

Abstract
Background
Maternal diabetes increases risk for congenital malformations, particularly cardiac outflow tract defects. Maternal diabetes inhibits expression of Pax3 in neuroepithelium through hyperglycemia-induced oxidative stress. The neuroepithelium gives rise to the neural crest, and Pax3 expression in cardiac neural crest (CNC) is required for CNC migration to the heart and for outflow tract septation. Here we tested whether maternal diabetes, through hyperglycemia-induced oxidative stress, before the onset of CNC delamination, impairs CNC migration and cardiac outflow tract septation.
Methods
CNC migration was mapped in mouse embryos whose mothers were diabetic, or transiently hyperglycemic, or in which oxidative stress was transiently induced, using reporters linked to Pax3 expression. CNC apoptosis was examined by TUNEL assay. Outflow tract septation was examined histologically and by gross inspection.
Results
Few, if any, migrating CNC cells were observed in embryos of diabetic mice, and this was associated with increased apoptosis along the path of CNC migration. Outflow tract defects were significantly increased in fetuses of diabetic mice. Notably, induction of hyperglycemia or oxidative stress on the day prior to the onset of Pax3 expression and CNC migration also impaired CNC migration, increased apoptosis, and caused outflow tract defects. However, antioxidants administered on the day prior to the onset of Pax3 expression and CNC migration prevented these effects of hyperglycemia or oxidative stress.
Conclusions
In diabetic pregnancy, oxidative stress, which inhibits expression of genes required for CNC viability, causes subsequent CNC depletion by apoptosis during migration, which leads to outflow tract defects.
Keywords: Cardiac neural crest, outflow tract, Pax3, diabetic pregnancy, oxidative stress, diabetic teratogenesis
INTRODUCTION
Maternal diabetes significantly increases the risk for embryonic malformations (Evers and others, 2004; Loffredo and others, 2001; Macintosh and others, 2006; Schaefer-Graf and others, 2000; Suhonen and others, 2000; Towner and others, 1995; Wren and others, 2003). These malformations arise at the early stages of organogenesis, approximately the first eight weeks of pregnancy (Hanson and others), and so, are related to pre-gestational, rather than gestational, diabetes. Virtually any organ system can be affected, although malformation of the neural tube, which gives rise to the brain and spinal cord, or of the heart and great vessels, are among the most frequently observed (Chung and Myrianthopoulos, 1975; Farrell and others, 2002; Macintosh and others, 2006; Nielsen and others, 2005; Schaefer-Graf and others, 2000; Yang and others, 2006). The most common defects affecting the heart are conotruncal, pharyngeal arch artery, and septal defects (Ferencz and others; Loffredo and others, 2001).
The neural crest is a population of cells that derives from the neuroectoderm. They make an epithelial to mesenchymal transformation and migrate to various locations in the body where they contribute to the formation a wide variety of tissues, including the peripheral nervous system, the jaw, and the adrenal medulla (Anderson, 1997; Sauka-Spengler and Bronner-Fraser, 2006). The cardiac neural crest (CNC) arise between the mid-otic placode and the third somite and migrate around and between the somites through pharyngeal arches 3, 4, and 6 to the cardiac outflow tract (Chan and others, 2004; Conway and others, 1997; Kirby, 1987; Kirby and others, 1983). In the mouse embryo, migration of CNC begins on day 8.5 of gestation, at the 7 somite stage, and the CNC cells reach the outflow tract by the 32 somite stage on day 9.5. They associate with conotruncal cushions which will give rise to the conotruncal septum, and truncus arteriosus. By day 12.5 CNC cells can be identified within the outflow tracts and around the third, fourth, and left sixth aortic arch arteries (Jiang and others, 2000). Using quail-chick chimeras and CNC ablation experiments, it has been shown that migration of CNC cells to the heart is essential for septation of the primitive single cardiac outflow tract into the aorta and pulmonary arteries and for formation of the aortic arch arteries (Besson and others, 1986; Bockman and others, 1987; Kirby, 1987; Kirby and others, 1983; Kirby and others, 1985; Nishibatake and others, 1987).
The Pax genes encode paired box-containing transcription factors that are expressed during embryonic development (Gruss and Walther, 1992; Robson and others, 2006). Pax3 is expressed in the neural tube, neural crest, and somites (Goulding and others, 1991). A 1.6 kb element 5’ of the Pax3 coding sequence is sufficient for transcription in most of the neural tube and in neural crest, while additional sequences between −14 kb and −1.6 kb are required for expression in somites (Natoli and others, 1997). Expression of Pax3 begins in the neuroectoderm of the neural folds on day 8.5 and declines in a tissue-specific fashion during development (Goulding and others, 1991; Gruss and Walther, 1992). Expression of Pax3 in CNC diminishes before they arrive at the heart, although some Pax3-expressing cells may still be detected in the aorta after septation (Conway and others, 1997; Epstein and others, 2000). In mouse strains with null Splotch Pax3 alleles (Pax3Sp/Sp, Pax3Sp1H/Sp1H, and Pax3Sp2H/ Sp2H) CNC migration is impaired and disorganized (Conway and others, 1997; Epstein and others, 2000) and outflow tract defects like those caused by CNC ablation occur with 100% penetrance (Conway and others, 1997; Franz, 1989). However, introduction of a Pax3 transgene under the control of the 1.6 kb Pax3 5′ flanking element rescues Splotch embryos from outflow tract defects (Li and others, 1999). In humans, mutant PAX3 alleles are responsible for some forms of Waardenburg syndrome, DiGeorge syndrome, and Hirchsprung disease, which may include defects such as aortic interruption, double outlet right ventricle, and persistent truncus arteriosus (Creazzo and others, 1998), indicating that abnormal expression of PAX3-dependent genes in the human embryo can lead to defective development of CNC and its derivatives.
We have previously shown in a mouse model of diabetic pregnancy that maternal diabetes inhibits expression of Pax3 beginning on day 8.5 and significantly increases the incidence of neural tube defects (NTDs) (Phelan and others, 1997). The NTDs, primarily exencephaly, resemble the NTDs that occur in Pax3-deficient Splotch embryos, suggesting that inhibition of Pax3 expression below a critical threshold phenocopies the effect of Pax3 null mutation. In humans and in mouse models, the incidence of malformations is related to severity of hyperglycemia (Fine and others, 1999; Greene and others, 1989), and the elevated circulating glucose resulting from diabetes is necessary and sufficient to inhibit Pax3 expression and to cause NTD (Fine and others, 1999). Excess glucose transported to, and metabolized by, the embryo induces oxidative stress, and oxidative stress mediates the adverse effects of maternal diabetes on Pax3 expression and NTD (Chang and others, 2003; Fine and others, 1999). Hyperglycemia or oxidative stress must be induced on the day prior to the onset of Pax3 expression, day 7.5, in order for reduced Pax3 expression and increased NTD to occur, and hyperglycemia occurring after the onset of Pax3 expression has no effect on Pax3 or NTD (Chang and others, 2003; Fine and others, 1999). Notably, when levels of Pax3 are insufficient, either due to maternal diabetes or null Splotch alleles, apoptosis that is specifically limited to Pax3-expressing cells is observed (Phelan and others, 1997). This suggests that the NTDs caused by Pax3 insufficiency result from elimination of cells forming the neural tube. The apoptosis is p53-mediated, and inactivation of p53 prevents both apoptosis and NTDs in Pax3-deficient embryos (Pani and others, 2002). Steady-state levels of p53 protein, but not mRNA, are increased in Pax3Sp/Sp embryos (Pani and others, 2002), suggesting that Pax3 is not required to regulate expression of genes leading to neural tube closure, but it is required to inhibit the synthesis or stability of p53 protein in order to maintain neuroectoderm viability during closure of the neural tube. Abnormal expression of many genes may occur in diabetic pregnancy, depending on the timing and severity of maternal hyperglycemia. However, because of the absolute requirement for Pax3 for neural tube closure, insufficient production of Pax3 may be all that is necessary to impair development of the neural tube.
