complex I content. Also, the fact that ETS CII is defective despite similar CII protein and mRNA levels suggest that the decrease in CIII is rate‐limiting for CII‐related respiration.
We next explored whether Mfn2 deletion led to similar consequences in the WAT, where endogenous Mfn2 expression in much lower. The size of epididymal WAT adipocytes was not affected by Mfn2 deficiency (Fig EV2A). However, lipolysis and coupled respiration were decreased in the eWAT from Mfn2‐adKO mice compared to control mice (Fig EV2B and C), albeit in a more modest way than in BAT. In line with this, the effect of Mfn2 deficiency on Complex I and III protein levels was also milder in eWAT compared to BAT (Figs EV2D and 2H). Altogether, these data suggest that disrupted Mfn2 expression in BAT leads to mitochondrial dysfunction and impaired fat utilization. While these effects are also manifested in WAT, their magnitude is lower than in BAT, probably due to the poor presence of Mfn2 in white adipocytes.
Figure EV2. eWAT function is not affected in Mfn2‐adKO mice and ER stress is increased in Mfn2‐adKO BAT on low‐fat diet.
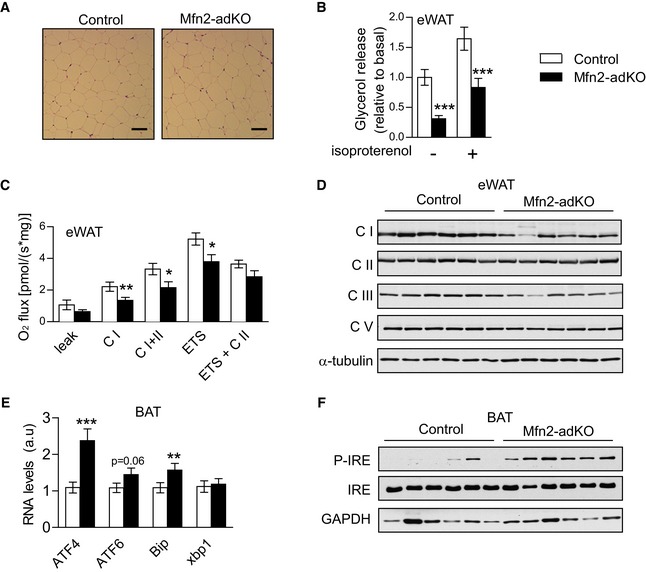
-
AH&E staining of eWAT (scale bar, 100 μm).
-
BLipolysis was evaluated by measuring glycerol release in isolated eWAT stimulated or not with 1 μM isoproterenol for 5 h.
-
COxidative phosphorylation and electron transfer capacity in eWAT.
-
DMitochondrial proteins levels in eWAT.
-
E, FmRNA (E) and proteins levels (F) for ER stress markers in BAT.
Mfn2 facilitates mitochondria–lipid droplet interaction
Mfn2 has been proposed to influence the interaction between mitochondria and other organelles, such as the endoplasmic reticulum (ER) (de Brito & Scorrano, 2008; Cosson et al, 2012; Filadi et al, 2015). In line with this, Mfn2 deficiency has been shown to trigger ER stress at least in liver, brain, and muscle (Sebastian et al, 2012; Schneeberger et al, 2013). Confirming these observations, Mfn2 deficiency also increased ER stress markers and p‐IRE phosphorylation in BAT (Fig EV2E and F).
Given the above observation, we hypothesized that Mfn2 in the BAT might also link mitochondria to other membrane organelles. Several groups have proposed that mitochondria and LDs might also closely collaborate, and that the connectivity between LDs and mitochondria would facilitate fatty acid transfer during lipolysis (Pidoux et al, 2011; Rambold et al, 2015). We hence hypothesized that Mfn2 protein could influence mitochondria–LD interactions. To answer this question, we performed electron microscopy (EM) analyses on BAT from control and Mfn2‐adKO mice (Fig 3A). First, these tests unraveled how mitochondrial shape was altered in Mfn2‐adKO genotype. The mitochondria from Mfn2‐adKO mice had more rounded appearance, and cristae content was severely diminished (Figs 3A and EV3A for a lower magnification). As expected from the role of Mfn2 as a mitochondrial fusion protein, mitochondrial length was decreased in Mfn2‐adKO BAT (Fig 3B). Interestingly, EM images revealed that mitochondria predominantly positioned themselves in contact to the LDs, with approximately 60% of mitochondria being in immediate contact to LD membranes (Fig 3A and C). In Mfn2‐adKO BAT, however, while mitochondria positioned in the vicinity of LD, direct contacts between mitochondrial and LD were up to 50% less frequent (Fig 3A and C). In line with the EM ultrastructural analyses, the association of mitochondria and LDs was further corroborated using subcellular fractionation techniques. LD‐associated proteins, such as perilipin 1 and perilipin 3 (PLIN1 and PLIN3), could be observed in mitochondrial fractions from the BAT of control animals (Fig 3D). The presence of PLIN1 and PLIN3 in mitochondrial extracts was drastically reduced in mitochondrial fractions from the BAT of Mfn2‐adKO mice (Fig 3D), further consolidating the impairment in mitochondria–LDs interaction. These differences were not due to differential contamination from cytosolic, plasma membrane, or intracellular vesicle components, as testified by the borderline detectable GLUT4 and LDH levels in our mitochondrial fractions (Fig 3D).
Figure 3. Mfn2 enhances mitochondria/lipid droplet interactions.
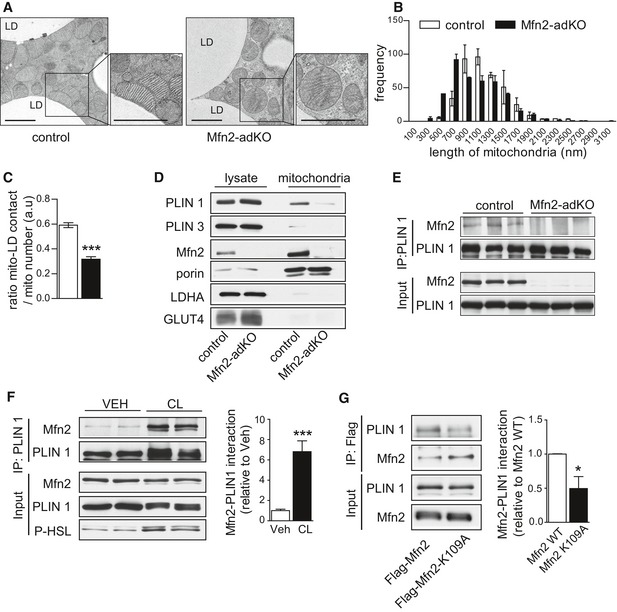
- Electron microscopy (EM) images from BAT (scale bar, 2 μm) of 20‐week‐old male mice.
- Mitochondrial length quantifications from EM images, corresponding to 12 independent images per BAT sample (n = 3 per genotype).
- Mitochondria–lipid droplet interaction was evaluated by measuring the ratio between mitochondria displaying direct contact with lipid droplet membranes and the total number of mitochondria (12 independent images per BAT sample; n = 3 per genotype).
- Mitochondrial fractions and crude protein extract material from BAT were used to evaluate the presence of lipid droplet (PLIN1, PLIN3), mitochondrial (Mfn2, porin), cytosolic (LDH), or membrane (GLUT4) proteins.
- The interaction between Mfn2 and PLIN1 was evaluated by immunoprecipitating PLIN1 from BAT of control and Mfn2‐adKO mice.
- Differentiated brown adipocytes were stimulated with vehicle or 1 μM CL316,243 (CL) for 5 h. Then, total proteins were extracted and immunoprecipitated against PLIN1. Quantifications for the increase in Mfn2‐PLIN1 interaction after CL treatment are shown on the right.
- Brown adipocytes were transfected with either FLAG‐tagged wild‐type Mfn2 or with a FLAG‐tagged GTPase dead Mfn2 mutant (K109A). Then, adipocytes were differentiated for 3 days, and total protein homogenates were obtained to test the interaction between Mfn2 and PLIN1 via FLAG immunoprecipitation. The graph on the right displays quantification for the decrease in Mfn2‐PLIN1 interaction observed when the K109A mutant is present.
Figure EV3. Mfn2 influences mitochondria–lipid droplet connectivity and specifically interacts with PLIN1.
