

ABSTRACT
Erwinia amylovora is the causative agent of fire blight, a devastating plant disease affecting members of the Rosaceae. Alternatives to antibiotics for control of fire blight symptoms and outbreaks are highly desirable, due to increasing drug resistance and tight regulatory restrictions. Moreover, the available diagnostic methods either lack sensitivity, lack speed, or are unable to discriminate between live and dead bacteria. Owing to their extreme biological specificity, bacteriophages are promising alternatives for both aims. In this study, the virulent broad-host-range E. amylovora virus Y2 was engineered to enhance its killing activity and for use as a luciferase reporter phage, respectively. Toward these aims, a depolymerase gene of E. amylovora virus L1 (dpoL1-C) or a bacterial luxAB fusion was introduced into the genome of Y2 by homologous recombination. The genes were placed downstream of the major capsid protein orf68, under the control of the native promoter. The modifications did not affect viability of infectivity of the recombinant viruses. Phage Y2::dpoL1-C demonstrated synergistic activity between the depolymerase degrading the exopolysaccharide capsule and phage infection, which greatly enhanced bacterial killing. It also significantly reduced the ability of E. amylovora to colonize the surface of detached flowers. The reporter phage Y2::luxAB transduced bacterial luciferase into host cells and induced synthesis of large amounts of a LuxAB luciferase fusion. After the addition of aldehyde substrate, bioluminescence could be readily monitored, and this enabled rapid and specific detection of low numbers of viable bacteria, without enrichment, both in vitro and in plant material.
IMPORTANCE Fire blight, caused by Erwinia amylovora, is the major threat to global pome fruit production, with high economic losses every year. Bacteriophages represent promising alternatives to not only control the disease, but also for rapid diagnostics. To enhance biocontrol efficacy, we combined the desired properties of two phages, Y2 (broad host range) and L1 (depolymerase for capsule degradation) in a single recombinant phage. This phage showed enhanced biocontrol and could reduce E. amylovora on flowers. Phage Y2 was also genetically engineered into a luciferase reporter phage, which transduces bacterial bioluminescence into infected cells and allows detection of low numbers of viable target bacteria. The combination of speed, sensitivity, and specificity is superior to previously used diagnostic methods. In conclusion, genetic engineering could improve the properties of phage Y2 toward better killing efficacy and sensitive detection of E. amylovora cells.
KEYWORDS: recombinant phage, reporter, depolymerase, luciferase, bacteriophage, fire blight, reporter phage
INTRODUCTION
Fire blight, caused by Erwinia amylovora, is a devastating plant disease. The Gram-negative pathogen can infect several species within the Rosaceae, representing a global threat to commercial apple and pear production, with significant economic impact due to control measures and compensation costs for plants that need to be destroyed (1–3). Therefore, reliable diagnostics and effective control measures are paramount to minimize loss and prevent spread of the disease.
Effective disease-control may be achieved by application of antibiotics (e.g., streptomycin) during the flowering period. However, regulatory restrictions, public health concerns, and the development of resistant strains demand alternative control measures (4, 5). Diagnostics of E. amylovora include cultural, molecular, and immunological tests (6). A rapid lateral flow immunosorbent assay (Ea AgriStrip), which detects >5 × 105 CFU/ml within 15 min, was developed for the latter (7). However, such antibody-based assays often lack the required sensitivity. A preenrichment step may compensate for low sensitivity to avoid false-negative results, which, however, compromises the speed of this method (6). Other tests are either time-consuming (cultures) or do not discriminate between live and dead bacteria (PCR). A detection system, which combines the desirable 4S properties (simplicity, speed, sensitivity, and specificity), would improve the management of fire blight infections and outbreaks.
Bacteriophages are ubiquitous bacterial viruses, representing the most abundant group of biological entities on Earth (8). In general, they infect only a specific range of host bacteria, which renders them useful and environmentally friendly alternatives for both detection and control of bacterial pathogens. Their specificity avoids infection of commensal environmental bacteria and reduces the risk of false-positive diagnostic results. Virulent phages are preferred, since they generally feature a much broader host range within a species or genus, and, in contrast to temperate phages, they are mostly unable to transduce host DNA (9). Phage-based control of pathogens has been shown a promising alternative for several plant diseases (10).
Using genetic engineering, properties of phages may be specifically tailored toward certain goals, such as enhanced killing or use as a reporter phage. The efficacy of Escherichia coli virus T7 to disperse biofilms could be increased by introduction of dspB, which encodes an enzyme for biofilm degradation (11). Molecular engineering can also be used to alter the host specificity of bacteriophages. The introduction or exchange of tail-associated genes enabled E. coli viruses T2 and T7 to infect a broader range of host bacteria (12–14). Likewise, the tail fiber gene of Pseudomonas virus PaP1 was replaced by the one of phage JG004, which caused a change in host specificity (15). For detection purposes, reporter phages induce production of an easily traceable enzyme or protein. Common reporters are luciferases from bacteria or insects, fluorescent proteins, glycosidases, and others (16). Reporter phages have been designed for specific detection of various Gram-negative and Gram-positive species, such as E. coli (17–20), Salmonella (21, 22), Yersinia pestis (23), Shigella spp. (24), Listeria monocytogenes (25, 26), Bacillus anthracis (27, 28), and Mycobacterium spp. (29, 30). Recent reviews summarized both pathogen detection with bioluminescent reporter phages and the various described phage-based detection methods (16, 31). The first luciferase reporter phage for detection of a plant pathogen (Pseudomonas cannabina pv. alisalensis), PBSPCA1::luxAB, was described by Schofield et al. (32). PBSPCA1::luxAB could detect P. cannabina pv. alisalensis in plant material and differentiate it from Pseudomonas syringae pv. maculicola, causing a less severe disease (32).
The availability of genome sequences is a prerequisite for phage engineering, and completely sequenced E. amylovora viruses permit construction of recombinant phages for improvement of biocontrol and rapid diagnostics, respectively. In this study, we aimed to introduce different heterologous genes into the virulent E. amylovora phage Y2 (33), to enhance its killing activity, and to create a bioluminescent reporter phage, respectively. Y2 is a perfect candidate for both purposes, due to its broad host range and high specificity for E. amylovora. The first approach is based on our observations that the exopolysaccharide (EPS) capsule of E. amylovora impedes Y2 infection, which could be overcome by supplementation with a recombinant depolymerase enzyme from phage L1 (34). Here, we report insertion of depolymerase dpoL1 gene into the genome of Y2 and demonstrate its EPS-degrading properties and its positive effect on phage infection and killing. With respect to the reporter phage, the reporter of choice was a bacterial luciferase from Vibrio harveyi, provided as a luxAB fusion. The recombinant reporter phage allowed rapid, specific, and sensitive bioluminescent detection of viable E. amylovora cells.
RESULTS
Recombinant phage Y2::dpoL1-C produces functional DpoL1.
The synergistic activity between the broad host range of phage Y2 and the depolymerase enzyme of phage L1 was combined in a single recombinant phage. Here, an N-terminally truncated version of the depolymerase gene (dpoL1-C) was introduced into the genome of phage Y2 by homologous recombination during infection of E. amylovora 4/82 carrying pBlue::Y2-dpoL1-C. Screening of phage lysate enabled identification of plaques produced by recombinant phage Y2::dpoL1-C. Expanding haloes indicated the activity of depolymerase released from lysed bacterial hosts (Fig. 1). In addition, Y2::dpoL1-C produced clearer plaques than the parental phage. In total, two plaques with a surrounding halo could be identified among approximately 15,000 plaques screened, corresponding to a recombination rate of approximately 1:7,500 or 1.3 × 10−4. PCR amplification of the region encompassing orf68 and dpoL1-C confirmed correct integration into the Y2 genome. The PCR product generated with the Y2-ctrl primers was of the expected size (2,296 bp), ∼2 kbp larger than the wild-type (wt) fragment (349 bp) (Fig. 2A). Sequencing confirmed the 1,947-bp sequence introduced and that no deletions, insertions, or frameshifts had occurred. In conclusion, dpoL1-C preceded by an artificial ribosome-binding site (RBS) region was introduced at the expected locus (nucleotide [nt] 35636).
