

A chemokine receptor type 2–binding peptide adapted as a PET probe can help detect lung inflammation in a mouse model and human tissues.
Abstract
Purpose
To characterize a chemokine receptor type 2 (CCR2)–binding peptide adapted for use as a positron emission tomography (PET) radiotracer for noninvasive detection of lung inflammation in a mouse model of lung injury and in human tissues from subjects with lung disease.
Materials and Methods
The study was approved by institutional animal and human studies committees. Informed consent was obtained from patients. A 7-amino acid CCR2 binding peptide (extracellular loop 1 inverso [ECL1i]) was conjugated to tetraazacyclododecane tetraacetic acid (DOTA) and labeled with copper 64 (64Cu) or fluorescent dye. Lung inflammation was induced with intratracheal administration of lipopolysaccharide (LPS) in wild-type (n = 19) and CCR2-deficient (n = 4) mice, and these mice were compared with wild-type mice given control saline (n = 5) by using PET performed after intravenous injection of 64Cu-DOTA-ECL1i. Lung immune cells and those binding fluorescently labeled ECL1i in vivo were detected with flow cytometry. Lung inflammation in tissue from subjects with nondiseased lungs donated for lung transplantation (n = 11) and those with chronic obstructive pulmonary disease (COPD) who were undergoing lung transplantation (n = 16) was evaluated for CCR2 with immunostaining and autoradiography (n = 6, COPD) with 64Cu-DOTA-ECL1i. Groups were compared with analysis of variance, the Mann-Whitney U test, or the t test.
Results
Signal on PET images obtained in mouse lungs after injury with LPS was significantly greater than that in the saline control group (mean = 4.43% of injected dose [ID] per gram of tissue vs 0.99% of injected dose per gram of tissue; P < .001). PET signal was significantly diminished with blocking studies using nonradiolabeled ECL1i in excess (mean = 0.63% ID per gram of tissue; P < .001) and in CCR2-deficient mice (mean = 0.39% ID per gram of tissue; P < .001). The ECL1i signal was associated with an elevated level of mouse lung monocytes. COPD lung tissue displayed significantly elevated CCR2 levels compared with nondiseased tissue (median = 12.8% vs 1.2% cells per sample; P = .002), which was detected with 64Cu-DOTA-ECL1i by using autoradiography.
Conclusion
64Cu-DOTA-ECL1i is a promising tool for PET-based detection of CCR2-directed inflammation in an animal model and in human tissues as a step toward clinical translation.
© RSNA, 2017
Introduction
Lung inflammation is a result of the recruitment of inflammatory cells, guided along chemokine gradients by their cognate receptors (1,2). In some lung conditions (eg, asthma, eosinophilic pneumonia), patterns of inflammatory cell recruitment can be used to diagnose diseases and direct therapeutic decisions (3,4). However, the development of a detection strategy for identifying immune population signatures has been difficult because many inflammatory cells are localized within the lung parenchyma, out of reach of conventional diagnostic tools. Consequently, the understanding of the contribution of immune cell subsets in disease is limited and the clinical development of specific antagonists for lung disease is stalled. Noninvasive detection could ultimately be used to characterize an individual’s molecular status, disease activity, and response to established or new therapies (5).
Chemokine (C-C motif) ligand type 2 (also called monocyte chemoattractant protein-1) and its receptor chemokine (C-C motif) receptor type 2 (CCR2) are often elevated in the lung tissue of subjects with pulmonary disease (6–10). CCR2 is a surface receptor found on most inflammatory monocytes and macrophages, as well as on some dendritic cells and lymphocytes (1,2). CCR2-positive inflammatory monocytes are essential early responding immune cells, and excessive or prolonged recruitment impairs the resolution of inflammation and propagates disease progression (3,11,12). Experimentally, endotoxin triggers CCR2-dependent migration of monocytes and macrophages to the lung and influences subsequent neutrophil recruitment (13–15). CCR2-positive signaling in monocytes provides a secondary source of proinflammatory cytokines and proteases, contributing to lung injury (12,16). Recruitment of CCR2-dependent leukocytes affects the magnitude and duration of acute respiratory distress syndrome (17), and elevated numbers of CCR2-positive monocytes are associated with ongoing inflammation in chronic obstructive pulmonary disease (COPD) (6,7), which supports the use of CCR2-positive cells as a marker of disease activity. CCR2-positive cells may also serve as a therapeutic target because CCR2 blockade improves outcome in animal models of disease and has steered considerable effort toward the development and testing of CCR2 antagonists (18,19).
