

Abstract
Protecting genome integrity against transposable elements is achieved by intricate molecular mechanisms involving PIWI proteins, their associated small RNAs (piRNAs), and epigenetic modifiers such as DNA methylation. Eusocial bees, in particular the Western honeybee, Apis mellifera, have one of the lowest contents of transposable elements in the animal kingdom, and, unlike other animals with a functional DNA methylation system, appear not to methylate their transposons. This raises the question of whether the PIWI machinery has been retained in this species. Using comparative genomics, mass spectrometry, and expressional profiling, we present seminal evidence that the piRNA system is conserved in honeybees. We show that honey bee piRNAs contain a 2'-O-methyl modification at the 3' end, and have a bias towards a 5' terminal U, which are signature features of their biogenesis. Both piRNA repertoire and expression levels are greater in reproductive individuals than in sterile workers. Haploid males, where the detrimental effects of transposons are dominant, have the greatest piRNA levels, but surprisingly, the highest expression of transposons. These results show that even in a transposon-depleted species, the piRNA system is required to guard the vulnerable haploid genome and reproductive castes against transposon-associated genomic instability. This also suggests that dosage plays an important role in the regulation of transposons and piRNAs expression in haplo-diploid systems.
Keywords: piRNA, PIWI proteins, transposons, honeybee, haplodiploid, mass spectrometry
Introduction
Uncontrolled movement of transposable elements poses a threat to genomic stability (Werren 2011). PIWI proteins and Piwi-associated RNAs (piRNAs) play a critical role in maintaining the genome integrity of animals by preventing transposon activity (Thomson and Lin 2009; Mani and Juliano 2013). piRNAs are a class of small non-coding RNAs, between 26 and 31 nucleotides (nt) in length (Zheng et al. 2010) that are often derived from mobile elements (Biryukova and Ye 2015). They bind to proteins of the PIWI family and act as guides to silence transposons via epigenetic changes, such as histone modification or DNA methylation, or through post-transcriptional degradation and cleavage (Castaneda et al. 2011). The control of transposable elements by piRNAs is especially important in the germline (Carmell et al. 2007; Chen et al. 2007; Klattenhoff and Theurkauf 2008), and mutations in the piRNA pathway often result in defective gametogenesis (Carmell et al. 2007; Chen et al. 2007). piRNAs are also involved in germline stem cell maintenance, DNA damage repair, sex determination, and modulation of gene expression associated with learning and memory, as well as development (Yin and Lin 2007; Aravin and Bourc'his 2008; Yin et al. 2011; Rajasethupathy et al. 2012; Kiuchi et al. 2014).
The biogenesis of piRNAs in Drosophila, the primary organism of comparison in this study, has been reviewed in detail (Luteijn and Ketting 2013; Mani and Juliano 2013; Ross et al. 2014). In short, the biogenesis of piRNAs can occur through either a primary or secondary pathway. It is believed that the primary pathway is associated with the initial generation of piRNAs, while the secondary pathway (otherwise known as the ‘ping-pong’ amplification pathway) matches/maintains the total pool of piRNAs relative to transcriptional output of their targets. Most primary piRNAs are generated from single-stranded precursors, which are transcribed canonically from long uni-directional genomic clusters located predominantly within pericentromeric and telomeric heterochromatin regions; however, they can also be transcribed non-canonically as dual-stranded clusters (Brennecke et al. 2007). Primary piRNAs show a bias toward the presence of a uracil residue at their 5′ end, which generates a bias for an adenosine residue at the tenth position in secondary derived piRNAs, while the 3′ end of piRNAs commonly contains a 2′-O-methyl modification. In Drosophila, piRNAs are loaded into one of three Argonaute protein complexes, that of Argonaute 3 (Ago3), aubergine (Aub), or Piwi, which show distinct expression patterns. piRNAs generated via the primary pathway are normally loaded into Aub and Piwi, with Piwi localizing to the nucleus, while Aub remains within the cytoplasm. piRNAs loaded into Ago3 are normally generated by the secondary ‘ping-pong’ pathway (Luteijn and Ketting 2013; Mani and Juliano 2013). In a number of insects, including the honeybee, instead of the two orthologs of Aub and Piwi, only a single protein is present, referred to as Piwi/Aub from herein (Liao et al. 2010). Several factors other than PIWI proteins are involved in piRNAs biogenesis and are critical for uni-strand/dual-strand piRNA cluster processing, nuclear export, cytoplasmic processing (cleavage, trimming, and methylation), PIWI-protein loading as well as cellular localization (fig. 1, for review see Czech and Hannon 2016, for the protein Squash see Haase et al. 2010).
Fig. 1.

—Identification of piRNA biogenesis proteins in insects. D.mel: Drosophila melanogaster, A.mel: Apis mellifera (honeybee), A.flo: Apis florea, A.dor: Apis dorsata, B.imp: Bombus impatiens, B.ter: Bombus terrestris, N.vit: Nasonia vitripennis (Wasp), B.mor: Bombyx mori (Silkworm), A.pis: Acyrthosiphon pisum, T.cas: Tribolium castaneum (Beetle). *Piwi/Aub represents the ortholog of the Drosophila Piwi and Aub proteins. # Yb BoYb and SoYb are the orthologs of TDRD12#.
Although Piwi/Aub protein has been identified in the honeybee (Liao et al. 2010), piRNAs have not yet been characterized in this species. This social insect is an attractive model in which novel insights into the epigenomic mechanisms controlling transposable elements could be obtained. Indeed, the transposable element content in the genomes of complex eusocial bees is amongst the lowest in the animal kingdom, and the genome of the European honeybee is a notable case of transposon depletion (Elsik et al. 2014; Kapheim et al. 2015). Specifically, transposable elements make up roughly 3% of the honeybee genome (Elsik et al. 2014) compared with 6% in Drosophila melanogaster (Drosophila 12 Genomes Consortium et al. 2007), 45% in humans (Lander et al. 2001), and over 80% in some plant species such as maize (Schaack et al. 2010). As such, the honeybee provides a valuable system to investigate the function, evolution, and regulation of these pervasive genetic mobile elements.
The evolutionary origins of transposon depletion in the honeybee genome are unclear but a number of hypotheses have been put forward. One argument is that transposons are efficiently eliminated from haploid males where their negative effect on fitness is inevitably dominant (The Honeybee Genome Sequencing Consortium 2006). However, this idea is not supported by the fact that the genome of the wasp Nasonia vitripenis, another haplo-diploid hymenopteran species, has a substantially larger transposon content (Werren et al. 2010). Other explanations have been put forward (Kapheim et al. 2015), including the high recombination rates typically seen in social Hymenoptera (Wilfert et al. 2007) or a decreased exposure to transposon vectors such as pathogens and parasites (Schaack et al. 2010). However, high recombination rates could be a consequence rather than a cause of low transposable element content, as it has been observed that genomic regions with high transposon density have lower recombination rates (Dooner and He 2008).
A striking characteristic of transposons in the honeybee is that they appear to be not methylated (Lyko et al. 2010; Foret et al. 2012) despite the fact that the bee genome encodes a fully functional DNA methylation machinery (Lyko and Maleszka 2011; Foret et al. 2012; Maleszka 2016; Wedd and Maleszka 2016). This is very unusual in the animal kingdom, where the presence of a DNA methylation gene complement typically implies the methylation of transposons (Feng et al. 2010). For instance, the high methylation level of mammalian genomes has been attributed to their large transposon content (Yoder et al. 1997). One possibility is that the low-transposon content in the honeybee does not require DNA methylation. In the honeybee, DNA methylation is therefore exclusively a marker of gene activity and never a silencing mechanism (Foret et al. 2009). Some animal species such as Drosophila have lost DNA methyltransferases (Raddatz et al. 2013), but are nonetheless able to regulate their transposable elements using piRNAs. We therefore sought to understand if the honeybee genome encodes a functional piRNA pathway, and if so, to evaluate its role in the regulation of transposable elements. We report orthologs for most Drosophila genes involved in piRNA biosynthesis and show, for the first time in a social insect, the presence of RNA sequences with a size distribution and nucleotide composition typical of functional piRNAs (e.g., a bias toward a 5′ terminal U and the presence of a 2′-O-methyl modification at the 3′ end, a characteristic of their biogenesis). A subset of these sequences maps to transposable elements and displays signatures of the ping-pong amplification pathway.