FIG 1.
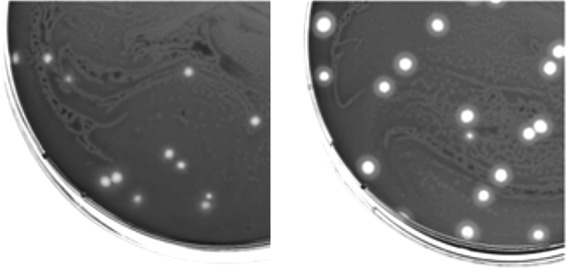
Plaques of Y2 (left) and Y2::dpoL1-C (right) on a lawn of E. amylovora CFBP 1430. The depolymerase produces a distinct halo surrounding the plaques of Y2::dpoL1-C and gives them a clearer appearance.
FIG 2.
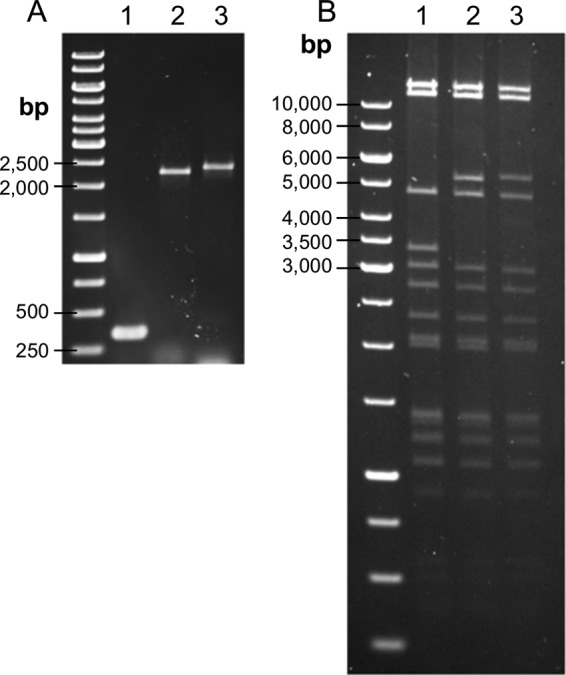
Genomic analyses of the recombinant phages and comparison to the parental phage. (A) PCR analysis of the genome region, where the heterologous genes were integrated. (B) VspI restriction digestion of phage DNA. Lanes: 1, Y2; 2, Y2::dpoL1-C; 3, Y2::luxAB. The sizes are indicated.
Introduction of additional sequence did not cause loss of other information.
Restriction fragment length polymorphism analysis was applied to confirm the integration of the heterologous gene and to determine whether other regions of the genome, particularly the genome ends, were affected. Comparison of the restriction patterns (Fig. 2B) confirmed the presence of additional DNA stretches in the genome of Y2::dpoL1-C. All VspI fragments but one were the same size. The fragments bearing orf68 (Y2, 3,365 bp; Y2::dpoL1-C, 5,312 bp) differed by the number of nucleotides integrated into the phage genome. The terminal fragments (2,745 and 11,580 bp) remained unchanged.
Y2::dpoL1-C features an identical host range and a similar latency period, but differs in burst size from native Y2.
The host range of the recombinant phage was identical to the parental phage Y2 (see Table S1 in the supplemental material). In a one-step growth experiment, phage-infected E. amylovora cells began releasing Y2::dpoL1-C progeny approximately 30 min after infection, which is very similar to the latency period of the parental phage (Fig. 3). The calculated burst size of Y2::dpoL1-C was 10 phage per cell. This is lower than the burst size of Y2, which produces an average of 18 progeny per infected cell.
FIG 3.
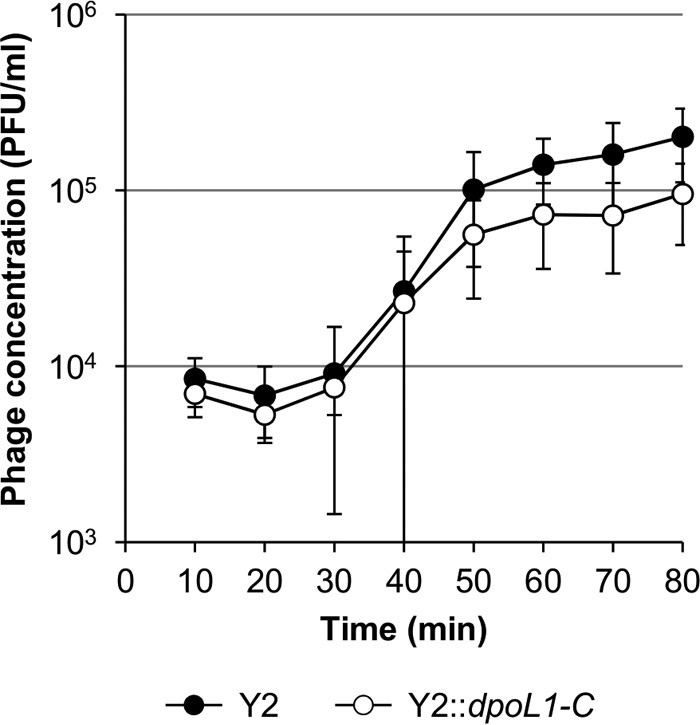
One-step growth curves of phages Y2 and Y2::dpoL1-C.
Y2::dpoL1-C features enhanced infectivity and killing efficacy.
Infection experiments were performed to test whether Y2::dpoL1-C has an increased potential to lyse E. amylovora cultures. It is important to note that bacterial growth was not affected by treatment with DpoL1-C alone (Fig. 4). We show that efficacy of phage Y2 could be strongly increased by the external addition of DpoL1-C. The phage/enzyme cocktail reduced the number of viable cells by almost 4 log10 within 2 h. The enhanced killing efficacy of the recombinant Y2::dpoL1-C compared to the parental phage became apparent during longer incubation periods. Although the reduction of bacteria was similar within the first 8 h of postinfection, viable cell counts continued in opposite directions between 8 and 24 h of postinfection. After 24 h, the number of cells infected by Y2::dpoL1-C dropped by ∼3 log10 compared to wild-type Y2. Interestingly, the final cell counts in cultures treated with either separate phage and enzyme (Y2/DpoL1-C) or enzyme-producing recombinant Y2::dpoL1-C were similar.
FIG 4.
