

Abstract
Over 50% of patients who survive neuroinvasive infection with West Nile virus (WNV) exhibit chronic cognitive sequelae1,2. Although thousands of cases of WNV-mediated memory dysfunction accrue annually3, the mechanisms responsible for these impairments are unknown. The classical complement cascade, a key component of innate immune pathogen defence, mediates synaptic pruning by microglia during early postnatal development4,5. Here we show that viral infection of adult hippocampal neurons induces complement-mediated elimination of presynaptic terminals in a murine WNV neuroinvasive disease model. Inoculation of WNV-NS5-E218A, a WNV with a mutant NS5(E218A) protein6,7 leads to survival rates and cognitive dysfunction that mirror human WNV neuroinvasive disease. WNV-NS5-E218A-recovered mice (recovery defined as survival after acute infection) display impaired spatial learning and persistence of phagocytic microglia without loss of hippocampal neurons or volume. Hippocampi from WNV-NS5-E218A-recovered mice with poor spatial learning show increased expression of genes that drive synaptic remodelling by microglia via complement. C1QA was upregulated and localized to microglia, infected neurons and presynaptic terminals during WNV neuroinvasive disease. Murine and human WNV neuroinvasive disease post-mortem samples exhibit loss of hippocampal CA3 presynaptic terminals, and murine studies revealed microglial engulfment of presynaptic terminals during acute infection and after recovery. Mice with fewer microglia (Il34−/− mice with a deficiency in IL-34 production) or deficiency in complement C3 or C3a receptor were protected from WNV-induced synaptic terminal loss. Our study provides a new murine model of WNV-induced spatial memory impairment, and identifies a potential mechanism underlying neurocognitive impairment in patients recovering from WNV neuroinvasive disease.
Studies in humans and rodents indicate that WNV targets neurons within the hippocampus8, which is essential for spatial and contextual memory formation9. Patients that survive WNV neuroinvasive disease often exhibit impaired visuospatial processing and memory1,10. In post-mortem samples of patients with WNV neuroinvasive disease and in rodent models with low survival rates (<50%), significant neuronal loss, inflammation, and microglial activation occur within infected brain regions11–14. However, the extent of viral burden and neuron loss may be much lower in individuals who survive WNV neuroinvasive disease and may vary between brain regions. In addition, host-pathogen interactions could explain the range of cognitive sequelae experienced by WNV neuroinvasive disease survivors1.
Mechanisms underlying cognitive impairments in patients recovering from West Nile neuroinvasive disease are unknown, mainly due to lack of murine recovery models. Current models that use virulent WNV strains yield either 100% death following intracranial WNV infection or 10–70% survival and variable CNS viral burdens following peripheral routes of infection15. To circumvent this in order to develop a model of recovery from WNV neuroinvasive disease, we used a strain of WNV with a point mutation in nonstructural protein 5 NS5(E218A), which lacks functional 2′-O methyltransferase that generates a 5′ cap on viral RNA to evade type I interferon-mediated restriction of viral translation6,16. While WNV-NS5-E218A replicates in permissive cells, 90% of 8-week-old mice survive intracranial inoculation (Extended Data Fig. 1a), with uniform viral brain burdens peaking between 6–8-days post-infection (dpi)7, followed by viral clearance at 15 dpi (Extended Data Fig. 1b, c). Intracranial infection with WNV-NS5-E218A induces neuroinflammation within 7 dpi, with numbers and phenotypes of infiltrating leukocytes similar to intracranial infection with virulent strain WNV-NY99 (Extended Data Fig. 1d), and consistent with data demonstrating early reversion of WNV-NS5-E218A to wild-type virus within the central nervous system7. WNV-NS5-E218A intracranial infection results in few apoptotic neurons (0.5–1.5%) at 7 dpi (Extended Data Fig. 1e), similar to 8-week-old mice after footpad infection with WNV-NY99 (ref. 12). Thus, WNV-NS5-E218A may be used to examine behavioural, cellular, and molecular mechanisms of recovery from WNV neuroinvasive disease.
The Barnes maze behavioural task was used to determine whether WNV-NS5-E218A-recovered mice exhibit neurocognitive deficits17. At 46 dpi, WNV-infected mice exhibit slower learning, commit more errors (Fig. 1a) and require more time (Fig. 1b) before locating the target hole than mock-infected controls (Supplementary Videos 1 and 2). Studies performed in mice at 22 dpi showed similar effects (Fig. 1e and Extended Data Fig. 1f). WNV-NS5-E218A-recovered animals improve over the 5 days of training, although they continue to make significantly more errors than controls on each day. Recovered animals do not exhibit impairments in motor activity or exploratory anxiety in an open-field behavioural assessment at 45 dpi (Fig. 1c, d) or 21 dpi (Extended Data Fig. 1g), indicating that impairments in Barnes maze solving are specific to spatial learning.
Figure 1. WNV-mediated spatial learning and memory impairments and activated microglia persist beyond 45 days post-infection.
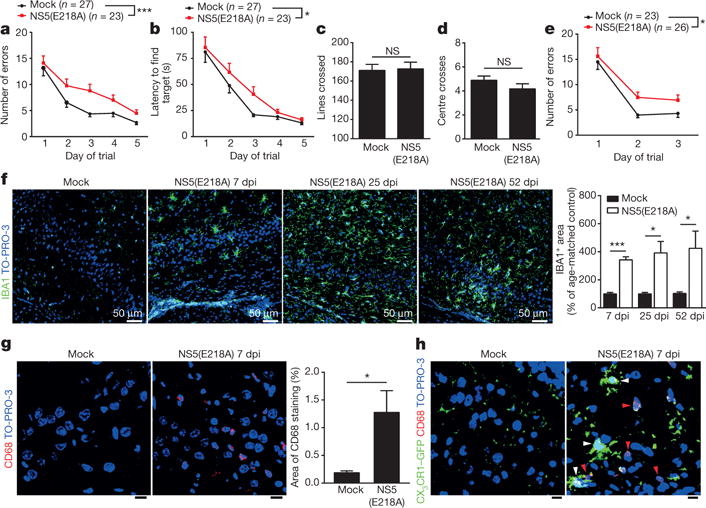
a, b, At 46 days post-infection (dpi), mock or WNV-NS5-E218A-infected mice underwent 5 days of testing on the Barnes maze spatial learning task. Errors (a) and latency (b) before finding target hole were scored daily (mean of 2 trials per day, ***P < 0.001, *P < 0.05 by repeated measures two-way ANOVA). c, d, At 45 dpi, mice were observed on the open-field test and assessed for locomotor activity (c) and anxiety (d). a–d, Mock (n = 27) and WNV-NS5-E218A-infected (n = 23) mice. e, Mock (n = 23) and WNV-NS5-E218A-infected (n = 26) mice were tested at 22 dpi on a 3-day version of the Barnes maze, and evaluated as in a. f, Immunostaining for IBA1 in control and WNV-NS5-E218A-infected mice at 7 dpi (n = 6 or 7 per group for control or WNV, respectively), 25 dpi (n = 3 or 4 for control or WNV, respectively), and 52 dpi (n = 6 or 4 for control or WNV, respectively) (mean of 2 technical replicates used). g, h, Immunostaining shows increased levels of CD68, a microglial/macrophage lysosomal activation marker, in WNV-NS5-E218A-infected wild-type mice (g) (n = 4 mice per group) and CX3CR1–GFP+/− (h) (n = 3 mice per group) mice. h, CD68 is present within CX3CR1-positive microglia (white arrowheads) and infiltrating macrophages (red arrowheads). Images are representative of at least 3 mice per group. All panels, ***P < 0.001, *P < 0.05, NS, not significant by two-tailed t-test, and scale bars, 10 μm, unless otherwise noted. Error bars, s.e.m. Immunostaining and quantification were performed within the hippocampal CA3 region.