As noted above, along with NTDs, cardiac outflow tract defects are among the most common to occur during diabetic pregnancy (Ferencz and others; Loffredo and others, 2001). Maternal diabetes has been shown to cause defects of cranial and cardiac neural crest-derived structures in the rat, and vitamin E may diminish the severity of many of these defects (Molin and others, 2004; Siman and others, 2000). Injection of 30 mM/l glucose into the neural tubes of chick embryos increases defects of the aorta and pharyngeal arch arteries, and the effect of high glucose is suppressed by co-injection of N-acetylcysteine (Roest and others, 2007). Because oxidative stress inhibits Pax3 expression before the neural crest delaminate from the neuroepithelium, deficient Pax3 in CNC may be responsible for defective CNC and outflow tract development.
Recognizing that CNC migration and cardiac outflow tract septation are dependent on Pax3, and that CNC derive from neuroectoderm, where Pax3 expression is reduced following oxidative stress induced on day 7.5, in this study we tested whether oxidative stress induced on day 7.5 could lead to defective CNC migration, thereby being responsible for cardiac outflow tract defects during diabetic pregnancy.
MATERIALS AND METHODS
Mouse strains
Two mouse lines, in which LacZ or Green Fluorescent Protein (GFP) served as transcriptional reporters for the endogenous Pax3 gene, have been previously described (Relaix and others, 2004). Briefly, the LacZ line (Pax3IRESnLacZ/+, referred to here as Pax3LacZ/+) was generated by homologous recombination to insert LacZ within the Pax3 coding sequence (Relaix and others, 2003). Thus, LacZ interrupts the Pax3 coding sequence; the single intact Pax3 allele in Pax3LacZ/+ embryos is sufficient for normal development. The GFP strain was generated by homologous recombination to insert a GFP coding sequence with an internal ribosome entry site (IRES) within the 3′ untranslated region of Pax3 (Morgan, et al., submitted). Thus, both Pax3 and GFP proteins are generated when Pax3 is transcribed. Both lines are on a FVB background. To generate embryos, mice were mated using genetic crosses as described in the text, and noon on the day on which a copulation plug was found was determined to be day 0.5 of pregnancy.
Induction of diabetes
Insulin-deficient diabetes was induced in 4- to 6-wk-old female FVB mice (Jackson Laboratory, Bar Harbor, ME) using 100 mg/kg streptozotocin (STZ, Sigma Chemicals, St. Louis, MO), and treated with insulin pellets implanted subcutaneously (Linshin, Scarborough, Ontario) as described previously (Phelan and others, 1997). Blood glucose was monitored with a Glucometer Elite (Bayer, Mishawaka, IN).
Transient induction of hyperglycemia or oxidative stress
Transient hyperglycemia was induced on day 7.5 of pregnancy by subcutaneous (s.c.) injection of 2 cc of 12.5% glucose dissolved in phosphate-buffered saline (PBS) at approximately hourly intervals to maintain blood glucose >17 mmol/l over a 10-hour period as described (Fine and others, 1999). Control animals were injected with PBS. Oxidative stress was induced at noon on day 7.5 of pregnancy with a single s.c. injection with Antimycin A (AA, Sigma Chemicals, St. Louise, MO). The AA was dissolved in 25% (vol./vol.) propylene glycol and administered at 3 mg/kg body as described (Chang and others, 2003).
Antioxidants
Glutathione ethyl ester (GSH-EE; Sigma Chemicals, St. Louis, MO) dissolved in PBS was administered at 1 mmol/kg, in three doses on day 7.5 as described (Horal and others, 2004). Vitamin E succinate ((+)-α-tocopherol succinate, Sigma Chemicals, St. Louis, MO) was added at 0.125%, w/w to control chow (Lab Diet 5020, Purina, St. Louis, MO), which supplemented dietary vitamin E 21-fold. The vitamin E-enriched chow was fed to pregnant mice beginning on day 0.5 of pregnancy as described (Chang and others, 2003).
In all cases embryos were recovered on either day 9.5 to assess CNCC migration and apoptosis, or day 16.5 to assess cardiac outflow tract septation.
All procedures using animals conformed to the principles of laboratory animal care set forth by the National Institutes of Health (NIH) and were approved by the Joslin Diabetes Center Institutional Animal Care and Use Committee.
X-GAL staining
Dissected embryos were fixed for 15 min with 4% paraformaldehyde in PBS at 4° C and stained with X-GAL as described (Relaix and others, 2003). Briefly, embryos were washed three times with PBS supplemented with 0.2% NP-40, then stained at 37° C for 16 h with a solution containing 0.4 mg/ml X-GAL (Roche Applied Science Indianapolis, IN), 2 mmol/l MgCl2, 0.2% NP-40, 20 mmol/l K4Fe(CN)6, and 20 mmol/l K3Fe(CN)6, dissolved in PBS. Embryos were extensively washed with PBS prior to visualization.
TUNEL assay
Whole-mount TUNEL assay of day 9.5 embryos was performed as described (Phelan and others, 1997) except that sheep anti-digoxigenin FAB fragment conjugated to fluorescein isothiocyanate (FITC, Roche Applied Science) was used to detect digoxigenin (excitation 494 nm emission 523 nm).
Microscopy
X-GAL-stained day 9.5 embryos and day 16.5 fetuses were examined using a Nikon SMZ800 stereomicroscope. Images were captured using an attached SPOT RT CCD camera Model 2.2.1. and SPOT software Ver.3.0.5 (Diagnostics Instruments Inc). Pax3GFP/+ and TUNNEL-stained embryos were captured using a Zeiss LSM META 510 upright confocal microscope and Zeiss LSM software.
Hematoxylin and Eosin Staining
Whole fetuses recovered on day 16.5 were fixed overnight in 4% phosphate-buffered paraformaldehyde, then stored at 4°C in PBS. Before embedding, the thorax was dissected from the rest of the fetus, was submerged in 20% sucrose (in PBS) for 4 hours at 4°C, and then embedded in Tissue-Tek O. C. T. compound (Ted Pella, Inc., Redding, CA). Embedded tissues were stored at −80° C. Five μm serial sections from the atria through approximately the top half of the ventricles (approximately 40–50 sections per embryos) were obtained using a cryostat. Slides were stained with hematoxylin and eosin, and imaged using the stereomicroscope and SPOT camera described above.
Statistical Analyses
Data were analyzed by unpaired student’s t test or one way-ANOVA with Neuman Keuls post hoc test, or by Chi square analysis, as indicated in legends of the tables, using Prism 4 software (GraphPad Software).