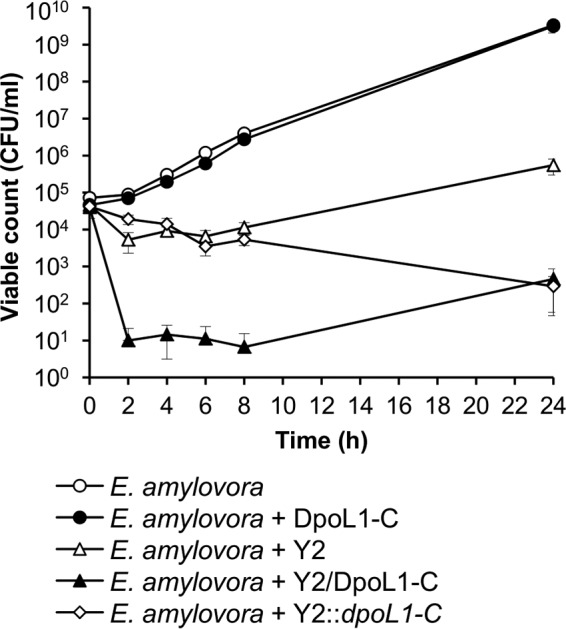
In vitro phage infection of E. amylovora CFBP 1430. Bacteria were treated with phage and/or DpoL1-C, and the numbers of viable cells were monitored over time. Means of three experiments performed in triplicate ± the standard deviations (SD) are shown.
Y2::dpoL1-C reduces E. amylovora contamination on flowers.
Detached flowers were used as an in planta model to study the biocontrol efficacy of Y2::dpoL1-C. Bacteria could readily grow on the plant surfaces. After 48 h, bacterial counts in the untreated control groups were as high as 8.8 × 107, 2.2 × 108, and 4.1 × 108 CFU/flower, respectively, depending on the inoculation levels (Fig. 5). Viable bacteria were reduced by Y2::dpoL1-C in an inoculum-dependent manner. On flowers inoculated with 102 CFU/ml, Y2::dpoL1-C caused a reduction of the final cell number by ∼6 logs. It needs to be highlighted that no bacteria could be recovered from 95% of the flower samples of this group. The efficacy of Y2::dpoL1-C was lower on flowers inoculated with higher bacterial numbers, where the average bacterial concentrations reached 5.4 × 104 CFU/flower (inoculum, 104 CFU/ml) and 5.1 × 105 CFU/flower (106 CFU/ml). This corresponded to reductions of 3.6 and 2.9 log10, respectively, compared to the untreated controls. Again, the average cell numbers were strongly affected by outliers. No viable cells were recovered from 81% of the phage-treated flowers inoculated with 104 CFU/ml, whereas this rate was different on flowers inoculated with 106 CFU/ml, where only 19% were apparently free of E. amylovora cells. These findings indicate a critical threshold in the bacterium/phage ratio, above which the efficacy of phage drops significantly.
FIG 5.
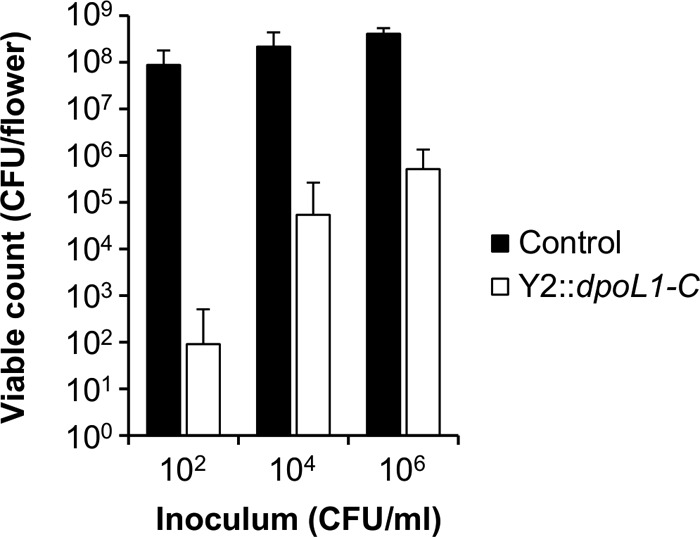
Activity of Y2::dpoL1-C on detached flowers. Flowers were inoculated with various concentrations of E. amylovora CFBP 1430 NalR (as indicated) and treated with Y2::dpoL1-C. Cells were enumerated at 48 h postinfection. Bars represent means plus the SD of triplicate experiments, which were repeated twice.
Construction, isolation, and characterization of Y2::luxAB.
The luxAB fusion gene was introduced into the genome of Y2 to generate a reporter phage Y2::luxAB. Y2::luxAB construction followed the same strategy as for Y2::dpoL1-C. Because plaque morphology of Y2::luxAB is identical to Y2, screening and isolation were performed by using luciferase production and bioluminescence of cells infected by putative luciferase-encoding phages as a read-out. For this, phage particles were extracted from semiconfluent lysed agar plates, and subsequent measuring of relative light unit (RLU) values in diluted samples was performed. Those samples producing RLU values above the background were used to produce new lysis plates, with continuously lower numbers of plaques per plate until single plaques could be tested by PCR.
After isolation and propagation of identified luciferase-transducing phages, correct integration of luxAB into the Y2 genome was confirmed by PCR. Y2-ctrl primers generated a PCR product of the expected size (2,404 bp) (Fig. 2A), and sequencing confirmed integration of 2,055 bp at the expected site without deletions, insertions, or frameshifts. The insertion site of luxAB preceded by an artificial RBS region was located at position 35636, directly downstream of the major capsid gene (orf68) to achieve high expression of luxAB from the promoter of orf68.
Fragment analysis of VspI-digested DNA confirmed that the fragment of Y2::luxAB bearing orf68 was larger (5,420 bp) than that of Y2 (3,365 bp) (Fig. 2B) and that the physical genome termini did not change, i.e., the fragments were the same sizes (2,745 and 11,580 bp).
As expected, the host ranges of Y2::luxAB and the parental phage were identical. Additional Gram-negative bacteria were tested and found to be resistant to infection by Y2::luxAB (see Table S1 in the supplemental material).
Luciferase expression is increased in DpoL1-treated cells.
Luciferase expression and bioluminescence signal emission from Y2::luxAB-infected cells was dependent on the incubation temperature and peaked at 70 min at 17°C and at 50 min (22°C) and 40 min (27°C) postinfection (Fig. 6A). Thereafter, the reporter signal generally decreased rapidly, probably due to cell lysis. The values were highest at 22°C. Although the addition of DpoL1 did not cause a temporal shift of light emission, the luciferase expression was generally higher when the cells were exposed to the depolymerase enzyme. DpoL1-treated cultures expressed 25% (17°C), 12% (22°C), and 18% (27°C) higher peak luciferase activities, respectively, than the untreated cultures.
FIG 6.

Expression of luciferase by Y2::luxAB-infected E. amylovora CFBP 1430 cells grown under different conditions. (A) Temperature and time dependency. Cells of E. amylovora CFBP 1430 were grown in LB broth (●) or LB broth supplemented with depolymerase (○) and infected with Y2::luxAB. The expression of luciferase was measured at regular intervals, and the mean values of three independent experiments ± the SD are displayed. (B) Sensitivity of Y2::luxAB-based detection. A dilution series of E. amylovora CFBP 1430 was infected with Y2::luxAB, and the light emission was measured after 50 min. Mean values of three independent experiments ± the SD are displayed and are compared to controls with bacteria only. The background levels were generally around 100 RLU (horizontal line), and asterisks indicate a significant increase compared to the uninfected control (P < 0.05; Student t test).
Y2::luxAB detects low numbers of viable E. amylovora cells.
Serial dilutions of a E. amylovora culture were infected with the luciferase reporter phage to determine the lower detection limit. A concentration of 3.8 × 103 CFU/ml was the lowest to directly produce RLU values that were significantly higher than the controls (P < 0.05; Student t test) (Fig. 6B). Above this limit, numbers of infected cells and light outputs was directly correlated over approximately 4 orders of magnitude. Thus, RLU values may also be used to estimate the concentration of E. amylovora cells in an unknown sample. Again, the application of external DpoL1 enzyme for the removal of the EPS capsule material slightly increased the overall sensitivity of the assay and further reduced the detection limit.