Caspase-3-dependent apoptosis of neurons occurs in animals that succumb to WNV neuroinvasive disease12. TUNEL and NeuN-staining did not detect ongoing apoptosis (Extended Data Fig. 2a) or loss of NeuN+ neurons within the circuitry of the entorhinal cortex and hippocampus that serve spatial learning (Extended Data Fig. 2b). Similarly, hippocampal and total brain volumes at 52 dpi do not differ between mock-infected and WNV-NS5-E218A-infected animals (Extended Data Fig. 2c), and GFAP staining within the hippocampus was unchanged (Extended Data Fig. 2d). However, microglial nodules were detected within the hippocampus of WNV-NS5-E218A-recovered animals at 7, 25, and 52 dpi (Extended Data Fig. 3b). These nodules contained increased numbers of IBA1-positive cells with activated cell morphology (Fig. 1e). At 7 dpi, increased levels of CD68, a marker of microglial/macrophage lysosomal activation, were observed within both microglia and infiltrating macrophages of CX3CR1–GFP+/− mice (Fig. 1g, h), however macrophage infiltration was absent by 25 dpi (Extended Data Fig. 2f).
Whole-transcriptome microarray of hippocampi RNA from mock-infected versus WNV-NS5-E218A-infected mice at 25 dpi enabled detection of differential expression of 1,364 transcripts. Pathway analysis identified signatures associated with the generation and maintenance of synapses, activation of innate immune responses, and microglial proteins involved in sensing endogenous ligands and microbes18 (Supplementary Table 1). In the latter category, we identified genes associated with microglial-mediated phagocytosis (Cx3cr1 which encodes CX3CR1; Dap12 which encodes Dap12 (also known as Tyrobp); Fcer1g which encodes FcεR1G; Fcgr2b which encodes FcγR2; Rac2 which encodes Rac2 and Was which encodes WAS) and the classical complement pathway (C1qa which encodes C1QA; C2 which encodes C2; C3 which encodes C3; C4b which encodes C4b and Serp1 which encodes Serping1) (Fig. 2a), which were validated using quantitative PCR (qPCR) (Fig. 2b). C1q and C3 are required for retinogeniculate and cortical synaptic pruning during murine CNS development4,5,19. Although complement contributes to control of WNV dissemination following peripheral infection20, complement expression within the brain during WNV neuroinvasive disease has not been investigated.
Figure 2. Transcriptional profile of good and poor spatial learners during WNV recovery.

a, Heat maps show relative expression of significantly altered genes (see Methods) generated from hippocampal microarray of mock vs WNV-NS5-E218A-recovered mice at 25 dpi, each column represents individual mice. b, Validation of select genes and pathways in a unique set of mice by qPCR (mock (n = 5) and WNV-NS5E218A (n = 6) mice). c, Scatter plot depicting number of errors committed on day 2 of Barnes maze testing, showing good (blue) and poor (green) learners among WNV-NS5-E218A-infected and mock-infected (red) controls. d, Principle component analysis of microarray samples separated by groups as in c. WNV, West Nile virus. e, Relative expression heat map showing the top 50 upregulated and 50 downregulated genes by microarray comparing WNV-recovery good and poor learners; each row represents individual mice. f, Venn diagram of microarray data showing number of genes significantly altered from mock-infected controls (P < 0.05, fold change>1.5) in WNV-recovery good or poor learners. g, Validation by qPCR of select genes altered between WNV good learners and WNV poor learners using a separate cohort of mice (mock (n = 5), WNV good (n = 3), and WNV poor (n = 3) mice). All panels, ***P <0.001, *P <0.05, NS, not significant bytwo-tailed t-test. Error bars, s.e.m.
To identify genetic signatures specific to spatial learning defects, we categorized WNV-NS5-E218A-recovered mice into those that perform similar to mock-infected animals (good learners categorized as <8 errors) and those that exhibit severe learning deficits (poor learners categorized as >9.5 errors), on day 2 of Barnes maze testing (Figs 1e and 2c–e). A total of 747 genes were increased in the WNV poor learners, 45 genes altered in WNV good learners, and 572 genes altered in both groups compared to mock-infected littermates (Fig. 2f) (Supplementary Tables 2 and 3). WNV-NS5-E218A-recovered animals with poor learning exhibited increased levels of CRRY (also known as Cr11), Dap12, FcγR2, Rac2, and C1QA and decreased levels of Dlg2, a synaptic scaffolding protein, and the metabotropic glutamate receptor, Grm5, compared with good learners and mock-infected animals (Fig. 2g). Grm5 is downregulated in mouse brain during acute WNV, Japanese encephalitis virus, and reovirus infections21. KEGG pathway analysis of upregulated genes in WNV poor memory compared to WNV good memory mice revealed top pathways of cytokine signalling, calcium signalling, and B-cell receptor signalling (Supplementary Table 4), whereas top pathways of downregulated genes include long-term potentiation, axon guidance, and Wnt signalling (Supplementary Table 5).
Loss in neurons or persistence of virus could contribute to cognitive dysfunction. Evaluation of neuronal numbers throughout the hippocampus and entorhinal cortices of mock-infected and WNV-infected good and poor learners revealed no differences (Extended Data Fig. 2b). Analyses of WNV envelope protein positive and negative strand RNA from the hippocampi of WNV-NS5-E218A-recovered mice showed high levels of both strands at 7 dpi, which decreased by 25 and 52 dpi, with no differences in levels of either strand between good and poor learners at either time point (Extended Data Fig. 1h–j). These data suggest that persistence of replicating WNV-NS5-E218A does not contribute to alterations in learning.
Given the alterations in genes related to synaptic function, we quantified synaptic terminals within the hippocampus of WNV-NS5-E218A-infected mice. Numbers of colocalized presynaptic and postsynaptic puncta within the stratum lucidum of the hippocampal CA3 (mossy fibre terminals) were decreased at 7 dpi in WNV-NS5-E218A-infected animals compared to mock-infected controls (Fig. 3a). The decrease in colocalization was traced to a 40% reduction in number, but not size, of presynaptic terminals (Extended Data Fig. 3a), with no change in numbers of postsynaptic terminals (Fig. 3a). Altered expression of the presynaptic glutamatergic vesicular transporter, VGlut1 (also known as Slc17a7), in the hippocampus has been linked to cognitive impairment in rodents22. Evidence of hippocampal glutamatergic synapse loss was detectable at 25 dpi and WNV-NS5-E218A-recovered mice with poor spatial learning exhibited fewer VGlut1-positive synaptic puncta than WNV-NS5-E218A-infected good learners, which were fewer than in mock-infected controls (Fig. 3b). WNV-NY99 infection led to similar reductions in synaptic terminals (Extended Data Fig. 3b). Of note, mock-infected and WNV-NS5-E218A-recovered mice (all learners) display similar levels of phosphorylated neurofilament heavy chain (SMI-31) within hippocampal mossy fibre tracts (Extended Data Fig. 3c). These results indicate that axons are preserved despite elimination of synapses, suggesting that synapse elimination does not lead to neuronal death.