Notably, the diversity and expression level of piRNAs is greater in haploid males (drones) than in diploid females (queens and workers), and greater in reproductive individuals (drones and queens) than in sterile workers. This is consistent with the PIWI system playing a critical role in protecting genome integrity in the germline, especially against dominant effects in haploid individuals. Our data also suggest that piRNAs may play a role in modulating the development of caste-specific morphological and reproductive phenotypes. In honeybees, such developmental plasticity is known to be driven by dietary influences on the epigenome (Maleszka 2014; Wedd et al. 2016). Interestingly, a number of differentially expressed piRNAs appear to be active outside the gonads and to target non-transposable elements of the genome.
Our analysis of the piRNA system in a haplo-diploid organism, with a reduced range of transposable elements, expands our knowledge about these important, but poorly understood epigenomic mechanisms in a previously unexplored context.
Materials and Methods
Sample Collection
For all experiment presented, age-matched honey bee larvae (Apis mellifera var. ligustica) were collected for each caste in the spring of 2013 from a single hive, located at the Australian National University (ANU) in Canberra, Australia, that was founded by a queen mated with multiple drones (male). In total, 100 worker, 100 drone and 50 queen larvae were collected. Five biological replicates for each caste, each composed of 5 pooled larval samples, were removed from the initial pool of 100 larvae to undertake piRNA analysis and transcriptomics by high-throughput sequencing. For each caste, a further five biological replicates, each composed of five pooled larval samples, were removed from the initial pools for piRNAs analysis by StemLoop-PCR, Northern Blot and LC/MS. Each replicate, therefore, represents a selection of genetically diverse individuals. Both queen and worker larvae were collected at 96 ± 1 h after emergence, while drone larvae were 96 ± 5 h-old. Marking and assessing the age of drone larvae is less accurate than worker larvae because the haploid eggs from which drones emerge are laid in irregular patches around the edge of the brood frame.
RNA Library Preparation for Illumina High-Throughput Sequencing
For piRNA and transcriptomics analysis, we made use of a recently developed small RNA and transcriptional dataset generated by our laboratory (GEO NCBI database accession number GSE61253) (Ashby et al. 2016). Total RNA extraction and library preparations were carried out as previously detailed (Ashby et al. 2016). In brief, total RNA was extracted from larvae using Trizol reagent, following the manufacturer’s protocol (Life Technologies, Victoria, Australia). RNA integrity was determined by gel electrophoresis and quantified using a NanoDrop spectrophotometer. RNA libraries were constructed using NEBNext Multiplex RNA Library prep kit for Illumina Sequencing (#E7300S for small RNA and #E7420S for mRNA). Libraries were validated on a 2100 Bioanalyzer (Agilent Technologies, Integrated Sciences, Chatswood, Australia), using a high-sensitivity DNA LabChip. Small RNA and mRNA libraries were sequenced at the Biomolecular Research Facility (John Curtin School of Medical Research, Australian National University, Canberra, Australia) on an Illumina HiSeq 2500 platform (51 and 150 bp reads for the small RNA and mRNA libraries respectively). Transcriptome sequence information and raw counts have been submitted to the GEO NCBI database (accession number GSE61253).
Bioinformatic Analyses
piRNA Pathway Protein Identification
The piRNA biogenesis proteins were identified individually using bi-directional best BLASTP searches (NCBI blast version 2.2.29+ (Camacho et al. 2009)) with an e-value cutoff of 1e−5. Drosophila piRNA biogenesis protein sequences were used as queries, with the exception of the protein Trimmer, which is not present in Drosophila, and was therefore queried from the known sequence in Bombyx mori (Izumi et al. 2016). Using this query set, we predicted the presence of these piRNA biogenesis proteins in the honeybee, as well as a number of other insects, including: four close relatives of the European honeybee (two Apis species (Apis florea and Apis dorsata) and two bumblebees (Bombus impatiens and Bombus terrestris), the solitary wasp N.vitripenis, the silk moth B.mori (the only insect beside Drosophila where extensive work on piRNAs has been undertaken), the beetle Tribolium castaneum, and a hemimetabolous species: the pea aphid Acyrthosiphon pisum). The best hits from each species were then used as queries against the Drosophila piRNA pathway protein sequences (supplementary table S1, Supplementary Material online). The data related to Piwi/Aub proteins in N.vitripenis, B.mori, A.pisum, and T.castaneum was taken from the following publications (Kawaoka et al. 2008; Tomoyasu et al. 2008; Lu et al. 2011). The ortholog of TDRD12-like proteins (Yb/Brother of Yb (BoYb)/Sister of Yb (SoYb)) (Handler et al. 2011) can be found in all species listed above other than the three Apis species (A.mellifera, A.florea, and A.dorsata). We therefore used the TDRD12-like protein from B.mori as the query for bi-directional best BLASTP searches in these three Apis species. Differential expression of PIWI and piRNAs biogenesis proteins was assessed using the edgeR package (Robinson et al. 2010).
piRNA Identification
The 3′ adaptor sequences of small RNA reads were trimmed with a custom python script. Reads without adaptor sequences were discarded. After trimming, only the reads with a size of 24–35 nt were kept. tRNA, rRNA, and miRNA fragments were identified using Bowtie v1.0 (Langmead et al. 2009) allowing up to three mismatches and removed from further analyses. Bowtie v1.0 was used to identify putative piRNA by aligning the remaining reads to the honeybee genome (Amel_4.5), allowing up to three mismatches.
piRNA Target Analysis
Alignment to transposable elements (Elsik et al. 2014) was carried out using Bowtie v1.0, allowing up to three mismatches and up to 100 multiple mappings. Reads mapping to N different loci contributed 1/N counts to each locus. For instance, if a read mapped to ten different locations, each location received 0.1 counts. For the read overlap analysis of the ping-pong cycle, the reads mapping to the plus strand of transposon were used as seeds and searched against potential partners mapping to the reverse strand of the same transposon (Brennecke et al. 2007) using overlaps ranging from −25 to +25 bp.
In order to compare the expression of transposons and their corresponding piRNAs, we determined the expression of all known transposable elements (Elsik et al. 2014) by mapping them to our transcriptome data using Bowtie v2.2 (Langmead and Salzberg 2012).
Putative piRNAs were mapped to honeybee genes (amel_OGSv3.2, Elsik et al. 2016) and 12 honeybee viruses [Acute bee paralysis virus (Govan et al. 2000), Aphid lethal paralysis virus (Van Munster et al. 2002), Black queen cell virus (Leat et al. 2000), Chronic bee paralysis virus (Olivier et al. 2008), Deformed wing virus (Lanzi et al. 2006), Invertebrate iridescent virus 6 (Jakob et al. 2001), Israeli acute paralysis virus (Maori et al. 2007), Kashmir bee virus (de Miranda et al. 2004), Sacbrood virus (Ghosh et al. 1999), Slow bee paralysis virus (de Miranda et al. 2010), Tobacco ringspot virus (Zalloua et al. 1996), and Varroa destructor virus-1 (Ongus et al. 2004)] using Bowtie v1.0, allowing up to three mismatches and up to 100 multiple mappings. Reads mapping to N different loci contributed 1/N counts to each locus.
piRNA Cluster Identification
piRNA clusters were identified by two methods. We first used the method described in Brennecke et al. (2007). In brief, we used a 5-kb sliding window to identify regions with densities greater than one normalized count per kb using a custom-made python script. Normalized counts are defined by copy number of read/mapping sites. For instance, if a read has ten identical reads mapping to two different loci, the normalized count for each locus is five. Regions with lengths larger than 800 bp (the length of the smallest sequence in the genome assembly v4.5 is around 800 bp) and containing at least four different piRNAs were considered as a piRNA cluster candidate. In a second approach, we used the software piClust to predict piRNA cluster candidates (Jung et al. 2014) with a 5-kb sliding window, with at least five reads per cluster and a cutscore parameter of 3. Only clusters predicted by both methods are reported.