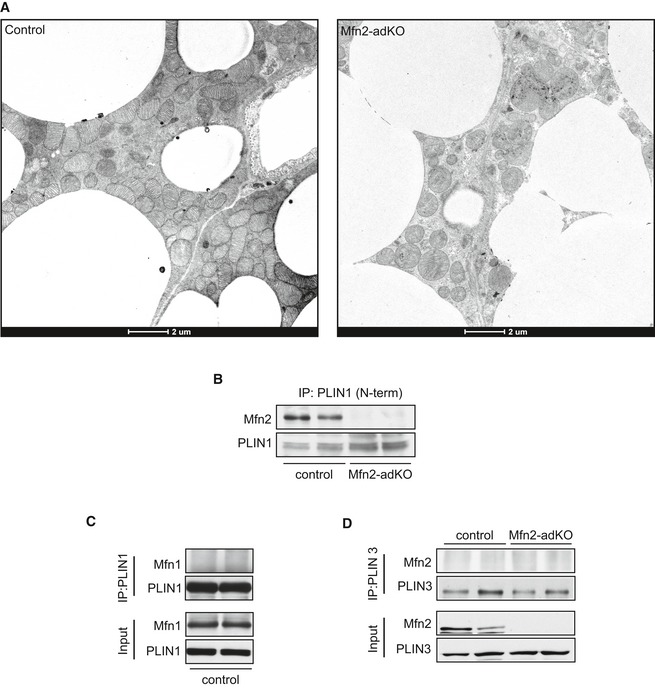
- Low‐magnification EM images of BAT from control and Mfn2‐adKO mice.
- Mfn2–PLIN1 interaction was evaluated by immunoprecipitating PLIN1 from BAT of control and Mfn2‐adKO mice using an antibody recognizing the N‐terminal region of PLIN1.
- Mfn1–PLIN1 interaction was evaluated by immunoprecipitating PLIN1 from BAT of control mice. The results illustrate the lack of co‐IP between these two proteins.
- The interaction between Mfn2 and PLIN3 was evaluated by immunoprecipitating PLIN3 from BAT of control and Mfn2‐adKO mice. The results illustrate the lack of co‐IP between these two proteins.
While mitochondria–LD interactions have been already documented in different studies (Jagerstrom et al, 2009; Pidoux et al, 2011; Rambold et al, 2015), the molecular mechanisms enabling these interactions are poorly understood. Immunoprecipitation experiments revealed that PLIN1, an adipose‐specific perilipin protein, co‐immunoprecipitated with Mfn2 using antibodies either against the C‐terminal (Fig 3E) or N‐terminal (Fig EV3B) region of PLIN1. In contrast, Mfn1 did not co‐immunoprecipitate with PLIN1 (Fig EV3C). The above results suggest that the interaction between PLIN1‐Mfn2 could be important for the LD–outer mitochondrial membrane interaction. Further, PLIN3 did not interact with Mfn2 (Fig EV3B), indicating that Mfn2 does not interact with all perilipin proteins. We then tried to understand whether this interaction was modulated by lipolytic stimuli, which are known to trigger mitochondria–LDs interaction (Yu et al, 2015). For this, we moved to differentiated primary brown adipocytes, where we confirmed that Mfn2 and PLIN1 co‐immunoprecipitated together, irrespective of the protein used for the pull‐down (Figs 3F and EV3D). Further, the incubation of brown adipocytes with CL enhanced the interaction between Mfn2 and PLIN1 by ~sevenfold. Finally, given that Mfn2 is a GTPase enzyme, we aimed to evaluate whether Mfn2 GTPase activity is required for the interaction. For this, we introduced wild‐type and GTPase dead (K109A; Chen et al, 2003) FLAG‐tagged forms of Mfn2 in differentiated brown adipocytes. The results (Fig 3G) illustrate that the introduction of GTPase dead Mfn2 mutants have an impaired ability to bind to PLIN1. Altogether, these results illustrate that Mfn2 and PLIN1 specifically bind each other, that this binding requires Mfn2 GTPase activity, and that it is modulated by adrenergic stimulation.
We previously showed that adrenergic‐induced respiration is impaired in brown adipocytes from Mfn2‐adKO mice. Whether the interaction between Mfn2 and PLIN1 plays a role in this process, however, is unclear, as Mfn2‐adKO adipocytes also display a marked mitochondrial dysfunction characterized by reductions in Complex I and Complex III levels (Fig 2I). To shed light into this issue, we used wild‐type (WT) or Mfn2‐deficient (Mfn2KO) mouse embryonic fibroblast (MEFs), which have comparable levels of Complex I and III levels (Fig EV4A). MEFs were loaded with oleic acid in order to generate lipid droplets. Then, MEFs were placed in nutrient‐deprived media in order to force the use their stored lipids as energy source. Given the absence of β3‐adrenergic receptors in MEFs cells, forskolin (Fsk) was used as a way to enhance cAMP levels, mimicking lipolytic stimuli. In WT MEFs, Fsk treatment did not significantly increase respiration. This changed, however, if PLIN1 was introduced in the cells, as Fsk treatment then translated into an increase in O2 consumption (Fig EV4B). This illustrates that the presence of PLIN1 in the LD membrane is key to mobilize fatty acids for mitochondrial respiration upon Fsk treatment. In contrast, Mfn2KO MEFs were unresponsive to Fsk, irrespective of PLIN1 expression (Fig EV4B), despite the fact that lipid droplets were present in Mfn2KO MEFs and that PLIN1 was properly phosphorylated upon Fsk treatment (Fig EV4C and D). The reintroduction of wild‐type Mfn2, but not the K109A mutant form, into PLIN1 expressing Mfn2KO MEFs was enough to recover Fsk responsiveness (Fig EV4B). These results illustrate that PLIN1 and Mfn2 are essential to provide cellular responsiveness to lipolytic stimuli, irrespective of mitochondrial respiratory complexes expression.
Figure EV4. PLIN1 and Mfn2 are both required for forskolin‐induced fat oxidation in MEF cells.
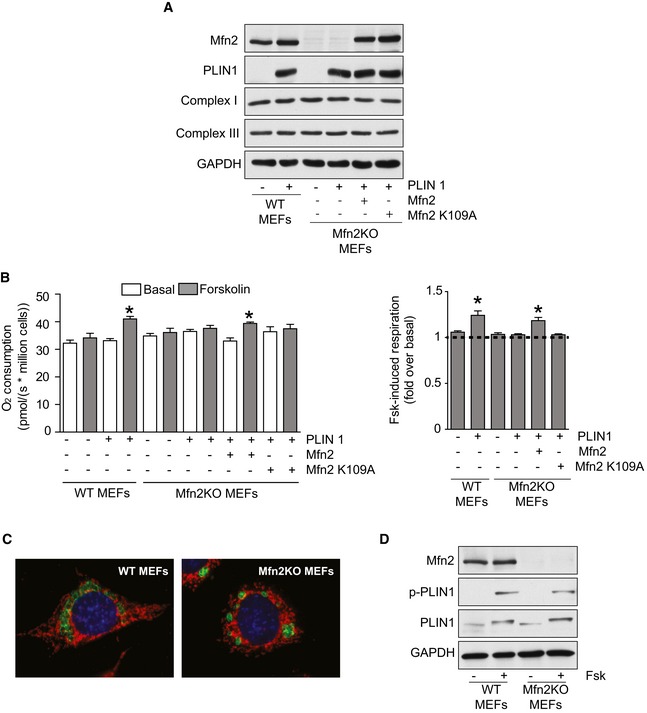
- MEF cells were transfected with PLIN1, wild‐type Mfn2, or Mfn2K109A as indicated. Then, 36 h later, cells were loaded with oleic acid (0.2 mM). After 12 h, protein extracts were obtained to test the markers indicated by Western blot.
- MEFs were treated as in (A). After 12 h of oleic acid loading, cells were trypsinized and counted. Then, two million cells were placed in respirometry chambers with air‐equilibrated serum‐free minimum essential medium. After evaluating basal respiration (basal; white bars), cells were treated with forskolin (Fsk; 1 μM; gray bars) and respiration rates were further evaluated. In the left graph, absolute respiration values are shown. In the right graph, the relative increase in respiration induced by Fsk is depicted. Values are presented as mean ± SEM of n = 6 independent experiments. * indicates statistically significant difference vs. the respective basal state at P < 0.05 (two‐tailed Student's t‐test). In the right panel, the ANOVA with Bonferroni post‐hoc test was used to evaluate the differences in Fsk‐induced respiration.
- MEFs were transfected with MitoDsRed to visualize mitochondria (in red) and with PLIN1 (in green) to visualize lipid droplets. Then, 36 h later, cells were loaded with oleic acid. After 12 h, cells were fixed with 4% paraformaldehyde and PLIN1 and mitochondria were visualized using fluorescence microscopy.
- Cells were treated as in (B). At the end of the treatment protein extracts were obtained and used for Western blot analysis of the indicated markers.