We have recently described the use of a peptide that binds the first extracellular loop of CCR2, called extracellular loop 1 inverso (ECL1i) (20), for noninvasive imaging in initial studies in a lung ischemia-reperfusion model (21), but the generalizability to common inflammatory conditions and the activity in human disease tissue is unknown. The goal of this study was to further characterize the ECL1i-based positron emission tomography (PET) radiotracer for the noninvasive detection of monocyte-related inflammation in a mouse model of endotoxin-induced lung injury and in human lung tissue.
Materials and Methods
This study was supported by National Heart, Lung, and Blood Institute Programs of Excellence in Nanotechnology: Integrated Nanosystems for Diagnosis and Therapy contract number HHN268201000046C (NIH BAA no. NHLBI-HV-10-08, Y.L., R.J.G., S.L.B.; R01 HL131908, Y.L., S.L.B.; U19 AI070489, M.J.H.; and R01 HL121791, (M.J.H.). S.L.B. is the Hubert S. and Dorothy R. Moog Professor of Pulmonary Medicine supported by the Barnes Jewish Hospital Foundation.
Human Tissues
The institutional review board approved these studies. Human COPD lung tissue samples were obtained at lung transplantation from individuals who provided consent before surgery (n = 16). All patients undergoing lung transplantation had very severe COPD according to clinical criteria. Nondiseased donated lungs were those not usable for transplantation or were tissues provided after lungs were downsized (n = 11). Samples were prospectively collected between 2005 and 2013, de-identified, and stored before use (Table E1 [online]). Samples were selected for analysis on the basis of tissue availability without preselection for specific clinical features.
Mice
The institutional animal care and use committee approved these studies. We evaluated 77 wild-type and four CCR2-deficient (CCR2−/−) mice (C57BL6/J strain) of both sexes (age, 8–12 weeks; weight, approximately 25 g). Mice were used for radiotracer stability studies (n = 3) and lung injury studies that included immunostaining for CCR2 (n = 6), in vivo immune cell peptide binding (n = 18), lung water weight measurements (n = 6), in vivo biodistribution analysis (n = 12), and PET/computed tomography (CT) (n = 36). For the lung injury studies, mice did not undergo treatment (treatment naive group, n = 8) or received intratracheal vehicle control (phosphate buffered saline [PBS], 1 μL/g, n = 26) or lipopolysaccharide (LPS) (endotoxin, Escherichia coli strain 055:B5; Sigma-Aldrich, St Louis, Mo) at a dose of 2.5 μg/g (n = 44). Other mice were administered a high dose (10 μg/g, n = 3) or a low dose (0.5 μg/g, n = 3) of LPS as described in Appendix E1 (online). All CCR2-deficient mice received an LPS dose of 2.5 μg/g. Lung water was quantified by using wet-to-dry weight ratios in mice treated with PBS or intermediate-dose LPS (three per group). Lungs were resected and weighed immediately. The weights obtained immediately after resection were compared with those obtained after drying at 65°C for 48 hours. The study flowchart is shown in Figure 1.
Figure 1a:

Flowcharts show study design of CCR2 imaging with ECL1i peptide. (a) Peptide labeling and stability. (b) Mouse imaging studies. The number of mice in each treatment group is indicated. Bold boxes indicate in vivo PET/CT studies. Time-activity analysis was performed on a subgroup of PET images obtained 24 hours after treatment. (c) Human lung tissue studies. A subgroup of lung tissues from subjects with COPD displaying elevated levels of CCR2-positive cells was analyzed for copper 64 (64Cu)–tetraazacyclododecane tetraacetic acid (DOTA)–ECL1i binding by using autoradiography followed by blocking (n = 6).
Figure 1b:

Flowcharts show study design of CCR2 imaging with ECL1i peptide. (a) Peptide labeling and stability. (b) Mouse imaging studies. The number of mice in each treatment group is indicated. Bold boxes indicate in vivo PET/CT studies. Time-activity analysis was performed on a subgroup of PET images obtained 24 hours after treatment. (c) Human lung tissue studies. A subgroup of lung tissues from subjects with COPD displaying elevated levels of CCR2-positive cells was analyzed for copper 64 (64Cu)–tetraazacyclododecane tetraacetic acid (DOTA)–ECL1i binding by using autoradiography followed by blocking (n = 6).
