

ABSTRACT
There has been an absence of an efficient method of gene knockdown in the important human pathogen Staphylococcus aureus like RNA interference in eukaryotes. The previously developed antisense RNA technology is mainly applied for forward genetic screening but is rather limited in specific gene knockdown because of the lack of rational antisense RNA design strategies. Here we report an efficient and specific system for gene knockdown in S. aureus based on the type II clustered regularly interspaced short palindromic repeat (CRISPR) system from Streptococcus pyogenes. We can achieve gene silencing with the coexpression of dCas9, an RNA-guided DNA binding protein, and a small guide RNA complementary to the target gene. With this system, we have successfully silenced diverse sets of genes varying in size and expression level in different S. aureus strains. This system exhibited high-efficiency knockdown of both essential and nonessential genes, and its effect is inducible and reversible. In addition, the system can repress the expression of multiple genes simultaneously and silence an entire operon or part of it. This RNA-guided DNA targeting system thus provides a simple, rapid, and affordable method for selective gene knockdown in S. aureus.
IMPORTANCE Staphylococcus aureus is an important human and animal pathogen that can cause a diversity of infectious diseases. Molecular genetic study of S. aureus has provided an avenue for the understanding of its virulence, pathogenesis, and drug resistance, leading to the discovery of new therapies for the treatment of staphylococcal infections. However, methodologies developed for genetic manipulation of S. aureus usually involve either low efficiency or laborious procedures. Here we report an RNA-guided system for gene knockdown in S. aureus and show its high efficiency and simplicity for selective gene silencing in different strains of S. aureus. This simple, rapid, and affordable system may serve as a promising tool for functional gene study in S. aureus, especially for the study of essential genes, thus facilitating the understanding of this pathogen and its interaction with its hosts.
KEYWORDS: CRISPRi, gene knockdown, Staphylococcus aureus, dCas9
INTRODUCTION
Staphylococcus aureus is an opportunistic pathogen that can cause a diversity of infectious diseases in humans and animals. The global emergence and spread of antibiotic-resistant S. aureus pose serious challenges to the treatment of infections it causes (1). Genetic studies of S. aureus have provided an avenue for molecular understanding of the virulence, pathogenesis, and drug resistance of the organism, contributing to the development of new strategies for the treatment of infections it causes. Most of the studies have centered on virulence and antibiotic resistance genes, and the functions of these genes were evaluated mainly by targeted gene inactivation (2). So far, a plethora of methodologies have been developed for the study of the molecular genetics of S. aureus (3), among which there are two main kinds of techniques for targeted gene inactivation, i.e., gene knockout, which directly deletes or disrupts a gene in the genome, and gene knockdown, which represses gene expression but does not destroy the gene (2, 4). Allelic exchange is now the technique most commonly used to achieve gene knockout in S. aureus (5). Unfortunately, natural allelic replacements occur at a rather low rate in S. aureus and a counterselection system or an extensive screening process is required to screen for the desired mutants, making this method time-consuming and costly (5, 6). Moreover, gene knockout cannot be employed for the study of essential genes, which play important roles in the survival and pathogenesis of S. aureus (7).
Several efficient strategies have been developed for the knockdown or conditional knockout of essential genes in S. aureus in the past 20 years. One strategy involves controllable expression of an essential gene by inserting the inducible promoter into the region upstream of its chromosomal site (8), which also requires arduous cloning like allelic exchange. The second strategy uses targetrons derived from a Lactococcus lactis Ll.LtrB group II intron to create conditional mutations of both essential and nonessential genes in S. aureus (6). The temperature sensitivity of these targetrons requires that the mutant phenotype can only be manifested at high temperatures, which may not be proper for studying the physiological function of the gene. The third strategy, also the only one that does not alter the genome sequence, can repress gene expression by producing antisense RNA (asRNA) complementary to the target gene mRNA. Gene silencing by asRNA has been adopted for the study of essential genes in both eukaryotes and prokaryotes (9–11). It has been used to construct a random asRNA library and screen for essential genes in S. aureus (7). However, there remain some problems when it is employed to study specific genes because of the absence of rational asRNA design strategies, leading to a high failure rate and laborious construction and screening for efficient asRNAs (12, 13). Therefore, new techniques that are efficient, simple, and economical are still needed for the functional study of both essential and nonessential genes in S. aureus.
Recent work on the clustered regularly interspaced short palindromic repeat (CRISPR) system, an adaptive immune system of prokaryotes, has led to the development of novel strategies for gene knockout and knockdown in both eukaryotes and prokaryotes (14, 15). The CRISPR system has been found in most archaea and bacteria, providing the host with immunity against foreign nucleic acids like phage and plasmid nucleic acids (16). Of the various CRISPR systems characterized so far, the type II system has been best studied and exploited for its simplicity and potential as a genome editing tool (17). It requires only a single protein, called Cas9, for target DNA cleavage, while other systems need a multimeric complex of proteins. A naturally occurring CRISPR-Cas9 system needs two RNAs, a CRISPR RNA (crRNA) for target DNA binding and a trans-activating crRNA (tracrRNA) for Cas9 binding, to form hybrids and bind with Cas9 to mediate the cleavage of target DNA. The engineered CRISPR-Cas9 system has circumvented the problem of expression of two RNAs by designing a single guide RNA (sgRNA) that mimics the crRNA-tracrRNA complex (18). Other than sequence complementarity between the sgRNA and the target DNA, a short sequence (5′-NGG-3′) that immediately follows the targeting DNA sequence is also essential for determining the specificity of the system (19, 20). Since its emergence, this engineered CRISPR-Cas9 system has been applied to perform genome editing across a wide variety of cell types and whole organisms (17).
Recently, this CRISPR-Cas9 system has been modified for target gene regulation rather than disruption. The key component of this modified system is a catalytically dead Cas9 (dCas9) lacking endonuclease activity, which contains two silencing mutations of the Cas9 nuclease domains (D10A and H841A) (14). When the dCas9-sgRNA complex binds to the specific gene target, it does not cleave the DNA like wild-type Cas9 but blocks RNA polymerase from binding to the promoter or moving forward on the DNA to achieve transcriptional repression. This modified system, known as CRISPR interference (CRISPRi), has shown high efficiency in gene silencing in both Escherichia coli (14) and mycobacteria (21). We consider this system a potentially powerful tool for functional studies of specific genes, especially essential genes in S. aureus, yet there have been no reports of its application in staphylococci.
Here we implemented dCas9 and sgRNA to construct an efficient system for gene knockdown in S. aureus. E. coli-S. aureus shuttle plasmid pSD1 was first constructed for constitutive expression of the sgRNA and conditional expression of dCas9 under the control of an anhydrotetracycline (ATc)-inducible promoter (22), making this system both inducible and reversible. To achieve specific gene knockdown, we only need to clone the gene-specific complementary sequences into the sgRNA expression cassette in the plasmid. Restriction enzyme SapI has been chosen to make this cloning quite simple. With this system, we have successfully silenced diverse sets of genes varying in size and expression level in different S. aureus strains with relatively high efficiency compared to that of the asRNA method. We have also demonstrated that this system can repress the expression of multiple genes simultaneously, silence an entire operon or part of it, and be used for functional studies of essential genes.
RESULTS
Construction of dCas9 and sgRNA expression plasmid pSD1.