Mfn2‐adKO mice are protected against high‐fat diet‐induced insulin resistance
Given the alterations in lipid metabolism in the BAT and WAT from Mfn2‐adKO mice, we next aimed to understand how Mfn2‐adKO mice handled dietary lipid overload in the shape of a high‐fat diet (HFD). Surprisingly, body weight and composition were similar between genotypes after 8 weeks of HFD (Fig 4A and B). There were, however, important fat distribution changes. There was a marked increase in eWAT weight (Fig 4C), despite no difference was observed in white adipocytes size (Fig EV5A), which suggests a higher eWAT expandability. Also, the fatty appearance of the BAT of Mfn2‐adKO mice was even more prominent after HFD (Fig 4D). In line, defective response to thermogenic stimuli (Figs 4E and EV5B) and mitochondrial dysfunction in the BAT and eWAT were further aggravated by HFD in the Mfn2‐adKO mice (Fig EV5C and D).
Figure 4. Improved insulin sensitivity in Mfn2‐adKO mice fed high‐fat diet (HFD).
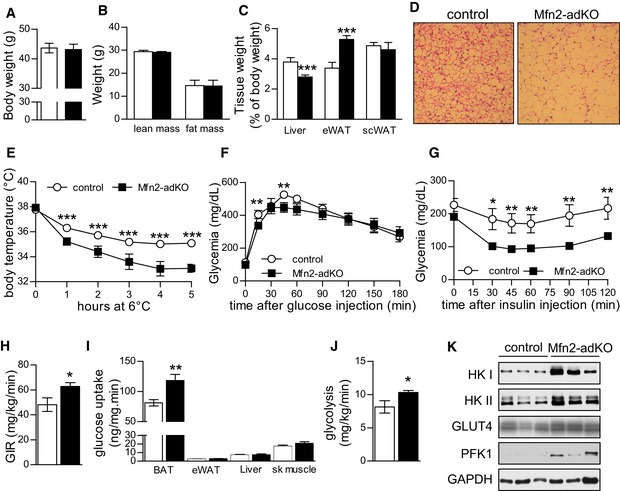
-
A, BBody weight (A) and composition (B) were measured through EchoMRI.
-
CAfter a 12‐h fast, mice were euthanized and tissue weights were measured.
-
DH&E staining from BAT (scale bar, 100 μm).
-
EThermogenic capacity was evaluated by placing WT and Mfn2‐adKO mice at 6°C for 5 h.
-
F, GAn intraperitoneal glucose tolerance test (F) and an intraperitoneal insulin tolerance test (G) were performed on HFD‐fed control and Mfn2‐adKO mice.
-
H–JHyperinsulinemic–euglycemic clamps were performed on HFD‐fed control and Mfn2‐adKO mice. Glucose infusion rate (GIR) (H), tissues glucose utilization (I), and glycolysis rates (J) are represented.
-
KProtein levels from total BAT homogenates from HFD‐fed mice.
Figure EV5. Mfn2‐adKO mice display brown adipose tissue dysfunction on high‐fat diet.
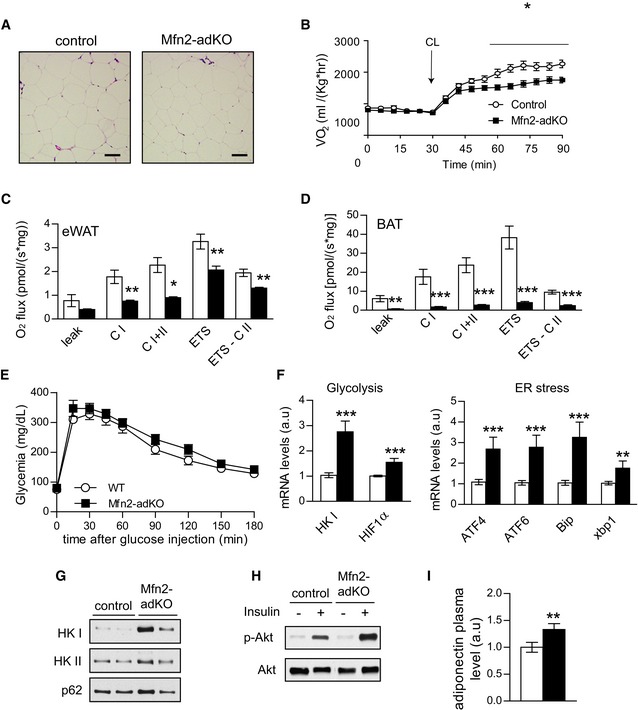
-
AH&E staining of eWAT on HFD (scale bar, 100 μm).
-
BNon‐shivering thermogenesis was evaluated by measuring baseline and CL (1 mg/kg)‐induced O2 consumption in anesthetized mice at 30°C (n = 8 mice per genotype).
-
C, DOxidative phosphorylation and electron transfer system (ETS) capacity in eWAT (C) and BAT (D) from HFD‐fed control and Mfn2‐adKO mice.
-
EAn intraperitoneal glucose tolerance test was performed on low‐fat diet‐fed control and Mfn2‐adKO mice.
-
FGlycolytic and ER stress markers gene expression in BAT of HFD‐fed control and Mfn2‐adKO mice.
-
GProtein levels of glycolytic markers in mitochondrial fractions from control and Mfn2‐adKO BAT.
-
HInsulin signaling in ex vivo incubated BAT with or without insulin (50 nM) from control and Mfn2‐adKO mice fed on HFD.
-
IAdiponectin plasma level from HFD‐fed control and Mfn2‐adKO mice.
On LFD, glucose tolerance was similar between control and Mfn2‐adKO mice (Fig EV5E). However, given the widely reported positive effects of BAT thermogenic function in the protection against glucose intolerance, we predicted that the defective thermogenesis in Mfn2‐adKO mice would make them more susceptible to HFD‐induced perturbations in glucose homeostasis. Surprisingly, this was not the case. On HFD, Mfn2‐adKO displayed a better glucose tolerance and remained more insulin sensitive, as shown by glucose and insulin tolerance tests, respectively (Fig 4F and G). To better understand the origin of these effects, we performed hyperinsulinemic–euglycemic clamps. In line with the observations from the insulin tolerance tests, Mfn2‐adKO mice required a significantly higher GIR under the hyperinsulinemic clamp conditions (18 mU insulin/kg/min) (Fig 4H). Strikingly, the BAT was a major contributor to the higher insulin‐stimulated glucose clearance in Mfn2‐adKO mice (Fig 4I). Given the mitochondrial dysfunction in the BAT of Mfn2‐aKO mice, it was not surprising that this higher glucose turnover was linked to increased glycolytic rates (Fig 4J). In order to substantiate the higher glycolytic capacity in the BAT of Mfn2‐adKO mice, we examined the levels of glycolytic enzymes at the protein and mRNA levels (Figs 4K and EV5F, respectively). Protein levels of different glycolytic enzymes, such as hexokinase I and II (HKI and HKII) as well as phopho‐fructokinase I, were increased in the BAT of Mfn2‐adKO mice (Fig 4K). Interestingly, we also observed an increase in HKI and HKII protein levels in mitochondrial fraction of the BAT from Mfn2‐adKO mice (Fig EV5G). This is important in order to enhance glycolytic fluxes, as mitochondria‐bound HKI and HKII display increased specific activity and decreased feedback inhibition relative to their soluble monomeric counterparts in the cytosol (Robey & Hay, 2006). These changes at the protein level were in line with changes at the mRNA levels of glycolysis‐related genes, including the transcription factor HIF‐1α, a master regulator of glycolytic gene expression (Fig EV5F). Besides glycolytic enzymes, the levels of the insulin‐sensitive glucose transporter GLUT4 were also increased in Mfn2‐adKO compared to control BAT (Fig 4K). Further, ex vivo experiments in BAT from HFD‐fed control and Mfn2‐adKO mice illustrated that insulin‐stimulated Akt phosphorylation was markedly increased in Mfn2‐deficient BAT (Fig EV5H).
Altogether, these results explain the higher insulin‐stimulated glucose uptake in BAT from Mfn2‐adKO, where glycolytic capacity is massively increased as an adaptation to the impaired ability to use fat as energy substrate. To further sustain this point, we reasoned that if the glycolytic rewiring in Mfn2‐adKO is aimed to feed basal non‐shivering thermogenesis, housing Mfn2‐adKO mice at thermoneutrality should prevent it. In agreement, when HFD‐fed Mfn2‐adKO mice were acclimated to thermoneutrality, the levels of HKI and HKII in the BAT were comparable to that of control littermates (Fig 5A). Importantly, there were no differences in insulin sensitivity between HFD‐fed control and Mfn2‐adKO at thermoneutrality (Fig 5B). This testifies that the higher insulin sensitivity of Mfn2‐adKO mice kept at regular housing temperatures derives from the enhanced glycolytic capacity in BAT. Further, when acclimated at thermoneutrality, Mfn2‐adKO mice displayed an even more exaggerated sensitivity to cold (Fig 5C). In fact, most Mfn2‐adKO mice had to be removed from the cold room 4 h after the initiation of the test. These observations indicate that the metabolic rewiring in the BAT of HFD‐fed Mfn2‐adKO is an adaptive response to sustain non‐shivering thermogenesis when mice are below thermoneutrality.