Figure 3. West Nile virus causes a loss in hippocampal CA3 synaptic terminals in mice and humans.
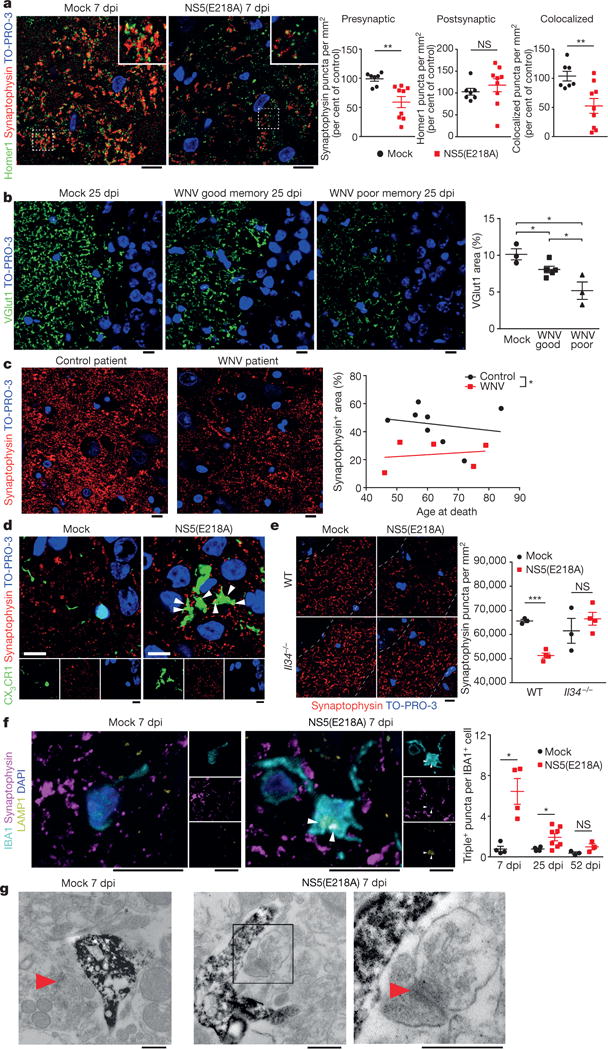
a, Immunostaining and quantification of colocalized presynpatic and postsynaptic puncta using the markers synaptophysin and Homer1, respectively, at 7 dpi in wild-type mice (mock (n = 7) and WNV-NS5-E218A (n = 9) mice). Data are the mean of 2 staining experiments. b, Immunostaining and quantification of staining area for glutamatergic presynaptic marker, VGlut1, at 25 dpi in mock or WNV-NS5-E218A-recovered mice with good or poor spatial memory performance (mock (n = 3), WNV good (n = 5), and WNV poor (n = 3) mice). c, Immunostaining and quantification of synaptophysin+ area in acute WNV encephalitis patients with age- and sex-matched controls. d, CX3CR1–GFP heterozygous mice were immunostained for synaptophysin and GFP with arrowheads depicting colocalization. Images shown are representative of 3 mice per group. e, Synaptic terminal elimination at 7 dpi is absent in WNV-NS5-E218A-infected Il34−/− mice, which have reduced numbers of microglia. f, Immunostaining showing the number of colocalized synaptophysin, LAMP-1 (lysosomal marker), and IBA1 puncta per IBA1+ cell at 7, 25 and 52 dpi. g, Electron micrographs from mock or WNV-NS5-E218A-infected hippocampus at 7 dpi with immune-DAB enhancement of IBA1. An IBA1+ cell is shown surrounding a nearby synapse (right panels, boxed area, red arrowhead). Images shown are representative of 3 mice per group. All panels, ***P <0.001,**P < 0.01, *P< 0.05, NS, not significant, by two-tailed t-test. Error bars, s.e.m. Immunostaining, electron microscopy, and quantification performed within the hippocampal CA3 region. Scale bars, 10 μm unless otherwise noted.
Acute post-mortem WNV neuroinvasive disease patient specimens similarly show reduced numbers of CA3 presynaptic terminals compared to age-matched patient controls (Fig. 3c and Supplementary Table 5). In 3 out of 5 WNV cases, WNV antigen was detected within the CA2/CA3 region, but absent from neighbouring regions (Supplementary Table 6 and Extended Data Fig. 4a). Hippocampal CA1 and entorhinal cortex in the human samples also displayed presynaptic terminal loss (Extended Data Fig. 4b, c). These data show that synaptic pathology is observed in areas without detectable viral antigen. Thus, WNV infection may alter synapse homeostasis both in affected and connected brain regions.
Given their phagocytic appearance in WNV-recovered mice, we wondered if microglia were driving synapse loss. Three-dimensional reconstructions of microglia from CX3CR1–GFP+/− WNV-NS5-E218A-infected mice (Fig. 3d) revealed synaptophysin-positive puncta within GFP+ cells (Supplementary Videos 3 and 4). Additionally, presynaptic terminals from mice with fewer and less proliferative microglia (Il34−/−)23 were not eliminated during WNV-NS5-E218A acute infection (Fig. 3e). Colocalization of synaptophysin, lysosomal-associated membrane protein 1 (LAMP1), and IBA1, revealed increased numbers of presynaptic terminals within the lysosomes of IBA1-positive cells, but not S100β+ astrocytes (Extended Data Fig. 3d), in the CA3 of WNV-NS5-E218A-infected mice at 7 and 25 dpi compared with mock-infected animals (Fig. 3f). Numbers of engulfed presynaptic terminals recover to baseline levels by 52 dpi, suggesting that presynaptic elimination eventually abates. Electron microscopy of the CA3 and molecular layer (Fig. 3g) during acute WNV neuroinvasive disease identified microglia enriched in phagosomes (Extended Data Fig. 3e) and with processes that surround synapses (Fig. 3g).
Macrophage-mediated phagocytosis in the periphery often requires antibody and complement deposition, however, no differences in the amount of endogenous mouse IgG coating VGlut1-positive or Homer1-positive, CA3 synaptic terminals at 7 and 25 dpi following WNV-NS5-E218A infection were observed compared to mock-infected controls (Extended Data Fig. 3f, g). Furthermore, μMT−/− mice, which lack mature B cells, eliminate synaptic terminals during acute WNV-NS5-E218A infection at similar levels as in wild-type mice (Extended Data Fig. 3h). Hippocampal upregulation of C1QA, the initiating factor of the classical complement cascade, was detected at 7, 25 and 52 dpi compared to mock-infected controls (Fig. 4a). Hippocampal C1qa mRNA in conjunction with IBA1 expression was detected in mock-infected and WNV-NS5-E218A-infected mice at 7 dpi, the latter of which was increased (Fig. 4b). C1QA protein was detected in IBA1+ cellular processes adjacent to or surrounding neurons (Fig. 4b, c).
Figure 4. Classical complement activation in neurons and microglia drives WNV-mediated synaptic terminal elimination.