StemLoop PCR
StemLoop PCR was carried out as previously detailed (Ashby et al. 2016). Total RNA was isolated from independent larval samples (n = 5 per caste) collected from the same hive and at the same time as those processed for sequencing. In brief, StemLoop qRT-PCR validation of piRNA expression was adapted from the protocol (Varkonyi-Gasic et al. 2007). Reverse transcription was performed in a 15-µl reaction volume containing; 100 ng of total RNA, 50-nM Stem-Loop RT Primer (supplementary table S4, Supplementary Material online) and 0.5-mM dNTPs. To investigate the localization of piRNAs within the honeybee, total RNA was extracted from the following tissues and investigated by StemLoop PCR: ovaries (obtained from 2-week-old, mature mated queens), testes (from 2-week-old, mature drones), sperm (from 2-week-old, mature drones), and brains (pollen foragers). Semi-quantitative RT-PCR was performed on a StepOnePlus Real-Time PCR System (Applied Biosystems (Life Technologies), Melgrave, Victoria, Australia). The 15-µl PCR reaction mixture consisted of; 1 µl of cDNA, 50 nM forward and universal reverse primers (supplementary table S4, Supplementary Material online), and 1×Fast SYBR Green Master Mix (Life Technologies, Melgrave, Victoria, Australia). For analysis of piRNA expression, the Mean Normalized Expression (MNE) of each target piRNA was calculated separately for each caste (drone, queen worker) using the methods previously described (Ashby et al. 2010) using ame-mir-263b as a reference (control) sequence as previously validated (Ashby et al. 2016).
Periodate Treatment and Beta-Elimination
A beta-elimination assay, modified from Alefelder et al. (1998), was undertaken to investigate the presence of a 2′-O methyl addition at the 3′ end of predicted piRNAs (supplementary table S2, Supplementary Material online). Twenty micrograms of enriched small RNA (17–200 nt in length) were obtained by passing 1 mg of total RNA through an RNA Clean & Concentrator column system (Zymo Research), following the manufactures instructions. Fifteen micrograms of enriched small RNA or synthetic piRNA (Sigma-Aldrich) was dissolved in 37.5 µl of distilled H2O, 12.5 µl freshly made sodium periodate (NaIO4, final concentration 25 mM), 50 µl of 2× borax/boric acid buffer (final concentration 60 mM, pH 8.6). The solution was incubated in the dark for 30 min at room temperature. Following incubation, glycerol (100%, 10 µl) was added to each reaction tube to quench unreacted sodium periodate, with the reaction mixture incubated for a further 10 min in the dark at room temperature. The pH was then raised to 9.5 using 1 M NaOH before the samples were incubated at 45 °C for 90 min before precipitation with 100% EtOH. For northern blots, both beta-eliminated and untreated RNA were resolved on a 20% acrylamide:bis-acrylamide PAGE, 8 M urea gel with the aid of a microRNA Marker (#N2102S, NEB). Following electrophoresis, RNA was transferred to an Hbond-N+ membrane (Amersham) using a Trans-Blot SD semi-dry transfer cell (Bio-Rad) following the manufacture’s protocol. Following electro-blotting, RNA was fixed to the membrane by UV cross-linking for 1 min, followed by baking at 65 °C for 1 h. Membranes were pre-hybridized in ExpressHyb hybridization solution (Clontech) for 1 h at 45 °C. During pre-hybridization, 5′-labeled probes against a predicted piRNA and miRNA sequence were prepared as follows: 5 µl of 10× T4 DNA polynucleotide kinase buffer (NEB), 5 µl of 10× T4 DNA polynucleotide (NEB), 0.5 µl of DNA oligonucleotide (100 µM, antisense strand, supplementary table S2, Supplementary Material online), 2 µl of gamma [32P]-ATP (6,000 Ci/mmol, PerkinElmer) and 37.5 µl of distilled H2O. Labeled probes were cleaned and unbound gamma [32P]-ATP removed using a G-25 sephadex column. Membranes were hybridized with the labeled probe in ExpressHyb hybridization solution for 12 h before being washed in 2× SSC, 0.2% SDS and visualized using a PhosphorImager. To demonstrate specificity, each probe was pre-incubated with a synthetic version of their target sequence. Pre-incubation caused a loss of the hybridization signal, indicating target specificity (data not shown). The use of a scrambled probe demonstrated a lack of non-specific binding (data not shown). As we were not comparing piRNA expression levels, an internal control gene was not probed for.
Analysis of 2′-O Methyl Modifications by LC/MS
For LC/MS analysis of 3′-end modifications, small RNAs were size selected and purified from 10 µg of total RNA (n = 5 per caste) using a 15% acrylamide:bis-acrylamide PAGE, 8-M urea gel with the aid of a microRNA Marker (#N2102S, NEB). The size selected small RNAs were eluted from the gel by incubation in a 0.3-M NaCl solution overnight at 4 °C, before precipitation using isopropanol/ethanol. The precipitated pellet was dissolved in 6.5 µl RNAase free, DEPC-treated, distilled water. Enriched small RNA samples, as well as synthetic piRNAs (positive control, supplementary table S2, Supplementary Material online), were enzymatically digested with RNase T2 (#LS01501, Worthington Biochemical Corporation). As RNase T2 cleaves the 3′ side of a phosphodiester bond it will generate free 3′-phosphoribonucleotides from internal residues and free ribonucleosides from 3′ terminal residues. Thus, the detection of 2′-O methyl modified ribonucleosides will represent the 3′ end terminal modifications of the small RNAs. Synthetic piRNAs were used to determine the efficiency of the RNase T2 enzyme digest, as well as acting as standards for LC/MS analysis. Each synthesized piRNA represented a predicted piRNA and were chosen to represent the four possible 2′-O methyl modified bases that could be observed at the 3′ terminal end (Am, Gm, Cm, and Um, supplementary table S2, Supplementary Material online). For the RNase T2 digestion, the following components were mixed and incubated at 37 °C for 3 h (pH 4.5): 5 µg of enriched small RNA larval sample or 5 µg synthetic piRNA sample, 2 µl of 200 mM ammonium acetate (pH 4.5, final conc. 20 mM), 100 U/ml of RNase T2, with the final volume adjusted to 20 µl with dH2O.
Modified ribnucleosides were analyzed using an Agilent 6530 High Resolution Accurate-Mass LC/MS Q-TOF (Agilent Technologies, Inc. Santa Clara, CA, USA) at the Joint Mass Spectrometry Facility (Research School of Biology, Australian National University). Samples were subjected to electrospray ionization (ESI) in the dual Jetstream interface in the positive polarity under the following conditions: gas temperature 250 °C, drying gas 5 l/min, nebulizer 30 psig, sheath gas temperature 350 °C, and flow rate of 11 l/min, capillary voltage 2,500 V, fragmentor 138 V, and nozzle voltage 500 V. Samples and standards (7 µl) were injected onto an Agilent ZORBAX Eclipse XDB-C18 column (2.1 × 50 mm; 1.8 µm) held at 35 ± 0.5 °C and analytes eluted with a linear gradient from 0% to 1% solvent B over 6 min, 1–6% solvent B from 6–9.25 min (then held at 75% from 12 to 21 min) at a flow rate of 100 μl/min. Mobile phase A consisted of water containing 0.1% formic acid and mobile phase B consisted of 0.2% acetonitrile/water and 0.1% acetic acid. The QTOF was operated in targeted MS/MS mode using collision-induced dissociation [N2 collision gas supplied at 18 psi (124.1 kPa), m/z 1.3 isolation window] where the MS extended dynamic range was set from m/z 100 to 1,000 at 2 spectra/s, and MS/MS m/z 50–1,000 at 3 spectra/s. Synthetic standards were used to optimize the LC-MS/MS to determine their retention times, and acceptable collision energies to produce signature product ions relating to the precursor ions. Data were acquired and analyzed using Agilent Technologies Masshunter software (ver. B.5.0).
Results
Identification of piRNA Pathway Proteins
To identify if the honeybee genome contains the repertoire of genes required for piRNA biogenesis, we looked for Drosophila orthologs in the honeybee by bi-directional BLASTP best searches. Orthologs of piRNA pathway proteins in Drosophila were also searched for within eight other fully sequenced insect genomes using the same method (supplementary table S1, Supplementary Material online). Since Trimmer, a protein involved in piRNA 3′ end trimming, has been identified in B.mori (Izumi et al. 2016), while Nibbler possesses this function in Drosophila (Hayashi et al. 2016), we used B.mori Trimmer as the query for the ortholog search in the honeybee and other insects. Similarly, TDRD12-like protein from B.mori was used as a query for the blast search in all three Apis species investigated (see Methods section).