Figure 1c:

Flowcharts show study design of CCR2 imaging with ECL1i peptide. (a) Peptide labeling and stability. (b) Mouse imaging studies. The number of mice in each treatment group is indicated. Bold boxes indicate in vivo PET/CT studies. Time-activity analysis was performed on a subgroup of PET images obtained 24 hours after treatment. (c) Human lung tissue studies. A subgroup of lung tissues from subjects with COPD displaying elevated levels of CCR2-positive cells was analyzed for copper 64 (64Cu)–tetraazacyclododecane tetraacetic acid (DOTA)–ECL1i binding by using autoradiography followed by blocking (n = 6).
Synthesis, Labeling, and Stability of ECL1i
The ECL1i peptide (LGTFLKC) was synthesized from d-form amino acids by CPC Scientific (Sunnyvale, Calif). DOTA-ECL1i was prepared by conjugating DOTA, which was modified as maleimido-monoamide-DOTA (1.573 mg, 0.2 µmol; Macrocyclics, Dallas, Tex), to the cysteine residue of ECL1i by using established methods (22). The crude conjugate was purified by means of high-performance liquid chromatography to reach 99% chemical purity and verified with mass spectrometry. 64Cu was selected as an initial radiolabel for ECL1i on the basis of a high specific activity that enabled the administration of a trace amount and provided a decisive PET signal if present, straightforward radiochemistry by means of the conjugation of the DOTA chelator on the peptide, on-site availability, and previous experience with this radionuclide (22). The DOTA-ECL1i conjugate was radiolabeled with 64CuCl2 as previously described (22). 64Cu-DOTA-ECL1i was tested for stability by means of incubation in mouse serum at 37°C for 1 hour and, in vivo, in blood and lung 1 hour after injection (n = 3) with radio–high-performance liquid chromatography analysis as described in Appendix E1 (online). Maleimide-modified amine-reactive dye (Dylight 550; ThermoFisher Scientific, Waltham, Mass) was conjugated to ECL1i on the cysteine residue by using the same protocol for DOTA conjugation and purified with high-performance liquid chromatography.
Biodistribution Analysis
For biodistribution analysis, mice in the treatment-naive group and those treated with intratracheal PBS or LPS (four in each group) received a bolus injection of 100 μL (0.37 MBq per mouse) 64Cu-DOTA-ECL1i via the tail vein 24 hours after treatment. After euthanasia, which was performed 1 hour after radiotracer injection, organs of interest were collected, weighed, and assayed by means of a gamma counter (Beckman, Brea, Calif) as previously described (23). Standards were prepared and measured in parallel to calculate the percentage of injected dose (ID) per gram of tissue.
PET/CT and Image Analysis
Dynamic PET image acquisition (from 0 to 60 minutes) and corresponding computed tomographic (CT) images were obtained by using cross-calibrated scanners (Inveon microPET/CT [Siemens, Malvern, Pa] or Focus 220 PET [Concorde Microsystems, Knoxville, Tenn]) immediately after tail vein injection of 64Cu-DOTA-ECL1i (3.7 MBq per mouse) at 24, 48, and 144 hours after LPS treatment. Organ uptake was calculated as the percentage ID per gram of tissue in three-dimensional regions of interest from PET images, without correction for partial volume effect, by using software (Inveon Research Workplace, Siemens) (23). Time-activity curves were calculated from the regions of interest from PET images obtained in a subgroup of the PBS- and LPS-treated mice (three per group) undergoing imaging at 24 hours. Competitive PET blocking studies were performed in the LPS mouse model with coinjection of an excess amount of nonradiolabeled ECL1i (900.9 pmol) and 64Cu-DOTA-ECL1i (1.8 pmol) (ECL1i-to-64Cu-DOTA-ECL1i molar ratio = 500:1) followed by a 0–60-minute dynamic scan. PET/CT images of naive (n = 4) and PBS-treated (n = 5) mice at 24 hours were used as controls for all LPS-treatment studies performed at 24 hours, including ECL1i blocking studies (n = 4), CCR2−/− mice (n = 4), low-dose (n = 3) and high-dose (n = 3) LPS, and images obtained at 48 (n = 3) and 144 (n = 3) hours after LPS.