To implement the CRISPRi system in S. aureus, ATc-inducible expression plasmid pRMC2 (22) was used to express the dcas9 gene derived from Streptococcus pyogenes cas9 and custom-designed sgRNA in S. aureus. The expression of dcas9 is under the control of the ATc-inducible promoter PtetO, while the expression of sgRNA is controlled by the constitutive promoter PpflB from S. aureus (23) (Fig. 1). sgRNA consists of mainly two regions, the 5′ variable region for target DNA binding and the 3′ constant region for dCas9 binding (Fig. 1A). The constant region can also work as its own transcription terminator. Target-specific sgRNA expression can be achieved by cloning the coding sequence of the variable region into the sgRNA expression cassette (Fig. 1A). To avoid the introduction of extra nucleotides during the assembly of the sgRNA expression cassette, a special restriction enzyme, SapI, that produces editable overhangs was used (Fig. 1B). In the modular sgRNA expression cassette, there was a cloning box with two SapI sites between the promoter and coding sequence of the sgRNA constant region (Fig. 1C). The overhangs of the two SapI sites upon digestion have been designed to coincide with the sequence of the promoter (5′-TAG-3′) and the coding sequence of the sgRNA constant region (5′-GTT-3′) (Fig. 1C), so that the inserted fragment with two 5′ overhangs of 5′-CTA-3′ and 5′-AAC-3′ can ligate with the promoter and coding sequence of the constant region without leaving a scar (Fig. 1D). Since pRMC2 already contains a SapI site outside the multiple cloning sites, a single nucleotide mutation was first introduced into the SapI site to destroy it. The modular sgRNA expression cassette and the dcas9 gene fragment with a ribosome binding site (RBS) and a transcription terminator were then cloned into the multiple cloning sites (Fig. 1E). The resulting plasmid was designated pSD1. For specific gene knockdown, target-specific complementary oligonucleotide sequences were synthesized, annealed, and cloned into SapI-digested pSD1 as described in Materials and Methods (Fig. 1D). Of note, the pSD1 plasmid without the insertion of the target-specific fragment can also express an sgRNA whose variable region is coded by the SapI cloning box. BLAST analysis showed that this sgRNA has no specific target in S. aureus (consecutive identity sequence of <10 bases), which makes it a proper negative control for analysis of target-specific sgRNA knockdown efficiency.
FIG 1.

Schematic outline of the construction of dCas9 and sgRNA expression plasmid pSD1. (A) Schematic representation of sgRNA and organization of sgRNA expression cassette. The black line represents the sgRNA variable region for target DNA binding, and the gray line represents the constant region for dCas9 binding. (B) Recognition sequence and SapI cutting sites. The arrows indicate the cutting sites, and N represents any one of the four nucleotide bases. (C) Partial sequence of the modular sgRNA expression cassette. The gray bold nucleotides are the SapI recognition sequences, and arrows indicate the SapI cutting sites. Two 5′ overhang regions of 3 nt each (black bold) are produced upon SapI digestion. (D) Target-specific oligonucleotides coding for the sgRNA variable region. The gray sequences of the two oligonucleotides are complementary to each other, about 23 nt. A hybrid form of the two oligonucleotides has two 5′ overhangs (in black) for cloning into the SapI-digested sgRNA expression cassette. (E) Schematic diagram of plasmid pSD1 construction. Abbreviations: T, transcription terminator; MCS, multiple cloning site; V-sgRNA, coding sequence of the sgRNA variable region; C-sgRNA, coding sequence of the sgRNA constant region.
Knockdown efficiency of CRISPRi in different S. aureus strains.
Next, we detected the efficiency of this CRISPRi-mediated gene knockdown system in different S. aureus strains. First, the alpha-toxin gene (hla) in S. aureus strain NCTC 8325 was chosen as the target and four sgRNAs that bind to different regions of the gene were designed (Fig. 2A). The alpha-toxin virulence of the cell would decrease upon hla expression reduction. We determined the Hla protein level and alpha-hemolytic activity by Western blot and alpha hemolysis assays, respectively. The results showed that in the absence of ATc, in which case dCas9 is not supposed to be expressed, strains expressing sgRNAs targeting hla showed no observable difference in the Hla protein level and alpha-hemolytic activity from the wild-type strain and the control strain expressing the invalid control sgRNA; but in the presence of 100 ng/ml ATc, strains expressing sgRNA binding to the nontemplate strand of the hla gene (sgRNA1, sgRNA2, and sgRNA3) showed a remarkable decrease in their Hla protein and alpha-hemolytic activity levels compared to those of the wild-type and control strains, whereas the strain expressing sgRNA binding to the template strand (sgRNA4) showed no obvious decrease (Fig. 2B). These data indicate that the CRISPRi system is inducible, and only sgRNA binding to the nontemplate DNA strand exhibited strong silencing efficiency, which is consistent with an earlier report on E. coli (14). Since the repression of gene expression by dCas9 occurs at the transcriptional level, we detected the hla mRNA levels in both the knockdown and control strains by quantitative reverse transcription (qRT)-PCR. Consistent with the Western blot and alpha hemolysis assay results, strains expressing sgRNA1, sgRNA2, and sgRNA3 exhibited a >100-fold decrease in the hla mRNA level after treatment with 100 ng/ml ATc, while the strain expressing sgRNA4 showed a much less extensive decrease (∼2.5-fold) (Fig. 2C). Meanwhile, it was also observed that the three high-efficiency knockdown strains showed an ∼3-fold decrease in hla mRNA even without ATc induction (Fig. 2C). We speculate that this was caused by leaky expression of dCas9 since inducible promoters like PtetO always have some basal expression (24). Another two genes, sarT and spa, with different sizes and expression levels in NCTC 8325 have also been successfully silenced by CRISPRi (Fig. 2D).
FIG 2.

Knockdown efficiency of CRISPRi in S. aureus NCTC 8325. (A) Genetic organization of hla gene and binding sites of different sgRNAs. Arrows represent a pair of primers for qRT-PCR. (B) Hla protein levels and alpha-hemolytic activities of knockdown and control strains with or without ATc induction were measured by Western blot and alpha hemolysis assays, respectively. (C) Relative transcription level of hla in knockdown strains with or without ATc induction. (D) Knockdown of sarT and spa in NCTC 8325 by CRISPRi. (E) The knockdown effect of the CRISPRi system is reversible. The first generation of NSD0 and NSD1 was grown in the absence of ATc to an OD600 of 2 and then diluted 1:100 in ATc-containing TSB to obtain the second generation. The second generation was grown to an OD600 of 2 and then washed with TSB and diluted 1:100 in TSB lacking ATc to obtain the third generation. The third generation was also grown to an OD600 of 2. The hla expression levels in the different generations were measured by qRT-PCR. NSD0 was used as a control. The error bars indicate the standard errors of the means of three biological replicates. NS, not significant (P > 0.05); ***, P < 0.001. Abbreviations: WT, wild-type NCTC 8325; NSD0, NCTC 8325 carrying plasmid pSD1; NSD1, NCTC 8325 with the hla sgRNA1; NSD2, NCTC 8325 with the hla sgRNA2; NSD3, NCTC 8325 with the hla sgRNA3; NSD4, NCTC 8325 with the hla sgRNA4; NSD5, NCTC 8325 expressing the sarT sgRNA1; NSD6, NCTC 8325 expressing the sarT sgRNA2; NSD7, NCTC 8325 expressing the spa sgRNA1; G1 to G3, first, second, and third generations of bacteria.
Given that the CRISPRi system is inducible, its knockdown effect should be induced in the presence of the inducer and reversed when the inducer is removed. To test whether our CRISPRi system is reversible, we cultured hla knockdown strain NSD1 in the absence of ATc to the mid-exponential phase (regarded as the first generation). The bacterial cells were then diluted 1:100 with ATc-containing medium and allowed to grow to the mid-exponential phase again (second generation). The second generation of NSD1 was washed with tryptic soy broth (TSB) to remove ATc and then diluted 1:100 with the medium lacking ATc and also allowed to grow to the mid-exponential phase (third generation). As expected, hla expression was significantly repressed in the second generation of NSD1 when ATc was present, but after ATc was removed, hla expression was restored to the normal level in the third generation of NSD1 (Fig. 2E). This result demonstrates that the knockdown effect of this CRISPRi system is inducible and reversible.