Y2::luxAB for the detection of E. amylovora in field samples.
Y2::luxAB was used to detect viable E. amylovora cells in excised plant material displaying fire blight symptoms. Light emission from all samples (n = 24) was significantly higher than from the control (P > 0.05; Student t test) (Fig. 7). The RLU values of several samples exceeded the values determined for the samples with the highest contamination rate in the spiking experiment (see Fig. 6), indicating that the sampled plant material was heavily infected. This finding was further confirmed by using an Ea AgriStrip detection system, which is less sensitive and features a detection limit of 5.0 × 105 CFU/ml (7). Here, this enzyme-linked immunosorbent assay (ELISA)-based method gave positive results for all 24 samples. The presence of E. amylovora was further confirmed by plating on King's B agar, where typical E. amylovora colonies could be identified in every case.
FIG 7.

Detection of E. amylovora in plant tissue samples by Y2::luxAB. Values indicate means of two measurements. The minus (“−”) indicates the negative control.
DISCUSSION
The broad-host-range, virulent, and yet host-specific E. amylovora phage Y2 was modified for enhanced biocontrol and rapid diagnostics by the introduction of a dpoL1-C depolymerase or a luxAB luciferase gene, respectively. Infection and killing by recombinant phage Y2::dpoL1-C is no longer impeded by an EPS capsule, as is the case for Y2, and infection by Y2::luxAB enables rapid and sensitive detection of viable cells of E. amylovora in <1 h.
The genes of interest dpoL1-C and luxAB should be expressed at the highest possible levels. Therefore, we followed a strategy we have used earlier for the construction of recombinant Listeria phage (26) and inserted them immediately downstream of the major capsid protein orf68. The expression of dpoL1-C and luxAB in Y2::dpoL1-C and Y2::luxAB, respectively, is now under the control of a strong, late promoter. However, the introduction of new genetic material into a phage genome without removing any other sequence may potentially exceed the limitations dictated by the capsid size and lead to the uncontrolled loss of other genes and possibly reduce the infectivity or even compromise the viability of the phage. Analyses of the recombinant phages produced here confirmed not only that the heterologous genes were introduced at the target position but also, more importantly, that no sequence information was lost.
The introduction of dpoL1-C into the genome of Y2 combined the synergistic activity of the phage and the capsule-degrading enzyme in a single agent. This strategy was postulated earlier for disruption of bacterial biofilms (35, 36). In an elegant approach, Escherichia coli phage T7 was equipped with a biofilm-degrading enzyme (dspB) for an enhanced dispersion of biofilms (11). However, dspB is not of phage origin, in contrast to dpoL1-C. To our knowledge, the approach used here to achieve synergism by combination of the activities of a phage and a heterologous enzyme encoded by another phage from a different morphotype (Y2 myovirus and L1 podovirus) is novel.
It is also noteworthy that the enhanced infection and killing efficacy of Y2::dpoL1-C should be considered a secondary effect, since DpoL1-C is not present as a structural component of the recombinant phage. In fact, the depolymerase was not present when bacteria are first infected by Y2::dpoL1-C but was expressed only during this first round of infection, and the enzyme is released only after the cells begin to lyse. Then, its activity facilitates the subsequent infection of any remaining bacterial cells. Thus, while the effect mediated by the enzyme is somewhat delayed, we have shown here that it is nonetheless sufficient to significantly improve the potential to infect encapsulated E. amylovora host cells. The EPS of E. amylovora is at least partially removed by the enzyme and no longer acts as a barrier, enhancing the exposure of bacterial cells to phage attack.
The efficacy of phage-based pathogen control on detached flowers was found to be dependent on the starting inoculum. Above a certain threshold, the efficacy of Y2::dpoL1-C was drastically reduced. It is likely that the majority of bacteria were infected immediately after phage application. However, the remaining (uninfected) cells may have been able to regrow because the diffusion of phage particles as a major determinant of phage infection is hampered on plant surfaces. Under natural environmental conditions, UV inactivation may also complicate phage-based biocontrol (37, 38). Therefore, an early intervention before the bacteria reach a critical in vivo concentration might be the best strategy, and repeated application of phage is expected to further increase efficacy.
E. amylovora cells could rapidly be detected by measuring the bacterial luciferase activity transduced by Y2::luxAB. The temperature dependency was in agreement with the growth characteristics of the host bacteria. The highest RLU values were reached earliest at 27°C, which is close to the optimal temperature for E. amylovora growth (39). At this temperature, the peak was observed after 40 min, which seems faster than with most other luxAB reporter phages (23, 26–28, 32, 40, 41). Temperatures higher than 27°C were not tested because of the inherent instability of the fusion enzyme at elevated temperatures (26, 42). The rapid decline of the RLU values at all temperatures (flash-type bioluminescence) is most likely due to the limited supply of FMNH2, which is also caused by phage-mediated cell lysis (26).
The limit for direct detection (without enrichment) of E. amylovora cells following infection with Y2::luxAB was found to be as low as 3.8 × 103 CFU/ml. Detection limits of other luciferase reporter phages ranged from a few cells per milliliter to approximately 103 to 104 CFU/ml (23, 24, 26, 27, 32, 40, 41). Of these, systems including temperate phages were shown to have lowest detection limits based upon long-term continuous production of the reporter protein (20, 27, 40). However, Y2::luxAB is a virulent phage, and temperate E. amylovora viruses have not yet been found (43). Therefore, the best option to increase sensitivity of the Y2::luxAB assay is a preenrichment step, which not only increases the number of bacteria to detectable numbers but also enables metabolic recovery of the target cells. This is in line with earlier studies, which also demonstrated the utility of an enrichment step prior to detection with reporter phage (26, 44).
Bioluminescence was found directly correlated to bacterial cell numbers. Interestingly, the light emission of cells treated with DpoL1 was higher in every case. Since cell counts in DpoL1-treated and -untreated samples were identical, this finding suggests that in the presence of DpoL1 the infection by Y2::luxAB is more effective. A certain fraction of wild-type (untreated) cells may remain uninfected due to their extensive EPS capsule, which acts as a barrier against phages not featuring a depolymerase, such as wild-type Y2. The addition of exogenous recombinant DpoL1 is one way to assist phages without a depolymerase for both the detection and the control of E. amylovora. However, we demonstrate here that this cumbersome approach may be circumvented by modification of the phage to express their own soluble depolymerase.
Y2::luxAB was highly efficient and accurate in the detection of E. amylovora from actual field samples, and phage activity was unaffected by other microorganisms present on the tissue or plant extracts. The experiments demonstrated that random wild-type E. amylovora field isolates are all sensitive to Y2::luxAB infection, which is attributed to the broad host range of Y2, and minimizes possible false-negative results. Because of the extreme host specificity of Y2, the risk for false-positive assays is also extremely low. Furthermore, the sensitivity of the Y2::luxAB assay appears generally sufficient for in vivo detection purposes. Although the field samples tested here were heavily infected (>5.0 × 105 CFU/ml), the bacterial load on average symptomatic field samples usually lies in the range of 105 to 106 CFU/g (6). Altogether, luciferase reporter phage Y2::luxAB offers a useful combination of viable cell specificity, high sensitivity, and speed and appears to be superior to ELISA-based tests.