a, qPCR analysis of hippocampal C1qa mRNA normalized to Gapdh in mock (n = 8) or 7 dpi (n = 3), 25 dpi (n = 8) or 52 dpi (n = 7) after WNV-NS5-E218A infection. b, Fluorescent in situ hybridization using RNA probes for neuron specific enolase (NSE) with sense or antisense C1QA coupled with immunostaining for IBA1 in WNV-NS5-E218A-infected or control mice with high magnification insets. Images are representative of 3 mice per group. Scale bars, 50 μm. c, Immunostaining for C1QA protein and IBA1 with high magnification insets. Arrowheads depict colocalization. d, Immunostaining for C1QA protein and WNV antigen at 7 dpi with a WNV-infected neuron shown in high magnification inset. e, Immunostaining for C1QA with neuronal marker, Map2. f, Immunostaining shows colocalization of presynaptic marker, VGlut1, with C3d at 7 dpi (mock (n = 4), WNV-NS5-E218A (n = 6)).g, Immunostaining showing colocalization of C1QA and VGlut1 with representative super-resolution micrographs shown. Scale bars, 5 μm. h, Synaptophysin (Syp) immunostaining in WNV-NS5-E218A-infected wild-type, complement C3-null, complement receptors C3aR-null, CR1/2-null, and CR3-null mice at 7 dpi, normalized to age and genotype-matched, mock-infected controls. i, Immunostaining and quantification of number of colocalized (arrowheads) synaptophysin+, LAMP-1+ (lysosomal marker), and IBA1+ puncta per IBA1+ cell in WNV-NS5-E218A-infected wild-type, C3-null and C3aR-null mice at 7 dpi (fold-change over control). All panels, ***P <0.001, **P< 0.01, *P< 0.05, NS, not significant, by two-tailed t-test. Error bars, s.e.m. Immunostaining and quantification performed within the hippocampal CA3 region. Scale bars, 10 μm unless otherwise noted.
C1QA also colocalized within WNV antigen-positive cells with neuronal morphology (Fig. 4d) and with Map2-positive neurites (Fig. 4e). Increased levels of the complement C3 cleavage product, C3d, also colocalized with VGlut1-positive terminals at 7 dpi (Fig. 4f). Levels of colocalization of C1QA with VGlut1-positive presynaptic terminals were significantly increased in WNV-NS5-E218A-recovered animals compared with mock-infected controls at 7 and 25, but not 52 dpi (Fig. 4g). WNV-NS5-E218A-infected complement C3−/− and C3ar1−/−, but not CR1/2−/−or Itgam−/− (Itgam is also known as Cr3), mice showed no differences in synaptophysin-positive synaptic terminals compared to mock-infected controls (Fig. 4h). Presynaptic puncta within microglial (IBA1+) lysosomes were also decreased in WNV-NS5-E218A-infected complement C3−/− and C3ar1−/− compared to wild-type mice (Fig. 4i), indicating that WNV-mediated elimination of synaptic terminals requires C3 and C3aR.
Many pathogens and pathogen-associated molecular patterns induce complement activation within the central nervous system, including WNV24, HIV25, and amyloid plaques26. Our study suggests that complement C3 and C3aR mediate presynaptic terminal loss in the hippocampi of mice that exhibit spatial learning defects during recovery from West Nile neuroinvasive disease. Microglia and recognition of C3 cleavage products by complement receptor C3aR, which is expressed by both neurons and microglia27, are required for this process. Although astrocytes did not exhibit increased colocalization with synaptic terminals, we cannot completely rule out their contribution to this process, as their rates of lysosomal digestion differs from microglia28. Furthermore, alterations to NMDA receptor-mediated long-term potentiation could also contribute to altered synapse homeostasis and memory as several genes in this pathway are differentially expressed in WNV-poor learners (Supplementary Table 4). It is unknown whether complement labelled and eliminated terminals are connected to WNV infected or healthy neurons. In the context of neurotropic viral infection, elimination of presynaptic terminals may prevent trans-synaptic viral spread or aberrant signalling of infected neurons. Further studies will determine if complement- and microglial-dependent synapse elimination prevents neuron-to-neuron spread of WNV and other neurotropic viruses.
METHODS
Animals
At the outset of all experiments, 8–10-week-old male and female mice were used. C57BL/6J and CD11b−/− (CR3−/−) mice were obtained from Jackson Laboratories. C3−/−,CR1/2−/− (in mice, CR1 and CR2 are splice variants both derived from the mouse Cr2 gene), and C3aR−/− mice (> 10 generations back-crossed to C57BL/6) were obtained from John Atkinson (Washington University), CX3CR1–GFP+/− mice (> 10 generations backcrossed to C57Bl/6) were obtained from Richard Ransohoff (Lerner Research Institute, Cleveland Clinic Foundation), μMT−/− mice (C57BL/6 background) were obtained from Michael Diamond (Washington University), and Il34−/− mice (C57BL/6 background) were obtained from Marco Colonna (Washington University). All mice were randomly assigned to control or experimental groups at the beginning of each experiment. All experimental protocols were performed in compliance with the Washington University School of Medicine Animal Studies Committee (protocol number 20140122).
Mouse models of WNV infection
The WNV strain 3000.0259 was isolated in New York and passaged once in C6/36 Aedes albopictus cells to generate an insect-cell-derived stock. Then 100 plaque-forming units (pfu) of WNV-NY99 were delivered in 50 μl to the footpad of anaesthetized mice. WNV-NS5-E218A, which harbours a single point mutation in the 2′ O-methyl-transferase gene, was obtained from Michael Diamond (Washington University) and passaged in Vero cells as described previously6. Deeply anaesthetized mice were administered with 104 pfu of WNV-NS5-E218A or 10 pfu of WNV-NY99 in 10 μl into the brain’s third ventricle via a guided 29 gauge needle.
Stock titres of all viruses were determined by using BHK21 cells for viral plaque assay as previously described14.
Leukocyte isolation and flow cytometry
Cells were isolated from brains of wild-type mice at day 7 post-infection and stained with fluorescently conjugated antibodies to CD4, CD8, CD11b, and CD45 as previously described14. Data collection and analysis were performed with an LSRII flow cytometer using FlowJo software.
Behavioural testing
Test for anxiety and locomotor behavior
The open-field test was used to assess baseline differences in anxiety or locomotor behaviour, before Barnes maze experiments. A standard Open Field arena (54 × 54 cm, custom built) was used, consisting of a simple square box with a grid (6 squares per side) along the base. Animals were placed in the centre of the arena and allowed to explore for 5 min. The arena was decontaminated with 70% ethanol between each trial. Locomotor activity was assessed by counting the number of lines the animal crossed during the testing period, and anxiety was assessed by counting the number of times the animal crossed through the centre of the field. Behaviour was recorded using a camera (Canon PowerShot SD1100 IS), and a blinded experimenter scored the trial. Any mice that jumped out of the open-field maze were excluded from analyses.
Test for visual spatial memory
The Barnes maze was used to assess visual spatial memory. An elevated Barnes maze (91.4 cm diameter, custom built) containing 19 empty holes and 1 target hole with a hidden escape chamber was used for testing (5 cm diameter holes were evenly spaced around the table, 6.35 cm from the edge). Visual cues were placed around the room and remained in the same location during the entire testing period. Mice were tested on the Barnes maze over the course of 5 consecutive days. Each mouse received two trials per day, spaced exactly 30 min apart. For each trial, the mouse was placed in the centre of the maze in a covered start box for 10 s, and removal of the box signalled the start of a trial. Each mouse was given 3 min to explore the maze and find the target hole. Mice that did not enter the target hole within 3 min were gently guided into the hole. After each trial, the mouse remained in the target hole for exactly 1 min, and then was returned to its home cage. The maze was decontaminated with 70% ethanol between each trial. The numbers of errors (nose pokes over non-target holes) and the latency to find the target hole (amount of time elapsed before nose poke over target hole) were measured. Behaviour was recorded using a camera (Canon Powershot SD1100IS), and a blinded experimenter scored the trials. Any mice which fell off the Barnes maze table during any trial were excluded from analyses. No randomization was required for these studies.