Flow Cytometry
Mice received intratracheal PBS (n = 4) or LPS (n = 5), and 24 hours later were injected intravenously with ECL1i labeled with amine-reactive dye (Dylight 550, 100 μg) to determine the type of inflammatory cell binding ECL1i. Other mice (four in the PBS group and five in the LPS group) were treated in parallel but were not given ECL1i-Dylight 550. One hour after injection, a single cell suspension was produced from the lungs, immunostained with immune cell markers (Table E2 [online]), and analyzed with flow cytometry as described in Appendix E1 (online).
Immunostaining and Microscopy
Tissue sections were immunostained by using a CCR2 monoclonal antibody (Novus Biologicals, Littleton, Colo) in mouse and human tissue (mice, n = 6; human tissue, n = 27). In each human tissue section, CCR2-staining cells were assessed from five photomicrographs obtained at ×200 magnification. Images were analyzed by investigators who were unfamiliar with the hypothesis. The percentage of CCR2 expression was determined as a ratio of the area of CCR2 signal relative to 49,6-diamidino-2-phenylindole (DAPI) by using software (ImageJ, v1.50; National Institutes of Health, Bethesda, Md). Details are provided in Appendix E1 (online).
Autoradiography
Fixed human lung tissue sections that were deparaffinized and hydrated in PBS were incubated with 64Cu-DOTA-ECL1i for 5 minutes (donor tissue, n = 4; COPD tissue, n = 6). Blocking studies were performed in lung sections from subjects with COPD demonstrating levels of CCR2 immunostaining above the median (n = 6) by means of coincubation with 500-fold nonradioactive ECL1i. Slides were washed 30 times with water and then placed in an instant imager (Packard, Meriden, Conn) for 30 minutes. Images were postprocessed by using software (Imager, Packard), and the percentage of blocked signal was calculated.
Statistical Methods
Data were analyzed by using software (Prism, version 6.07; GraphPad, La Jolla, Calif). Differences between groups were compared by using the two-tailed Student t test. Means of nonparametric data from human samples were compared with the Mann-Whitney U test. Multiple means were compared by using a one- or two-way analysis of variance with the Tukey test. P < .05 was indicative of a statistically significant difference.
Results
64Cu-DOTA-ECL1i Radiochemistry and Stability
Results of mass spectrometry of the conjugate confirmed that one DOTA conjugated to one ECL1i peptide (mass/charge ratio calculated: 1306.65, observed: 1306.69) (Fig E1, A [online]). The radiochemical purity of 64Cu-DOTA-ECL1i used for animal studies was 98% or greater, and this was confirmed with radio–high-performance liquid chromatography (Fig E1, B [online]). The mean specific activity (±standard deviation) of 64Cu-DOTA-ECL1i was 55.5 MBq/nmol ± 1.11 (n = 20), which enabled trace amounts (approximately 70 pmol) to be injected for in vivo studies. 64Cu-DOTA-ECL1i was stable in mouse serum in vitro at 37°C for 1 hour (mean, 98.2% ± 2.1) and in vivo in serum (96.5% ± 1.1) and lung (95.6% ± 3.0) recovered from treatment-naive mice 1 hour after injection by using high-performance liquid chromatography analyses and gamma counting (Fig E2 [online]).
Biodistribution of 64Cu-DOTA-ECL1i in the LPS Mouse Lung Injury Model
We assayed 64Cu-DOTA-ECL1i in a well-characterized lung injury model in which endotoxin (LPS) activates the accumulation of CCR2-expressing cells in the lung (13,24). As expected, CCR2-expressing cells were detected in mouse lungs 24 hours after intratracheal delivery of LPS (Fig 2a). The in vivo pharmacokinetic evaluation of 64Cu-DOTA-ECL1i acquired 1 hour after intravenous injection was compared in naive mice and 24 hours after the delivery of intratracheal control vehicle PBS or LPS (Fig 2b). Accumulation of tracer in the lung of mice given LPS was 2.5 times higher than that in the PBS group, although there was no difference in blood retention. 64Cu-DOTA-ECL1i showed renal clearance, as evidenced by the kidney accumulation in both groups. Liver and bone marrow uptake of 64Cu-DOTA-ECL1i in LPS-treated mice was significantly higher than that in PBS-treated mice.