Of the 32 genes proteins involved in the piRNA pathway in Drosophila, 23 have an ortholog in the honeybee (fig. 1), and were detected in our RNA-seq libraries (data not shown). Most of the genes proteins missing from the bee are also absent in the other insects that we inspected, and probably fulfill functions specific to Drosophila, or represent Drosophila specific expansions (supplementary table S1, Supplementary Material Online).
As in most insects, the honeybee genome encodes two proteins of the PIWI family: AmAgo3 (GB49909) and AmAub (GB54204) (supplementary table S1, Supplementary Material Online). AmAub is orthologous to the two Drosophila paralogs Piwi and Aub (denoted as Piwi/Aub). In a number of species, significant gene duplication of Aub and Piwi has been observed (Lewis et al. 2016). For example, eight Piwi/Aub paralogs have been identified in A.pisum (Lu et al. 2011), whereas only one has been found in B.mori (Kawaoka et al. 2008) and T.castaneum (Tomoyasu et al. 2008). Like B.mori and T.castaneum, the honeybee has only a single copy of Piwi/Aub (Liao et al. 2010).
The insect genomes examined also encode a complement of Tudor-domain containing proteins (TDRD) very similar to Drosophila. Similar to that previously reported in the silkworm (Xiol et al. 2014), a notable difference is that the honeybee, like other insects, has only one protein corresponding to the Drosophila TDRD12-like proteins: Yb/BoYb/SoYb (supplementary table S1, Supplementary Material online). These proteins are involved in the transport of piRNA precursors from the nucleus to cytoplasm in Drosophila. Yb is expressed exclusively in somatic cells while BoYb and SoYb are specific to germ cells (Handler et al. 2011). We can thus speculate that the expansion of these genes in Drosphila was followed by a cell type specific sub-functionalization, and that the original function is still carried out by a single gene in other insects. The Drosophila TDRD protein Krimper plays an important role in strengthening the ping-pong cycle (Sato et al. 2015), but no ortholog was detected in other insect species.
We also failed to detect orthologous sequences of the Drosophila proteins Cutoff, Deadlock, and Rhino in other insects. These three proteins form a complex implicated in dual-strand, but not uni-strand, cluster transcription (Mohn et al. 2014). This finding raises the possibility that piRNAs in the honeybee are processed primarily as uni-strand clusters. The Shutdown protein, involved in loading piRNA into proteins of the PIWI family (Preall et al. 2012), appears to be missing from the honeybee, but is present in other insect species.
In summary, the complement of piRNA pathway genes appears to be near identical between the honeybee, other Hymenoptera, Lepidoptera, and Coleoptera, and, to a lesser extent, Hemiptera. As the main differences in the piRNA pathway between these insects and Drosophila are caused by Drosophila-specific expansions we conclude that a functional piRNA pathway is present in the honeybee.
Identification of Expressed piRNA Candidates
In order to identify potential piRNAs in the honeybee genome, we used a dataset, recently developed in our laboratory (GEO NCBI database accession number GSE61253) of small RNAs expressed in drone, queen, and worker honeybees after 96 h of larval development (Ashby et al. 2016). This dataset has the distinct advantage that it is well replicated (n = 5 per type of larva), deeply sequenced (around 10 million reads per library), and contains the three honeybee castes: haploid males (drones) and the diploid females (fertile queens and sterile workers). After initial quality check and adapter trimming, reads were mapped to the known honeybee miRNAs, tRNAs, and rRNAs. Around 40% (male larvae) and 20% (female larvae) of these sequences do not align to any of these RNA species (supplementary fig. S1, Supplementary Material online) and could include piRNA molecules. Depending on the sample, 32–52% of these unannotated sequences map to the honeybee genome and display a peak between 26 and 31 bp in length (fig. 2A and supplementary fig. S2, Supplementary Material online). This size distribution suggests the presence of genuine piRNAs (Mani and Juliano 2013). This putative piRNA peak is more pronounced in drones relative to females, and more marked in queens than in workers, suggesting that piRNAs are more abundant in drones than queens and workers, and have higher expression in reproductive than sterile females. The diversity of piRNA species observed in each caste mirrored that of expression levels, with drones showing the greatest repertoire followed by queens than workers (fig. 3 and supplementary fig. S4, Supplementary Material online).
Fig. 2.

—Candidate piRNAs in the honeybee genome. Shaded regions show the minimum/maximum values observed between biological replicates. (A) Size distribution of small RNAs of 24 bp and larger mapped to the honeybee genome. A peak between 26 and 31 nt can be observed, consistent with the size of piRNAs in other species. (B) Percentage of sequences with a 5′ U. The majority of sequences between 26 and 31 nt have the 5′ U characteristic of piRNAs. (C) Size distribution of small RNAs mapped to transposons, the typical piRNA peak between 26 and 31 nt is more pronounced than for the total small RNA population.
Fig. 3.

—Nucleotide compositions at each position of putative piRNAs mapped sense and antisense to transposons. The number of reads is indicated (n). In drones and queens, A is over-represented at the 10th position of sequences mapping sense to transposons, a signature of the ping-pong pathway.
In order to further investigate whether the size peak corresponds to real piRNAs, we analyzed the nucleotide content of these sequences (fig. 2B). Primary piRNAs often start with a U ribonucleotide at their 5′ end (Brennecke et al. 2007). A clear 5′ U bias is noticeable in all samples for sequences between 26 and 31 nt. This bias is particularly evident in drones. These results strongly suggest that piRNAs are present in the honeybee with a length in the range of 26–31 nt with a higher expression and diversity in drones, followed by queens and then workers.
Predicted piRNAs Display Characteristic 3′ Terminal 2′-O Methyl Modification
A hallmark feature of piRNA biogenesis is the addition of a 2′-O-methyl modification to the 3′ terminal ribonucleotide performed by the Hen1 enzyme, a protein conserved in all species investigated (supplementary table S1, Supplementary Material online). To investigate whether putative piRNAs in the honeybee display this characteristic modification, we undertook a beta-elimination assay and liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) analysis.
Beta-Elimination
Treatment of RNA with sodium periodate followed by borate (pH 9.5) causes removal of the terminal 3′ ribonucleotide via beta-elimination. However, RNA species in which the terminal ribonucleotide has been modified at its 2′ or 3′ hydroxyl group are protected from this reaction. Northern blot analysis, with antisense probes to the putative piRNA P4 (supplementary table S2, Supplementary Material online) illustrates that its migration rate is unaffected by beta-elimination treatment (fig. 4). In contrast, an antisense probe for ame-mir-71, which does not contain a 3′ terminal 2′-O methyl modification, shows a faster migration rate in larval samples following beta-elimination. This result indicates that the 3′ terminal sugar ring of piRNAs contains a modification to either the 2′ or 3′ hydroxyl group, while ame-mir-71, as noted, does not contain such a modification. As a control, we synthesized piRNA P4 with a 2′-O methyl modified 3′ terminal ribonucleotide, and ame-mir-71 miRNA with no modification to its 3′ terminal ribonucleotide (supplementary table S2, Supplementary Material online). As observed in our larval samples, beta-elimination does not affect the migration rate of the synthetic piRNA, but does increase the migration rate of the unmodified ame-mir-71 (data not shown).
Fig. 4.