Figure 5. Thermoneutrality blunts the glycolytic rewiring in the BAT of HFD‐fed Mfn2‐adKO mice and sensitizes them to cold.
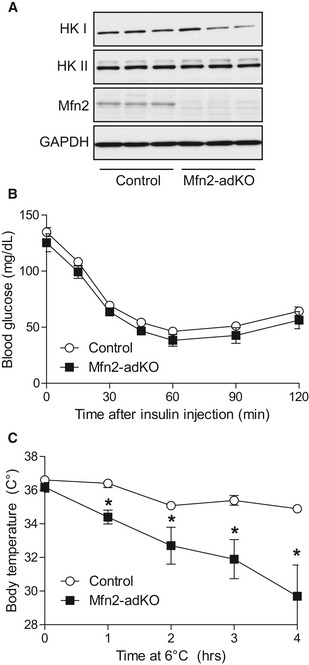
- High‐fat diet (HFD)‐fed control and Mfn2‐adKO littermates were housed for 8 weeks at thermoneutrality. Then, mice were sacrificed and protein homogenates were obtained from their BAT in order to test the indicated markers.
- Mice were housed as in (A). After 6 weeks at thermoneutrality, mice were fasted for 6 h and injected with insulin (1 U/kg). Then, blood glucose levels were measured at the indicated times.
- Mice were housed as in (A). After 7 weeks at thermoneutrality, mice were placed on a cold chamber at 6°C and body temperature was evaluated using a rectal thermometer at the indicated times. Mice were excluded from the test if their body temperature was ≤ 30°C.
High‐fat diet‐induced hepatic steatosis is improved in Mfn2‐adKO mice
Mfn2‐adKO and control mice displayed important fat distribution changes upon HFD. Particularly, liver weight was decreased in HFD‐fed Mfn2‐adKO mice compared to control mice (Fig 4C). This suggested that the enhanced lipid accumulation in eWAT and interscapular depots could protect liver from steatotic events. To evaluate liver function, we initially performed blood biochemistry analyses on LFD‐ and HFD‐fed mice. No differences between genotypes were observed on LFD (Table 1). However, Mfn2‐adKO mice displayed marked decreases in cholesterol levels, as well as in two markers of hepatic damage, the alanine aminotransferase (ALT) and the aspartate aminotransferase (AST) (Table 1). Together with the decreased liver weight, the decrease in these blood parameters constituted a second line of evidence, indicating that Mfn2‐adKO mice were protected against HFD‐induced damage on hepatic function.
Table 1.
Blood biochemistry analyses for control and Mfn2‐adKO mice on low‐ or high‐fat diet
| Parameters | Low‐fat diet | High‐fat diet | ||
|---|---|---|---|---|
| Control | Mfn2‐adKO | Control | Mfn2‐adKO | |
| Total cholesterol (mmol/l) | 2.88 ± 0.13 | 3.02 ± 0.25 | 4.83 ± 0.20 | 4.13 ± 0.30* |
| HDL cholesterol (mmol/l) | 2.72 ± 0.13 | 2.81 ± 0.27 | 4.35 ± 0.11 | 3.87 ± 0.22* |
| LDL cholesterol (mmol/l) | 0.16 ± 0.03 | 0.13 ± 0.02 | 0.48 ± 0.06 | 0.23 ± 0.04*** |
| ALT (U/l) | 70.33 ± 20.90 | 40.50 ± 2.32 | 213.33 ± 38.40 | 72.33 ± 7.97*** |
| AST (U/l) | 151.33 ± 43.14 | 94.33 ± 17.37 | 132.33 ± 14.24 | 83.33 ± 12.63*** |
| Triglycerides (mmol/l) | 0.91 ± 0.08 | 0.74 ± 0.10 | 0.88 ± 0.78 | 1.00 ± 0.10 |
| Free fat acids (mmol/l) | 1.02 ± 0.07 | 0.88 ± 0.07 | 0.62 ± 0.07 | 0.69 ± 0.08 |
Control and Mfn2‐adKO mice were randomized into either a low‐fat diet or a high‐fat diet at 8 weeks of age. Four months later, plasma levels of the markers indicated were measured. All values are expressed as mean ± SEM for n = 9 mice per group.
* and ***indicate statistical significant difference vs. respective control group at P < 0.05 and P < 0.01, respectively.
In addition to regular blood biochemistry analyses, and given that Mfn2 deficiency is restricted to adipose tissue, we also measured the plasma levels of a panel of 38 adipokines in the plasma of the mice. Among them, the FGF21 was the most highly upregulated, with a fourfold increase in the plasma of Mfn2‐adKO mice compared to control (Fig 6A). The second most highly upregulated adipokine was the adiponectin, which was 1.5‐fold more abundant in the plasma of Mfn2‐adKO mice (Fig EV5I). To evaluate whether the elevated plasma levels of FGF21 originated from adipose tissue, we assessed FGF21 expression in both eWAT and BAT depots. Whereas FGF21 gene expression was only ~twofold increase in eWAT (data not shown), the expression was ~30‐fold increased in the BAT of Mfn2‐adKO mice (Fig 6B).
Figure 6. Hepatic steatosis is reduced in Mfn2‐adKO mice on high‐fat diet (HFD).
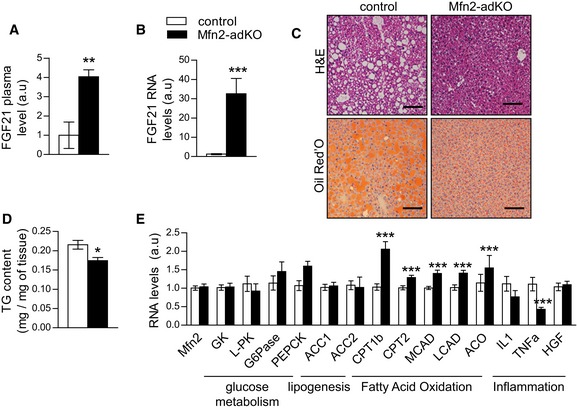
- FGF21 plasma levels in HFD‐fed male control and Mfn2‐adKO mice.
- FGF21 mRNA level in BAT from HFD‐fed control and Mfn2‐adKO mice.
- H&E and Oil Red O staining of liver from HFD‐fed control and Mfn2‐adKO mice (scale bar, 100 μm).
- Liver triglycerides (TG) content from HFD‐fed control and Mfn2‐adKO mice.
- mRNA levels in liver from HFD‐fed control and Mfn2‐adKO mice.
FGF21 has marked effects on hepatic lipid metabolism. In line with that and the lower liver tissue weight, H&E and Oil Red O staining revealed a much lower lipid content in the livers of Mfn2‐adKO mice (Fig 6C). This went in line with a significant decrease in the hepatic triglycerides content (Fig 6D). Gene expression analyses further indicated that, whereas glucose metabolism and lipogenesis pathways were not modulated, genes involved in FAO were upregulated in the livers of Mfn2‐adKO mice (Fig 6E). Inflammation markers were also decreased in Mfn2‐adKO liver, testifying for the protection against HFD‐induced metabolic damage (Fig 6E). These results suggest that the higher lipid‐buffering capacity of Mfn2‐adKO fat tissues and the higher FGF21 release from the BAT improve hepatic lipid oxidation and prevent hepatic damage upon high‐fat feeding.
Discussion
The role of mitochondrial dynamics in regulation of whole‐body metabolism has gained much attention during the last decade. Mitochondrial morphology differs sharply between tissues and is believed to participate in the fine‐tuning of energy availability to energy demand (Liesa & Shirihai, 2013). Notably, Mfn2 is highly expressed in the BAT, despite being a tissue whose mitochondrial network has a very fragmented appearance. In this work, we demonstrate for the first time that Mfn2 expression plays a major role in BAT metabolism by coupling the mitochondria with the LD and maintaining mitochondrial oxidative capacity. We have shown that Mfn2‐adKO mice display decreased thermogenic capacity due to impaired lipolytic and oxidative capacity. Surprisingly, when these mice are challenged with a HFD, their BAT metabolism rewires in order to compensate for their impaired ability to use fat as energy source. This way, they enhance adipose tissue expandability and their ability to obtain energy from glycolytic paths. This grants Mfn2‐adKO mice with protection against HFD‐induced insulin resistance and liver steatosis. Altogether, this study illustrates the high flexibility and adaptability of adipose tissues and their dramatic impact in mouse energy metabolism (Fig 7).