RESULTS
Effects of maternal diabetes on CNC migration and cardiac outflow tract septation
The effect of maternal diabetes on CNC cell migration and outflow tract septation was examined using embryos carrying a LacZ reporter gene within the coding sequence of one of the Pax3 alleles. The LacZ reporter allows the fate of Pax3-expressing cells to be mapped but interrupts production of a functional Pax3 protein product from the recombined allele. Male Pax3LacZ/+ mice were mated with wild type FVB females that had been made diabetic with STZ and treated with subcutaneous insulin pellets, or with nondiabetic control females. The blood glucose concentration in STZ diabetic mice was 24.72 ± 0.99 mmol/l before insulin pellet insertion, and then it was normalized to 8.47 ± 1.7 mmol/l by the insulin pellets. Beginning on day 4.5 of pregnancy, blood glucose levels rose to 16.43 ± 0.18 mmol/l in the diabetic mice, whereas it remained at 8.98 ± 0.40 mmol/l in nondiabetic mice. STZ diabetic mice remained hyperglycemic until they were killed to recover embryos (Table 1).
Table 1.
Effect of maternal diabetes on CNC cell migration and outflow tract septation
| Blood glucose (mmol/l) | CNC migration | Blood glucose (mmol/l) | Outflow tract septation | |||
|---|---|---|---|---|---|---|
| normal | defective | normal | defective | |||
| Nondiabetic | 8.83 ± 0.31 | 28 | 0 | 9.11 ± 0.31 | 20 | 0 |
| Diabetic | 26.17 ± 2.17a | 0 | 19a | 23.73 ± 1.93a | 3 | 6b |
Day 9.5 embryos or day 16.5 fetuses were obtained from diabetic and non-diabetic FVB female mice that had been mated with Pax3LacZ/+ males. Embryos were processed with X-GAL, and normal or defective CNC cell migration of Pax3LacZ/+ embryos was scored blindly. Cardiac outflow tracts were examined and scored as normal or defective. Only data from embryos subsequently genotyped as Pax3LacZ/+ were included. Blood glucose concentrations are the morning fed glucose readings on the day of dissection ± S.E.M. Differences between blood glucose concentrations were analyzed by unpaired student’s t test, and effects of treatments on CNC migration were analyzed by χ2 test.
p < 0.0001 vs. nondiabetic
p = 0.002 vs. nondiabetic
To study the effects of maternal diabetes on CNC migration, embryos were recovered on day 9.5, when CNC cells can be observed migrating from between the somites and the otic pit toward the heart. CNC migration of Pax3LacZ/+ embryos was scored blindly as “normal” or “defective”. Normal CNC cell migration was exhibited by all of the Pax3LacZ/+ embryos of nondiabetic mice, but in all of the embryos of diabetic mice, migrating CNC cells were either scarce or undetectable (Figure 1A and B and Table 1). Of note, few, if any, β-galactosidase-positive CNC cells were observed emerging from the region between the upper somite and the otic pit in the embryo of the diabetic mouse.
Figure 1. CNC cell migration, apoptosis and outflow tract septation in Pax3LacZ/+ embryos of nondiabetic and diabetic mice.
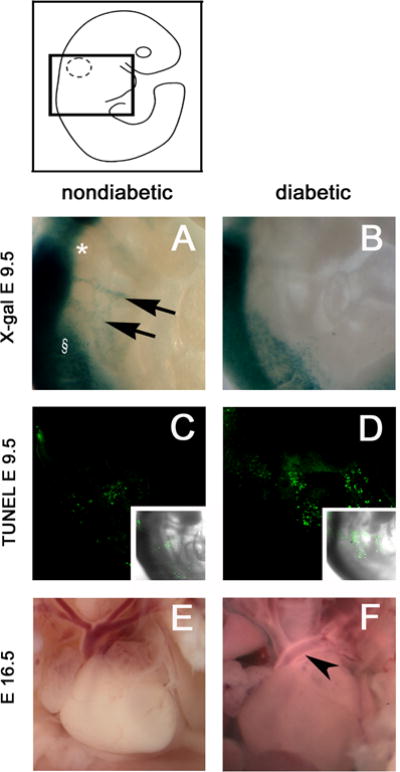
The approximate area of embryos shown in figures 1–4 is indicated within the boxed area of the embryo diagram. (A-D) Pax3LacZ/+ embryos were recovered on day 9.5 from diabetic or nondiabetic FVB females that had been crossed with Pax3LacZ/+ males. Embryos were reacted with X-GAL (A, B) to visualize CNC cells, or reacted with a whole-mount TUNEL procedure (C, D) to visualize apoptotic nuclei. The fluorescence overlay of a differential interference contrast (DIC) image of the embryos are shown in the insets. (A, C) Pax3LacZ/+ embryos of nondiabetic mice. (B, D) Pax3LacZ/+ embryos of diabetic mice. In image A the otic pit is marked by *, the first somite is marked by §, and the migrating CNC are marked by arrows. (E-F) Pax3LacZ/+ fetuses were recovered on day 16.5 from crosses of diabetic or nondiabetic FVB females with Pax3LacZ/+ males and cardiac outflow tracts were examined. Images of fetal hearts from (E) non-diabetic pregnancy shows the normal outflow tract arrangement, whereas (F) from a diabetic pregnancy shows a typical outflow tract defects in which the aorta and left pulmonary artery have not correctly separated into distinct vessels (see arrow head).
The scarcity of β-galactosidase-positive CNC on day 9.5 appears to be due to CNC apoptosis. As shown in Figure 1C, there were very few TUNEL-positive cells along the path of CNC cell migration in the embryo of the nondiabetic mouse, but, as shown in Figure 1D, there were a large number of TUNEL-positive cells in the embryo of the diabetic mouse.
Maternal diabetes also caused outflow tract defects in day 16.5 Pax3LacZ/+ fetuses of diabetic mice (Figure 1E and F, Table 1). By day 16.5, the aorta, pulmonary arteries, and aortic arch arteries are established in normally developing fetuses, but Pax3-deficient fetuses die from cardiac insufficiency and are rarely recovered after day 16.5. Typically, the ascending aorta was not flexed correctly due to failure to separate from one or both pulmonary arteries, and the 4th pharyngeal arch artery (aortic arch) was interrupted or absent. These types of defects may also occur during diabetic pregnancy of humans (Ferencz and others; Loffredo and others, 2001), and are seen in animal models as a result of CNC ablation (Bockman and others, 1987; Kirby and others, 1985) or Splotch Pax3 alleles (Franz, 1989).
Maternal hyperglycemia-induced oxidative stress is responsible for disturbed CNC migration
In the experiments above, the mothers were diabetic throughout the pregnancy until embryos were recovered. Therefore, it cannot be determined whether the defective CNC migration, apoptosis, and outflow tract septation was caused by the diabetic milieu while these processes were taking place, or whether a prior disturbance was responsible. Maternal hyperglycemia, occurring within an approximately 10 hour time interval on day 7.5 of pregnancy, is sufficient to inhibit Pax3 expression and to induce NTDs, even though Pax3 expression does not begin until day 8.5, by which time euglycemia has been restored (Fine and others, 1999). Furthermore, induction of oxidative stress with a single administration of antimycin A, a mitochondrial complex III inhibitor (Boveris, 1977; Turrens and others, 1985), at noon on day 7.5 has the same effect on Pax3 expression and NTDs as maternal hyperglycemia, and administration of antioxidants blocks the effects of hyperglycemia (Chang and others, 2003). If induction of maternal hyperglycemia or oxidative stress on day 7.5 inhibits CNC migration and induces outflow tract defects, this would indicate that the defective CNC migration and outflow tract septation in embryos of diabetic mice were due to adverse effects on CNC induction, such as processes that lead to induction of Pax3 expression, that occur on day 7.5, rather than adverse effects while CNC are migrating or outflow tracts are septating.