Figure 2a:

Biodistribution of 64Cu-DOTA-ECL1i in mice with lung injury. Mice were not treated (naive) or received intratracheal PBS or LPS. (a) Photomicrographs (original magnification, ×400) illustrate immunostaining for CCR2 24 hours after intratracheal delivery of PBS or LPS. Tissues were counterstained with hematoxylin. Bar = 50 μm. (b) Chart shows biodistribution of 64Cu-DOTA-ECL1i at 24 hours in treatment-naive, PBS, and LPS groups. Data are means ± standard deviations. There were four mice per group. * = P < .001 (two-way analysis of variance with Tukey test).
Figure 2b:

Biodistribution of 64Cu-DOTA-ECL1i in mice with lung injury. Mice were not treated (naive) or received intratracheal PBS or LPS. (a) Photomicrographs (original magnification, ×400) illustrate immunostaining for CCR2 24 hours after intratracheal delivery of PBS or LPS. Tissues were counterstained with hematoxylin. Bar = 50 μm. (b) Chart shows biodistribution of 64Cu-DOTA-ECL1i at 24 hours in treatment-naive, PBS, and LPS groups. Data are means ± standard deviations. There were four mice per group. * = P < .001 (two-way analysis of variance with Tukey test).
CCR2 Imaging in LPS Lung Injury
On the basis of the biodistribution studies, we administered intratracheal PBS or LPS and determined if intravenous 64Cu-DOTA-ECL1i could be used as a PET agent to image lung inflammation. Compared with PBS delivery, there was a prominent signal in the lungs of LPS-treated mice at 24 hours (PBS mean: 0.99% ID per gram of tissue ± 0.29, n = 5; LPS mean: 4.43% ID per gram of tissue ± 1.44, n = 7; P < .001) (Fig 3). Consistent with biodistribution studies, LPS also increased activity in the liver and renal clearance was indicated by enhanced kidney and bladder activity in all mice. The time-activity curve acquired 24 hours after injury demonstrated a higher and consistent accumulation of 64Cu-DOTA-ECL1i in the LPS-treated lung compared with the PBS-treated control lung, which was significantly lower, and further diminished over the imaging period. 64Cu-DOTA-ECL1i uptake in the lung peaked at 24 hours, followed by a loss of signal in mice injected and imaged at 48 hours (mean: 1.04% ID per gram of tissue ± 0.17, n = 5) or 144 hours (mean: 1.29% ID per gram of tissue ± 0.32, n = 5) after LPS (Fig 3c). Mice were administered high and low doses of LPS to determine the sensitivity of 64Cu-DOTA-ECL1i detection with PET. The lung signal in mice treated with a low dose of LPS (mean: 1.82% ID per gram of tissue ± 0.17, n = 5) was decreased compared with that in mice treated with intermediate and high doses (Fig 3d).
Figure 3a:

PET 64Cu-DOTA-ECL1i imaging in mouse lung injury model. Mice received intratracheal PBS or LPS followed by intravenous injection of 64Cu-DOTA-ECL1i. (a) Representative PET images of maximum intensity projection reconstructed PET/CT scans (center) and from indicated planes acquired 24 hours after treatment and after injection with 64Cu-DOTA-ECL1i . (b) Time-activity curves of 64Cu-DOTA-ECL1i in heart and lungs from 0 to 60 minutes after injection (n = 3 per group from subgroup described in c). * = P < .001 compared with lung PBS and LPS groups (unpaired two-tailed Student t test). Data are means ± standard deviations. (c) Bar chart shows 64Cu-DOTA-ECL1i lung uptake after treatment (PBS: n = 5; LPS: n = 7 at 24 hours, n = 3 at 48 hours, n = 3 at 144 hours). * = P < .001 compared with PBS. Data are means ± standard deviations. (d) Uptake in lung after delivery of low (n = 3), intermediate (Int) (n = 7), and high (n = 3) dose of LPS. Intermediate dose activity is same data as shown in c for comparison. Data are means ± standard deviations. * = P < .001 and ** = P = .006 compared with low-dose LPS (one-way analysis of variance with Tukey test).