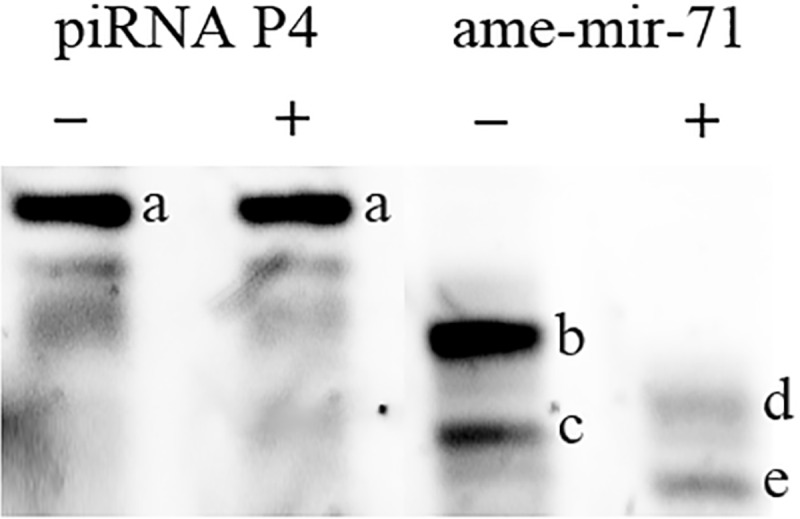
—Northern blot analysis of 3′ terminal ribonucleotide modifications. A small RNA sample purified from 96-h drone larvae was either subjected (+) or not subjected (−) to beta-elimination. Northern blots were probed for the predicted piRNA P4 (supplementary table S2, Supplementary Material online) and ame-mir-71. Beta-elimination did not affect the migration rate of the predicted piRNA, but did induce a faster migration of ame-mir-71 indicating that the former is modified at the 3′ end. (A) piRNA P4, unaltered after beta-elimination, (B) ame-mir-71, (C) short ame-mir-71 isomiR, (D) shortened ame-mir-71 after beta-elimination, (E) shortened isomiR.
LC-ESI-MS/MS Analysis
To elucidate the type and localization of the 3′ terminal modification observed in the putative honeybee piRNAs, we undertook targeted LC-ESI-MS/MS analysis. Small RNAs were size selected and purified from larvae samples (n = 5 per caste) using a 15% acrylamide:bis-acrylamide PAGE, 8-M urea gel before being digested with RNase T2. Such digestion generates free 3′-phosphoribonucleotides from internal residues and free ribonucleosides from 3′ terminal residues. Thus, detected ribonucleosides represent the 3′ terminal nucleotide of these small RNAs. As shown in figure 5, precursor ions [M+ H]+ for each of the four possible 2′-O methyl ribonucleosides (Am = 282.119 m/z, Gm = 298.114 m/z, Cm = 258.108 m/z, and Um = 259.092 m/z), along with their characteristic product ions [base peak product ions (BP) for Am= 136.060 m/z, Gm= 152.056 m/z, Cm = 112.050 m/z, and Um = 113.039 m/z] were detected in all three honeybee castes. To distinguish between 2′-O methyl and 3′-O methyl modifications on the 3′ terminal nucleotide, retention times (tR, min) for our synthetic piRNAs standards (supplementary table S2, Supplementary Material online) with their related MS/MS product ions were used to identify and confirm the presence of a methyl group on the 2′ position of the sugar ring backbone. Taken together, beta-elimination and LC-ESI-MS/MS results demonstrate that our putative honeybee piRNAs display the characteristic 2′-O methyl 3′ terminal modification that is a hallmark of their biogenesis. In accordance with the sequencing data, count levels for each of the modified bases were greater in the haploid male relative to either of the diploid females (fig. 5), suggesting a higher expression level of piRNAs in drone larvae.
Fig. 5.

—Chemical structures, LC-ESI-QTOF targeted MS/MS parameters and respective characteristic MS/MS spectra for both synthetic and bee larval 2′-O-methyl modified ribonucleosides. Ribonucleosides were generated by treatment with ribonuclease T2 of synthetic piRNAs and piRNA-enriched larval samples. The blue diamond in each MS/MS spectrum indicates the position of the targeted precursor ion. For all four modified bases, greater count levels were observed in drones relative to both queens and workers. For 2′-O-methyladenosine and 2′-O-methylguanosine, greater count levels were seen in queens relative to workers, while for 2′-O-methylcytidine and 2′-O-methyluridine, similar count levels were observed in both female castes.
Analysis of Transposon-Derived Putative piRNAs
The primary function of piRNAs is to silence transposable elements. We thus assessed whether this is also the case in the honeybee despite the very low transposon content of its genome. To this end, we mapped putative piRNAs to the transposable elements of the honeybee genome. In Drosophila, around 68–78% of piRNAs are derived from transposons, covering nearly all known types of transposons (Brennecke et al. 2007). In the honeybee, however, the majority of putative piRNAs (∼70%) align to intra and intergenic regions, with only 11.89% of putative piRNAs mapping to transposons in drones, and 0.82% and 0.19% in queens and workers, respectively (supplementary table S3, Supplementary Material online). This relatively low mapping rate to transposons could be associated with some of these sequences not being genuine piRNAs, or because piRNAs in the honeybee might predominantly undertake functions unrelated to transposon silencing. Alternatively, some of these piRNAs might have originated from transposons that have now mutated beyond recognition. The discrepancy in mapping rate between the honeybee and Drosophila could also stem from the fact that the honeybee libraries were made from whole larvae while the Drosophila sequences came from gonads or germline cells (Brennecke et al. 2007). The size distribution of the sequences mapping to transposons has a very distinct peak at 28 nt in all castes (fig. 2C and supplementary fig. S3, Supplementary Material online). This size is consistent with that of piRNAs in other animals and therefore suggests that these sequences are genuine piRNAs involved in transposon silencing.
The difference in the percentage/diversity of piRNAs mapped to transposons between male and female larvae suggests that drones recruit more piRNAs to protect their haploid genome, which is more susceptible to the deleterious consequences of mobile elements. Furthermore, the higher percentage/diversity of piRNAs in fertile females (queens), relative to their sterile sisters (workers), also indicates that piRNAs are critical in protecting the castes containing a functional germline, as rampant transposition may damage germline integrity.
We found that putative piRNAs map to mobile elements from six different transposon families (fig. 6). Mariner, the main transposon in the honeybee genome (The Honeybee Genome Sequencing Consortium 2006), is the primary target of piRNAs in all samples (57.09–63.25%), followed by PiggyBac (18.61–20.44%), and R2 (13.90–18.36%). The remaining three transposons, Copia, Bel-Pao, and I, account for <5% of the total transposon-derived putative piRNAs. Note that piRNAs targeting Bel-Pao, Copia, and I elements were not detected in worker larvae (fig. 6). piRNAs mapping to transposons display a strong antisense bias (fig. 6B), supporting the idea that they are involved in the repression of these mobile elements.
Fig. 6.

—Mapping of piRNAs to transposons. (A) Distribution of piRNAs mapped to different families of transposons in drones, queens, and workers. (B) Strand-specificity of piRNAs mapping to transposons. Left: log2 of the ratio of piRNAs mapping to the plus and minus strand of transposons in the different small RNA libraries; right: summary of transposon abundance. NC: no count. The majority of candidate piRNAs tend to map to the minus strand of transposons.
Secondary piRNAs generated by the ping-pong amplification pathway generally have a 10-nt overlap with primary piRNAs at their 5′ end. Due to the binding properties of the Argonaute protein Aub, the secondary piRNA often contains an A ribonucleotide at the 10th position (Wang et al. 2014). To investigate whether piRNAs are amplified by the ping-pong cycle in the honeybee, we examined if this signature is present in the piRNA candidates mapping to transposons. Figure 3 shows that the majority of sequences antisense to transposons (primary piRNA candidates) start with U while sequences mapping to the sense strand of transposons (secondary piRNA candidates) have a distinct prevalence of an A ribonucleotide at the 10th position. This strongly suggests that the secondary piRNA biogenesis pathway is present in the honeybee.
In order to gain further evidence for the presence of a ping-pong cycle in the honeybee, we looked for another of its signatures: the 10-nt overlap between the 5′ end of primary piRNAs and their secondary partners. A clear overlap with a 10-nt offset is visible in drones and queens (fig. 7). This signature is markedly weaker in workers, which may be due to the lower number of transposon-derived putative piRNAs mapped in this caste. Overall, 47% (drones), 13% (queens), and 6% (workers) of the piRNAs mapping antisense to transposons had a potential sense ping-pong partner. It is noteworthy that the bias toward a 5′ U on antisense piRNAs and an A at the 10th position on sense piRNAs were also observed in candidate piRNAs without any identified transposon partners (supplementary fig. S4, Supplementary Material online). A similar observation has been reported in Hydra (Lim et al. 2014), suggesting that the pairing of ping-pong partners might sometimes follow rules less stringent than our mapping parameters (up to three mismatches).