Immunohistochemistry
Following perfusion with ice-cold PBS and 4% PFA, brains were immersion-fixed overnight in 4% PFA, followed by cryoprotection in two exchanges of 30% sucrose for 72 h, then frozen in OCT (Fisher). 9μm-thick fixed-frozen coronal brain sections were washed with PBS and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich), and nonspecific antibody was blocked with 5–10% normal goat serum (Santa Cruz Biotechnology) for 1 h at room temperature. Mouse on mouse kit (MOM basic kit, Vector) was used as per the manufacturer’s protocol when detecting synaptophysin to reduce endogenous mouse antibody staining. After block, slides were exposed to primary antibody or isotype matched IgG overnight at 4 °C, washed with 1 × PBS and incubated with secondary antibodies for 1 h at room temperature. Nuclei were counterstained with TO-PRO-3 (Invitrogen) and coverslips were applied with vectashield (Vector). Immunofluorescence was analysed using a Zeiss LSM 510 laser-scanning confocal microscope and accompanying software (Zeiss). Positive immunofluorescent signals were quantified by a blinded experimenter using the NIH Image analysis software, ImageJ.
TUNEL staining was performed using the TMR-red in situ cell death detection kit (Roche) as per manufacturer’s instructions. C1QA staining was performed as previously described29.
Antibodies
C1QA (undiluted, described previously29), WNV (1:100, described previously14), rat anti-GFAP (1:200, Invitrogen catalogue number 13-0300), rabbit anti-IBA1 (1:100, WAKO catalogue number 019-19741), mouse anti-NeuN-biotin (1:100, Millipore catalogue number MAB 377B), chicken anti-GFP (1:1,000, Abcam catalogue number 13970), rabbit anti-Synapsin1 (1:200, Millipore catalogue number Ab1543), mouse anti-synaptophysin (1:50, DAKO catalogue number M0776 or 1:50 Abcam catalogue number ab8049), guinea-pig anti-VGlut1 (1:300, Synaptic Systems catalogue number 135304), rabbit anti-Homer1 (1:200, Synaptic Systems 160003), rabbit anti-C3d (1:500, DAKO catalogue number A0063), rabbit anti S100β (1:300, Abcam catalogue number ab52642), rat anti-Lamp1 (1:50, BD Pharmingen catalogue number 553792), and rat anti-CD68 (1:200, Serotec catalogue number MCA1957).
Secondary antibodies conjugated to Alexa-488, Alexa-555, or Alexa-633 (Invitrogen) were used at 1:400 dilution.
MRI
Mice were intracardially perfused, first with ice-cold PBS and then with a mixture of 4% PFA and 10% Multihance (gadobenate dimeglumine, Bracco Diagnostics, Princeton, NJ). Heads were further fixed in 4% PFA for 24 h before being trimmed of extraneous tissue around the skull (to minimize the field of view). Heads were then placed in 1% Multihance in PBS until being imaged several days later. Ex vivo, whole-head MR imaging experiments were performed at 4.7 T using an Agilent/Varian (Santa Clara, CA) DirectDrive1 small-animal scanner. Data were collected with a custom-made RF foil coil that fits tightly around the head using a 3D, T1-weighted gradient echo sequence with the following parameters: TR = 105 ms, TE = 6 ms, flip angle = 90°, isotropic resolution = (0.0625 mm)3, and scan time ~ 11 h. Regions of interest were manually drawn for the whole brain and hippocampus in ITK-SNAP (http://www.itksnap.org) from which volumes were calculated.
Collection and purification of hippocampal RNA
Mice were perfused with ice-cold PBS then hippocampi were dissected and snap-frozen in Tri-reagent (Ambion). Hippocampal tissue was then homogenized, and RNA purified as previously described30 using the RNA Ribopure kit (Ambion). RNA was precipitated with 25 mM ammonium acetate in 100% ethanol at −80 °C overnight, resuspended in RNase-free H2O, and checked for purity. RNA was then treated with RNase-out and DNase I (invitrogen) as per the manufacturer’s protocol.
Microarray
Hippocampal RNA was isolated as described above and submitted to the Washington University Genome Technology Access Center. The total RNA quality and quantity were then determined by Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA) and NanoDrop ND-1000 Spectrophotometer (Thermo Scientific NanoDrop, Wilmington, DE), according to manufacturer’s recommendations, respectively. A total of 400 ng of RNA transcripts from each sample were amplified by T7 linear amplification with the MessageAmp TotalPrep Amplification kit (Life Technologies-Ambion, Austin, TX). Then 1.5 μg of each amplified and biotinylated RNA (aRNA) sample was hybridized onto Illumina MouseWG-6 v2 expression beadchips, followed by cy3 steptavidin-based staining, washing, and scanning, according to Illumina standard protocol. The iScan scanner-created image data were loaded into Illumina GenomeStudio (v2011) for generation of expression values and data normalizations. Only those probes that were detected at P < 0.05 in at least one of the samples were used in downstream statistical analysis. Background subtracted and quantile normalized data were used in statistical analysis for identification of differentially expressed genes with one-way ANOVA test using Partek Genomics Suite (v6.6, St. Louis, MO). All original P values in the ANOVA analysis were adjusted by q-value based multiple test correction31. KEGG pathway analysis was performed using DAVID bioinformatics database functional annotation tool v.6.7 (ref. 32). Microarray data has been deposited in the Gene Expression Omnibus (accession number GSE72139)
Real-time quantitative RT-PCR
cDNA was synthesized using random hexamers, oligodT15, and MultiScribe reverse transcriptase (Applied Biosystems). A single reverse transcription master mix was used to reverse transcribe all samples to minimize differences in reverse transcription efficiency. The following conditions were used for reverse transcription reactions: 25 °C for 10 min, 48 °C for 30 min, and 95 °C for 5 min.
For all primer sets except for the strand-specific WNV PCR reaction (detailed below), PCR reactions were prepared using Power SYBR Green PCR mastermix and calculated copies were normalized against copies of the housekeeping gene, Gapdh.
Primers are listed in Supplementary Table 6.
WNV strand specific real-time RT-PCR
Two-step strand specific RT-PCR was performed using GVA and T7 tagged primers during cDNA synthesis as a modification to a procedure previously described33. First, cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) per the manufacturer’s instruction, with the addition of 1 pmol of the positive-strand primer GVA_TxsspE1229 and 1 pmol of the negative strand primer T7_TxsspE1160 to the 10 μl reaction mixture. Strand specific cDNA was then amplified in a qPCR reaction using primers directed against the strand specific WNV sequence and the tag sequence. Each 12.5 μl qPCR reaction mixture contained 6.25 μl TaqMan Gene Expression Master mix, 10 pmol tag primer (positive mix: GVA, negative mix: T7), 10 pmol strand specific primer (positive mix: TXsspE1160, negative mix: TXsspE1229), 2.5 pmol strand specific probe (WNVsspEProbe) and 50 ng strand specific cDNA. Thermal cycling was performed using Applied Biosystems ViiA 7 Real-Time PCR system with a 384-well block. Copies were calculated based on a standard curve generated from purified PCR product from positive strand or negative strand reactions, and normalized to amount of Gapdh in each sample.