Then we detected the efficiency of the CRISPRi system in methicillin-resistant S. aureus (MRSA) strain N315. The drug resistance gene mecA was chosen as the target, and an sgRNA binding to the nontemplate strand of mecA near the start codon was designed (Fig. 3). Since mecA is mainly responsible for beta-lactam resistance in MRSA strains, knockdown of this gene should enhance the susceptibility of MRSA strains to beta-lactam antibiotics such as oxacillin. We used the disk diffusion method to measure the oxacillin susceptibility levels of the knockdown and control strains and used a non-beta-lactam antibiotic, daptomycin, as a control. The growth inhibition zone around oxacillin on the knockdown strain plate was significantly larger than that on the control strain plate, while the growth inhibition zones around daptomycin on both strain plates were similar in size (Fig. 3), indicating that the reduction of the beta-lactam resistance of N315 was caused by specific knockdown of mecA. Taken together, these data demonstrate that CRISPRi can be widely used for gene knockdown in different S. aureus strains.
FIG 3.
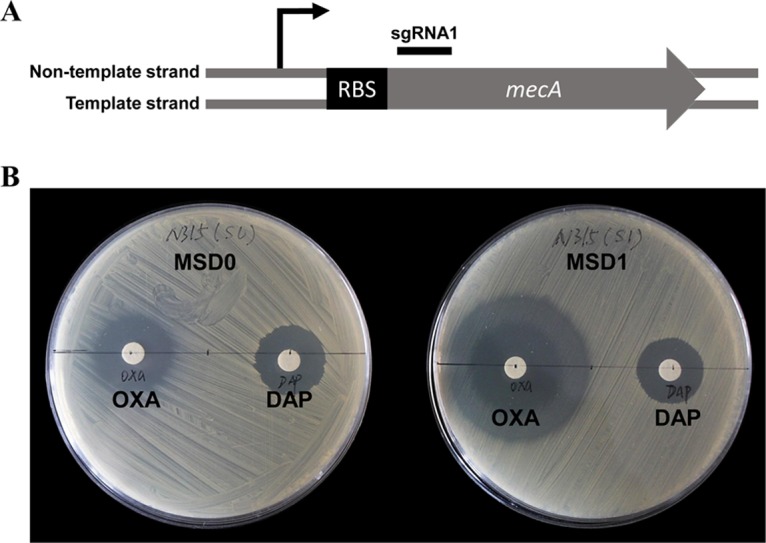
Knockdown of mecA in S. aureus N315 by CRISPRi. (A) Genetic organization of the mecA gene and the sgRNA binding site. (B) Oxacillin susceptibilities of knockdown (MSD1) and control (MSD0) strains detected by disk diffusion test. Daptomycin (a non-beta-lactam antibiotic) was used as a control. Abbreviations: OXA, oxacillin; DAP, daptomycin; MSD0, N315 carrying plasmid pSD1; MSD1, N315 expressing the mecA sgRNA1.
Knockdown of multiple genes simultaneously by CRISPRi.
The targeting specificity of CRISPRi is totally determined by sgRNA, and one knockdown plasmid can harbor multiple sgRNA expression cassettes. We explored whether this system can knock down multiple genes simultaneously in S. aureus. sgRNA1 targeting spa and sgRNA2 targeting hla were expressed separately or together in NCTC 8325 (Fig. 4A). When expressed alone, each of these two sgRNAs can specifically silence its target gene but not affect the expression of the other gene. When the two sgRNAs were expressed together, both genes were efficiently repressed, though at a lower rate than when they were expressed alone (Fig. 4A), which may have resulted from the competitive binding of two sgRNAs to dCas9. These results suggest that sgRNA targeting is specific and this system can be used to knock down multiple genes simultaneously in S. aureus. We also found that by expressing multiple sgRNAs targeting the same gene, enhanced gene knockdown can be achieved (Fig. 4B).
FIG 4.

The CRISPRi system can simultaneously target multiple genes or sites. (A) Knockdown of hla and spa simultaneously by CRISPRi. Both genes are efficiently silenced in the double-knockdown strain. (B) Enhanced knockdown of hla by expression of both sgRNA1 and sgRNA2. The hla knockdown levels in the single- and double-knockdown strains were determined by alpha hemolysis and qRT-PCR. The alpha hemolysis data are representative of three independent experiments. The error bars indicate the standard errors of the means of three biological replicates. *, P < 0.05; **, P < 0.01. Abbreviations: NSD0, NCTC 8325 carrying plasmid pSD1; NSD1, NCTC 8325 expressing the hla sgRNA1; NSD2, NCTC 8325 expressing the hla sgRNA2; NSD7, NCTC 8325 expressing the spa sgRNA1; NSD8, NCTC 8325 expressing both the hla sgRNA1 and the hla sgRNA2; NSD9, NCTC 8325 expressing both the hla sgRNA2 and the spa sgRNA1.
Knockdown of operon genes by CRISPRi.
Since dCas9-sgRNA represses gene expression by blocking RNA polymerase from binding or moving forward on the DNA, in an operon where several genes are cotranscribed, sgRNA targeting the first gene or the promoter should silence the expression of the whole operon, while sgRNA targeting other positions should strongly repress the expression of genes downstream of the position but have a less pronounced effect on upstream genes because transcription stops at the targeting site. To determine the efficacy of the CRISPRi system on operon gene knockdown in S. aureus, we first chose the ccrAB operon in MRSA strain N315 as the target. The ccrAB operon contains only two genes, ccrA and downstream gene ccrB, which are cotranscribed from a promoter upstream of ccrA (25). We designed four sgRNAs, i.e., two pairs each of which targeted the same gene, and detected the two genes' expression levels in the knockdown and control strains (Fig. 5A). The qRT-PCR result showed that sgRNAs targeting ccrA (sgRNA1, sgRNA2) can remarkably repress the expression of both genes. Meanwhile, sgRNAs targeting ccrB (sgRNA3, sgRNA4) can significantly repress ccrB expression but exhibited rather slight repression of ccrA (Fig. 5A).
FIG 5.

Whole or partial operon knockdown by CRISPRi. The knockdown effect of CRISPRi on the ccrAB operon in S. aureus N315 (A) and the era-containing operon in S. aureus NCTC 8325 (B) was determined by selectively targeting different operon genes. The relative transcription levels of different operon genes in different knockdown strains were measured by qRT-PCR. The error bars indicate the standard errors of the means of three biological replicates. Abbreviations: MSD0, N315 carrying plasmid pSD1; MSD2, N315 expressing the ccrAB sgRNA1; MSD3, N315 expressing the ccrAB sgRNA2; MSD4, N315 expressing the ccrAB sgRNA3; MSD5, N315 expressing the ccrAB sgRNA4; NSD0, NCTC 8325 carrying plasmid pSD1; NSD10, NCTC 8325 expressing the era sgRNA1; NSD11, NCTC 8325 expressing the era sgRNA2; NSD12, NCTC 8325 expressing the era sgRNA3; NSD13, NCTC 8325 expressing the era sgRNA4.