In conclusion, the desired properties of E. amylovora phage Y2 could be further improved in two directions by introducing heterologous genes. Both recombinant phages combine the amenities of the parental phage with an additional benefit. Recombinant Y2::dpoL1-C induces the production and the release of an EPS depolymerase, which renders other host cells more accessible to phage attack and collateral damage. In addition, the phage origin of dpoL1-C renders Y2::dpoL1-C a hybrid rather than a transgenic phage particle, a potentially important argument for legislation of phage application in agriculture. The Y2::luxAB reporter phage represents an ideal tool for rapid diagnosis of fire blight in plant material. It specifically detects E. amylovora and discriminates between live and dead cells, in contrast to molecular methods or immunological assays. The assay is faster than any culture-based method. Although the assay's sensitivity is already better than that of a conventional ELISA, its detection limit can be further reduced by introducing an enrichment step.
MATERIALS AND METHODS
Vector pBlue::Y2-dpoL1-C for site-specific integration into phage Y2.
The native dpoL1 gene of phage L1 was modified for insertion into the intergenic region between orf68 and orf69 (nt 35636) of the Y2 genome, where transcriptional control is provided by the strong promoter upstream of the major capsid gene. Since the depolymerase would not constitute a structural component of Y2::dpoL1-C, the N-terminal domain not required for enzyme activity (34) was omitted. An artificial ribosome-binding site (RBS; GGAGGT), a spacer of 6 nt, and a start codon preceding dpoL1-C were introduced for proper translation. The final sequence at the integration site of the recombinant phage was 5′-TAAGTTGATGGAGGTGAATCTATG-3′ (underlined, stop and start codons of orf68 and dpoL1-C, respectively; boldfacing, RBS). The dpoL1-C (ΔN1–180) sequence was amplified from L1-DNA using the primer pair Y2-Dpo-C/D (Table 1), and the upstream and downstream regions of the targeted insertion site were amplified with the primer pairs Y2-Dpo-A/B and Y2-Dpo-E/F using Y2-DNA as the template. Primers B/C and D/E contained complementary sequences for the subsequent overlap PCR. Primer C introduced the artificial RBS region and the start codon. Primer F introduced a BamHI restriction site. A native EcoRI restriction site is located 7 nt downstream of the annealing region (3′ end) of primer A. PCRs were performed using the Phusion high-fidelity DNA polymerase (Finnzymes Oy, Vantaa, Finland) according to the manufacturer's instructions, and the fragments were purified by gel extraction (QIAquick gel extraction kit; Qiagen, Hilden, Germany). The final PCR product was generated through extension overlap PCR, using primers Y2-Dpo-A and Y2-Dpo-F and the three fragments (AB, CD, and EF) as the templates. In the first five cycles, the annealing of the overlapping regions was ensured through a touchdown of the annealing temperature (initial, 64°C; final, 60°C; 1 cycle each). The correct length PCR product was excised from an electrophoresis gel and purified by gel extraction (QIAquick gel extraction kit). It was composed of dpoL1-C with the translation signals as described above (total size, 1,947 bp) and flanking regions upstream (304 bp) and downstream (278 bp) complementary to the genome of phage Y2 (Fig. 8). The sequence and the cloning vector pBluescript II SK(−) (Agilent Technologies, Santa Clara, CA) were prepared for ligation by restriction digestion with the HF endonucleases EcoRI and BamHI (New England BioLabs, Ipswich, MA). Ligation was performed using T4 ligase (Fermentas, St. Leon-Rot, Germany), resulting in pBlue::Y2-dpoL1-C.
TABLE 1.
Primers used for the construction and confirmation of recombinant phages Y2::dpoL1-C and Y2::luxAB
| Name | Sequencea |
|---|---|
| Y2-Dpo-A | CCA GTC TGA CAT GAC CAT CCT GCA ATT CTT CAT TGC |
| Y2-Dpo-B | CAT AGA TTC ACC TCC ATC AAC TTA GAT ACC TGA GTT GAT AG |
| Y2-Dpo-C | CTA AGT TGA TGG AGG TGA ATC TAT GAT TGG CGG CTA CAT CTC GTT TG |
| Y2-Dpo-D | GCT TTC ACC CGC CGA CAT TTA TCC TTG TGA AGA TAC AGA ACC |
| Y2-Dpo-E | ATG TCG GCG GGT GAA AGC CCG CCT CAG |
| Y2-Dpo-F | GAT CGG ATC CGG ACA TTC TTC TTA TCC ATC TGG |
| Y2-lux-A | CCA GTC TGA CAT GAC CAT CCT GCA ATT CTT CAT TGC |
| Y2-lux-B | TTT CAT AGA TTC ACC TCC ATC AAC TTA GAT ACC TGA GTT GAT AG |
| Y2-lux-C | AGT TGA TGG AGG TGA ATC TAT GAA ATT TGG AAA CTT CCT TCT CAC TTA TCA GCC ACC |
| Y2-lux-D | TTT CAC CCG CCG ACA TTT ACG AGT GGT ATT TGA CG |
| Y2-lux-E | GTA AAT GTC GGC GGG TGA AAG CCC GCC TCA G |
| Y2-lux-F | GAT CGG ATC CGG ACA TTC TTC TTA TCC ATC TGG |
| Y2-lux-seq2 | CTG GTC AAC CAA AAT GTA GAT GG |
| Y2-lux-seq3 | TTT GAA CAG GTT AAC CAC TTT GG |
| Y2-ctrl-fw | GTT ATC TGC CGT ACT GTT AAT GG |
| Y2-ctrl-rev | GTC ATC CAG TTC AAC AGT TGC |
Boldfacing indicates annealing regions, italics indicate overlap regions, single underlining indicates artificial ribosome-binding sites, and double underlining indicates restriction sites.
FIG 8.
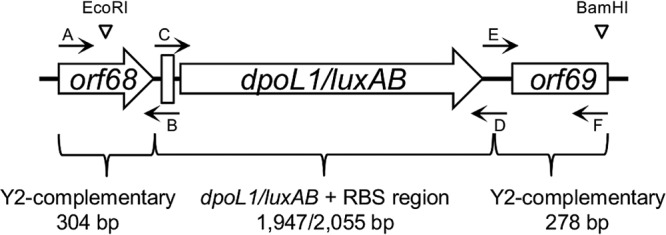
Schematic representation of the construct generated by overlap PCR for cloning into pBluescript and subsequent homologous recombination with Y2 genomic DNA. Annealing regions of the primers are depicted by small black arrows, and the restriction sites of EcoRI and BamHI, as well as the RBS (thick vertical line), are indicated. The image is not drawn to scale.
pBlue::Y2-luxAB for recombination of bacterial luciferase.
Bacterial luciferase from V. harveyi was constructed for integration at the same locus as described above for dpoL1-C. The native bacterial lux operon consists of five genes (luxCDABE). Although the luciferase itself is a heterodimer, composed of an α subunit (LuxA) and a β subunit (LuxB), the luxCDE genes encode fatty acid reductases required for production of the long-chain aldehyde (45). Due to space limitations in the capsid of Y2 and the need for a strong signal following phage infection, a luxAB fusion gene was used (26). The pBlue::Y2-luxAB vector was constructed using the Y2-lux primers listed in Table 1 according to the protocol described above, with modifications in the extension overlap PCR. Fragments AB and CD, as well as fragments CD and EF, were separately linked together prior to the final overlap PCR. No touchdown of the annealing temperature was required due to the large overlaps of fragments AD and CF. The final fragment AF encompassed the luxAB gene, preceded by the same RBS (total size, 2,055 bp) and flanked by the Y2 complementary regions described above (Fig. 8).