For the following sequences, the bold sequence represents the tag sequence, while the underlined sequence represents the strand specific sequence: GVA_TxsspE1229: 5′-TTTGCTAGCTTTAGGACCTACTATATCTACCTGGTCAGCACGTTTGTCATTG-3′; T7_TxsspE1160: 5′-GCGTAATACGACTCACTATATCAGCGATCTCTCCACCAAAG-3′ ; TXsspE1229: 5′-GGGTCAGCACGTTTGTCATTG-3′; TXsspE1160: 5′-TCAGCGATCTCTCCACCAAAG-3′; T7tag: 5′-GCGTAATACGACTCACTATA-3′; GVAtag:5′-TTTGCTAGCTTTAGGACCTACTATATCTACCT-3′; WNVsspEProbe: FAM-TGCCCGACCATGGGAGAAGCTC-TAMRA.
Quantification of synaptic terminals
ImageJ was used to threshold single-plane confocal images, draw a region of interest encompassing the CA3 mossy fibres, and to quantify the number of synaptophysin or VGlut1+ puncta containing between 0.5 and 25 square μm2 in area. For each mouse, at least 12 images at 63 × magnification were counted, which were derived from at least 4 fixed-frozen coronal sections spaced 50 μM apart.
Quantification of synaptic terminal engulfment by IBA1+ cells
ImageJ was used to threshold single-plane confocal images, draw a region of interest encompassing each IBA1+ cell within the hippocampal CA3, and to quantify the number of synaptophysin+, Lamp1+, IBA1+ puncta between 0.2 and 25 μm2 in area within each IBA1+ cell. For each mouse, at least 8 images at 63 × magnification were counted, which were derived from at least 4 fixed-frozen coronal sections spaced 50 μM apart.
Electron microscopy
Mice were perfused with 4% PFA, the brain was removed, and immersion fixed for 24 h at room temperature. Tissue was washed in PBS, and 100 μm sections were cut from regions of interest using a vibratome. Sections were incubated in 0.5% gelatin, 5% horse serum, and 0.01% saponin in PBS for 5 h on a rotator at room temperature. Sections were then incubated for 48 h at 4 °C on a rotator with rabbit anti-IBA1 (1:600; Wako). After washing in dPBS, sections were incubated overnight at 4 °C on rotator with donkey anti-rabbit biotinylated secondary antibody, (1:500; Rockland) in 0.5% gelatin and 5% horse serum in dPBS. Sections were again washed, and then incubated with streptavidin-HRP (1:1000; Rockland, S000-03) for 3 h at room temperature, followed by another wash. HRP was visualized using the DAB Substrate Kit (Cell Marque,957D) for 5 min, washed and then fixed with 2% PFA, 2.5% glutaraldehyde, in PBS for 30 min, followed by a wash in PBS. Sections were post-fixed in 1% osmium tetroxide in PBS for 30 min at room temperature, washed in PBS, and then dehydrated in sequential concentrations of ethanol for 30 min each. Sections were then infiltrated with 1:1 Spurr’s resin and 100% ethanol overnight on a rotator, followed by two changes in pure Spurr’s resin over 24 h. Sections were embedded with Aclar film (EMS, 50425), and polymerized at 60 °C for 48 h. The hippocampus was trimmed from the polymerized section, glued to a previously prepared block of Spurr’s resin, and allowed to cure for at least 24 h. Using a Diatome ultra 45° diamond knife and a LEICA Ultracut UC7, blocks were sectioned at 500 nm to confirm location of the tissue and the positivity of antibodies. Once confirmed, 90 nm sections were cut and picked up onto 200 hex mesh, formvar-carbon coated copper grids (Ted Pella, 01800-F). Images were digitally captured using a JEOL 1200 EX II transmission electron microscope with AMT digital camera.
Three-dimensional reconstruction of confocal z-stack images
Confocal z-stack images taken with a Zeiss LSM 510 META microscope at 63 × magnification, consisting of at least 10 images were transformed into 3D reconstruction videos using Volocity 3D image analysis software (PerkinElmer).
Super-resolution microscopy of presynaptic terminals
Sections were imaged using a Zeiss ELYRA PS1 microscope. 0.101 μm optical slices of the CA3 region of the hippocampus were captured and images subsequently processed using Zeiss SIM algorithms to generate structured illumination images. Zeiss Zen software was used to generate orthogonal viewpoints showing colocalization of VGlut1 and C1q in the x, y and z planes.
Human tissue
Human autopsy hippocampal tissue embedded in paraffin was obtained from St. Louis University Medical Center (St. Louis, MO) and Presbyterian / St. Luke’s Medical Center (Denver, CO). Sections were first deparaffinized and then boiled in 10 mM sodium citrate buffer for 30 min, for antigen retrieval before staining with anti-synaptophysin antibody (DAKO, 1:50).
In situ hybridization
Fluorescent in situ hybridization was performed on 9 μm coronal brain sections that were 4% PFA-fixed and frozen. C1QA and NSE were used as double mRNA staining and IBA1 as immune-staining. C1qA and NSE anti-sense RNA was made and labelled with cyanine and fluorescein, respectively, using an RNA labelling kit (Roche). C1QA was amplified by PCR from pCMV SPORT6 C1qA plasmid (Openbiosystem MMM1013-63584), using forward 5′-GGCATCCGGACTGGTATCCGAGG-3′ and reverse 5′-GGTAAATGCGACCCTTTGCGGGG-3′ primers, which was digested with SalI and transcribed with T7 promoter. The RNA probes were incubated overnight at 64 °C, and then detected with antibodies against fluorescein and cyanine (Roche). The staining reaction was then amplified with a TSA staining kit (PerkinElmer). A rabbit anti-IBA1 (Wako) antibody was used to label macrophages and microglia and detected with a donkey anti-rabbit 647 antibody (Life technologies). Stained sections were then imaged and analysed on a Zeiss AX10 fluorescent microscope. Statistical analysis. To determine mouse group sizes for virological or immunological studies, power analysis was performed using the following values: probability of type I error = 0.05, power = 80%, fivefold hypothetical difference in mean, and population variance of 25-fold (virological studies) or 12-fold (immunological studies). Results from Barnes maze spatial learning and memory studies were compared by repeated measures two-way ANOVA. Microarray data was analysed by one-way ANOVA and fold change greater than 1.5, false discovery rate q < 0.05 to correct for multiple hypotheses for mock vs WNV-all comparison, P < 0.05 for WNV good learners vs WNV poor learners. Variance between groups was equivalent except for cases noted within figure legends and the comparison for number of presynaptic terminals in Fig. 3 a and the comparisons at 7 and 25 dpi in Fig. 3f, in which a Welch’s correction on the two-tailed t-test was used to correct for unequal variance. All other experiments were compared by Student’s two-tailed t-test, with *P < 0.05 considered significant. Power calculations using results observed in a pilot study in which WNV-recovered mice exhibit a two-fold increase in peak errors compared with mock-infected animals indicate that at least 15 recovered mice per group will be required to obtain statistical significance (P < 0.05) on Barnes maze testing.