Figure 7. Representation of the effects of Mfn2 deficiency in BAT .
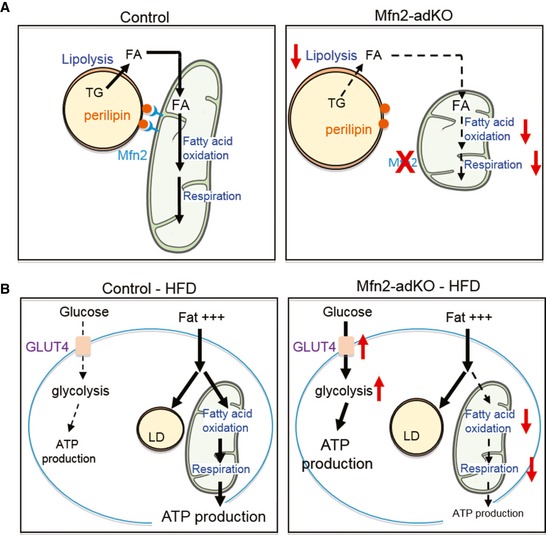
- Schematic representation of Mfn2 deficiency in BAT on mitochondria–lipid droplet (LD) interaction. In the left graph, Mfn2 allows the docking of mitochondria to LDs, ensuring an efficient transfer of fatty acids to the mitochondria for beta‐oxidation. On the right, Mfn2‐deficient BAT displays defects in mitochondrial oxidative capacity and LD docking, prompting the accumulation of fat within the LD.
- Representation of the metabolic adaptation of the HFD‐BAT from Mfn2‐adKO mice to compensate for the impaired ability to use fat as energy source. In control mice, fat is used as main energy source through oxidative paths. In the right, the Mfn2‐deficient BAT fails to oxidize fat, forcing the adaptation to massively use glycolytic paths for energy production.
Mitochondrial fission has been previously described as a physiological response in brown adipocytes in order to increase energy dissipation (Wikstrom et al, 2014). However, neither Mfn1‐adKO nor Mfn2‐adKO BAT display higher uncoupling capacity. This suggests that mitochondrial fission per se is not enough to promote uncoupling. While Mfn1 deficiency did not alter thermogenic capacity, Mfn2 defective BAT is characterized by an inability to maintain body temperature upon cold exposure. This could be due, at least in part, to a marked decrease in Complex I and III levels in the mitochondria from the BAT of Mfn2‐adKO animals, which dampened respiratory capacity in BAT homogenates. Mitochondrial dysfunction and reduced Complex I respiratory activity in Mfn2 defective tissues has been previously reported in conditional knockout models for POMC neurons and liver tissue (Sebastian et al, 2012; Schneeberger et al, 2013), albeit never as severe as in our Mfn2‐adKO mice. Decreased Complex I activity upon Mfn2 deficiency has also been observed in skeletal muscle, brain, and liver tissues (Sebastian et al, 2012; Schneeberger et al, 2013). The specificity of the decrease of Complex I in Mfn2 defective tissues might be linked to a potential direct interaction between Mfn2 and Complex I, as recently described in heart (Segales et al, 2013). Similarly, the decrease in Complex III might be linked to the ability of Complex I and Complex III to assemble into respiratory supercomplexes (Enriquez, 2016). Given that the decreases in Complex I and III levels were not mirrored by comparable decreases at the mRNA level of their subunits, our work suggests that Mfn2 might help stabilizing Complex I/Complex III supercomplexes. These observations, together with the reduced expression of fatty acid oxidation genes, are in line with the higher fat accumulation within the LD of the BAT of Mfn2‐adKO mice.
On a second level, mitochondrial dynamics have also been proposed to allow mitochondrial motility across the cell and to modulate the interaction of mitochondria with other cellular organelles. Our work indicates that Mfn2, in fact, might be a key player in the interaction between the mitochondria and LDs. The existence of mitochondria–LD interactions has been previously documented (Pu et al, 2011; Yu et al, 2015). It has been proposed that the close localization of these organelles might facilitate the exchange of FA and couple lipolytic and fatty acid oxidation processes. In line with this, our experiments in MEF cells demonstrate that the effectiveness of lipolytic stimuli to trigger fat oxidation requires the combined expression of PLIN1 and Mfn2. Importantly, Mfn2KO cells do not display reduced Complex I levels. This indicates that, in addition to the reduced expression of respiratory complexes, the lack of Mfn2‐PLIN1 interaction is another important molecular layer explaining the defective lipolytic response of Mfn2‐deficient adipose tissues.
The LD has specialized proteins to facilitate interactions with the cellular environment, including mitochondria. While initial studies identified SNAP23, a SNARE protein implicated in LD fusion, as a key element for LD–mitochondria interactions (Jagerstrom et al, 2009), a number of recent studies have also demonstrated that perilipins, one of the canonical LD membrane proteins, could act as modulators of LD–mitochondria interactions (Wang et al, 2011; Ramos et al, 2014; Yu et al, 2015). Accordingly, and as supported by our results, perilipins were often found in mitochondrial extracts from muscle, heart, and BAT (Wang et al, 2011; Bosma et al, 2012; Yu et al, 2015). Further, experiments using perilipin 5 (PLIN5) demonstrate that perilipin overexpression is enough to alter the docking of mitochondria to the lipid droplet (Wang et al, 2011). In particular, the C‐terminal domain of perilipin proteins seems to be key for LD–mitochondria interactions (Wang et al, 2011). Perilipin 1 (PLIN1) is a predominant perilipin in adipose tissues, and its expression is almost exclusive to WAT and BAT (for review, see Bickel et al, 2009). Our analyses unveil PLIN1 as a strongly interacting protein with Mfn2. The interaction between Mfn2 and PLIN1 is enhanced upon adrenergic stimulation, in line with observations indicating that mitochondria are targeted to the lipid droplet in the BAT upon cold exposure (Yu et al, 2015). The ability of PLIN1 to become phosphorylated by PKA, including three residues in the C‐terminal domain (Bickel et al, 2009), suggests that perilipin phosphorylation might modulate the interaction with Mfn2. The fact that Mfn1 does not interact with PLIN1 and that mutation of the GTPase activity of Mfn2 abolishes this binding further testifies for the specificity and relevance of the Mfn2‐PLIN1 interaction. It has been previously proposed that OPA1 could assemble a supramolecular complex containing PKA and perilipin and would be responsible for LD–mitochondria interactions (Pidoux et al, 2011). OPA1, however, generally localizes to the inner mitochondrial membrane, which might force the need for molecular adaptors in order to interact with proteins residing outside of the outer mitochondrial membrane. Interestingly, Mfn2 has been shown to interact with OPA1 (Guillery et al, 2008), which could reconcile the above observations.
Given the wide range of data illustrating how improved BAT thermogenic capacity might benefit glucose homeostasis, we hypothesized that Mfn2 deletion in BAT would sensitize against the metabolic damage promoted by lipid overload. Surprisingly, whereas lipid catabolism and thermogenesis were still impaired in HFD‐fed Mfn2‐adKO mice, glucose tolerance and insulin sensitivity were increased. This was due, in great part, to a metabolic rewiring in BAT aimed to maximize glycolytic capacity. Such rewiring upon Mfn2 depletion has been previously described in skeletal muscle cells, where Mfn2 deletion‐related reductions in oxidative capacity were compensated by a higher rate of glucose uptake and glycolysis (Pich et al, 2005). This dramatic metabolic shift was most robustly manifested under HFD, suggesting that it is consequent to their need to sustain basal thermogenic needs despite their inability to use lipids, the main dietary energy source when fed a HFD. This is further supported by our experiments at thermoneutrality, where the glycolytic shift in BAT from HFD‐fed Mfn2‐adKO mice was fully prevented. The higher glucose uptake capacity, however, is not sufficient to sustain non‐shivering thermogenesis when mice are exposed to cold. Nonetheless, in the absence of the glycolytic shift, such as when mice are housed at thermoneutrality, Mfn2‐adKO mice become dramatically sensitive to cold exposure. Our results support the view that reduced BAT thermogenic function and oxidative capacity in adipose tissues can also be beneficial for overall insulin sensitivity in certain scenarios, such as those allowing for improved adipose tissue expandability. Our results are in line with recent evidence from mouse models for adipose‐specific impairments in fatty acid catabolism, either through the deletion of the adipose triglyceride lipase (ATGL) (Schoiswohl et al, 2015), the TCA cycle enzyme fumarate hydratase (Yang et al, 2016), or the epigenetic regulator lysine‐specific demethylase 1 (Lsd1) (Duteil et al, 2016), all of which displayed impaired thermogenesis yet prevention against diet‐induced insulin resistance. Nevertheless, there is a remarkable variability on how the genetic ablation of mitochondrial proteins specifically in adipose tissues influences diet‐induced metabolic damage (Vernochet et al, 2014; Lee et al, 2015), likely reflecting the different impacts of these genes on the multiple functions of adipose tissue mitochondria beyond thermogenesis. Of note, the BAT of Mfn2‐adKO is more insulin sensitive despite harboring significantly higher levels of ER stress (Fig EV5F), which is generally conceived as detrimental for insulin action (Ozcan et al, 2004).