The sequence at the insertion site of the recombinant phage was 5′-TAAGTTGATGGAGGTGAATCTATG-3′ (underlining, stop and start codons of orf68 and luxAB, respectively; boldfacing, RBS). AF was subsequently cloned into pBluescript II SK(−) via BamHI and EcoRI restriction sites to yield pBlue::Y2-luxAB.
Transformation.
Electrocompetent E. amylovora 4/82 cells were transformed with pBlue::Y2-dpoL1-C or pBlue::Y2-luxAB by electroporation (GenePulser [Bio-Rad, Hercules, CA]; settings: 2.5 kV, 25 μF, and 200 Ω). After the addition of 1 ml of SOC medium and 1 h of incubation at 30°C, a dilution series was plated on Luria-Bertani (LB) plates containing ampicillin (100 μg/ml), which were incubated overnight at 27°C. Insert-bearing clones were identified by PCR, and the constructs were sequenced.
Homologous recombination.
Portions (25 ml) of LB medium supplemented with ampicillin (100 μg/ml) were inoculated with E. amylovora 4/82 carrying the construct pBlue::Y2-dpoL1-C or pBlue::Y2-luxAB, followed by incubation under constant shaking at 30°C. At an optical density at 600 nm of 0.1 (∼1.5 × 108 CFU/ml), phage Y2 was added to a final concentration of 1.5 × 106 PFU/ml (MOI = 0.01), and the cultures were further incubated at 30°C. After 24 h, the remaining bacterial cells and debris were removed by centrifugation (10,000 × g, 10 min) and filtration (0.2-μm-pore-size filter; Sarstedt, Nümbrecht, Germany), and the lysate was stored at 4°C.
Screening and identification of recombinant phages.
The recombinant phages Y2::dpoL1-C and Y2::luxAB were identified using the depolymerase or luciferase functions as convenient readouts in specifically adapted soft-agar overlay assays. Phage lysates (10 μl of an appropriate dilution) were mixed with 100 μl of a fresh overnight culture of E. amylovora CFBP 1430 (Y2::dpoL1-C) or 4/82 (Y2::luxAB) in 4 ml of molten and tempered (47.5°C) LB soft agar (LB medium supplemented with 10 mM CaCl2 and 2 mM MgSO4 on 0.4% agar). The mixture was then poured onto agar plates (Y2::dpoL1-C, LB agar with 1% glucose; Y2::luxAB, LB agar). After overnight incubation at 27°C, plaques (ca. 300 per plate) produced by wild type and recombinant Y2::dpoL1-C were visually screened for surrounding halo formation, the hallmark of Dpo-producing phages. The use of strain CFBP 1430 instead of strain 4/82 facilitated the screening because the former produces larger amounts of EPS. Clear single plaques with a halo could then be isolated using a Pasteur pipette and resuspended in 500 μl of SM buffer (50 mM Tris, 100 mM NaCl, 8 mM MgSO4 [pH 7.4]). This step was repeated at least twice to ensure the purity of the phage isolate. For Y2::luxAB screening and isolation, 5-ml portions of SM buffer were poured onto plates showing a semiconfluent lysis (approximately 500 to 1,000 PFU/plate) following initial plating of the phage mixture. The SM buffer was collected after 2 h, and the luciferase activity was expressed in RLU was measured immediately after the injection of 50 μl of aldehyde substrate (0.25% [vol/vol] nonanal in 70% ethanol) into 1-ml samples, using a tube luminometer with a programmable injector (Lumat LB9501; Berthold Technologies GmbH, Regensdorf, Switzerland), as described earlier (26). Samples with an RLU of >250 were considered positive, and the presence of Y2::luxAB was demonstrated by PCR using the primer pair Y2-lux-seq2/3 (Table 1). The primers annealed in the luxAB gene and in the Y2 backbone, respectively, enabling the detection of the recombinant phage and avoiding false-positive results, i.e., detection of wild-type phage or the pBlue::Y2-luxAB recombination plasmid. The SM buffer from a plate with a positive PCR result was used for further rounds of enrichment and testing. Plates with fewer PFU were produced and screened by the same method. By doing so, the ratio between recombinant and wild-type phage was continuously increased during several rounds of plating. At a ratio of >1:50, single plaques were picked as described above and tested by PCR. Finally, the purity of the phage was guaranteed by three consecutive single plaque isolations. The integrations of dpoL1-C and luxAB, respectively, were analyzed by PCR with the primer pair Y2-ctrl-fw/rev (Table 1), annealing up- and downstream of the integrated DNA (Fig. 8). The PCR products were sequenced for confirmation. Finally, the phages were propagated to a high titer and purified with CsCl step gradients, and phage genomic DNA was isolated through phenol-chloroform extractions, as described previously (33). Restriction digestions were performed according to the manufacturer's instructions.
Host range analysis.
Host ranges of the recombinant phages were tested as described previously (33), using the spot-on-the-lawn technique on agar plates.
One-step growth curves.
The growth parameters of phage Y2::dpoL1-C were determined by a classical one-step growth curve. To this end, LB broth supplemented with CaCl2 (10 mM) and MgSO4 (2 mM) was inoculated with E. amylovora CFBP 1430. The culture was grown under constant shaking at 27°C until an optical density of 0.5 had been reached. Ten milliliters of the culture was transferred to a prewarmed (27°C) 50-ml centrifuge tube, and phage was added to a final concentration of 5 × 105 PFU/ml. The tube was allowed to stand for 5 min at 27°C for phage adsorption. Thereafter, 1 ml of the culture was centrifuged (1 min, 7,000 × g), and the supernatant containing unadsorbed phage was removed. The pellet was resuspended in 10 ml of fresh prewarmed (27°C) LB broth containing CaCl2 (10 mM) and MgSO4 (2 mM), and the culture was further incubated at 27°C with shaking at regular intervals. The phage concentration was determined every 10 min by using soft agar overlays. The experiment was repeated twice.
In vitro activity of Y2::dpoL1-C.
The efficacy of the phages Y2 and Y2::dpoL1-C, as well as DpoL1-C and a phage/enzyme cocktail to control E. amylovora, was tested in vitro as described previously (34). Briefly, E. amylovora CFBP 1430 (105 CFU/ml) was infected with 108 PFU/ml and/or 5 μg/ml enzyme. The cultures were incubated at 30°C under constant shaking, and the cell numbers were monitored over time. Each experiment consisted of three technical replicates per treatment and was independently repeated twice.
Killing efficacy of Y2::dpoL1-C on flower leaf surfaces.
Experiments on detached flowers were performed to determine the biocontrol efficacy of Y2::dpoL1-C as a function of the initial cell numbers. Newly opened apple flowers of the cultivar ‘Galaxy’ were collected and stored in 1% (wt/vol) sucrose medium. The flowers were spray inoculated with different concentrations of bacteria (102, 104, and 106 CFU/ml, respectively, E. amylovora CFBP 1430 Nalr in PBS plus 0.01% [vol/vol] Tween 20). After 1 h, phages (3.0 × 108 PFU/ml in SM plus 0.01% [vol/vol] Tween 20) were applied by spraying. The flowers were incubated in plastic boxes at 20°C. The boxes were equipped with wet paper towels to ensure a constant high humidity. After 48 h, the petals and peduncles were removed, the flowers were placed in 1 ml of phosphate-buffered saline/Tween 20 to wash off the bacterial cells, and the cell numbers were determined on LB plates containing nalidixic acid (50 μg/ml) and cycloheximide (50 μg/ml). The experiment was performed in groups of seven flowers each and independently repeated twice.
Parameters for luciferase expression.