Figure 3c:

PET 64Cu-DOTA-ECL1i imaging in mouse lung injury model. Mice received intratracheal PBS or LPS followed by intravenous injection of 64Cu-DOTA-ECL1i. (a) Representative PET images of maximum intensity projection reconstructed PET/CT scans (center) and from indicated planes acquired 24 hours after treatment and after injection with 64Cu-DOTA-ECL1i . (b) Time-activity curves of 64Cu-DOTA-ECL1i in heart and lungs from 0 to 60 minutes after injection (n = 3 per group from subgroup described in c). * = P < .001 compared with lung PBS and LPS groups (unpaired two-tailed Student t test). Data are means ± standard deviations. (c) Bar chart shows 64Cu-DOTA-ECL1i lung uptake after treatment (PBS: n = 5; LPS: n = 7 at 24 hours, n = 3 at 48 hours, n = 3 at 144 hours). * = P < .001 compared with PBS. Data are means ± standard deviations. (d) Uptake in lung after delivery of low (n = 3), intermediate (Int) (n = 7), and high (n = 3) dose of LPS. Intermediate dose activity is same data as shown in c for comparison. Data are means ± standard deviations. * = P < .001 and ** = P = .006 compared with low-dose LPS (one-way analysis of variance with Tukey test).
Figure 3d:
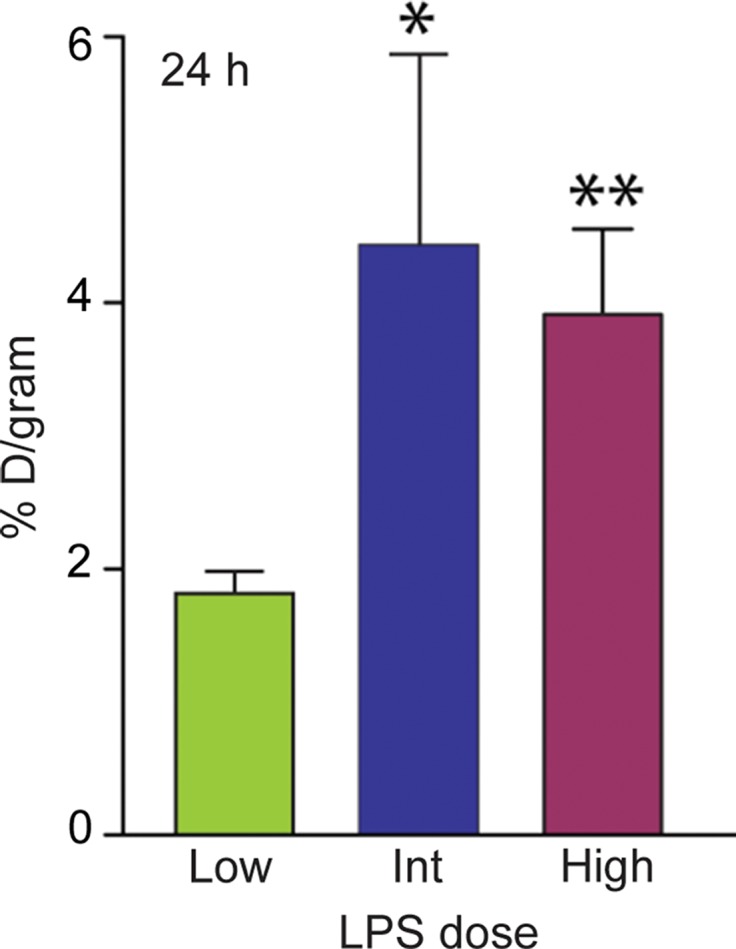
PET 64Cu-DOTA-ECL1i imaging in mouse lung injury model. Mice received intratracheal PBS or LPS followed by intravenous injection of 64Cu-DOTA-ECL1i. (a) Representative PET images of maximum intensity projection reconstructed PET/CT scans (center) and from indicated planes acquired 24 hours after treatment and after injection with 64Cu-DOTA-ECL1i . (b) Time-activity curves of 64Cu-DOTA-ECL1i in heart and lungs from 0 to 60 minutes after injection (n = 3 per group from subgroup described in c). * = P < .001 compared with lung PBS and LPS groups (unpaired two-tailed Student t test). Data are means ± standard deviations. (c) Bar chart shows 64Cu-DOTA-ECL1i lung uptake after treatment (PBS: n = 5; LPS: n = 7 at 24 hours, n = 3 at 48 hours, n = 3 at 144 hours). * = P < .001 compared with PBS. Data are means ± standard deviations. (d) Uptake in lung after delivery of low (n = 3), intermediate (Int) (n = 7), and high (n = 3) dose of LPS. Intermediate dose activity is same data as shown in c for comparison. Data are means ± standard deviations. * = P < .001 and ** = P = .006 compared with low-dose LPS (one-way analysis of variance with Tukey test).