We further applied this system to knock down a bigger operon with more genes in S. aureus NCTC 8325. Homology analysis and RT-PCR identified a conserved operon with six genes in S. aureus. The operon contains a predicted essential gene, era, and five other genes whose functions in S. aureus have not been revealed (26, 27). Four sgRNAs targeting different genes in the operon were designed, and it was observed that they could strongly repress the expression of genes downstream of their target sites but exhibited limited inhibition of the expression of upstream genes, and the inhibition was even weaker with longer distances between the upstream gene and the target site (Fig. 5B). Taken together, these results indicate that the CRISPRi system can be used flexibly to knock down an entire operon or part of it by targeting different positions of the operon. Theoretically, an essential gene knockdown should lead to a growth defect of the bacterial cell, but in our experiment, no growth inhibition was observed in all four era-containing operon knockdown strains, which suggests that era may not be essential in S. aureus NCTC 8325.
Study of essential genes by CRISPRi.
One principal advantage of gene knockdown over gene knockout is that it can also be used to study essential genes. We thus used the CRISPRi system for knockdown of essential genes in S. aureus to determine its efficacy in the functional study of essential genes. Three experimentally confirmed essential genes, murE, rpsC, and walR, in S. aureus RN4220 have been targeted (7, 8, 28). Growth of these knockdown strains was detected at first. As expected, the knockdown strains showed appreciable growth inhibition in the presence of 100 ng/ml ATc (Fig. 6B), whereas in the absence of ATc, only marginal inhibition was observed compared with the control strain (Fig. 6A). This result proves that the CRISPRi system can also be used for efficient knockdown of essential genes and further demonstrates that the knockdown efficiency of the system is inducible. We then conducted a functional analysis of the walR knockdown strain. WalR and WalK compose a highly conserved two-component system that positively regulates autolysin synthesis and biofilm formation in S. aureus (29). To prevent the severe growth defect of the knockdown strain from hindering the functional study of essential genes, we used a lower concentration (20 ng/ml) of ATc to mitigate the growth inhibition of the walR knockdown strain (Fig. 6C). We examined the Triton X-100-induced autolytic activities of the walR knockdown strain and the control strain. Consistent with the function of the walKR two-component system, the walR knockdown strain exhibited strikingly high resistance to Triton X-100-induced autolysis compared with the control strain (Fig. 6D). Therefore, CRISPRi can serve as a powerful method for the functional study of essential genes.
FIG 6.

Functional study of essential genes in S. aureus by CRISPRi. (A) Growth curves of control and essential gene (murE, rpsC, and walR) knockdown strains without ATc induction. (B) Growth curves of control and essential gene knockdown strains with the induction by 100 ng/ml ATc. (C) Growth curves of a walR knockdown strain (RSD3) with the induction by different concentrations of ATc. A low concentration of ATc is enough to induce high-level gene knockdown. (D) Triton X-100-induced autolysis of a walR knockdown strain (RSD3) and a control strain (RSD0). The error bars indicate the standard errors of the means of four biological replicates. NS, not significant (P > 0.05); *, P < 0.05; **, P < 0.01; ***, P < 0.001. Abbreviations: RSD0, RN4220 carrying plasmid pSD1; RSD1, RN4220 expressing sgRNA targeting murE; RSD2, RN4220 expressing sgRNA targeting rpsC; RSD3, RN4220 expressing sgRNA targeting walR.
DISCUSSION
The absence of an efficient gene knockdown method in prokaryotes like RNA interference in eukaryotes severely hindered the study of essential genes in bacteria, especially in the important human pathogen S. aureus. The functions of many essential genes in S. aureus are still unknown, which strongly hampers vaccine development and drug discovery. asRNA technology has been adapted for bacteria as a gene silencing method since the end of last century (30, 31). While antisense regulation by small noncoding RNAs has been found to be a ubiquitous natural phenomenon in bacteria, the mechanisms of action are largely unknown (32, 33). Even in the best-studied species, E. coli, the mechanisms are not fully understood, which makes rational asRNA design quite challenging (13). Moreover, the asRNA design principle developed from E. coli was not applicable for gene silencing in Gram-positive bacteria (12), so the only approach by which to get optimal asRNA for a target gene in a Gram-positive pathogen like S. aureus is screening of a library of randomly produced asRNAs (13), making this method both time-consuming and costly. Despite this, the asRNA technique was still broadly used in S. aureus since there are no better options for gene knockdown in that organism (34–36).
In this study, we provide a much more efficient and simpler tool for selective gene silencing in S. aureus by using sgRNA-guided dCas9. With a well-characterized mechanism of action and a general blueprint for sgRNA design, the CRISPRi system can theoretically be easily and efficiently used to inactivate any target gene. We showed that this system can successfully silence various genes in different S. aureus strains, with a much higher success rate and better efficiency than the asRNA method. By using this system, we achieved >100-fold repression of hla expression in S. aureus NCTC 8325 (Fig. 2), while early studies using asRNAs targeting hla could only downregulate the expression of hla approximately 16-fold (30, 37). Moreover, the CRISPRi system is inducible and reversible (Fig. 2E), allowing efficient control of its knockdown effect. We also demonstrated that CRISPRi can serve as a powerful tool for the identification and functional study of essential genes in S. aureus (Fig. 6). We believe that it will greatly promote research on genes essential for bacterial growth and pathogenesis.
Another significant advantage of the CRISPRi system over other RNA-guided silencing systems is the low off-target effect, which has been confirmed by whole-transcriptome analyses of both bacteria and eukaryotes (14, 15). Since dCas9-sgRNA requires no host factors for function, this naturally low off-target effect is supposed to be constant in different organisms, including S. aureus. It has been revealed that the specificity of the CRISPRi system is determined jointly by the 5′-NGG-3′ sequence and at least a 12-bp consecutive sgRNA-DNA stretch (14). For bacteria with a small genome like S. aureus (∼2.8 Mb), this 14-nucleotide (nt)-long region is enough to determine a unique target site, but to avoid potential off-target effects, a BLAST analysis of sgRNA in the genome is always recommended to rule out additional potential binding sites (with 14-nt base pairing with sgRNA including 5′-NGG-3′).
The CRISPRi system can also knock down multiple genes simultaneously, which is vitally valuable for molecular study of S. aureus. Since most cellular processes associated with drug resistance and pathogenicity in S. aureus involve a series of genes, the effect of a particular gene on cellular activity might be limiting (2). While inactivation of multiple genes by allelic replacement or asRNA is laborious, it can be easily achieved by CRISPRi by expressing multiple sgRNAs together. Since a single sgRNA expression cassette is only ∼300 bp, it is possible for one knockdown plasmid to carry several sgRNA expression cassettes. Although there is, to some extent, competition between different sgRNAs, each of them still retains high knockdown efficiency (Fig. 4A). For genes that are difficult to silence by a single sgRNA, enhanced knockdown can also be achieved by expressing multiple sgRNAs targeting different sites of the same gene (Fig. 4B).
Operons play important roles in the regulation of metabolism and virulence in pathogenic bacteria. In fact, it has been recognized that 62% of the genes in S. aureus are located within operons (38). Therefore, genetic study of operons is crucial to understand the molecular mechanisms of the virulence and pathogenesis of S. aureus. The CRISPRi system has been proved to be a powerful tool for the study of operon genes. It can be used flexibly to knock down an entire operon or part of it (Fig. 5). Moreover, it can also serve as a simple method for the confirmation of operons predicted by in silico or experimental approaches.
Despite all of these advantages of the CRISPRi system, it is also worth noting that a leaky effect may exist for sgRNAs with high knockdown efficiency. In our study, we found that sgRNAs with low repression rates did not display an obvious leaky effect, while sgRNAs with high repression rates usually showed a significant leaky effect (Fig. 2C and 6A; see Fig. S1 in the supplemental material). However, we also found that the leaky efficiency of an sgRNA is very limited compared to the induced knockdown efficiency (usually <10%), so it will not influence the inducibility of the knockdown effect and functional study of the target gene.