Fig. 7.

—Overlap between candidate primary and secondary piRNAs mapping to transposons. Sequences in drones and queens show a clear prevalence of 10-nt overlap, characteristic of the ping-pong pathway.
To examine the relationship between putative piRNAs and their target transposons, we compared their expression level in each sample (fig. 8, supplementary table S5, Supplementary Material online). Modeling the amount of piRNA as a function of the amount of corresponding transposable element (quantitative variable) and the caste (factor), we found that piRNAs are positively correlated to their targets (ANCOVA, F = 44.7, P = 2.21 × 10−9). These results indicate that piRNA synthesis could be upregulated by the expression of transposable elements. However, workers have more transposons than queens and fewer piRNAs, indicating a distinct regulation mechanism in sterile females.
Fig. 8.

—Expression levels of transposons and their corresponding piRNAs (both sense and antisense). Drones have higher levels of both transposons and piRNAs than queens and workers, while queens have more piRNAs and fewer transposons than workers.
In mosquitoes, piRNAs have been implicated in antiviral defense (Vodovar et al. 2012). Therefore, due to the low mapping rate of piRNAs to mobile elements in the honeybee, we investigated whether piRNAs were instead mapping to viruses. We found <0.01% of putative piRNAs mapped to viruses (data not shown), suggesting that piRNAs may not make a significant contribution to antiviral defense in the honeybee, similar to that seen in Drosophila (Petit et al. 2016).
A large number of piRNA candidates map to protein coding genes. Supplementary figure S5, Supplementary Material online shows the five putative piRNA with the highest counts that map to genes. These piRNA often map to very narrow and well-defined regions of these genes. Moreover, they predominantly map antisense to these genes. Further research is required to clarify if these piRNAs regulate the genes present on the opposite strand or if they act in trans. However, their precise mapping pattern and their high level of expression suggest that these molecules could conceivably play an important role in regulating the epigenetic state of the cell.
Characterization of Putative piRNA Clusters
Using the program piClust (Jung et al. 2014) and the method of Brennecke et al. (2007), we investigated the presence of piRNA clusters in the honeybee genome. We identified a total of 67 putative piRNA clusters (58 in drones, 25 in queens, and 14 in workers) encoding 7,967 distinct piRNA species (fig. 9A). Only 11 piRNA clusters were found in common between drone, queen, and worker larvae. Of the 67 putative piRNA clusters, 28 are located in unmapped scaffolds (scaffolds of the honeybee assembly are typically short and do not contain any genetic marker enabling them to be placed on chromosomes). This suggests that as in other species (Brennecke et al. 2007), piRNA clusters are predominantly located within heterochromatic regions that are harder to sequence accounting for many of the honeybee unmapped scaffolds. Most of the inferred piRNA clusters are located on a single strand, while only five appear to be dual-strand (fig. 9A). This might be explained by the lack of the Deadlock–Cutoff–Rhino complex, which is required for dual-strand piRNA cluster activity in Drosophila (Mohn et al. 2014). The few detected dual-strand clusters might be different in nature from those in Drosophila, since these honeybee sequences include some splicing events, whereas the Drosophila dual-strand clusters are not spliced (Zhang et al. 2014). On average, 63.56%, 52.07%, and 54.75% of transposon-derived piRNAs can be aligned to piRNA clusters in drones, queens, and workers, respectively, while the rest are scattered individually across the genome. The clusters expressed in the drone include piRNA mapping to all six types of transposons described above. The clusters identified in queens, however, only encode piRNAs mapping to Mariner, PiggyBac, and R2, and in workers the clusters only map to Mariner and PiggyBac.
Fig. 9.

—piRNA clusters and phasing in the honeybee. (A) Number of clusters in the different castes. Significantly more clusters were identified in the drone, and most clusters were classified as single-strand. (B) Distribution of the distances between the 3′ end an upstream piRNA and the 5′ end of the next downstream piRNA for all piRNA pairs present in clusters. A peak distance around 20 nt can be observed. (C) An exemplary piRNA cluster.
Discrete peaks can be seen within the honeybee piRNA clusters (fig. 9B and C). This phased organization has been recently reported in Drosophila and mouse, where it results from a “tertiary” piRNA pathway dependent on the Zucchini endonuclease (Han et al. 2015; Mohn et al. 2015). Unlike Drosophila, where phased piRNAs are typically immediately adjacent to each other, the honeybee sequences are spaced by around 20 bp (fig. 9B), suggesting that the corresponding pre-piRNA have a size of ∼50 bp. These long pre-piRNAs are likely processed to their mature size by the honeybee ortholog of the Trimmer exonuclease (supplementary table S1, Supplementary Material online) recently discovered in the silk moth but missing from Drosophila (Izumi et al. 2016).
Confirmation of Differential Expression of piRNAs in Different Castes
We employed StemLoop real-time PCR to further test the hypothesis that the expression level of piRNAs are highest in drones and lowest in sterile workers at this early larval stage. We tested the expression of six putative piRNAs, chosen from sequences that were detected in all larval samples (supplementary table S4, Supplementary Material online). As predicted, each of the putative piRNAs shows higher levels of expression in the haploid male drone, followed by the fertile female queen and finally the sterile female worker (fig. 10A).
Fig. 10.
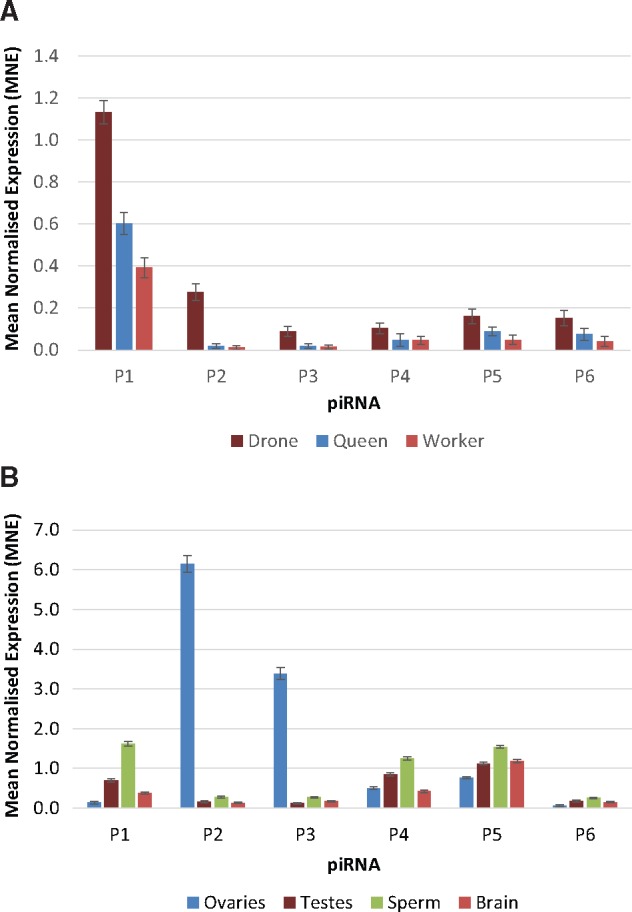
—Relative expression levels of six putative piRNAs estimated by stem-loop PCR in (A) drones queens and workers, and (B) ovaries, testes, sperm, and worker brain.
To investigate the localization of piRNAs within the honeybee, total RNA was extracted from ovaries, testes, sperm, and worker brains. All six putative piRNAs were detected in each of the tested tissue types (fig. 10B). Putative piRNAs P1, P4-P6 showed greatest expression in sperm, while putative piRNAs P2 and P3 showed extremely high expression levels in ovaries relative to all other tissues. At least two of these piRNAs (P2 and P4) are likely targeting PiggyBac transposons. It is noteworthy that the tissues containing germline cells (ovaries, testes, and sperm) have the highest piRNA levels. However, the brain expresses piRNA P5 at fairly high levels. Further experiments are required to establish if the role of piRNA in the brain is associated with the control of transposons or with other functions. Further research is also needed to elucidate whether this tissue specificity of piRNAs reflects tissue-specific expression of transposable elements or if they are associated with other tissue-specific functions unrelated to transposons.