The time-dependent expression of luciferase by E. amylovora cells infected with Y2::luxAB was investigated at three different temperatures (17, 22, and 27°C). E. amylovora strain CFBP 1430 was grown in LB broth or LB broth supplemented with DpoL1 at 5 μg/ml to a concentration of approximately 108 CFU/ml. The culture was infected with 3.0 × 108 PFU/ml of Y2::luxAB, mixed, and immediately split into aliquots of 1 ml. The RLU were measured using a luminometer at t0 and in 10-min intervals for 90 min. Two additional measurements were made after 120 and 150 min. The experiments consisted of three technical replicates and were independently repeated twice.
Detection limit using Y2::luxAB reporter phage.
Experiments with various cell concentrations were carried out to determine the sensitivity of Y2::luxAB. E. amylovora CFBP 1430 was grown in LB broth or in LB broth supplemented with DpoL1 (5 μg/ml) at 22°C to an approximate concentration of 108 CFU/ml. A 1:10-dilution series in a total volume of 5 ml was infected with Y2::luxAB (3.0 × 108 PFU/ml) and further incubated at 22°C. The RLU of 1-ml aliquots were measured 50 min after infection. The background RLU level was measured in controls with phage or bacteria only, and the CFU/ml of uninfected cultures were determined. The experiments were carried out three times in three replicates.
Detection of E. amylovora in field isolates.
Tissue samples displaying putative fire blight symptoms (necrosis, ooze) were collected from a Swiss apple orchard. Flowers with peduncles and small fruits (samples 1 to 10), leaves (samples 11 to 20), and twigs (samples 21 to 24) were cut into pieces and macerated with 5 ml PBS per gram of plant tissue for 15 min in centrifuge tubes. PBS samples (500 µl) were mixed with the same volume of 2× LB broth containing 6.0 × 108 PFU/ml of phage Y2::luxAB and 10 μg/ml DpoL1. The RLU were measured after 50 min of incubation at 22°C. Control samples did not contain phage. For comparison, the same PBS samples were analyzed with an Ea AgriStrip (7) and by plating a dilution series on King's B medium (46) containing 100 μg/ml cycloheximide.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by the Swiss Federal Office for Agriculture (BLW Fire Blight Project–Biocontrol) and was conducted within the European Union Cooperation in Science and Technology (COST) Action 864 and supported by the Swiss ProfiCrops Research network.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00341-17.
REFERENCES
- 1.Bonn WG, van der Zwet T. 2000. Distribution and economic importance of fire blight, p 37–53. In Vanneste JL. (ed), Fire blight: the disease and its causative agent, Erwinia amylovora. CAB International, Wallingford, United Kingdom. [Google Scholar]
- 2.Duffy B, Schärer HJ, Bünter M, Klay A, Holliger E. 2005. Regulatory measures against Erwinia amylovora in Switzerland. EPPO Bull 35:239–244. doi: 10.1111/j.1365-2338.2005.00820.x. [DOI] [Google Scholar]
- 3.Thomson S. 2000. Epidemiology of fire blight, p 9–36. In Vanneste, JL (ed), Fire blight: the disease and its causative agent, Erwinia amylovora. CAB International, Wallingford, United Kingdom. [Google Scholar]
- 4.McManus PS, Stockwell VO, Sundin GW, Jones AL. 2002. Antibiotic use in plant agriculture. Annu Rev Phytopathol 40:443–465. doi: 10.1146/annurev.phyto.40.120301.093927. [DOI] [PubMed] [Google Scholar]
- 5.Stockwell VO, Duffy B. 2012. Use of antibiotics in plant agriculture. Rev Sci Tech 31:199–210. [DOI] [PubMed] [Google Scholar]
- 6.EPPO. 2013. PM 7/20 (2) Erwinia amylovora. EPPO Bull 43:21–45. doi: 10.1111/epp.12019. [DOI] [Google Scholar]
- 7.Braun-Kiewnick A, Altenbach D, Oberhansli T, Bitterlin W, Duffy B. 2011. A rapid lateral-flow immunoassay for phytosanitary detection of Erwinia amylovora and on-site fire blight diagnosis. J Microbiol Methods 87:1–9. doi: 10.1016/j.mimet.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 8.Cobián Güemes AG, Youle M, Cantú VA, Felts B, Nulton J, Rohwer F. 2016. Viruses as winners in the game of life. Annu Rev Virol 3:197–214. doi: 10.1146/annurev-virology-100114-054952. [DOI] [PubMed] [Google Scholar]
- 9.Hagens S, Loessner MJ. 2007. Application of bacteriophages for detection and control of foodborne pathogens. Appl Microbiol Biotechnol 76:513–519. doi: 10.1007/s00253-007-1031-8. [DOI] [PubMed] [Google Scholar]
- 10.Buttimer C, McAuliffe O, Ross RP, Hill C, O'Mahony J, Coffey A. 2017. Bacteriophages and bacterial plant diseases. Front Microbiol 8:34. doi: 10.3389/fmicb.2017.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu TK, Collins JJ. 2007. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci U S A 104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahichi F, Synnott AJ, Yamamichi K, Osada T, Tanji Y. 2009. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol Lett 295:211–217. doi: 10.1111/j.1574-6968.2009.01588.x. [DOI] [PubMed] [Google Scholar]
- 13.Scholl D, Adhya S, Merril C. 2005. Escherichia coli K1's capsule is a barrier to bacteriophage T7. Appl Environ Microbiol 71:4872–4874. doi: 10.1128/AEM.71.8.4872-4874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoichi M, Abe M, Miyanaga K, Unno H, Tanji Y. 2005. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157:H7. J Biotechnol 115:101–107. doi: 10.1016/j.jbiotec.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Le S, He X, Tan Y, Huang G, Zhang L, Lux R, Shi W, Hu F. 2013. Mapping the tail fiber as the receptor binding protein responsible for differential host specificity of Pseudomonas aeruginosa bacteriophages PaP1 and JG004. PLoS One 8:e68562. doi: 10.1371/journal.pone.0068562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anany H, Chou Y, Cucic S, Derda R, Evoy S, Griffiths MW. 2017. From bits and pieces to whole phage to nanomachines: pathogen detection using bacteriophages. Annu Rev Food Sci Technol 8:305–329. doi: 10.1146/annurev-food-041715-033235. [DOI] [PubMed] [Google Scholar]
- 17.Oda M, Morita M, Unno H, Tanji Y. 2004. Rapid detection of Escherichia coli O157:H7 by using green fluorescent protein-labeled PP01 bacteriophage. Appl Environ Microbiol 70:527–534. doi: 10.1128/AEM.70.1.527-534.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brigati JR, Ripp SA, Johnson CM, Iakova PA, Jegier P, Sayler GS. 2007. Bacteriophage-based bioluminescent bioreporter for the detection of Escherichia coli O157:H7. J Food Prot 70:1386–1392. doi: 10.4315/0362-028X-70.6.1386. [DOI] [PubMed] [Google Scholar]