Extended Data
Extended Data Figure 1. Murine intracranial infection with attenuated WNV-NS5-E218A induces similar viral loads and inflammatory response as wild-type WNV-NY99, but greater overall survival.

a, Plaque assay for infectious virus (measured in plaque-forming units per g of tissue) performed on dissected brain tissue at various days post-infection with either footpad infection with 102 pfu of WNV-NY99 or intracranial infection with 104 pfu of WNV-NS5-E218A. Each point represents an individual mouse. b, Survival curves of mice infected at 8-weeks-old by the footpad with WNV-NY99 or intracranially with WNV-NY99 or WNV-NS5-E218A. c, Flow cytometric analysis of dissected cortex, hippocampus and cerebellum at 6 dpi with WNV-NY99 and WNV-NS5-E218A with plots for CD45 and CD11b. d, Quantification of flow cytometry data from c. Shown are numbers of leukocytes (CD45high), lymphocytes (CD45high, CD11blow), and activated macrophages and microglia (CD45high, CD11bhigh) compared to mock-infected controls (n = 4 mice per group). e, Immunostaining and counts for TUNEL staining for apoptotic cells with co-staining for the neuronal marker, NeuN, during peak infection (7 dpi) of WNV-NS5-E218A (n = 5) compared to mock-infected controls (n = 4). DG, dentate gyrus, CTX, entorhinal, perirhinal, and visual cortex. f, Some mice were tested at 22 dpi on a three-day version of the Barnes maze, and evaluated for latency to find target hole (*P < 0.05 by repeated measures two-way ANOVA). g, Prior to Barnes maze testing, mice were tested on open field for total lines crossed in 2 min at 21 dpi. h, qPCR for positive strand (non-replicating strand) and negative strand (replicating) WNV envelope protein message remaining in hippocampal tissue at 7, 25 and 52 dpi (n = 13, 4, and 14 mice per group for 7, 25, and 52 dpi, respectively), measured in copies per Gapdh. i, qPCR for positive strand WNV envelope protein at 52 dpi in WNV good learners (fewer than 8 errors on day 2 of Barnes maze, n = 5) and WNV poor learners (greater than 9.5 errors on day 2 of Barnes maze, n = 9). j, qPCR for negative strand WNV envelope protein at 52 dpi in WNV good learners (fewer than 8 errors on day 2 of Barnes maze, n = 5) and WNV poor learners (greater than 9.5 errors on day 2 of Barnes maze, n = 9). Result was not significant by Student’s two-tailed t-test.
Extended Data Figure 2. At 25–52 days post-WNV-NS5-E218A infection, mice do not show any appreciable loss in brain volume, neuron or astrocyte numbers, or macrophage infiltration.

a, Immunostaining for the neuronal marker, NeuN, with TUNEL staining for apoptotic cells within the hippocampus at 52 dpi. Quantification of the number of TUNEL+ neurons and total TUNEL+ cells is shown in mock (n = 3) and WNV-NS5-E218A (n = 6). Scale bar, 20 μm. b, Immunostaining and quantification of the number of NeuN+ neurons per mm2 within the CA1, CA3, dentate gyrus and entorhinal cortex at 25 days after mock (n = 4) or WNV-NS5-E218A infection. WNV-infected animals were subdivided into good (n = 5) and poor (n = 3) learners. Scale bar, 100 μm. c, Post-mortem mouse brains were imaged by MRI at 52 dpi to determine tissue volume of the hippocampus (outlined in red) and total brain (n = 5 mice per group). Scale bar, 1 mm. Not significant by Student’s two-tailed t-test (P < 0.05 considered significant). d, Immunostaining for the reactive astrocyte marker, GFAP, shows that WNV-NS5-E218A-infected mice do not exhibit greater hippocampal astrocyte activation than mock-infected controls at 52 dpi. NS, not significant by Student’s two-tailed t-test. e, Haematoxylin and eosin (H&E) staining was performed at 52 dpi in WNV-NS5-E218A-recovered and mock-recovered mice. Occasional microglial nodules (arrowhead) surrounded by lymphocytes were observed within the hippocampus. CA1 pyr, CA1 pyramidal layer. f, Flow cytometric analysis of whole brain from mock and WNV-NS5-E218A-infected mice at 8 and 25 dpi was performed to determine numbers of microglia (CD45low, CD11blow), macrophages (CD45high, CD11bhigh), and lymphocytes (CD45high, CD11bnegative). Note the decrease in macrophage population from 7 to 25 dpi.
Extended Data Figure 3. Despite synaptic terminal loss, no changes to synaptic terminal size, axons, or astrocyte or antibody association with terminals during WNV infection.

a, Immunostaining for the presynaptic marker, synaptophysin, at 7 dpi comparing mock (n = 7) with WNV-NS5-E218A-infected (n = 5) mice. Quantification of synaptophysin+ puncta size was performed within the hippocampal CA3. Scale bar, 10 μm. b, Immunostaining for the presynaptic marker, synapsin1, within the hippocampal CA3 in uninfected controls (n = 3) and footpad-infected WNV-NY-1999 (n = 4) at 8 dpi. Quantification was performed on the numbers of synapsin1+ puncta per mm2 with *P < 0.05 considered significant. c, Immunostaining within the hippocampal CA3 for SMI-31, which detect phosphorylated neurofilament and marks axons at 25 dpi (n = 5–6 mice per group). Quantification of the area of SMI-31 per mm2 (not significant by Student’s t-test). d, Immunostaining within the hippocampal CA3 for the presynaptic marker, synaptophysin, co-labelled with the astrocyte marker, S100β at 7 dpi (n = 3 mice per group). Quantification of the percentage of total S100β+ area and synaptophysin+ area colocalized with S100β (not significant by Student’s t-test). e, Electron microscopy was performed on hippocampal CA3 sections from day 7 after mock (left panel) or WNV-NS5-E218A (right panels) infection, with immune-DAB enhancement of IBA1. Note the presence of many phagosomes and cytoplasmic inclusions within the WNV-E218A-infected microglia. Electron micrographs shown are representative of n = 3 mice per group. Scale bars, 1 μm. f, Immunostaining for the presynaptic marker, VGlut1, and endogenous murine IgG (mIgG) at 7 days after mock (n = 4) or WNV-NS5-E218A (n = 4) infection. Quantification was performed on the total per cent of mIgG staining area as well as the per cent of VGlut1+ staining area colocalized with mIgG. g, Immunostaining for the postsynaptic marker, Homer1, and endogenous mIgG at 25 days after mock (n = 4) or WNV-NS5-E218A-infection, which were divided into WNV-infected mice which made fewer than 8 errors on day 2 of the Barnes maze (WNV good learners, n = 5) and WNV-infected mice which made greater than 9.5 errors on day 2 of the Barnes maze testing (WNV poor learners, n = 3). Quantification was performed on the total per cent of mIgG staining area as well as the percent of Homer1+ staining area colocalized with mIgG. Significance was determined by Student’s two-tailed t-test with P < 0.05 considered as significant. NS, not significant. h, Immunostaining and quantification of number of VGlut1 hippocampal CA3 presynaptic terminals at 7 dpi in wild-type and μMT−/− mice. (*P < 0.05, NS, not significant, by Student’s two-tailed t-test). Scale bars, 10 μm.
Extended Data Figure 4. WNV infection of human hippocampal CA2/CA3 neurons with loss of synapses within the hippocampal CA1 and the entorhinal cortex.