Blood biochemistry and liver analyses also demonstrated that hepatic function is improved in Mfn2‐adKO mice under HFD. These mice were protected against hypercholesterolemia and hepatic steatosis. This could be due to, at least, two major contributors. First, liver weight is reduced in HFD‐fed Mfn2‐adKO animals, associated with an increase of eWAT size. This increased lipid‐buffering capacity in eWAT and interscapular fat and their blunted lipolytic capacity might protect liver from steatosis. Together, our observations agree with the concept that increased fat tissue expandability might be protective against diet‐induced metabolic damage (Tan & Vidal‐Puig, 2008). A second key factor influencing the protection against hepatic steatosis in Mfn2‐adKO mice is their differential adipokine expression. Circulating FGF21 and adiponectin levels were markedly increased in the blood from Mfn2‐adKO mice compared to control mice, linked to a 30‐fold higher FGF21 expression in BAT from Mfn2‐adKO mice. Circulating FGF21 is positively correlated with liver insulin sensitivity, and FGF21 administration prevents hepatic steatosis under HFD condition (Xu et al, 2009). This great upregulation of FGF21 in BAT might be consequent to mitochondrial dysfunction, as recently proposed (Kim et al, 2013). Interestingly, mitochondrial dysfunction‐induced FGF21 expression is triggered via Atf4 (Kim et al, 2013), a master regulator of the integrated stress response, and Atf4 is highly upregulated in the BAT of Mfn2‐adKO mice (Figs EV2F and EV5F).
One key aspect for discussion is the role of WAT in the overall phenotype of the Mfn2‐adKO mice. Interestingly, a parallel work (K. Mahdaviani, M. Liesa, O.S. Shirihai, personal communication) has generated mice where the Mfn2 gene has been deleted exclusively on BAT tissue (BAT‐Mfn2 KO mice). BAT‐Mfn2 KO mice recapitulate many of the phenotypes observed in our Mfn2‐adKO mice, including impaired thermogenesis and protection against diet‐induced insulin resistance (K. Mahdaviani, M. Liesa, O.S. Shirihai, personal communication). This suggests that a great burden of the Mfn2‐adKO phenotypes stem from the BAT. Nonetheless, these two mouse models also show some differences. For example, eWAT expansion under HFD was only seen in the Mfn2‐adKO model. Similarly, despite comparable Complex I and Complex III deficiencies in BAT at the protein level, mitochondrial dysfunction was only overtly manifested in our mouse model. Interestingly, Mfn2‐adKO mice display higher circulating levels of adiponectin, which has been shown to negatively impact on thermogenic capacity and mitochondrial function in BAT (Qiao et al, 2014). This suggests that WAT has an important non‐negligible role in the metabolic phenotypes of Mfn2‐adKO mice, not only through increasing adipose tissue expandability, but also by exacerbating the negative effects of the Mfn2 ablation in BAT at the levels of lipolytic and respiratory capacity.
Altogether, our Mfn2‐adKO mice model is the first in vivo model demonstrating the importance of Mfn2 in adipose tissue and the direct role of this protein in mitochondria–LD interactions. This, in turn, consolidates the role of mitochondrial dynamics in relating mitochondria to other cellular organelles. Finally, this study also illustrates the adaptive nature of the BAT metabolism in response to nutrient challenges and how it can impact on whole‐body energy homeostasis.
Materials and Methods
Animal care
Mfn1‐adKO and Mfn2‐adKO mice were generated by crossing Mfn1loxP/loxP or Mfn2loxP/loxP mice (Chen et al, 2007) backcrossed to a C57BL/6 background, with mice expressing the Cre recombinase under the adiponectin promoter (adipoQ‐Cre mice). In the article, Mfn2loxP/loxP and Mfn2loxP/loxP‐adipoQCRE/− mice will be respectively called control and Mfn2‐adKO. Mfn1loxP/loxP or Mfn2loxP/loxP were bred, respectively, with Mfn1loxP/loxP‐adipoQCRE/− and Mfn2loxP/loxP‐adipoQCRE/−. Mice genotyping was performed on earpiece to distinguish CRE/− from −/− mice. DNA was extracted following the procedure of the DNA express extract kit from KapaBiosystems (#KK7151). Then, genomic DNA were amplified for CRE gene (forward primer 5′‐CTGGAAAATGCTTCTGTCCGTTTGC‐3′ and reverse primer 5′‐AATCCATCGCTCGACCAGTTTAGTTACC‐3′) and an internal PCR control gene (forward primer 5′‐TTTCAGTTGTGGTCTTCAGAGAGC‐3′ and reverse primer 5′‐CAACAACAAAAAAACCCTGCC‐3′) by PCR and load on an agarose gel to observe the CRE transgene expression. Mice were kept in a standard temperature‐ and humidity‐controlled environment with a 12‐h:12‐h light–dark cycle. Mice had nesting material and ad libitum access to water and commercial LFD or HFD (D12450J and D12492, respectively, from Research Diets Inc.). Unless otherwise stated, male and female mice were used for the experiments. All animal experiments were carried according to national Swiss and EU ethical guidelines and approved by the local animal experimentation committee under licence 2570.
Animal phenotyping
All clinical tests were carried out according to standard operational procedures established within the Eumorphia program (http://empress.har.mrc.ac.uk/). Animals were systematically randomized for the tests, ensuring similar numbers per genotype in the cohorts, and the experimenters were blinded to the genotype until the data were processed. Mice that showed any sign of severity, predefined by the Veterinary Office of the Canton of Vaud, Switzerland (authorization no. 2570), were euthanized. These animals, together with those who died spontaneously during the experiments, were excluded from the data analyses. Body composition was determined by Echo‐MRI (Echo Medical Systems, Houston, TX, USA). Oxygen consumption (VO2 and VCO2), food intake, and activity were monitored by indirect calorimetry using the comprehensive laboratory animal monitoring system (CLAMS; Columbus Instruments, Columbus, OH, USA). EE was estimated using VO2 and VCO2 values from indirect calorimetry, using the following equation EE (in kJ/h) = (15.818 × VO2) + (5.176 × VCO2) (Virtue & Vidal‐Puig, 2013). Food intake and activity were also monitored using the CLAMS during a 24‐h period. Glucose and insulin tolerance was analyzed by measuring blood glucose following intraperitoneal injection of 2 g/kg glucose or 0.3 U insulin/kg (human insulin actrapid, Lilly), respectively, after a 12‐h and 6‐h fast. For HFD‐fed mice, the insulin dose was increased to 0.75 U/kg. The cold test experiment was performed by placing mice at kept at 6°C during 5 h and measuring rectal body temperature every hour. Unless otherwise specified, animals were sacrificed at 8 a.m. after a 12‐h fast, in order to stabilize systemic parameters and to allow the measurement of blood biochemistry in the fasting state. Blood samples were collected in EDTA‐coated tubes, and plasma was isolated after centrifugation. Plasma parameters were measured using Dimension® Xpand Plus (Siemens Healthcare Diagnostics AG, Dudingen, Switzerland). FGF21 and adiponectin plasma levels were measured using the Mouse adipokine array kit (ARY013, R&D systems). Tissues were collected upon sacrifice and flash‐frozen in liquid nitrogen.
In vivo measurement of brown adipose tissue activity
In order to evaluate non‐shivering thermogenesis (BAT function), we measured is whole‐body O2 consumption in response to a specific β3‐adrenergic agonist, CL, in anesthetized mice as previously described (Cannon & Nedergaard, 2011). Briefly, mice, housed either at 22°C or 30°C for at least 1 month, were anesthetized using pentobarbital (60 mg/kg) and placed on a calorimetric chamber at 30°C. After 30 min, CL was subcutaneously injected (1 mg/kg) and mice were placed back in the chamber to follow up O2 consumption measurements.
Hyperinsulinemic–euglycemic clamp
Hyperinsulinemic–euglycemic clamps were performed at Physiogenex (Physiogenex SAS, France) according to standardized procedures, following the Guide for the Care and Use of Laboratory animals and French laws. Mice (n = 8 per genotype) underwent a surgery procedure to insert a catheter in the femoral vein. Clamp was performed 5 days post‐surgery on 6‐h fasted mice. Following a first blood collection, insulin was at 18 mU/kg/min for 110 min. Glucose infusion rate was adjusted according to blood glucose levels until the euglycemic steady state was reached. Moreover, 1 h before the end of the clamp, a bolus of 14C‐2Deoxy‐Glucose was performed. At the end of the 210‐min perfusion, mice were euthanized, several tissues were collected, and 14C‐radioactivity was measured to determine glucose utilization in BAT, eWAT, liver, and vastus lateralis skeletal muscle.