In conclusion, here we report an efficient gene knockdown method based on dCas9 in S. aureus. We believe that this simple, rapid, and affordable selective gene knockdown system will serve as a promising tool for the study of molecular genetics in S. aureus, thus facilitating understanding of the pathogen and the fight against the infections it causes.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. The E. coli strain was grown in Luria-Bertani (LB) medium with shaking at 220 rpm or on LB agar plates at 37°C. S. aureus strains were grown in TSB (Difco) with shaking at 220 rpm or on TSB agar plates at 37°C. When needed, 150 μg/ml ampicillin sodium salt for the E. coli strain or 15 μg/ml chloramphenicol (Chloromycetin) for S. aureus strains was added to the culture medium.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| E. coli Trans1-T1 | Clone host strain, F− ϕ80(lacZ)ΔM15 ΔlacX74 hsdR (rK− mK+) ΔrecA1398 endA1 tonA | TransGen |
| S. aureus | ||
| RN4220 | 8325-4, restriction-negative strain | Laboratory stock |
| NCTC 8325 | Wild type | Laboratory stock |
| N315 | HA-MRSA | Laboratory stock |
| RSD0 | RN4220 carrying plasmid pSD1 | This study |
| RSD1 | RN4220 carrying plasmid pSD1-murE | This study |
| RSD2 | RN4220 carrying plasmid pSD1-rpsC | This study |
| RSD3 | RN4220 carrying plasmid pSD1-walR | This study |
| NSD0 | NCTC 8325 carrying plasmid pSD1 | This study |
| NSD1 | NCTC 8325 carrying plasmid pSD1-hla1 | This study |
| NSD2 | NCTC 8325 carrying plasmid pSD1-hla2 | This study |
| NSD3 | NCTC 8325 carrying plasmid pSD1-hla3 | This study |
| NSD4 | NCTC 8325 carrying plasmid pSD1-hla4 | This study |
| NSD5 | NCTC 8325 carrying plasmid pSD1-sarT1 | This study |
| NSD6 | NCTC 8325 carrying plasmid pSD1-sarT2 | This study |
| NSD7 | NCTC 8325 carrying plasmid pSD1-spa1 | This study |
| NSD8 | NCTC 8325 carrying plasmid pSD1-hla1&2 | This study |
| NSD9 | NCTC 8325 carrying plasmid pSD1-spa1hla2 | This study |
| NSD10 | NCTC 8325 carrying plasmid pSD1-era1 | This study |
| NSD11 | NCTC 8325 carrying plasmid pSD1-era2 | This study |
| NSD12 | NCTC 8325 carrying plasmid pSD1-era3 | This study |
| NSD13 | NCTC 8325 carrying plasmid pSD1-era4 | This study |
| MSD0 | N315 carrying plasmid pSD1 | This study |
| MSD1 | N315 carrying plasmid pSD1-mecA1 | This study |
| MSD2 | N315 carrying plasmid pSD1-ccrAB1 | This study |
| MSD3 | N315 carrying plasmid pSD1-ccrAB2 | This study |
| MSD4 | N315 carrying plasmid pSD1-ccrAB3 | This study |
| MSD5 | N315 carrying plasmid pSD1-ccrAB4 | This study |
| Plasmids | ||
| pRMC2 | Shuttle vector, ATc inducible, Apr Cmr | 22 |
| pSD1 | pRMC2 derivative with dCas9 and sgRNA expression cassette | This study |
| pSD1-hla1 | pSD1 with sgRNA1 targeting hla | This study |
| pSD1-hla2 | pSD1 with sgRNA2 targeting hla | This study |
| pSD1-hla3 | pSD1 with sgRNA3 targeting hla | This study |
| pSD1-hla4 | pSD1 with sgRNA4 targeting hla | This study |
| pSD1-hla1&2 | pSD1 with sgRNA1 and sgRNA2 targeting hla | This study |
| pSD1-sarT1 | pSD1 with sgRNA1 targeting sarT | This study |
| pSD1-sarT2 | pSD1 with sgRNA2 targeting sarT | This study |
| pSD1-spa1 | pSD1 with sgRNA1 targeting spa | This study |
| pSD1-spa1hla2 | pSD1 with sgRNA1 targeting spa and sgRNA2 targeting hla | This study |
| pSD1-mecA1 | pSD1 with sgRNA1 targeting mecA | This study |
| pSD1-ccrAB1 | pSD1 with sgRNA1 targeting ccrAB | This study |
| pSD1-ccrAB2 | pSD1 with sgRNA2 targeting ccrAB | This study |
| pSD1-ccrAB3 | pSD1 with sgRNA3 targeting ccrAB | This study |
| pSD1-ccrAB4 | pSD1 with sgRNA4 targeting ccrAB | This study |
| pSD1-era1 | pSD1 with sgRNA1 targeting operon containing era | This study |
| pSD1-era2 | pSD1 with sgRNA2 targeting operon containing era | This study |
| pSD1-era3 | pSD1 with sgRNA3 targeting operon containing era | This study |
| pSD1-era4 | pSD1 with sgRNA4 targeting operon containing era | This study |
| pSD1-murE | pSD1 with sgRNA targeting murE | This study |
| pSD1-rpsC | pSD1 with sgRNA targeting rpsC | This study |
| pSD1-walR | pSD1 with sgRNA targeting walR | This study |
Abbreviations: Apr, ampicillin-resistant; Cmr, chloramphenicol-resistant.
Preparation of electrocompetent S. aureus cells.
S. aureus cells from a 15% glycerol stock were streaked onto a TSB agar plate and incubated at 37°C. A single colony was picked and incubated in 5 ml of TSB at 37°C overnight. One milliliter of the overnight culture was added to 100 ml of TSB in a 500-ml flask and shaken at 37°C until an optical density at 600 nm (OD600) of 0.4 was reached. The culture was put on ice for 5 min and then transferred to a sterile, round-bottom centrifuge tube. The cells were collected by centrifugation at 2,500 × g at 4°C for 10 min, the supernatant was discarded, the cells were resuspended gently in 10 ml of ice-cold 0.5 M sucrose, and the suspension was kept on ice for 5 min. The centrifugation and resuspension steps were repeated twice. The cells were then resuspended in 1 ml of ice-cold 0.5 M sucrose and kept on ice for 15 min. Finally, 100-μl aliquots were prepared in sterile microcentrifuge tubes and frozen in liquid nitrogen. The competent cells obtained were stored at −80°C.
Plasmid extraction and transformation in S. aureus.
Plasmids were isolated from all S. aureus strains with a plasmid purification kit (Sangon Biotech) in accordance with the manufacturer's instructions, except that the cells were pretreated with digestion buffer containing 40 U/ml lysostaphin, 10 mg/ml lysozyme, and 10% (vol/vol) glycerol for 30 to 60 min. Plasmids were transformed into all S. aureus strains by electroporation. Plasmid DNA (100 to 500 ng) and electrocompetent S. aureus cells (100 μl) were mixed and placed in a Gene Pulser cuvette with a 0.2-cm electrode gap. The settings for electroporation were as follows: voltage, 2.5 kV; capacitance, 50 μF; resistance, 200 Ω. After electroporation, 400 μl of TSB was added to the cuvette immediately and it was put on ice for about 15 min. The cells were then transferred into a 1.5-ml Eppendorf tube and incubated with shaking (220 rpm, 37°C) for 1 h before being spread onto a TSB plate.
Design and construction of the modular sgRNA expression cassette.