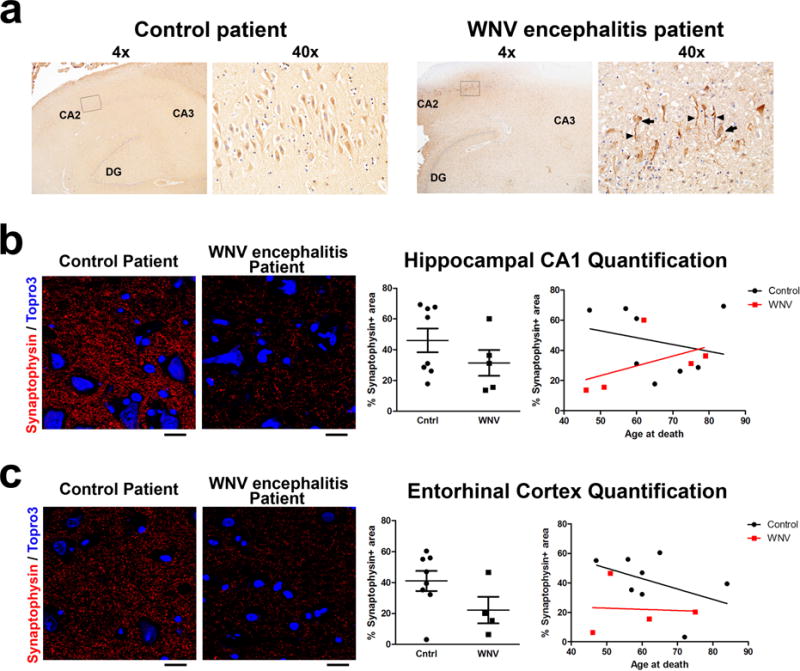
a, Immunostaining of human WNV encephalitis and control post-mortem hippocampal tissue for WNV-antigen. Shown at high magnification are neuron cell bodies (arrows) and neurites (arrowheads) within the hippocampal CA2/CA3 region. b, c, Immunostaining within the hippocampal CA1 (b) or entorhinal cortex (c) for the presynaptic marker, synaptophysin, within human WNV encephalitis and control autopsy cases. Quantification of the per cent of synaptophysin+ area (hippocampal CA1 P = 0.3, entorhinal cortex P = 0.11 by two-tailed Student’s t-test (not significant). Scale bar, 20 μm. In one WNV encephalitis patient sample, the entorhinal cortex could not be quantified because it was missing from the section.
Supplementary Material
Acknowledgments
Funding for this research was provided by the NIH F31 NS077640 (M.J.V), R01 NS052632 (R.S.K.), and U19 AI083019 (R.S.K. and M.S.S.). The authors would like to thank J. Atkinson and X. Wu for reagents and M. Diamond for critical reading of the manuscript.
Footnotes
Online Content Methods, along with any additional Extended Data display Items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions M.J.V. and R.S.K. contributed to the study design. M.J.V., C.G., D.D., D.M.D., B.B., A.S., J.Y, C.P-T, A.F., D.K.W., K.F., X.J., S.M., J.K.R., J.R.G., R.E.S., B.S. and R.S.K. contributed to data collection and/or interpretation. C.G., J.R.B. and M.S.S. developed single-strand PCR assays for WNV B.K.D., K.L.T identified, collected and provided patient samples. M.J.V. and R.S.K. wrote the paper. All authors discussed and commented on the manuscript.
Author Information Microarray data has been deposited in the Gene Expression Omnibus under the accession number GSE72139. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Sejvar JJ, et al. Neurologic manifestations and outcome of West Nile virus infection. J Am Med Assoc. 2003;290:511–515. doi: 10.1001/jama.290.4.511. [DOI] [PubMed] [Google Scholar]
- 2.Klee AL, et al. Long-term prognosis for clinical West Nile virus infection. Emerg Infect Dis. 2004;10:1405–1411. doi: 10.3201/eid1008.030879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen LR, et al. Estimated cumulative incidence of West Nile virus infection in US adults, 1999–2010. Epidemiol Infect. 2013;141:591–595. doi: 10.1017/S0950268812001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 5.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daffis S, et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szretter KJ, et al. 2′-O methylation of the viral mRNA cap by West Nile virus evades IfIt1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012;8:e1002698. doi: 10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armah HB, et al. Systemic distribution of West Nile virus infection: postmortem immunohistochemical study of six cases. Brain Pathol. 2007;17:354–362. doi: 10.1111/j.1750-3639.2007.00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarrard LE. On the role of the hippocampus in learning and memory in the rat. Behav Neural Biol. 1993;60:9–26. doi: 10.1016/0163-1047(93)90664-4. [DOI] [PubMed] [Google Scholar]
- 10.Sadek JR, et al. Persistent neuropsychological impairment associated with West Nile virus infection. J Clin Exp Neuropsychol. 2010;32:81–87. doi: 10.1080/13803390902881918. [DOI] [PubMed] [Google Scholar]
- 11.Clarke P, et al. Death receptor-mediated apoptotic signaling is activated in the brain following infection with West Nile virus in the absence of a peripheral immune response. J Virol. 2014;88:1080–1089. doi: 10.1128/JVI.02944-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samuel MA, Morrey JD, Diamond MS. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J Virol. 2007;81:2614–2623. doi: 10.1128/JVI.02311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCandless EE, Zhang B, Diamond MS, Klein RS. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc Natl Acad Sci USA. 2008;105:11270–11275. doi: 10.1073/pnas.0800898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durrant DM, Robinette ML, Klein RS. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med. 2013;210:503–516. doi: 10.1084/jem.20121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J Virol. 2008;82:8956–8964. doi: 10.1128/JVI.01118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Habjan M, et al. Sequestration by IFIT1 impairs translation of 2′ O-unmethylated capped RNA. PLoS Pathog. 2013;9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 18.Hickman SE, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu Y, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci USA. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mehlhop E, Diamond MS. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J Exp Med. 2006;203:1371–1381. doi: 10.1084/jem.20052388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarke P, Leser JS, Bowen RA, Tyler KL. Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. MBio. 2014;5:e00902–e00914. doi: 10.1128/mBio.00902-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ménard C, et al. Glutamate presynaptic vesicular transporter and postsynaptic receptor levels correlate with spatial memory status in aging rat models. Neurobiol Aging. 2015;36:1471–1482. doi: 10.1016/j.neurobiolaging.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13:753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehlhop E, et al. Complement activation is required for induction of a protective antibody response against West Nile virus infection. J Virol. 2005;79:7466–7477. doi: 10.1128/JVI.79.12.7466-7477.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ebenbichler CF, et al. Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J Exp Med. 1991;174:1417–1424. doi: 10.1084/jem.174.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veerhuis R, et al. Cytokines associated with amyloid plaques in Alzheimer’s disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Exp Neurol. 1999;160:289–299. doi: 10.1006/exnr.1999.7199. [DOI] [PubMed] [Google Scholar]
- 27.Lian H, et al. Astrocyte–microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci. 2016;36:577–589. doi: 10.1523/JNEUROSCI.2117-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung WS, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stephan AH, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33:13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun T, Vasek MJ, Klein RS. Congenitally acquired persistent lymphocytic choriomeningitis viral infection reduces neuronal progenitor pools in the adult hippocampus and subventricular zone. PLoS One. 2014;9:e96442. doi: 10.1371/journal.pone.0096442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.