To test this, FVB females that had been mated with Pax3LacZ/+ males were made hyperglycemic with glucose injections on day 7.5 as described (Fine and others, 1999). Control females were injected with PBS. Blood glucose concentrations in glucose-injected mice on day 7.5 were significantly elevated (Table 2) and were similar to those of mice that were diabetic throughout pregnancy (Table 1). Whereas none of the Pax3LacZ/+ embryos of control mice exhibited defective CNC cell migration, all of the Pax3LacZ/+ embryos of mice that had been made hyperglycemic exhibited defective CNC cell migration (Figure 2A and B, Table 2). The scarcity of LacZ-positive CNC cells on day 9.5 following transient maternal hyperglycemia on day 7.5 appears to be due to loss of CNC cells by apoptosis, as there were more TUNEL-positive cells along the path of CNC cell migration in embryos from glucose-treated compared to embryos of PBS controls (Figure 2G and H).
Table 2.
Effect of maternal hyperglycemia and antioxidants on embryonic CNC cell migration
| Treatment | Pax3LacZ/+ | Pax3GFP/+ | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Blood glucose (mmol/l) | normal | CNC defect | Blood glucose (mmol/l) | normal | CNC defect | |
| PBS | 7.8 ± 0.35 | 21 | 0 | 9.02 ± 0.3 | 15 | 0 |
| Glucose | 21.21 ± 0.55a | 0 | 36a | 21.14 ± 0.59a | 0 | 15a |
| PBS + GSH-EE | 8.63 ± 0.08b | 20 | 0 | 8.27 ± 0.06b | 16 | 0 |
| PBS + Vit E | 8.31 ± 0.07 b | 14 | 0 | 7.59 ± 0.16 b | 18 | 0 |
| Glucose + GSH-EE | 20.73 ± 0.4a | 15 | 5b | 20.49 ± 0.68a | 11 | 5b |
| Glucose + Vit E | 19.61 ± 0.57a | 14 | 5b | 20.67 ± 0.77a | 11 | 4b |
FVB female mice were mated with Pax3LacZ/+ or Pax3GFP/+ males and were made hyperglycemic with subcutaneous glucose injection on day 7.5. PBS was administered as a control. GSH-EE was administered in three doses on day 7.5 to PBS- or glucose-injected mice. Vitamin E succinate-supplemented chow was fed beginning on day 0.5 to PBS- or glucose-injected mice. Embryos were obtained on day 9.5. Pax3LacZ/+ embryos were processed with X-GAL, and Pax3GFP/+ embryos were imaged by confocal microscopy. Blood glucose concentrations are the mean hourly readings on day 7.5 ± S.E.M. Differences between blood glucose concentrations were analyzed by ANOVA and Neuman-Keul’s post test, and effects of treatments on CNC migration were analyzed by χ2 test.
p < 0.0001 vs. PBS
p < 0.0001 vs. glucose; NS vs. PBS
Figure 2. CNC cell migration and apoptosis in embryos of control and hyperglycemic mice, without or with antioxidant treatment.
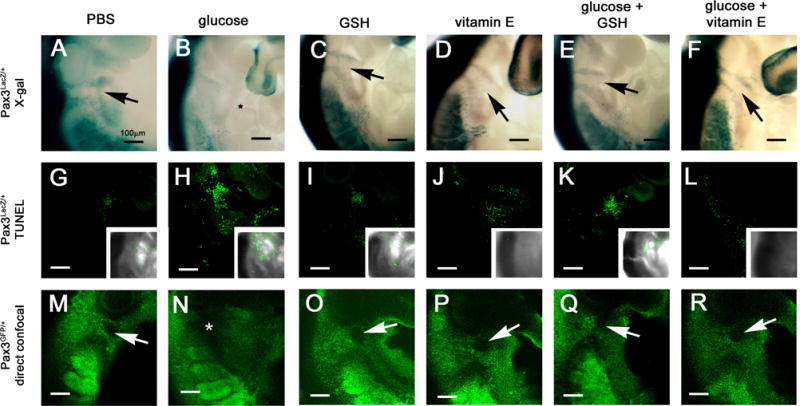
FVB females were crossed with either Pax3LacZ/+ or Pax3GFP/+ males and embryos were recovered on day 9.5. Pax3LacZ/+ embryos were reacted with X-GAL to visualize CNC migration (A-F), or subjected to a whole-mount TUNEL assay to visualize apoptosis (G-L). Pax3GFP/+ embryos were examined by confocal microscopy (M-R). (A, G, and M) embryos from pregnancies treated on day 7.5 with PBS at the same times that s.c. glucose was administered. (B, H, and N) embryos from pregnancies in which females were injected with glucose on day 7.5 to induce transient hyperglycemia. (C, I, O) embryos from pregnancies injected with GSH-EE on day 7.5. (D, J, P) embryos from pregnancies treated with supplemental vitamin E beginning on day 0.5. (E, K, Q) embryos from pregnancies treated with glucose and GSH-EE on day 7.5. (F, L, R) embryos from pregnancies treated with glucose on day 7.5 and supplemental vitamin E beginning on day 0.5 The location of absent CNC cells is marked by * in B and N. Arrows point to between the two streams of LacZ- or GFP-positive CNC cells.
The LacZ coding sequence interrupts the Pax3 coding sequence; consequently, Pax3LacZ/+ embryos are haploinsufficient for Pax3. To test whether similar defects in CNC migration in response to maternal hyperglycemia occur in embryos with two functional Pax3 alleles, Pax3GFP/+mice, in which a GFP reporter sequence was inserted downstream of the Pax3 coding sequence, were employed. Male Pax3GFP/+mice were crossed with female FVB mice, and the female mice were made hyperglycemic, or not, as above. The glucose-injected mice were significantly more hyperglycemic than PBS-injected mice (Table 2). As determined by confocal microscopy, normal migration of CNC was observed in all Pax3GFP/+ embryos of PBS-injected mice, whereas it was severely defective in all embryos of glucose-injected mice (Figure 2M and N, Table 2). These results indicate that the defective CNC migration in Pax3LacZ/+ embryos of diabetic or hyperglycemic mothers was not due to specific susceptibility of Pax3 haploinsufficient embryos to hyperglycemia-induced CNC maldevelopment.
To investigate whether oxidative stress was responsible for the adverse effects of maternal hyperglycemia on CNC migration, the effects of blocking oxidative stress in hyperglycemic pregnancies were tested. Pregnant FVB females that had been mated with either Pax3LacZ/+ or Pax3GFP/+ males were made hyperglycemic as above, and either injected with glutathione ethyl ester (GSH-EE), a membrane-permeable reduced glutathione precursor, on day 7.5, or fed chow supplemented with vitamin E succinate beginning on day 0.5 of pregnancy. Neither GSH-EE nor vitamin E had any effect on glucose concentrations in either PBS- or glucose-injected mice (Table 2). There was no difference in CNC cell migration in embryos of control mice treated with GSH-EE or vitamin E alone, but either treatment significantly increased the number of Pax3LacZ/+ or Pax3GFP/+ embryos of glucose-injected mice that displayed normal CNC migration (Table 2). Examples of CNC migration in embryos from glucose- and antioxidant-treated pregnancies can be seen in Figure 2. The normalization of CNC migration by antioxidants is associated with fewer apoptotic cells.