High‐resolution respirometry studies
Respirometry studies in homogenates from freshly extracted BAT and from pieces of eWAT were performed using high‐resolution respirometry (Oroboros Instruments, Innsbruck, Austria), as described previously (Boutant et al, 2015). For MEF cells, MEFs were trypsinized and 2 million cells were placed on the respirometry chamber filled with air‐equilibrated non‐supplemented minimum essential media. After ensuring a basal stable O2 consumption rate for at least 10 min, Fsk (1 μM) was injected into the chamber. Then, O2 consumption was measured for 15 min, ensuring at least 10 min of stable O2 consumption. Finally, antimycin A (Ama, 2.5 μM) was added to inhibit mitochondrial respiration, and the respiration values obtained were subtracted from the previous values. For experiments using mature brown adipocytes, BAT was collected from 30‐ to 35‐week‐old control or Mfn2‐adKO mice and brown adipocytes were isolated as previously described (Aune et al, 2013). Then, the adipocytes, in the top of the supernatant, were place in the respiratory chamber of the O2K Oxygraph, filled with air‐saturated mitochondrial respiration buffer (Kulkarni et al, 2016). After measuring respiration for 20 min, ensuring at least 10 min of stable O2 consumption rates, CL was injected into the chamber (1 μM) and O2 consumption rates were measured for another 20 min. Finally, first rotenone (1 μM) and then Ama were injected to the chamber in order to block Complex I and Complex III, respectively. The values obtained with Ama were substracted, and the relative increases in O2 consumption triggered by CL were measured respective to basal respiration for each adipocyte preparation. Respirometry analyses in isolated mitochondria were performed as recently described (Kulkarni et al, 2016).
Histology
H&E and Oil Red O stainings were performed using the fully automated Ventana Discovery XT (Roche Diagnostics, Rotkreuz, Switzerland). All steps were performed on the machine with Ventana solutions. EM was performed as described in (Bai et al, 2011).
Lipolysis
Lipolysis was determined on freshly extracted tissues using the ZenBio assay kit (LIP‐3‐NC), incubating the tissues in assay buffer supplemented with a vehicle or with isoproterenol (1 μM; 5 h) at 37°C under agitation, and then, glycerol release was measured.
Triglycerides content
Triglycerides were measured in adipose and liver tissues according to the protocol of Bioassay System Assay Kit (ETGA‐200).
Cell culture
MEFs (Kulkarni et al, 2016) and immortalized mouse brown pre‐adipocytes (Boutant et al, 2015) were grown and differentiated as previously described in mycoplasma‐free conditions. Adipocytes were considered fully differentiated at day 6 after the onset of the differentiation protocol, when they exhibited multilocular lipid droplets in their cytoplasm. For experiments, CL316,243 (C5976, SIGMA) was used at 1 μM concentrations unless otherwise specified.
Protein extraction and Western blotting
Protein extracts were isolated and quantified as previously described (Boutant et al, 2015), except for cellular fractionations studies (Frezza et al, 2007). For Western blotting, proteins were separated by SDS–PAGE and transferred onto nitrocellulose membranes. The following antibodies were used: antibodies against mouse Mfn1 (covering the amino acid span from 720 to 741) and Mfn2 (739–757) were generated by YenZym Antibodies LLC (San Francisco, USA) and validated internally using specific Mfn1 or Mfn2 overexpression and gene ablation models (see Figs 1B and EV4B and also Kulkarni et al, 2016); GAPDH (#2118), HK I (#2024), HK II (#2867), P‐HSL (#4139S), HSL (#4107S), ACC (#3676S), and PLIN1 (#9349) were purchased from Cell signaling; P‐ACC was obtained from Millipore (#07‐303); Antibodies against Complex I (NDUFA9, ab14713), Complex II (SDHA, ab14715), Complex III (UQCRC1, ab110252), Complex V (ATP5A, ab14748), GLUT4 (ab654), PFK1 (ab154804), IRE (ab37073), and P‐IRE (ab104157) were from Abcam, while α‐tubulin (T9026) was purchased from Sigma. For protein immunoprecipitation (IP) experiments, antibodies were pre‐incubated with magnetic beads (Invitrogen 10002D) and 300 μg of proteins extracts was pre‐cleared with clean beads during 1 h at 4°C under agitation. Then, the antibody‐coupled beads were washed and mixed with pre‐cleared extracts overnight at 4°C under constant agitation. Finally, the beads were washed and the immunoprecipitated material was extracted by adding Laemmli sample buffer (1×) and incubating for 5 min at 95°C. Densitometry quantifications were done using ImageJ software. All original full‐size images are available upon request.
mRNA analyses
Total mRNA extraction and cDNA conversion were performed as previously described (Boutant et al, 2015). Gene expression levels were analyzed using SYBR Green real‐time PCR (Roche). All primers used are listed in Appendix Table S1. Relative gene expression between genotypes was assessed through the ΔΔC t method, using β2‐microglobulin and cyclophillin as housekeeping genes.
Statistical analysis
Statistical analyses were performed with Prism software (GraphPad). The sample size was estimated on the basis of the known variability of the assays. For mouse studies, we were guided by the results obtained according to the standard operational procedures, established and validated within the Eumorphia program. Differences between two groups were analyzed using Student's t‐test (two‐tailed), and multiple comparisons were analyzed by ANOVA with a Bonferroni post hoc test. Group variances were similar in all cases. Data are expressed as means ± SEM.
Author contributions
MB, SSK, AZ, and CC conceived the project. MB, SSK, MJ, JR, MV‐A, RC, and CC performed animal experiments and molecular biology analyses. All authors interpreted results. MB and CC wrote the manuscript with contributions from SSK and AZ.
Conflict of interest
MB, SSK, MJ, JR, MV‐A, and CC are employees of the Nestlé Institute of Health Sciences S.A.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
The authors wish to thank the entire team of the Phenotyping Unit, Center of PhenoGenomics (CPG), School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne (EPFL), for their technical and scientific support in the performance and analysis of mouse phenotyping experiments. We thank Prof. Angela Valverde (IIBM/CSIC/UAM, Madrid, Spain) for providing the immortalized primary brown adipocytes. We are also grateful to Physiogenex S.A.S. for their help with hyperinsulinemic/euglycemic clamps. We would like to thank O. Shirihai for sharing data prior to publication. AZ is supported by research grants from the MINECO (SAF2013‐40987R), grant 2014SGR48 from the Generalitat de Catalunya, CIBERDEM (Instituto de Salud Carlos III), and Instituto de Salud Carlos III (PIE14/00045). A.Z. is a recipient of an ICREA “Academia” (Generalitat de Catalunya). MV‐A and CC are funded by the EU Marie Skłodowska‐Curie ITN—ChroMe (H2020‐MSCA‐ITN‐2015‐ChroMe—project number 675610).