Discussion
Our results provide seminal evidence that the honeybee has a fully functional piRNA system in spite of having a transposon-depleted genome. This is illustrated by the presence of key signatures of piRNA biogenesis, including: enrichment in small RNAs of appropriate size (26–31 ribonucleotides in length), sequence similarity with transposable elements, 5′-terminal U enrichment, 2′-O methylation on the 3′ nucleotide and a clustered organization. We also identified several characteristic signatures of the secondary ping-pong pathway, which include: 10-nt overlap between sense and antisense piRNAs and enrichment in A at the 10th position of piRNAs mapping to the sense strand of transposons.
Since most of the work on piRNAs has focused on Drosophila and mammalian models, it is still unclear how conserved the individual components of this pathway are across the animal kingdom. Our comparative analysis shows that the repertoire of genes involved in the piRNA pathway is very similar between insects. Interestingly, most of the Drosophila genes missing from the honeybee are also missing from the other insect investigated. Some of these discrepancies with the Drosophila model may lead to qualitative differences in piRNA pathways. For instance, the lack of Rhino, Cutoff, and Deadlock orthologs in the honeybee, as well as other insects, indicates that dual-strand clusters might be a trait specific to Drosophila or dipterans. It is also possible that the functions of some of the Drosophila genes involved in the piRNA pathway are fulfilled by other non-homologous or non-conserved genes in other insects.
Several lines of evidence (RNA-seq, LC-MS, and stem-loop PCR) congruently show striking differences in both the expression levels and repertoire of transposable elements and piRNAs seen between the three larval types. The reproductive castes, drones, and queens, have the highest level and greatest diversity of piRNAs. An intuitive explanation for this observation is that transposable elements need to be more tightly controlled in individuals whose germline is destined to be passed on to future generations.
While drones show the greatest diversity of piRNA species and the highest levels of expression, their transposable elements are also more highly expressed than in females. This finding points to a fundamental difference between drones and females in the nature of the negative feedback loop that controls transposons with piRNAs. Since drones are typically haploid and females are diploid, dosage is a possible explanation for this difference. Transposons hijack the transcriptional machinery of the cell and the transcription factor binding sites, therefore driving the expression of transposons as secondary targets of the organism’s transcription factors. Assuming that the concentration of transcription factors in haploid and diploid cells is similar, the primary targets of transcription factors will be more quickly saturated in haploid cells, and thus the secondary targets (transposons) will be more active. The high expression of piRNAs in drones might be an adaptive evolutionary response to this increased level of transposons or could be an intrinsic property of the piRNA-transposon feedback loop.
The current findings also have important consequences for our understanding of genome evolution. It has recently been proposed that the honeybee would be an ideal model to understand why polymorphic genomes appear to have high mutation rates (Lynch 2015). It is predicted that haploid drones should have lower mutation rates than diploid females. However, transposable elements are important factors affecting mutation and recombination (Dooner and He 2008). Our results indicate that the expression of transposable elements and their regulatory piRNAs should be factored into any predictions regarding the dynamics of genome evolution.
In accordance with prior studies implicating piRNAs in functions not related to transposon regulation in the germline (Ishizu et al. 2012), we observed piRNA species targeting regions antisense of protein-coding genes. While it would be premature to speculate on the possible roles of these piRNAs in controlling transcription units outside transposons, this finding clearly deserves a more detailed follow up study to unravel their biological significance.
Our study enables comparative analysis between mammals and a number of invertebrate species that express functional piRNA systems, but display different levels of transposable elements and the presence or absence of DNA methylation. Such comparative analyses are fundamental to unraveling the principles and evolutionary consequences of epigenetic mechanisms.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Spencer Whitney for his help with Northern blots.
Funding
This study was supported by the Australian Research Council DECRA grant DE130101 to SF and by the Australian Research Council Discovery grant DP160103053 to RM.
Literature Cited
- Alefelder S, Patel BK, Eckstein F.. 1998. Incorporation of terminal phosphorothioates into oligonucleotides. Nucleic Acids Res. 26:4983–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Bourc'his D.. 2008. Small RNA guides for de novo DNA methylation in mammalian germ cells. Genes Dev. 22:970–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby R, Foret S, Searle I, Maleszka R.. 2016. MicroRNAs in honey bee caste determination. Sci Rep. 6:18794.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby R, Kozulin P, Megaw PL, Morgan IG.. 2010. Alterations in ZENK and glucagon RNA transcript expression during increased ocular growth in chickens. Mol Vis. 16:11. [PMC free article] [PubMed] [Google Scholar]
- Biryukova I, Ye T.. 2015. Endogenous siRNAs and piRNAs derived from transposable elements and genes in the malaria vector mosquito Anopheles gambiae. BMC Genomics 16:278.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, et al. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128:1089–1103. [DOI] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmell MA, et al. 2007. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell. 12:503–514. [DOI] [PubMed] [Google Scholar]
- Castaneda J, Genzor P, Bortvin A.. 2011. piRNAs, transposon silencing, and germline genome integrity. Mutat Res. 714:95–104. [DOI] [PubMed] [Google Scholar]
- Chen Y, Pane A, Schupbach T.. 2007. Cutoff and Aubergine mutations result in upregulation of retrotransposons and activation of a checkpoint in the Drosophila germline. Curr Biol. 17:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Hannon GJ.. 2016. One loop to rule them all: the ping-pong cycle and piRNA-guided silencing. Trends Biochem Sci. 41:324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miranda JR, et al. 2004. Complete nucleotide sequence of Kashmir bee virus and comparison with acute bee paralysis virus. J Gen Virol. 85:2263–2270. [DOI] [PubMed] [Google Scholar]
- de Miranda JR, et al. 2010. Genetic characterization of slow bee paralysis virus of the honeybee (Apis mellifera L.). J Gen Virol. 91:2524–2530. [DOI] [PubMed] [Google Scholar]
- Dooner HK, He LM.. 2008. Maize genome structure variation: interplay between retrotransposon polymorphisms and genic recombination. Plant Cell 20:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium et al. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218. [DOI] [PubMed] [Google Scholar]
- Elsik CG, et al. 2014. Finding the missing honey bee genes: lessons learned from a genome upgrade. BMC Genomics 15:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsik CG, et al. 2016. Hymenoptera Genome Database: integrating genome annotations in HymenopteraMine. Nucleic Acids Res. 44:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, et al. 2010. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A. 107:8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foret S, et al. 2012. DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc Natl Acad Sci U S A. 109:4968–4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foret S, Kucharski R, Pittelkow Y, Lockett GA, Maleszka R.. 2009. Epigenetic regulation of the honey bee transcriptome: unravelling the nature of methylated genes. BMC Genomics 10:472.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh RC, Ball BV, Willcocks MM, Carter MJ.. 1999. The nucleotide sequence of sacbrood virus of the honey bee: an insect picorna-like virus. J Gen Virol. 80(Pt 6):1541–1549. [DOI] [PubMed] [Google Scholar]
- Govan VA, Leat N, Allsopp M, Davison S.. 2000. Analysis of the complete genome sequence of acute bee paralysis virus shows that it belongs to the novel group of insect-infecting RNA viruses. Virology 277:457–463. [DOI] [PubMed] [Google Scholar]
- Haase AD, et al. 2010. Probing the initiation and effector phases of the somatic piRNA pathway in Drosophila. Genes Dev. 24:2499–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, Zamore PD.. 2015. Noncoding RNA. piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348:817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler D, et al. 2011. A systematic analysis of Drosophila TUDOR domain-containing proteins identifies Vreteno and the Tdrd12 family as essential primary piRNA pathway factors. EMBO J. 30:3977–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R, et al. 2016. Genetic and mechanistic diversity of piRNA 3'-end formation. Nature 539:588–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizu H, Siomi H, Siomi MC.. 2012. Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 26:2361–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi N, et al. 2016. Identification and functional analysis of the Pre-piRNA 3' trimmer in silkworms. Cell 164:962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob NJ, Muller K, Bahr U, Darai G.. 2001. Analysis of the first complete DNA sequence of an invertebrate iridovirus: coding strategy of the genome of Chilo iridescent virus. Virology 286:182–196. [DOI] [PubMed] [Google Scholar]