Inducing oxidative stress replicates the effects of hyperglycemia on CNC migration
To test whether inducing oxidative stress independent of hyperglycemia was sufficient to induce abnormal CNC migration, antimycin A was administered on day 7.5 of pregnancy to FVB females that had been mated with either Pax3LacZ/+ or Pax3GFP/+ males. Antimycin A inhibits Pax3 expression without having any effect on blood glucose concentrations (Chang and others, 2003). There was a significant effect of antimycin A to cause defective CNC migration in both Pax3LacZ/+ and Pax3GFP/+ embryos, and this was correlated with an increase in apoptotic cells within the field of CNC migration (Figure 3, Table 3). There was a significant effect of the antioxidants, GSH-EE and vitamin E, to protect CNC migration from the adverse effects of antimycin A (Figure 3, Table 3).
Figure 3. CNC migration in embryos following induction of oxidative stress without or with antioxidants.
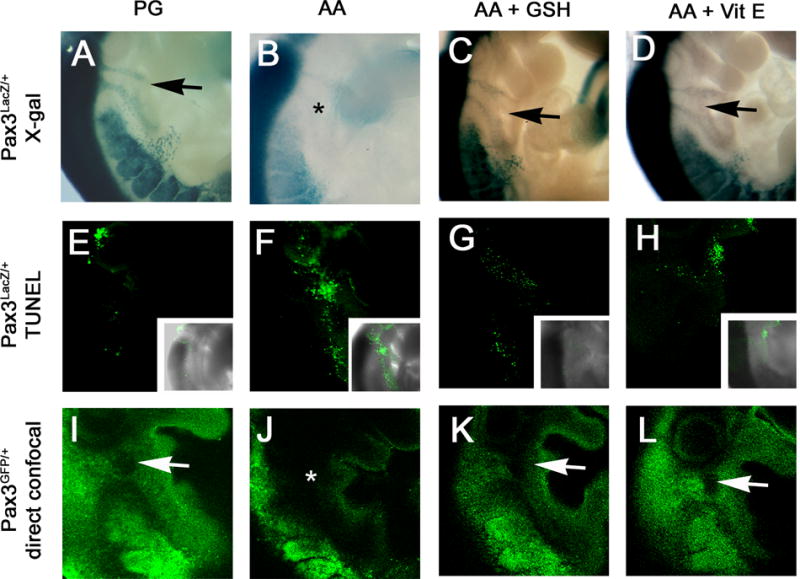
FVB females were crossed with either Pax3LacZ/+ or Pax3GFP/+ males. On day 7.5 of pregnancy, the females were injected with antimycin A (AA) or propylene glycol (PG) as a control. AA-treated pregnancies were also administered GSH-EE on day 7.5, or received supplemental dietary vitamin E succinate beginning on day 0.5. Embryos were obtained on day 9.5. Pax3LacZ/+ embryos were reacted with X-GAL to visualize CNC migration (A-D), or subjected to a whole-mount TUNEL assay to visualize apoptosis (E-H). Pax3GFP/+ embryos were examined by confocal microscopy (I-L). (A, E, and I) embryos from PG-treated pregnancies. (B, F and J) embryos from AA-treated pregnancies. (C, G, and K) embryos from AA plus GSH-EE-treated pregnancies. (D, H, and L) embryos from AA-treated pregnancies supplemented with vitamin E. The location of of absent CNC cell migration is marked by *. Arrows point to between the two streams of LacZ- or GFP-positive CNC cells.
Table 3.
Effect of oxidative stress and antioxidant treatment on embryonic CNC cell migration
| Treatment | Pax3LacZ/+ | Pax3GFP/+ | ||
|---|---|---|---|---|
| normal | CNC defect | normal | CNC defect | |
| PG | 19 | 1 | 12 | 0 |
| AA | 1 | 28a | 1 | 11a |
| AA + GSH-EE | 16 | 5b | 10 | 6c |
| AA + Vit E | 15 | 2 b | 13 | 3c |
Pax3LacZ/+ or Pax3GFP/+ embryos from control propylene glycol- (PG) or antimycin A- (AA) injected pregnancies were obtained on day 9.5 and examined for CNC migration defects. Antioxidants (GSH-EE or vitamin E succinate) were administered as in Table 2.
p < 0.0001 vs. PG
p < 0.0001 vs. AA; NS vs. PG
p = 0.003 vs. AA; NS vs. PG
Maternal hyperglycemia, which leads to oxidative stress, causes outflow tract defects
To test whether maternal hyperglycemia, leading to oxidative stress, caused cardiac outflow tract defects, in addition to defective CNC migration, fetuses of mice that had been made hyperglycemic, or treated with antimycin A, with or without GSH-EE or vitamin E, as above, were recovered on day 16.5, and their cardiac outflow tracts were examined.
The single primitive outflow tract had correctly septated into the aorta and the pulmonary arteries in all fetuses of control mice (those injected with PBS or PG vehicle alone on day 7.5, or administered GSH-EE or vitamin E) (Table 4). There was a significant increase in outflow tract defects, including partial or complete truncus arteriosus, and absence or misplacement of one or more of the great arteries, in fetuses of mice made hyperglycemic or administered antimycin A on day 7.5. Notably, GSH-EE or vitamin E significantly reduced outflow tract defects caused by maternal hyperglycemia or antimycin A. Examples of the defects caused by hyperglycemia or antimycin A, and protection by GSH-EE or vitamin E, in whole mount and in cross section are shown in Figure 4.
Table 4.
Effect of maternal hyperglycemia, oxidative stress and anti-oxidants on fetal outflow tract development
| Treatment | normal | OFT defect |
|---|---|---|
| Controls: | ||
| PBS | 26 | 0 |
| PG | 27 | 0 |
| GSH | 17 | 0 |
| Vit E | 26 | 0 |
| Hyperglycemia: | ||
| Glucose | 24 | 32a |
| Glucose + GSH | 21 | 4b |
| Glucose + Vit E | 22 | 4b |
| Oxidative Stress: | ||
| AA | 6 | 18a |
| AA + GSH | 20 | 2c |
| AA + Vit E | 22 | 6d |
Fetuses from control (PBS- or PG-injected), glucose-, or AA-injected FVB pregnancies were obtained on day 16.5 and the fetal hearts were examined for cardiac outflow tract (OFT) defects.
p < 0.0001 vs. PBS or PG
p = 0.0006 vs. glucose, NS vs. PBS
p < 0.0001 vs. AA, NS vs. PG
p = 0.0005 vs. AA, NS vs. PG
Figure 4. Effect of maternal hyperglycemia and oxidative stress on cardiac outflow tract septation.