To construct a modular sgRNA expression cassette that can be easily used for cloning and expression of target-specific sgRNA without any extra nucleotides, first the transcriptional start site-defined promoter PpflB was amplified by PCR from the S. aureus RN4220 genome with primers PpflB-SmaI-F and PpflB-R. A SapI cloning box with two SapI sites was amplified with primers sgRNA-SapI-F and sgRNA-SapI-R, and the coding sequence of the sgRNA constant region was amplified with primers sgRNA-F and sgRNA-R. The three DNA fragments were then assembled by SLiCE in one step (39). The overhangs of the two SapI sites upon digestion have been designed to coincide with the sequence of the promoter (5′-TAG-3′) and the coding sequence of the sgRNA constant region (5′-GTT-3′), so the two 5′ overhangs of the coding sequence of the sgRNA variable region, 5′-CTA-3′ and 5′-AAC-3′, can respectively ligate with the promoter and coding sequence of the constant region without leaving a scar. The sequences of the primers used in sgRNA expression cassette construction are shown in Table 2.
TABLE 2.
Sequences of primers used in this study
| Primer | Sequence (5′–3′) |
|---|---|
| PRMC2-NcoI-F | CATTCTCTGGTATTTGGACTCCTG |
| PRMC2-SapI-R | AGTCAGTGAGCGAGGAAGCGTAAGAGCGCCCAATAC |
| PRMC2-SapI-F | GTATTGGGCGCTCTTACGCTTCCTCGCTCACTGACT |
| PRMC2-EcoRI-R | AGCTTGATGGTACCGTTAACAGA |
| PpflB-SmaI-F | TAGCCCCGGGATATACTCCTAAATTAACTT |
| PpflB-R | TAGTCATTGTAGCATGTTTGTTGTG |
| sgRNA-SapI-F | CACAACAAACATGCTACAATGACTATGAAGAGC CATGTCAGGCTCTTCTGT |
| sgRNA-SapI-R | ACTTGCTATTTCTAGCTCTAAAACAGAAGAGCCT GACATGGCTCTTCATAG |
| sgRNA-F | GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAA |
| sgRNA-R | AAAAAAGCACCGACTCGGTGCCACTTTTTCAAG TTGATAACGGACTAGCCTTATTT |
| sgRNA-slice-BglII-F | GAAGATCTTAGCCCCGGGATATACTCC |
| sgRNA-slice-EcoRI-R | CGGAATTCAAAAAAGCACCGACTCGG |
| sgRNA-cassette-F | CGGAATTCATATACTCCTAAATTAACTT |
| sgRNA-cassette-R | CGGAATTCAAAAAAGCACCGACTCGG |
| dcas9-BglII-F | GAAGATCTGGGAGGCCGTTTCATGGATAA |
| dcas9-SmaI-R | TCCCCCGGGTATAAACGCAGAAAGGCCCAC |
| hla-sgRNA1-oligo1 | CTAGTTGAATAAATTCTTTATGAAACA |
| hla-sgRNA1-oligo2 | AACTGTTTCATAAAGAATTTATTCAAC |
| hla-sgRNA2-oligo1 | CTACATTAGCGACAGGATTCATTAATA |
| hla-sgRNA2-oligo2 | AACTATTAATGAATCCTGTCGCTAATG |
| hla-sgRNA3-oligo1 | CTACGGGTTCCAAGAATCTCTATCATA |
| hla-sgRNA3-oligo2 | AACTATGATAGAGATTCTTGGAACCCG |
| hla-sgRNA4-oligo1 | CTACTCAGTAACAACAACACTATTGCT |
| hla-sgRNA4-oligo2 | AACAGCAATAGTGTTGTTGTTACTGAG |
| mecA-sgRNA1-oligo1 | CTAACAACTACAACTATTAAAATAAG |
| mecA-sgRNA1-oligo2 | AACCTTATTTTAATAGTTGTAGTTGT |
| spa-sgRNA1-oligo1 | CTATCGTGTTGCGCAGCATTTGCAGC |
| spa-sgRNA1-oligo2 | AACGCTGCAAATGCTGCGCAACACGA |
| sarT-sgRNA1-oligo1 | CTATAACTCCAAATATATTTAGTTTG |
| sarT-sgRNA1-oligo2 | AACCAAACTAAATATATTTGGAGTTA |
| sarT-sgRNA2-oligo1 | CTATTACGCTTTTAACAATTCTAGAT |
| sarT-sgRNA2-oligo2 | AACATCTAGAATTGTTAAAAGCGTAA |
| ccrAB-sgRNA1-oligo1 | CTAATAGCCTATGACTTGTTTCATAT |
| ccrAB-sgRNA1-oligo2 | AACATATGAAACAAGTCATAGGCTAT |
| ccrAB-sgRNA2-oligo1 | CTAGATATGTTGAATATGATGTTTTT |
| ccrAB-sgRNA2-oligo2 | AACAAAAACATCATATTCAACATATC |
| ccrAB-sgRNA3-oligo1 | CTACGTTTATGCAATCGATGATTGCA |
| ccrAB-sgRNA3-oligo2 | AACTGCAATCATCGATTGCATAAACG |
| ccrAB-sgRNA4-oligo1 | CTAATATATCTTTAACTTCAAAATGA |
| ccrAB-sgRNA4-oligo2 | AACTCATTTTGAAGTTAAAGATATAT |
| era-sgRNA1-oligo1 | CTAATATCGTCTATTTGTATAATTCC |
| era-sgRNA1-oligo2 | AACGGAATTATACAAATAGACGATAT |
| era-sgRNA2-oligo1 | CTACCCTAAAATAACGCTATAAAATG |
| era-sgRNA2-oligo2 | AACCATTTTATAGCGTTATTTTAGGG |
| era-sgRNA3-oligo1 | CTAACAAATGTTGACTTTCCTACATT |
| era-sgRNA3-oligo2 | AACAATGTAGGAAAGTCAACATTTGT |
| era-sgRNA4-oligo1 | CTACTGCTTTGATGATAATCCCTTTT |
| era-sgRNA4-oligo2 | AACAAAAGGGATTATCATCAAAGCAG |
| murE-sgRNA-oligo1 | CTAATACTAGCTACTCTTAATGTGTC |
| murE-sgRNA-oligo2 | AACGACACATTAAGAGTAGCTAGTAT |
| rpsC-sgRNA1oligo1 | CTAATAATACCAACACGAAGTCCGAT |
| rpsC-sgRNA-oligo2 | AACATCGGACTTCGTGTTGGTATTAT |
| walR-sgRNA-oligo1 | CTATTAAATTCTAAAATATCAGCAAT |
| walR-sgRNA-oligo2 | AACATTGCTGATATTTTAGAATTTAA |
| RT-pta-F | AAAGCGCCAGGTGCTAAATTAC |
| RT-pta-R | CTGGACCAACTGCATCATATCC |
| RT-hla-F | AAAGTAGGCTGGAAAGTGAT |
| RT-hla-R | TAGCGAAGTCTGGTGAAAA |
| RT-spa-F | AAGATGGTAACGGAGTACATGTCG |
| RT-spa-R | CAAGTTCTTGACCAGGTTTGATC |
| RT-ccrA-F | GCACAGTTATTAGAAGAAGATA |
| RT-ccrA-R | GCCATATTGATTGTTGACA |
| RT-ccrB-F | CAATACCACGAATACACTT |
| RT-ccrB-R | CATCACATAATCTTCAATCAC |
| RT-era2-F | TGCTTTAGAAGAAGATGAGCCAGAG |
| RT-era2-R | GTCCGTAATTGTTTGCTTGTTCT |
| RT-era4-F | GTTAGAAAAGCACAACAAGAATC |
| RT-era4-R | TAGCTCGTTCAGCACATATCG |
| RT-era5-F | AAGGTGTTATGACAAGAGATGACG |
| RT-era5-R | AGCGACTTTCATCATATAGTCACC |
| RT-era6-F | GGTGCAAAAGTACCACTTATGGC |
| RT-era6-R | TTCCCATACCTCGCCACTGATT |
| pRMC2-seq-F | CACAGATGCGTAAGGAGA |
Design and construction of dCas9 and sgRNA expression plasmid pSD1.