- Jung I, Park JC, Kim S.. 2014. piClust: a density based piRNA clustering algorithm. Comput Biol Chem. 50:60–67. [DOI] [PubMed] [Google Scholar]
- Kapheim KM, et al. 2015. Social evolution. Genomic signatures of evolutionary transitions from solitary to group living. Science 348:1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaoka S, Minami K, Katsuma S, Mita K, Shimada T.. 2008. Developmentally synchronized expression of two Bombyx mori Piwi subfamily genes, SIWI and BmAGO3 in germ-line cells. Biochem Biophys Res Commun. 367:755–760. [DOI] [PubMed] [Google Scholar]
- Kiuchi T, et al. 2014. A single female-specific piRNA is the primary determiner of sex in the silkworm. Nature 509:633–636. [DOI] [PubMed] [Google Scholar]
- Klattenhoff C, Theurkauf W.. 2008. Biogenesis and germline functions of piRNAs. Development 135:3–9. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL.. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzi G, et al. 2006. Molecular and biological characterization of deformed wing virus of honeybees (Apis mellifera L.). J Virol. 80:4998–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leat N, Ball B, Govan V, Davison S.. 2000. Analysis of the complete genome sequence of black queen-cell virus, a picorna-like virus of honey bees. J Gen Virol. 81:2111–2119. [DOI] [PubMed] [Google Scholar]
- Lewis SH, Salmela H, Obbard DJ.. 2016. Duplication and diversification of dipteran argonaute genes, and the evolutionary divergence of Piwi and aubergine. Genome Biol Evol 8:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z, Jia Q, Li F, Han Z.. 2010. Identification of two piwi genes and their expression profile in honeybee, Apis mellifera. Arch Insect Biochem Physiol. 74:91–102. [DOI] [PubMed] [Google Scholar]
- Lim RS, Anand A, Nishimiya-Fujisawa C, Kobayashi S, Kai T.. 2014. Analysis of Hydra PIWI proteins and piRNAs uncover early evolutionary origins of the piRNA pathway. Dev Biol. 386:237–251. [DOI] [PubMed] [Google Scholar]
- Lu HL, et al. 2011. Expansion of genes encoding piRNA-associated argonaute proteins in the pea aphid: diversification of expression profiles in different plastic morphs. PLoS One 6:e28051.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn MJ, Ketting RF.. 2013. PIWI-interacting RNAs: from generation to transgenerational epigenetics. Nat Rev Genet. 14:523–534. [DOI] [PubMed] [Google Scholar]
- Lyko F, et al. 2010. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 8:e1000506.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F, Maleszka R.. 2011. Insects as innovative models for functional studies of DNA methylation. Trends Genet. 27:127–131. [DOI] [PubMed] [Google Scholar]
- Lynch M. 2015. Genetics: feedforward loop for diversity. Nature 523:414–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleszka R. 2014. The social honey bee in biomedical research: realities and expectations. Drug Disc Today: Dis Models. 12:7–13. [Google Scholar]
- Maleszka R. 2016. Epigenetic code and insect behavioural plasticity. Curr. Opin. Insect Sci. 15:45–52. [DOI] [PubMed] [Google Scholar]
- Mani SR, Juliano CE.. 2013. Untangling the web: the diverse functions of the PIWI/piRNA pathway. Mol Reprod Dev. 80:632–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maori E, et al. 2007. Isolation and characterization of Israeli acute paralysis virus, a dicistrovirus affecting honeybees in Israel: evidence for diversity due to intra- and inter-species recombination. J Gen Virol. 88:3428–3438. [DOI] [PubMed] [Google Scholar]
- Mohn F, Handler D, Brennecke J.. 2015. Noncoding RNA. piRNA-guided slicing specifies transcripts for Zucchini-dependent, phased piRNA biogenesis. Science 348:812–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Sienski G, Handler D, Brennecke J.. 2014. The rhino-deadlock-cutoff complex licenses noncanonical transcription of dual-strand piRNA clusters in Drosophila. Cell 157:1364–1379. [DOI] [PubMed] [Google Scholar]
- Olivier V, et al. 2008. Molecular characterisation and phylogenetic analysis of Chronic bee paralysis virus, a honey bee virus. Virus Res. 132:59–68. [DOI] [PubMed] [Google Scholar]
- Ongus JR, et al. 2004. Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J Gen Virol. 85:3747–3755. [DOI] [PubMed] [Google Scholar]
- Petit M, et al. 2016. piRNA pathway is not required for antiviral defense in Drosophila melanogaster. Proc Natl Acad Sci U S A. 113:E4218–E4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preall JB, Czech B, Guzzardo PM, Muerdter F, Hannon GJ.. 2012. shutdown is a component of the Drosophila piRNA biogenesis machinery. RNA 18:1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddatz G, et al. 2013. Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc Natl Acad Sci U S A. 110:8627–8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasethupathy P, et al. 2012. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 149:693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RJ, Weiner MM, Lin H.. 2014. PIWI proteins and PIWI-interacting RNAs in the soma. Nature 505:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, et al. 2015. Krimper enforces an antisense bias on piRNA pools by binding AGO3 in the Drosophila germline. Mol Cell. 59:553–563. [DOI] [PubMed] [Google Scholar]
- Schaack S, Gilbert C, Feschotte C.. 2010. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol. 25:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Honeybee Genome Sequencing Consortium 2006. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443:19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson T, Lin H.. 2009. The biogenesis and function of PIWI proteins and piRNAs: progress and prospect. Annu Rev Cell Dev Biol. 25:355–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoyasu Y, et al. 2008. Exploring systemic RNA interference in insects: a genome-wide survey for RNAi genes in Tribolium. Genome Biol. 9:R10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Munster M, et al. 2002. Sequence analysis and genomic organization of Aphid lethal paralysis virus: a new member of the family Dicistroviridae. J Gen Virol. 83:3131–3138. [DOI] [PubMed] [Google Scholar]
- Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP.. 2007. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3:12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovar N, et al. 2012. Arbovirus-derived piRNAs exhibit a ping-pong signature in mosquito cells. PLoS One 7:e30861.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, et al. 2014. The initial uridine of primary piRNAs does not create the tenth adenine that is the hallmark of secondary piRNAs. Mol Cell. 56:708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedd L, Kucharski R, Maleszka R.. 2016. Differentially methylated obligatory epialleles modulate context-dependent LAM gene expression in the honeybee Apis mellifera. Epigenetics. 11:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedd L, Maleszka R.. 2016. DNA Methylation and Gene Regulation in Honeybees: From Genome-Wide Analyses to Obligatory Epialleles. Adv. Exp. Med. Biol. 945:193–211. [DOI] [PubMed] [Google Scholar]
- Werren JH. 2011. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc Natl Acad Sci U S A. 108(Suppl 2):10863–10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH, et al. 2010. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327:343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfert L, Gadau J, Schmid-Hempel P.. 2007. Variation in genomic recombination rates among animal taxa and the case of social insects. Heredity 98:189–197. [DOI] [PubMed] [Google Scholar]
- Xiol J, et al. 2014. RNA clamping by Vasa assembles a piRNA amplifier complex on transposon transcripts. Cell 157:1698–1711. [DOI] [PubMed] [Google Scholar]
- Yin DT, et al. 2011. Germline stem cell gene PIWIL2 mediates DNA repair through relaxation of chromatin. PLoS One 6:e27154.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Lin H.. 2007. An epigenetic activation role of Piwi and a Piwi-associated piRNA in Drosophila melanogaster. Nature 450:304–308. [DOI] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH.. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13:335–340. [DOI] [PubMed] [Google Scholar]
- Zalloua PA, Buzayan JM, Bruening G.. 1996. Chemical cleavage of 5'-linked protein from tobacco ringspot virus genomic RNAs and characterization of the protein-RNA linkage. Virology 219:1–8. [DOI] [PubMed] [Google Scholar]
- Zhang Z, et al. 2014. The HP1 homolog rhino anchors a nuclear complex that suppresses piRNA precursor splicing. Cell 157:1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, et al. 2010. Mouse MOV10L1 associates with Piwi proteins and is an essential component of the Piwi-interacting RNA (piRNA) pathway. Proc Natl Acad Sci U S A. 107:11841–11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.