The EMBO Journal (2017) 36: 1543–1558
See also: K Mahdaviani et al (July 2017) and M Scheideler & S Herzig (July 2017)
References
- Aune UL, Ruiz L, Kajimura S (2013) Isolation and differentiation of stromal vascular cells to beige/brite cells. J Vis Exp 73: 50191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa‐Lhoret R, Wallberg‐Henriksson H, Laville M, Palacin M, Vidal H, Rivera F, Brand M, Zorzano A (2003) Mitofusin‐2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem 278: 17190–17197 [DOI] [PubMed] [Google Scholar]
- Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg‐Henriksson H, Manco M, Calvani M, Castagneto M, Palacin M, Mingrone G, Zierath JR, Vidal H, Zorzano A (2005) Expression of Mfn2, the Charcot‐Marie‐Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin‐6. Diabetes 54: 2685–2693 [DOI] [PubMed] [Google Scholar]
- Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier‐de Murcia J, Auwerx J (2011) PARP‐1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel PE, Tansey JT, Welte MA (2009) PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta 1791: 419–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette‐Mackie EJ, Scow RO (1983) Movement of lipolytic products to mitochondria in brown adipose tissue of young rats: an electron microscope study. J Lipid Res 24: 229–244 [PubMed] [Google Scholar]
- Bosma M, Minnaard R, Sparks LM, Schaart G, Losen M, de Baets MH, Duimel H, Kersten S, Bickel PE, Schrauwen P, Hesselink MK (2012) The lipid droplet coat protein perilipin 5 also localizes to muscle mitochondria. Histochem Cell Biol 137: 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutant M, Joffraud M, Kulkarni SS, Garcia‐Casarrubios E, Garcia‐Roves PM, Ratajczak J, Fernandez‐Marcos PJ, Valverde AM, Serrano M, Canto C (2015) SIRT1 enhances glucose tolerance by potentiating brown adipose tissue function. Mol Metab 4: 118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J (2011) Nonshivering thermogenesis and its adequate measurement in metabolic studies. J Exp Biol 214: 242–253 [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130: 548–562 [DOI] [PubMed] [Google Scholar]
- Cosson P, Marchetti A, Ravazzola M, Orci L (2012) Mitofusin‐2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS One 7: e46293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman SW (1970) Structure‐function relationships in the adipose cell. I. Ultrastructure of the isolated adipose cell. J Cell Biol 46: 326–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich MO, Liu ZW, Horvath TL (2013) Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet‐induced obesity. Cell 155: 188–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duteil D, Tosic M, Lausecker F, Nenseth HZ, Muller JM, Urban S, Willmann D, Petroll K, Messaddeq N, Arrigoni L, Manke T, Kornfeld JW, Bruning JC, Zagoriy V, Meret M, Dengjel J, Kanouni T, Schule R (2016) Lsd1 ablation triggers metabolic reprogramming of brown adipose tissue. Cell Rep 17: 1008–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos‐Flier E, Rosen ED (2011) Transcriptional control of adipose lipid handling by IRF4. Cell Metab 13: 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriquez JA (2016) Supramolecular organization of respiratory complexes. Annu Rev Physiol 78: 533–561 [DOI] [PubMed] [Google Scholar]
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum‐mitochondria coupling. Proc Natl Acad Sci USA 112: E2174–E2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L (2007) Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2: 287–295 [DOI] [PubMed] [Google Scholar]
- Guillery O, Malka F, Landes T, Guillou E, Blackstone C, Lombes A, Belenguer P, Arnoult D, Rojo M (2008) Metalloprotease‐mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell 100: 315–325 [DOI] [PubMed] [Google Scholar]
- Hernandez‐Alvarez MI, Thabit H, Burns N, Shah S, Brema I, Hatunic M, Finucane F, Liesa M, Chiellini C, Naon D, Zorzano A, Nolan JJ (2010) Subjects with early‐onset type 2 diabetes show defective activation of the skeletal muscle PGC‐1{alpha}/Mitofusin‐2 regulatory pathway in response to physical activity. Diabetes Care 33: 645–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagerstrom S, Polesie S, Wickstrom Y, Johansson BR, Schroder HD, Hojlund K, Bostrom P (2009) Lipid droplets interact with mitochondria using SNAP23. Cell Biol Int 33: 934–940 [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950 [DOI] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N et al (2013) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19: 83–92 [DOI] [PubMed] [Google Scholar]
- Kulkarni SS, Joffraud M, Boutant M, Ratajczak J, Gao AW, Maclachlan C, Hernandez‐Alvarez MI, Raymond F, Metairon S, Descombes P, Houtkooper RH, Zorzano A, Canto C (2016) Mfn1 deficiency in the liver protects against diet‐induced insulin resistance and enhances the hypoglycemic effect of metformin. Diabetes 65: 3552–3560 [DOI] [PubMed] [Google Scholar]
- Lee J, Ellis JM, Wolfgang MJ (2015) Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress‐induced inflammation. Cell Rep 10: 266–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS (2013) Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17: 491–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, Bengtsson T, Cannon B (2007) Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab 293: E444–E452 [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461 [DOI] [PubMed] [Google Scholar]
- Papanicolaou KN, Ngoh GA, Dabkowski ER, O'Connell KA, Ribeiro RF Jr, Stanley WC, Walsh K (2012) Cardiomyocyte deletion of mitofusin‐1 leads to mitochondrial fragmentation and improves tolerance to ROS‐induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol 302: H167–H179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Wiederkehr A, Kirkpatrick C, Mattenberger Y, Martinou JC, Marchetti P, Demaurex N, Wollheim CB (2008) Selective actions of mitochondrial fission/fusion genes on metabolism‐secretion coupling in insulin‐releasing cells. J Biol Chem 283: 33347–33356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirce V, Carobbio S, Vidal‐Puig A (2014) The different shades of fat. Nature 510: 76–83 [DOI] [PubMed] [Google Scholar]
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A (2005) The charcot‐marie‐tooth type 2A gene product, Mfn2, up‐regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet 14: 1405–1415 [DOI] [PubMed] [Google Scholar]
- Pidoux G, Witczak O, Jarnaess E, Myrvold L, Urlaub H, Stokka AJ, Kuntziger T, Tasken K (2011) Optic atrophy 1 is an A‐kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J 30: 4371–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J, Ha CW, Zhang S, Jung JP, Huh WK, Liu P (2011) Interactomic study on interaction between lipid droplets and mitochondria. Protein Cell 2: 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Yoo H, Bosco C, Lee B, Feng GS, Schaack J, Chi NW, Shao J (2014) Adiponectin reduces thermogenesis by inhibiting brown adipose tissue activation in mice. Diabetologia 57: 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, Lippincott‐Schwartz J (2015) Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32: 678–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos SV, MacPherson RE, Turnbull PC, Bott KN, LeBlanc P, Ward WE, Peters SJ (2014) Higher PLIN5 but not PLIN3 content in isolated skeletal muscle mitochondria following acute in vivo contraction in rat hindlimb. Physiol Rep 2: e12154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RB, Hay N (2006) Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 25: 4683–4696 [DOI] [PubMed] [Google Scholar]
- Schneeberger M, Dietrich MO, Sebastian D, Imbernon M, Castano C, Garcia A, Esteban Y, Gonzalez‐Franquesa A, Rodriguez IC, Bortolozzi A, Garcia‐Roves PM, Gomis R, Nogueiras R, Horvath TL, Zorzano A, Claret M (2013) Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155: 172–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoiswohl G, Stefanovic‐Racic M, Menke MN, Wills RC, Surlow BA, Basantani MK, Sitnick MT, Cai L, Yazbeck CF, Stolz DB, Pulinilkunnil T, O'Doherty RM, Kershaw EE (2015) Impact of reduced ATGL‐mediated adipocyte lipolysis on obesity‐associated insulin resistance and inflammation in male mice. Endocrinology 156: 3610–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian D, Hernandez‐Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Oresic M, Pich S, Burcelin R, Palacin M, Zorzano A (2012) Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segales J, Paz JC, Hernandez‐Alvarez MI, Sala D, Munoz JP, Noguera E, Pich S, Palacin M, Enriquez JA, Zorzano A (2013) A form of mitofusin 2 (Mfn2) lacking the transmembrane domains and the COOH‐terminal end stimulates metabolism in muscle and liver cells. Am J Physiol Endocrinol Metab 305: E1208–E1221 [DOI] [PubMed] [Google Scholar]
- Tan CY, Vidal‐Puig A (2008) Adipose tissue expandability: the metabolic problems of obesity may arise from the inability to become more obese. Biochem Soc Trans 36: 935–940 [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak‐Tarnopolsky SN, Devries MC, Hamadeh MJ (2007) Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol 292: R1271–R1278 [DOI] [PubMed] [Google Scholar]
- Toledo FG, Watkins S, Kelley DE (2006) Changes induced by physical activity and weight loss in the morphology of intermyofibrillar mitochondria in obese men and women. J Clin Endocrinol Metab 91: 3224–3227 [DOI] [PubMed] [Google Scholar]
- Vernochet C, Damilano F, Mourier A, Bezy O, Mori MA, Smyth G, Rosenzweig A, Larsson NG, Kahn CR (2014) Adipose tissue mitochondrial dysfunction triggers a lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications. FASEB J 28: 4408–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtue S, Vidal‐Puig A (2013) Assessment of brown adipose tissue function. Front Physiol 4: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C (2011) Perilipin 5, a lipid droplet‐associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52: 2159–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, Twig G, Petrovic N, Zingaretti C, Graham A, Cinti S, Corkey BE, Cannon B, Nedergaard J, Shirihai OS (2014) Hormone‐induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J 33: 418–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, Chen JL, Jung DY, Zhang Z, Ko HJ, Kim JK, Veniant MM (2009) Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet‐induced obese mice. Diabetes 58: 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wu JW, Wang SP, Severi I, Sartini L, Frizzell N, Cinti S, Yang G, Mitchell GA (2016) Adipose‐specific deficiency of fumarate hydratase in mice protects against obesity, hepatic steatosis and insulin resistance. Diabetes 65: 3396–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang S, Cui L, Wang W, Na H, Zhu X, Li L, Xu G, Yang F, Christian M, Liu P (2015) Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim Biophys Acta 1853: 918–928 [DOI] [PubMed] [Google Scholar]
- Zorzano A, Liesa M, Palacin M (2009) Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol 41: 1846–1854 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File