To construct plasmid pSD1 for constitutive expression of the sgRNA and conditional expression of dCas9, first the original SapI site in plasmid pRMC2 (22) was mutated by overlap PCR with primers PRMC2-NcoI-F, PRMC2-SapI-R, PRMC2-SapI-F, and PRMC2-EcoRI-R. The sgRNA expression cassette was then amplified by PCR from the SLiCE product with primers sgRNA-slice-BglII-F and sgRNA-slice-EcoRI-R and cloned into BglII-EcoRI double-digested plasmid pRMC2. This cloning also introduced an SmaI site upstream of the sgRNA expression cassette for the subsequent insertion of a dcas9 gene fragment. Finally, the dcas9 gene fragment with an RBS from S. aureus and a strong transcription terminator (BioBrick part BBa_B0015) was amplified from a previously constructed plasmid with primers dcas9-BglII-F and dcas9-SmaI-R and inserted into BglII-SmaI double-digested plasmid pRMC2-sgRNA. The final plasmid, pSD1, is about 11 kbp. The sequences of the primers used for plasmid construction are shown in Table 2.
Construction of the specific gene knockdown plasmid.
To achieve knockdown of an individual gene, the 5′-NGG-3′ sequence was first identified in the template strand of the target gene. The 23 nt immediately upstream of the 5′-NGG-3′ were then taken and added to the 3′ end of 5′-CTA-3′ to create sgRNA oligonucleotide 1, and the reverse complementary sequence of the 23 nt was taken and added to the 3′ end of 5′-AAC-3′ to create sgRNA oligonucleotide 2. A BLAST search was performed to detect the specificity of the sgRNA. Subsequently, the two oligonucleotides (20 μM) were annealed and then cloned into SapI-digested plasmid pSD1.
To construct a double-knockdown plasmid, two single-knockdown plasmids were first constructed. One sgRNA expression cassette was then amplified by PCR with promoter-specific forward primer sgRNA-cassette-F and terminator-specific reverse primer sgRNA-cassette-R. The PCR product was digested with EcoRI and then cloned into the other plasmid downstream of the existing sgRNA expression cassette.
The plasmids were first introduced into restriction-negative S. aureus strain RN4220 for modification and subsequently transformed into experimental strains. The sequences of the primers and sgRNA oligonucleotides used for plasmid construction are shown in Table 2.
Total RNA isolation and real-time qRT-PCR.
Total RNA was extracted by RNAiso Plus in accordance with the manufacturer's instructions (TaKaRa). Residual DNA was digested with RNase-free DNase I (TaKaRa). RT was carried out with the PrimeScript First Strand cDNA synthesis kit (TaKaRa), and real-time PCR was performed with SYBR Premix Ex Taq (TaKaRa) by using a StepOne real-time system (Applied Biosystems). The quantity of cDNA was normalized to the abundance of pta cDNA (40). All of the qRT-PCR assays were repeated at least three times.
Western blot assay of alpha-toxin.
Stationary-phase culture supernatant was collected and heated for 10 min at 95°C. The samples were then separated by 12% SDS-PAGE and electrotransferred onto a polyvinylidene difluoride membrane (GE, Piscataway, NJ). The protein was detected with a rabbit anti-alpha-toxin antibody (diluted 1:1,000; Sigma), followed by a horseradish peroxidase-conjugated sheep anti-rabbit antibody (diluted 1:10,000; Pierce). The images were obtained with ImageQuant LAS 4000 (GE).
Assay for alpha hemolysis.
The alpha-hemolytic activities of the S. aureus NCTC 8325 control strain and the hla knockdown strains were assayed on sheep blood agar. The OD600s of overnight culture were measured and adjusted to the same level. One-microliter culture volumes were then spotted onto sheep blood agar plates with or without ATc. The plates were left to dry for about 2 min and incubated at 37°C for 24 h.
Oxacillin susceptibility analysis.
The oxacillin susceptibilities of the S. aureus N315 control strain and the mecA knockdown strain were analyzed by disk diffusion test as previously described (41). Cultures of the knockdown and control strains were both diluted to an OD600 of 2.0. Sterile swabs were then used to streak the cultures onto Mueller-Hinton agar plates (Difco) with 100 ng/ml ATc to form bacterial lawns. The plates were left to dry for about 5 min. Disks containing oxacillin (128 mg/ml, 2.5 μl) and daptomycin (16 mg/ml, 2.5 μl) were then attached to the plates with sterile tweezers. The plates were incubated overnight at 37°C.
Growth curve measurement.
Overnight cultures of the RN4220 control strain and the essential gene knockdown strains in TSB without ATc were diluted to an OD600 of 0.05 into new TSB with or without ATc and allowed to grow in 96-well plates with shaking (220 rpm) at 37°C. Culture growth was monitored at 1-h intervals with a microplate reader (Elx800; Bio-Tek). For each sample, four biological replicates were analyzed.
Triton X-100-induced autolysis assay.
Bacterial cells grown overnight were diluted to an OD600 of 0.05 in TSB with or without 20 ng/ml ATc and allowed to grow with shaking (220 rpm) at 37°C to the early exponential phase (OD600 of 0.8). Cells were harvested by centrifugation at 5,000 × g for 10 min, washed three times with 50 mM Tris-HCl (pH 7.5), and resuspended in 50 mM Tris-HCl (pH 7.5). The resuspended cells (100 μl) were added to 96-well plates, and the same volume of 50 mM Tris-HCl (pH 7.5) containing 0.1% (vol/vol) Triton X-100 was added to each sample well quickly. The plates were incubated at 37°C with shaking (220 rpm). Cell autolysis was determined by measuring the progressive decrease in OD600 at half-hour intervals with a microplate reader (Elx800; Bio-Tek). For each sample, four biological replicates were analyzed.
Statistical analysis.
F tests of two samples for variance were performed. Unpaired two-tailed t tests for equal or unequal variance were then performed to calculate the significant differences (P values). All tests were performed by the data analysis tool in Microsoft Excel.
Accession number(s).
The annotated pSD1 sequence has been submitted to GenBank, and the accession number is KX685167.
Supplementary Material
ACKNOWLEDGMENTS
We thank WHU-China iGEM 2013 for distribution of dcas9 gene and G. Bai for technical assistance.
This work was supported by the National Natural Science Foundation of China (31670133).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00291-17.