Figure 3b:

PET 64Cu-DOTA-ECL1i imaging in mouse lung injury model. Mice received intratracheal PBS or LPS followed by intravenous injection of 64Cu-DOTA-ECL1i. (a) Representative PET images of maximum intensity projection reconstructed PET/CT scans (center) and from indicated planes acquired 24 hours after treatment and after injection with 64Cu-DOTA-ECL1i . (b) Time-activity curves of 64Cu-DOTA-ECL1i in heart and lungs from 0 to 60 minutes after injection (n = 3 per group from subgroup described in c). * = P < .001 compared with lung PBS and LPS groups (unpaired two-tailed Student t test). Data are means ± standard deviations. (c) Bar chart shows 64Cu-DOTA-ECL1i lung uptake after treatment (PBS: n = 5; LPS: n = 7 at 24 hours, n = 3 at 48 hours, n = 3 at 144 hours). * = P < .001 compared with PBS. Data are means ± standard deviations. (d) Uptake in lung after delivery of low (n = 3), intermediate (Int) (n = 7), and high (n = 3) dose of LPS. Intermediate dose activity is same data as shown in c for comparison. Data are means ± standard deviations. * = P < .001 and ** = P = .006 compared with low-dose LPS (one-way analysis of variance with Tukey test).
The specificity of the ECL1i radiotracer was examined in additional control conditions (Fig 4). The level of 64Cu-DOTA-ECL1i lung signal in treatment-naive mice (mean: 0.39% ID per gram of tissue, n = 3) was negligible and not significantly different from that in mice treated with PBS. Moreover, the signal in these conditions was similar to that in LPS-treated mice that were coinjected with excess nonradioactive ECL1i plus 64Cu-DOTA-ECL1i (blocked) (mean: 0.63% ID per gram of tissue ± 0.15, n = 4; P < .001). There was also nearly complete loss of lung signal in LPS-treated CCR2-deficient mice (mean: 0.39% ID per gram of tissue ± 0.04, n = 3; P < .001). We considered that blood flow may contribute to differences in the PET signal between LPS-treated and control mice; however, there were not significant differences in lung weight wet-to-dry ratios in PBS- compared with LPS-treated mice (5.20 g ± 0.20 vs 5.34 g ± 0.34, respectively, three per group; P = .650).
Figure 4a:

Specificity of 64Cu-DOTA-ECL1i imaging in LPS lung injury model. Mice were not treated or received intratracheal LPS and, 24 hours later, 64Cu-DOTA-ECL1i for PET/CT. Naive = treatment-naive wild-type mice, WT = LPS-treated wild-type mice, WT + Block = LPS-treated wild-type mouse coinjected with nonradiolabeled ECL1i, CCR2−/− = LPS-treated CCR2-deficient mice. (a) Representative transverse PET images of mouse lung from treatment-naive wild-type mice (n = 4), LPS-treated wild-type mice (n = 7 [from Fig 2b]), LPS-treated wild-type mouse coinjected with nonradiolabeled ECL1i (n = 4), and LPS-treated CCR2-deficient mice (n = 4). (b) Bar chart shows quantification of lung activity after treatment described in a. Data are means ± standard deviations. * = P < .001 compared with LPS-treated wild-type mice injected with 64Cu-DOTA-ECL1i (one-way analysis of variance with Tukey test).