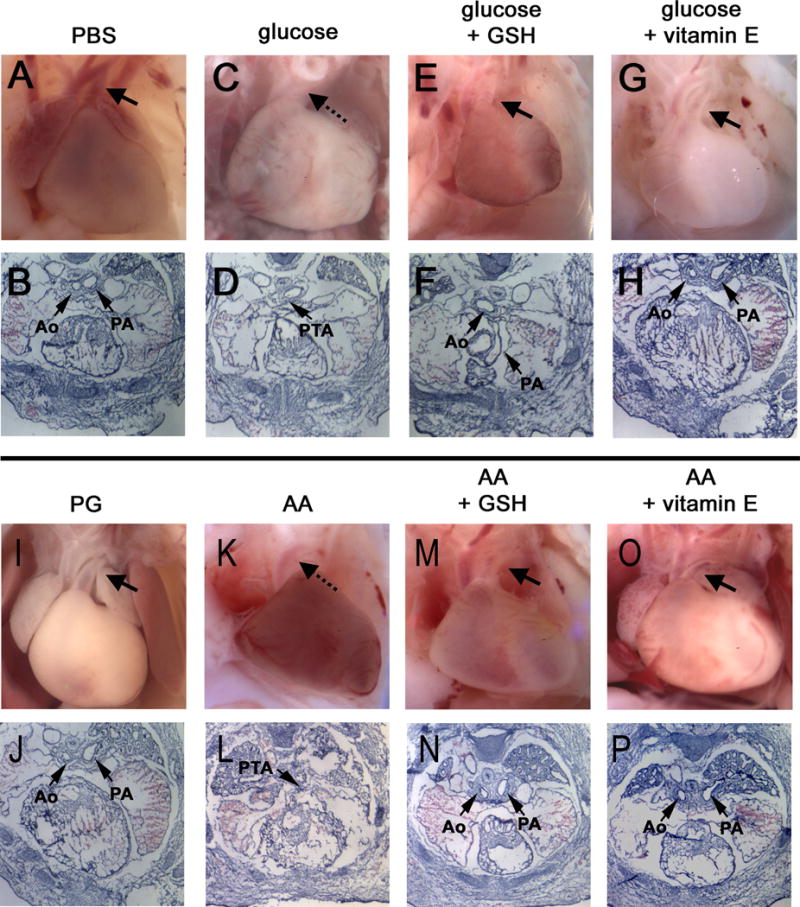
Hyperglycemia or oxidative stress was induced in pregnant FVB mice on day 7.5 as in Figures 2 and 3. GSH-EE was administered on day 7.5, and vitamin E was administered with chow beginning on day 0.5. Fetuses were recovered on day 16.5 and cardiac outflow tracts were examined either in situ in whole embryos, or in serial sections. In fetuses from glucose- (C, D) and AA- (K, L) injected pregnancies, ascending aortas are lying juxtaposed to the left pulmonary vessel, which has not correctly separated into a distinct vessel (see dotted arrows). Persistant truncus arteriosus (PTA) can be seen in sectioned hearts. Images of fetal hearts from glucose- (E, F, G, H) and AA- (M, N, O, P) injected pregnancies supplemented with GSH or vitamin E display proper separation of the aorta (Ao) from the pulmonary arteries (PA) and are indistinguishable from these structures from PBS- (A, B) and PG- (I, J) injected control pregnancies. In the whole mount images, in which the atria were dissected from whole mount hearts to aid visualization of the outflow tracts, the left pulmonary arteries can be seen passing under the aortas (arrows). In images of sections, the left pulmonary arteries are observed at the same level as the aortas (arrows).
DISCUSSION
Previous studies to investigate the molecular causes of NTDs in the embryos of diabetic mothers demonstrated that in mouse embryos, expression of Pax3 is suppressed beginning on embryonic day 8.5; subsequently, neuroepithelial cells undergo apoptosis and NTDs occur at increased frequency compared to embryos from nondiabetic pregnancies (Phelan and others, 1997). The neuroectoderm also gives rise to CNC cells, and CNC migration and outflow tract septation are dependent on Pax3 (Conway and others, 1997; Epstein and others, 2000; Franz, 1989). In this study we showed that hyperglycemia and oxidative stress on day 7.5 disturb subsequent CNC migration and cause cardiac outflow tract defects. Treatments which normalize oxidant status (GSH-EE and vitamin E) and which prevent inhibition of Pax3 expression (Chang and others, 2003; Horal and others, 2004) normalized CNC migration and outflow tract formation. Because oxidative stress occurring on day 7.5, well before the onset of CNC delamination and outflow tract septation, was responsible for the disruptive effects of maternal diabetes, this indicates that maternal diabetes does not directly inhibit epithelial to mesenchymal transformation, migration, or remodeling of the outflow tracts. Rather, diabetes-induced oxidative stress interferes with a process that is required for proper CNC migration on day 7.5. Oxidative stress on day 7.5 inhibits Pax3 expression beginning on day 8.5, whereas preventing oxidative stress prevents the inhibition of Pax3 expression (Chang and others, 2003; Fine and others, 1999). Pax3LacZ/LacZ mouse embryos, which carry two null Pax3 alleles, display defective CNC migration, apoptosis, and outflow tract defects (Morgan, et al., submitted) like the embryos of diabetic mice in this study. This suggests that inhibition of Pax3 expression prior to CNC migration is responsible for the defects observed here.
We had previously reported that Pax3 mRNA continued to be decreased at least up to day 11.5 in all tissue derivatives (neural tube, neural crest, somites) of embryos of diabetic mice as determined by in situ hybridization (Phelan and others, 1997). However, from in situ hybridization, we would not have been able to determine whether decreased Pax3 mRNA was due to continued suppression of transcription after day 8.5, or whether it was due to loss of Pax3-expressing cells due to apoptosis. There is a correlation between increased numbers of apoptotic cells within the field of CNC migration and decreased presence of reporter gene-expressing CNC cells on day 9.5 in Pax3LacZ/LacZ embryos (Morgan, et al., submitted) as observed here in embryos of diabetic or hyperglycemic mice. This suggests that CNC cell apoptosis resulting from insufficient production of Pax3 on day 8.5 is the primary explanation for the loss of Pax3 mRNA or β-galactosidase reporter. It is of note that the apoptotic cells were found distant from the neural tube, at locations along the pharyngeal arches where CNC cells would have migrated at the times the embryos were recovered on day 9.5. This indicates that CNC delamination and migration occurred on schedule. However, it was while the cells were migrating through the pharyngeal arches, about 24 hours after Pax3 expression should have been initiated, and approximately 48 hours after oxidative stress had occurred, that cells were eliminated by apoptosis.
It was noteworthy that the apoptosis was not exhibited throughout the embryo, but was limited to locations where Pax3-expressing neural crest cells (including cranial) should be found. This is consistent with our previous demonstration that apoptosis in day 10.5 embryos of diabetic mice or Pax3Sp/Sp embryos was limited to neuroepithelium where NTDs arose (Phelan and others, 1997), and that oxidative stress which is sufficient to inhibit Pax3 expression is not sufficient to directly cause cell death (Chang and others, 2003). Rather, prior insufficient production of Pax3 causes steady-state levels of the p53 tumor suppressor protein to increase, thereby causing apoptosis (Pani and others, 2002). Indeed, defective CNC migration and outflow tract septation in Pax3LacZ/LacZ embryos is prevented by inactivation of p53-dependent apoptosis (Morgan, et al., submitted). This suggests that inhibiting expression of Pax3 below a critical threshold by oxidative stress will lead to subsequent de-repression of p53-dependent apoptosis of CNC cells.