REFERENCES
- 1.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:2026–2027. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Lindsay JA. (ed). 2008. Staphylococcus: molecular genetics. Caister Academic Press, Poole, United Kingdom. [Google Scholar]
- 3.Prax M, Lee CY, Bertram R. 2013. An update on the molecular genetics toolbox for staphylococci. Microbiology 159:421–435. doi: 10.1099/mic.0.061705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakashima N, Miyazaki K. 2014. Bacterial cellular engineering by genome editing and gene silencing. Int J Mol Sci 15:2773–2793. doi: 10.3390/ijms15022773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Yao J, Zhong J, Fang Y, Geisinger E, Novick RP, Lambowitz AM. 2006. Use of targetrons to disrupt essential and nonessential genes in Staphylococcus aureus reveals temperature sensitivity of Ll.LtrB group II intron splicing. RNA 12:1271–1281. doi: 10.1261/rna.68706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji Y, Zhang B, Van Horn SF, Warren P, Woodnutt G, Burnham MK, Rosenberg M. 2001. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 293:2266–2269. doi: 10.1126/science.1063566. [DOI] [PubMed] [Google Scholar]
- 8.Jana M, Luong T-T, Komatsuzawa H, Shigeta M, Lee CY. 2000. A method for demonstrating gene essentiality in Staphylococcus aureus. Plasmid 44:100–104. doi: 10.1006/plas.2000.1473. [DOI] [PubMed] [Google Scholar]
- 9.Meng J, Kanzaki G, Meas D, Lam CK, Crummer H, Tain J, Xu HH. 2012. A genome-wide inducible phenotypic screen identifies antisense RNA constructs silencing Escherichia coli essential genes. FEMS Microbiol Lett 329:45–53. doi: 10.1111/j.1574-6968.2012.02503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Backer MD, Nelissen B, Logghe M, Viaene J, Loonen I, Vandoninck S, de Hoogt R, Dewaele S, Simons FA, Verhasselt P, Vanhoof G, Contreras R, Luyten WHML. 2001. An antisense-based functional genomics approach for identification of genes critical for growth of Candida albicans. Nat Biotechnol 19:235–241. doi: 10.1038/85677. [DOI] [PubMed] [Google Scholar]
- 11.Mizuta T, Fujiwara M, Hatta T, Abe T, Miyano-Kurosaki N, Shigeta S, Yokota T, Takaku H. 1999. Antisense oligonucleotides directed against the viral RNA polymerase gene enhance survival of mice infected with influenza A. Nat Biotechnol 17:583–587. doi: 10.1038/9893. [DOI] [PubMed] [Google Scholar]
- 12.Man SA, Cheng RB, Miao CC, Gong QH, Gu YC, Lu XZ, Han F, Yu WG. 2011. Artificial trans-encoded small non-coding RNAs specifically silence the selected gene expression in bacteria. Nucleic Acids Res 39:e50. doi: 10.1093/nar/gkr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho SH, Haning K, Contreras LM. 2015. Strain engineering via regulatory noncoding RNAs: not a one-blueprint-fits-all. Curr Opin Chem Eng 10:25–34. doi: 10.1016/j.coche.2015.07.008. [DOI] [Google Scholar]
- 14.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, Guilak F, Crawford GE, Reddy TE, Gersbach CA. 2013. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 17.Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. 2014. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marraffini LA, Sontheimer EJ. 2010. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11:181–190. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choudhary E, Thakur P, Pareek M, Agarwal N. 2015. Gene silencing by CRISPR interference in mycobacteria. Nat Commun 6:6267. doi: 10.1038/ncomms7267. [DOI] [PubMed] [Google Scholar]
- 22.Corrigan RM, Foster TJ. 2009. An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid 61:126–129. doi: 10.1016/j.plasmid.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Pagels M, Fuchs S, Pane-Farre J, Kohler C, Menschner L, Hecker M, McNamarra PJ, Bauer MC, von Wachenfeldt C, Liebeke M, Lalk M, Sander G, von Eiff C, Proctor RA, Engelmann S. 2010. Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol 76:1142–1161. doi: 10.1111/j.1365-2958.2010.07105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helle L, Kull M, Mayer S, Marincola G, Zelder M-E, Goerke C, Wolz C, Bertram R. 2011. Vectors for improved Tet repressor-dependent gradual gene induction or silencing in Staphylococcus aureus. Microbiology 157:3314–3323. doi: 10.1099/mic.0.052548-0. [DOI] [PubMed] [Google Scholar]
- 25.Katayama Y, Ito T, Hiramatsu K. 2000. A new class of genetic element, staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 44:1549–1555. doi: 10.1128/AAC.44.6.1549-1555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaudhuri RR, Allen AG, Owen PJ, Shalom G, Stone K, Harrison M, Burgis TA, Lockyer M, Garcia-Lara J, Foster SJ, Pleasance SJ, Peters SE, Maskell DJ, Charles IG. 2009. Comprehensive identification of essential Staphylococcus aureus genes using transposon-mediated differential hybridisation (TMDH). BMC Genomics 10:291. doi: 10.1186/1471-2164-10-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valentino MD, Foulston L, Sadaka A, Kos VN, Villet RA, Maria JS, Lazinski DW, Camilli A, Walker S, Hooper DC, Gilmore MS. 2014. Genes contributing to Staphylococcus aureus fitness in abscess- and infection-related ecologies. mBio 5:e01729-14. doi: 10.1128/mBio.01729-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin PK, Li T, Sun DX, Biek DP, Schmid MB. 1999. Role in cell permeability of an essential two-component system in Staphylococcus aureus. J Bacteriol 181:3666–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dubrac S, Boneca IG, Poupel O, Msadek T. 2007. New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J Bacteriol 189:8257–8269. doi: 10.1128/JB.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kernodle DS, Voladri RKR, Menzies BE, Hager CC, Edwards KM. 1997. Expression of an antisense hla fragment in Staphylococcus aureus reduces alpha-toxin production in vitro and attenuates lethal activity in a murine model. Infect Immun 65:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Good L, Nielsen PE. 1998. Antisense inhibition of gene expression in bacteria by PNA targeted to mRNA. Nat Biotechnol 16:355–358. doi: 10.1038/nbt0498-355. [DOI] [PubMed] [Google Scholar]
- 32.Romilly C, Caldelari I, Parmentier D, Lioliou E, Romby P, Fechter P. 2012. Current knowledge on regulatory RNAs and their machineries in Staphylococcus aureus. RNA Biol 9:402–413. doi: 10.4161/rna.20103. [DOI] [PubMed] [Google Scholar]
- 33.Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun JS, Zheng L, Landwehr C, Yang JS, Ji YD. 2005. Identification of a novel essential two-component signal transduction system, YhcSR, in Staphylococcus aureus. J Bacteriol 187:7876–7880. doi: 10.1128/JB.187.22.7876-7880.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubio A, Conrad M, Haselbeck RJ, Kedar GC, Brown-Driver V, Finn J, Silverman JA. 2011. Regulation of mprF by antisense RNA restores daptomycin susceptibility to daptomycin-resistant isolates of Staphylococcus aureus. Antimicrob Agents Chemother 55:364–367. doi: 10.1128/AAC.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forsyth A, Wang L. 2008. Techniques for the isolation and use of conditionally expressed antisense RNA to achieve essential gene knockdowns in Staphylococcus aureus. Methods Mol Biol 416:307–321. doi: 10.1007/978-1-59745-321-9_20. [DOI] [PubMed] [Google Scholar]
- 37.Ji YD, Marra A, Rosenberg M, Woodnutt G. 1999. Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. J Bacteriol 181:6585–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ten Broeke-Smits NJP, Pronk TE, Jongerius I, Bruning O, Wittink FR, Breit TM, van Strijp JAG, Fluit AC, Boel CHE. 2010. Operon structure of Staphylococcus aureus. Nucleic Acids Res 38:3263–3274. doi: 10.1093/nar/gkq058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Werling U, Edelmann W. 2012. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res 40:e55. doi: 10.1093/nar/gkr1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valihrach L, Demnerova K. 2012. Impact of normalization method on experimental outcome using RT-qPCR in Staphylococcus aureus. J Microbiol Methods 90:214–216. doi: 10.1016/j.mimet.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 41.Clinical and Laboratory Standards Institute. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 9th ed Approved standard. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.