- 19.Tanji Y, Furukawa C, Na SH, Hijikata T, Miyanaga K, Unno H. 2004. Escherichia coli detection by GFP-labeled lysozyme-inactivated T4 bacteriophage. J Biotechnol 114:11–20. doi: 10.1016/j.jbiotec.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Zhang D, Coronel-Aguilera CP, Romero PL, Perry L, Minocha U, Rosenfield C, Gehring AG, Paoli GC, Bhunia AK, Applegate B. 2016. The use of a novel NanoLuc-based reporter phage for the detection of Escherichia coli O157:H7. Sci Rep 6:33235. doi: 10.1038/srep33235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn J, Suissa M, Wyse J, Cohen I, Weiser I, Reznick S, Lubinsky-Mink S, Stewart G, Ulitzur S. 2002. Detection of bacteria using foreign DNA: the development of a bacteriophage reagent for Salmonella. Int J Food Microbiol 74:229–238. doi: 10.1016/S0168-1605(01)00683-3. [DOI] [PubMed] [Google Scholar]
- 22.Wolber PK, Green RL. 1990. Detection of bacteria by transduction of ice nucleation genes. Trends Biotechnol 8:276–279. doi: 10.1016/0167-7799(90)90195-4. [DOI] [PubMed] [Google Scholar]
- 23.Schofield DA, Molineux IJ, Westwater C. 2009. Diagnostic bioluminescent phage for detection of Yersinia pestis. J Clin Microbiol 47:3887–3894. doi: 10.1128/JCM.01533-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schofield DA, Wray DJ, Molineux IJ. 2015. Isolation and development of bioluminescent reporter phages for bacterial dysentery. Eur J Clin Microbiol Infect Dis 34:395–403. doi: 10.1007/s10096-014-2246-0. [DOI] [PubMed] [Google Scholar]
- 25.Hagens S, de Wouters T, Vollenweider P, Loessner MJ. 2011. Reporter bacteriophage A511::celB transduces a hyperthermostable glycosidase from Pyrococcus furiosus for rapid and simple detection of viable Listeria cells. Bacteriophage 1:143–151. doi: 10.4161/bact.1.3.16710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loessner MJ, Rees CED, Stewart GSAB, Scherer S. 1996. Construction of luciferase reporter bacteriophage A511::luxAB for rapid and sensitive detection of viable Listeria cells. Appl Environ Microbiol 62:1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharp NJ, Molineux IJ, Page MA, Schofield DA. 2016. Rapid detection of viable Bacillus anthracis spores in environmental samples by using engineered reporter phages. Appl Environ Microbiol 82:2380–2387. doi: 10.1128/AEM.03772-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schofield DA, Westwater C. 2009. Phage-mediated bioluminescent detection of Bacillus anthracis. J Appl Microbiol 107:1468–1478. doi: 10.1111/j.1365-2672.2009.04332.x. [DOI] [PubMed] [Google Scholar]
- 29.Sarkis GJ, Jr, RJW, Hatfull GF. 1995. L5 luciferase reporter mycobacteriophages: a sensitive tool for the detection and assay of live mycobacteria. Mol Microbiol 15:1055–1067. doi: 10.1111/j.1365-2958.1995.tb02281.x. [DOI] [PubMed] [Google Scholar]
- 30.Jacobs WJ, Barletta RG, Udani R, Chan J, Kalkut G, Sosne G, Kieser T, Sarkis GJ, Hatfull GF, Bloom BR. 1993. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science 260:819–822. doi: 10.1126/science.8484123. [DOI] [PubMed] [Google Scholar]
- 31.Klumpp J, Loessner MJ. 2014. Detection of bacteria with bioluminescent reporter bacteriophage. Adv Biochem Eng Biotechnol 144:155–171. [DOI] [PubMed] [Google Scholar]
- 32.Schofield DA, Bull CT, Rubio I, Wechter WP, Westwater C, Molineux IJ. 2012. Development of an engineered bioluminescent reporter phage for detection of bacterial blight of crucifers. Appl Environ Microbiol 78:3592–3598. doi: 10.1128/AEM.00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Born Y, Fieseler L, Marazzi J, Lurz R, Duffy B, Loessner MJ. 2011. Novel virulent and broad-host-range Erwinia amylovora bacteriophages reveal a high degree of mosaicism and a relationship to Enterobacteriaceae phages. Appl Environ Microbiol 77:5945–5954. doi: 10.1128/AEM.03022-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Born Y, Fieseler L, Klumpp J, Eugster MR, Zurfluh K, Duffy B, Loessner MJ. 2014. The tail-associated depolymerase of Erwinia amylovora phage L1 mediates host cell adsorption and enzymatic capsule removal, which can enhance infection by other phage. Environ Microbiol 16:2168–2180. doi: 10.1111/1462-2920.12212. [DOI] [PubMed] [Google Scholar]
- 35.Cornelissen A, Ceyssens PJ, T'Syen J, Van Praet H, Noben JP, Shaburova OV, Krylov VN, Volckaert G, Lavigne R. 2011. The T7-related Pseudomonas putida phage ϕ15 displays virion-associated biofilm degradation properties. PLoS One 6:e18597. doi: 10.1371/journal.pone.0018597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hughes KA, Sutherland IW, Jones MV. 1998. Biofilm susceptibility to bacteriophage attack: the role of phage-borne polysaccharide depolymerase. Microbiology 144:3039–3047. doi: 10.1099/00221287-144-11-3039. [DOI] [PubMed] [Google Scholar]
- 37.Balogh B, Jones JB, Momol MT, Olson SM, Obradovic A, King P, Jackson LE. 2003. Improved efficacy of newly formulated bacteriophages for management of bacterial spot on tomato. Plant Dis 87:949–954. doi: 10.1094/PDIS.2003.87.8.949. [DOI] [PubMed] [Google Scholar]
- 38.Iriarte FB, Balogh B, Momol MT, Smith LM, Wilson M, Jones JB. 2007. Factors affecting survival of bacteriophage on tomato leaf surfaces. Appl Environ Microbiol 73:1704–1711. doi: 10.1128/AEM.02118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hauben L, Swings J. 2005. Genus XIII. Erwinia, p 670–679. In Brenner DJ, Krieg NL, Staley JT, Garrity GM (ed), Bergey's manual of systematic bacteriology. Springer, New York, NY. [Google Scholar]
- 40.Kim S, Kim M, Ryu S. 2014. Development of an engineered bioluminescent reporter phage for the sensitive detection of viable Salmonella Typhimurium. Anal Chem 86:5858–5864. doi: 10.1021/ac500645c. [DOI] [PubMed] [Google Scholar]
- 41.Franche N, Vinay M, Ansaldi M. 2017. Substrate-independent luminescent phage-based biosensor to specifically detect enteric bacteria such as Escherichia coli. Environ Sci Pollut Res 24:42–51. doi: 10.1007/s11356-016-6288-y. [DOI] [PubMed] [Google Scholar]
- 42.Escher A, Okane DJ, Lee J, Szalay AA. 1989. Bacterial luciferase αβ fusion protein is fully active as a monomer and highly sensitive in vivo to elevated temperature. Proc Natl Acad Sci U S A 86:6528–6532. doi: 10.1073/pnas.86.17.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roach DR, Sjaarda DR, Sjaarda CP, Ayala CJ, Howcroft B, Castle AJ, Svircev AM. 2015. Absence of lysogeny in wild populations of Erwinia amylovora and Pantoea agglomerans. Microb Biotechnol 8:510–518. doi: 10.1111/1751-7915.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loessner MJ, Rudolf M, Scherer S. 1997. Evaluation of luciferase reporter bacteriophage A511::luxAB for detection of Listeria monocytogenes in contaminated foods. Appl Environ Microbiol 63:2961–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meighen EA. 1993. Bacterial bioluminescence: organization, regulation, and application of the lux genes. FASEB J 7:1016–1022. [DOI] [PubMed] [Google Scholar]
- 46.King EO, Ward MK, Raney DE. 1954. Two simple media for the demonstration of pyocyanin and fluorescein. J Lab Clin Med 44:301–307. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.