We noticed that there were some embryos with outflow tract defects, but not NTDs, and vice versa, although many embryos with one defect also exhibited the other. In contrast, embryos that are genetically Pax3-deficient (Pax3Sp/Sp or Pax3LacZ/LacZ) always display both cardiac outflow tract and NTDs. This suggests that the magnitude of inhibition of Pax3 expression by hyperglycemia-induced oxidative stress is not the same in all cells. Thus, if there are a critical number of CNC progenitors in which expression of Pax3 is inhibited below the threshold for viability, outflow tract defects will occur, and if there are a critical number of neuroepithelial progenitors in which expression of Pax3 is inhibited below the threshold for viability, NTDs will occur.
In summary, cardiac outflow tract defects in embryos of diabetic mothers appear to result from oxidative stress occurring on day 7.5, leading to subsequent CNC apoptosis. Because oxidative stress on day 7.5 also inhibits expression of Pax3, blocking oxidative stress prevents the inhibition of Pax3, and the same defective CNC migration, apoptosis, and outflow tract defects occur in Pax3-null embryos, this suggests that the molecular mechanism for the outflow tract defects is inhibition of Pax3 expression prior to delamination of CNC from neuroepithelium. It will be important in the future to elucidate the molecular mechanisms by which oxidative stress inhibits expression of Pax3, for example, whether it inhibits the induction or modification of transcription factors which must assemble on the Pax3 transcription control element, or perhaps chromatin modification.
It is of note that the window of opportunity for oxidative stress to cause defective CNC migration and outflow tract septation was not during the several days during which CNC migrate to their destination and the outflow tract is remodeled. Since this stage of development occurs in the human about the time of implantation (Moore and Persaud, 1993), when a woman may not yet be aware that she is pregnant, this indicates that it is critical to avoid episodes of hyperglycemia within this time period to avoid inducing outflow tract defects in the susceptible embryo.
Acknowledgments
We are grateful to Michelle Ocana, Mark Chafel and Daniel Tom of the Optical Imaging Facility at the Harvard Center for Neurodegeneration and Repair for help with confocal microscopy. We appreciate the contributions of John M. Lavorse and Shirley Tilghman for the generation of the Pax3GFP/+ mouse line, and of Margaret E. Buckingham for providing the Pax3nLacZ/+ (Pax3LacZ/+) mouse line. Resources in the Joslin Specialized Microscopy Core were supported by a DERC grant to the Joslin Diabetes Center. This work was supported by grants from the American Diabetes Association (1-04-RA-65) and the National Institutes of Health (RO1 DK52865) to MRL.
Research supported by 1-04-RA-65 from the American Diabetes Association and DK52865 from the National Institutes of Health (to MRL)
Abbreviations
- CNC
cardiac neural crest
- GSH
glutathione
- GSH-EE
glutathione ethyl ester
- GFP
Green Fluorescent Protein
- IRES
internal ribosome entry site
- NTD
neural tube defect
- STZ
streptozotocin
- s.c.
subcutaneous
- AA
antimycin A
- FITC
fluorescein isothiocyanate
Footnotes
Portions of this paper were presented at the 67th Annual Meeting of the American Diabetes Association, June 22–26, 2007, Chicago, IL
LITERATURE CITED
- Anderson DJ. Cellular and molecular biology of neural crest cell lineage determination. Trends Genet. 1997;13(7):276–280. doi: 10.1016/s0168-9525(97)01187-6. [DOI] [PubMed] [Google Scholar]
- Besson WT, Kirby ML, Mierop LHV, Teabeaut JR. Effects of the size of lesions of the cardiac neural crest at various embryonic ages on incidence and type of cardiac defects. Circulation. 1986;73(2):360–364. doi: 10.1161/01.cir.73.2.360. [DOI] [PubMed] [Google Scholar]
- Bockman DE, Redmond ME, Waldo K, Davis H, Kirby ML. Effect of neural crest ablation on development of the heart and arch arteries in the chick. Am J Anat. 1987;180(4):332–341. doi: 10.1002/aja.1001800403. [DOI] [PubMed] [Google Scholar]
- Boveris A. Mitochondrial production of superoxide radical and hydrogen peroxide. Adv Exp Med Biol. 1977;78:67–82. doi: 10.1007/978-1-4615-9035-4_5. [DOI] [PubMed] [Google Scholar]
- Chan WY, Cheung CS, Yung KM, Copp AJ. Cardiac neural crest of the mouse embryo: axial level of origin, migratory pathway and cell autonomy of the splotch (Sp2H) mutant effect. Development. 2004;131(14):3367–3379. doi: 10.1242/dev.01197. [DOI] [PubMed] [Google Scholar]
- Chang TI, Horal M, Jain S, Wang F, Patel R, Loeken MR. Oxidant regulation of gene expression and neural tube development: Insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia. 2003;46:538–545. doi: 10.1007/s00125-003-1063-2. [DOI] [PubMed] [Google Scholar]
- Chung CS, Myrianthopoulos NC. Factors affecting risks of congenital malformations. II. Effect of maternal diabetes on congenital malformations. Birth Defects. 1975;11(10):23–38. [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Copp AJ. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development. 1997;124(2):505–514. doi: 10.1242/dev.124.2.505. [DOI] [PubMed] [Google Scholar]
- Creazzo TL, Godt RE, Leatherbury L, Conway SJ, Kirby ML. Role of cardiac neural crest cells in cardiovascular development. Annu Rev Physiol. 1998;60:267–286. doi: 10.1146/annurev.physiol.60.1.267. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin F, Lu MM, Thomas M, Liu E, Wessels A, Lo CW. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127(9):1869–1870. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- Evers IM, de Valk HW, Visser GH. Risk of complications of pregnancy in women with type 1 diabetes: nationwide prospective study in the Netherlands. BMJ. 2004;328(7445):915–919. doi: 10.1136/bmj.38043.583160.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell T, Neale L, Cundy T. Congenital anomalies in the offspring of women with Type 1, Type 2 and gestational diabetes. Diabetic Medicine. 2002;19(4):322–326. doi: 10.1046/j.1464-5491.2002.00700.x. [DOI] [PubMed] [Google Scholar]
- Ferencz C, Rubin JD, McCarter RJ, Clark EB. Maternal diabetes and cardiovascular malformations: predominance of double outlet right ventricle and truncus arteriosus. Teratology. 1990;41(3):319–326. doi: 10.1002/tera.1420410309. [DOI] [PubMed] [Google Scholar]
- Fine E, Horal M, Chang T, Fortin G, Loeken M. Hyperglycemia is responsible for altered gene expression, apoptosis, and neural tube defects associated with diabetic pregnancy. Diabetes. 1999;48:2454–2462. doi: 10.2337/diabetes.48.12.2454. [DOI] [PubMed] [Google Scholar]
- Franz T. Persistent truncus arteriosus in the Splotch mutant mouse. Anat Embryol (Berl) 1989;180(5):457–464. doi: 10.1007/BF00305120. [DOI] [PubMed] [Google Scholar]
- Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991;10(5):1135–1147. doi: 10.1002/j.1460-2075.1991.tb08054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene MF, Hare JW, Cloherty JP, Benacerraf BR, Soeldner JS. First-trimester hemoglobin A1 and risk for major malformation and spontaneous abortion in diabetic pregnancy. Teratology. 1989;39:225–231. doi: 10.1002/tera.1420390303. [DOI] [PubMed] [Google Scholar]
- Gruss P, Walther C. Pax in development. Cell. 1992;69(5):719–722. doi: 10.1016/0092-8674(92)90281-g. [DOI] [PubMed] [Google Scholar]
- Hanson U, Persson B, Thunell S. Relationship between haemoglobin A1C in early type 1 (insulin-dependent) diabetic pregnancy and the occurrence of spontaneous abortion and fetal malformation in Sweden. Diabetologia. 1990;33(2):100–104. doi: 10.1007/BF00401047. [DOI] [PubMed] [Google Scholar]
- Horal M, Zhang Z, Virkamaki A, Stanton R, Loeken MR. Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: Involvement in diabetic teratogenesis. Birth Defects Res Part A Clin Mol Teratol. 2004;70:519–527. doi: 10.1002/bdra.20056. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127(8):1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kirby ML. Cardiac morphogenesis–recent research advances. Pediatr Res. 1987;21(3):219–224. doi: 10.1203/00006450-198703000-00001. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220(4601):1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Turnage KL, Hays BM. Characterization of conotruncal malformations following ablation of “cardiac” neural crest. Anat Rec. 1985;213(1):87–93. doi: 10.1002/ar.1092130112. [DOI] [PubMed] [Google Scholar]
- Li J, Liu KC, Jin F, Lu MM, Epstein JA. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development. 1999;126(11):2495–2503. doi: 10.1242/dev.126.11.2495. [DOI] [PubMed] [Google Scholar]
- Loffredo CA, Wilson PD, Ferencz C. Maternal diabetes: an independent risk factor for major cardiovascular malformations with increased mortality of affected infants. Teratology. 2001;64(2):98–106. doi: 10.1002/tera.1051. [DOI] [PubMed] [Google Scholar]
- Macintosh MCM, Fleming KM, Bailey JA, Doyle P, Modder J, Acolet D, Golightly S, Miller A. Perinatal mortality and congenital anomalies in babies of women with type 1 or type 2 diabetes in England, Wales, and Northern Ireland: population based study. BMJ. 2006;333(7560):177. doi: 10.1136/bmj.38856.692986.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin DGM, Roest PAM, Nordstrand H, Wisse LJ, Poelmann RE, Eriksson UJ, Groot ACG-D. Disturbed morphogenesis of cardiac outflow tract and increased rate of aortic arch anomalies in the offspring of diabetic rats. Birth Defects Research (Part A) 2004;70(12):927–938. doi: 10.1002/bdra.20101. [DOI] [PubMed] [Google Scholar]
- Moore KL, Persaud TVN. The Developing Human–Clinically Oriented Embryology. Philadelphil: W.B Saunders Co; 1993. [Google Scholar]
- Natoli TA, Ellsworth MK, Wu C, Gross KW, Pruitt SC. Positive and negative DNA sequence elements are required to establish the pattern of Pax-3 expression. Development. 1997;124:617–626. doi: 10.1242/dev.124.3.617. [DOI] [PubMed] [Google Scholar]
- Nielsen GL, Norgard B, Puho E, Rothman KJ, Sorensen HT, Czeizel AE. Risk of specific congenital abnormalities in offspring of women with diabetes. Diabetic Medicine. 2005;22(6):693–696. doi: 10.1111/j.1464-5491.2005.01477.x. [DOI] [PubMed] [Google Scholar]
- Nishibatake M, Kirby ML, Mierop LHV. Pathogenesis of persistent truncus arteriosus and dextroposed aorta in the chick embryo after neural crest ablation. Circulation. 1987;75(1):255–264. doi: 10.1161/01.cir.75.1.255. [DOI] [PubMed] [Google Scholar]
- Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3- dependent development and tumorigenesis. Genes Dev. 2002;16(6):676–680. doi: 10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan SA, Ito M, Loeken MR. Neural tube defects in embryos of diabetic mice: Role of the Pax-3 gene and apoptosis. Diabetes. 1997;46:1189–1197. doi: 10.2337/diab.46.7.1189. [DOI] [PubMed] [Google Scholar]
- Relaix F, Polimeni M, Rocancourt D, Ponzetto C, Schafer BW, Buckingham M. The transcriptional activator PAX3-FKHR rescues the defects of Pax3 mutant mice but induces a myogenic gain-of-function phenotype with ligand-independent activation of Met signaling in vivo. Genes Dev. 2003;17(23):2950–2965. doi: 10.1101/gad.281203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix F, Rocancourt D, Mansouri A, Buckingham M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 2004;18(9):1088–1105. doi: 10.1101/gad.301004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson EJD, He S-J, Eccles MR. A PANorama of PAX genes in cancer and development. Nat Rev Cancer. 2006;6(1):52–62. doi: 10.1038/nrc1778. [DOI] [PubMed] [Google Scholar]
- Roest PA, van Iperen L, Vis S, Wisse LJ, Poelmann RE, Steegers-Theunissen RP, Molin DG, Eriksson UJ, Gittenberger-De Groot AC. Exposure of neural crest cells to elevated glucose leads to congenital heart defects, an effect that can be prevented by N-acetylcysteine. Birth Defects Res A Clin Mol Teratol. 2007;79(3):231–235. doi: 10.1002/bdra.20341. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T, Bronner-Fraser M. Development and evolution of the migratory neural crest: a gene regulatory perspective. Current Opinion in Genetics & Development. 2006;16(4):360–366. doi: 10.1016/j.gde.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Schaefer-Graf UM, Buchanan TA, Xiang A, Songster G, Montoro M, Kjos SL. Patterns of congenital anomalies and relationship to initial maternal fasting glucose levels in pregnancies complicated by type 2 and gestational diabetes. Am J Obstet Gynecol. 2000;182(2):313–320. doi: 10.1016/s0002-9378(00)70217-1. [DOI] [PubMed] [Google Scholar]
- Siman CM, Gittenberger-De Groot AC, Wisse B, Eriksson UJ. Malformations in offspring of diabetic rats: morphometric analysis of neural crest-derived organs and effects of maternal vitamin E treatment. Teratology. 2000;61(5):355–367. doi: 10.1002/(SICI)1096-9926(200005)61:5<355::AID-TERA7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Suhonen L, Hiilesmaa V, Teramo K. Glycaemic control during early pregnancy and fetal malformations in women with type I diabetes mellitus. Diabetologia. 2000;43(1):79–82. doi: 10.1007/s001250050010. [DOI] [PubMed] [Google Scholar]
- Towner D, Kjos SL, Leung B, Montoro MM, Xiang A, Mestman JH, Buchanan TA. Congenital malformations in pregnancies complicated by NIDDM. Diabetes Care. 1995;18(11):1446–1451. doi: 10.2337/diacare.18.11.1446. [DOI] [PubMed] [Google Scholar]
- Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237(2):408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- Wren C, Birrell G, Hawthorne G. Cardiovascular malformations in infants of diabetic mothers. Heart. 2003;89(10):1217–1220. doi: 10.1136/heart.89.10.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Cummings EA, O’Connell C, Jangaard K. Fetal and Neonatal Outcomes of Diabetic Pregnancies. Obstet Gynecol. 2006;108(3):644–650. doi: 10.1097/01.AOG.0000231688.08263.47. [DOI] [PubMed] [Google Scholar]