Figure 4b:
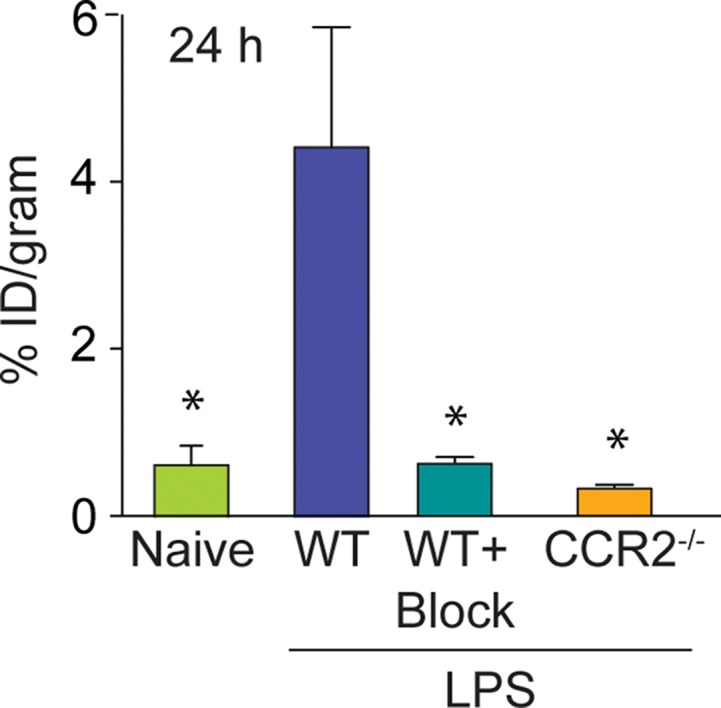
Specificity of 64Cu-DOTA-ECL1i imaging in LPS lung injury model. Mice were not treated or received intratracheal LPS and, 24 hours later, 64Cu-DOTA-ECL1i for PET/CT. Naive = treatment-naive wild-type mice, WT = LPS-treated wild-type mice, WT + Block = LPS-treated wild-type mouse coinjected with nonradiolabeled ECL1i, CCR2−/− = LPS-treated CCR2-deficient mice. (a) Representative transverse PET images of mouse lung from treatment-naive wild-type mice (n = 4), LPS-treated wild-type mice (n = 7 [from Fig 2b]), LPS-treated wild-type mouse coinjected with nonradiolabeled ECL1i (n = 4), and LPS-treated CCR2-deficient mice (n = 4). (b) Bar chart shows quantification of lung activity after treatment described in a. Data are means ± standard deviations. * = P < .001 compared with LPS-treated wild-type mice injected with 64Cu-DOTA-ECL1i (one-way analysis of variance with Tukey test).
ECL1i Tracer Binds Monocytes in Vivo
To determine the cell types that bound ECL1i in wild-type mice, the peptide was tagged with the fluorescent dye Dylight 550. At 24 hours after intratracheal delivery of PBS or LPS, ECL1i-Dylight 550 was injected intravenously and, 1 hour later, lungs were analyzed with flow cytometry. LPS induced significant binding of ECL1i-Dylight 550 to monocytes (Ly6Glo, Ly6Chi) (Fig 5). ECL1i-Dylight 550 also bound lung to macrophages (CD11bhi, CD11clow) and a very small group of “bright” dendritic cells (CD11bhi, CD11chi), consistent with known CCR2-positive populations (1,2).
Figure 5:

Detection of ECL1i Dylight 550 in lung immune cells after LPS-induced lung injury. Mice treated with intratracheal PBS or LPS were given intravenous fluorescent-labeled ECL1i (four mice in PBS group and five in LPS group) at 24 hours. Four mice in PBS group and four in LPS group were not injected with ECL1i and served as controls for flow cytometry. One hour later, lung cell digests were analyzed with flow cytometry for ECL1i Dylight 550–positive cells by using cell-type specific antibodies. PMN = neutrophils, Mono = monocytes, Mac = macrophages, DC = dendritic cells. Data are means ± standard errors of four independent experiments. * = P = .001 compared with PBS treatment (unpaired Student t test).
CCR2 Detection in Lung Tissue from Subjects with COPD
The percentage of CCR2-positive cells was significantly increased in lung tissues from subjects with COPD (median: 12.82% cells per sample; range: 1.16%–24.11% cells per sample) compared with lung donors (median: 1.66% cells per sample; range: 0.19%–23.67% cells per sample) (P = .002) (Fig 6, Table E1 [online]). However, there were a wide range of CCR2-positive cells, with the levels being similar in tissues from some subjects with and some subjects without COPD.
Figure 6a:

64Cu-DOTA-ECL1i binding in human lung samples from subjects with severe COPD. (a) Photomicrographs of CCR2 immunostaining in lung tissue from non-COPD lung donor and subject with severe COPD (original magnification, ×100) demonstrate a high number of CCR2-expressing cells. Bar = 100 μm. CCR2 is red and DAPI is blue. (b) Quantification of CCR2 staining area relative to DAPI in lung sections from donors (n = 11) and subjects with COPD (n = 16). Data were obtained from photomicrographs acquired at magnification of ×200. Bars indicate median values, which were significantly different between groups. * = P = .002 (Mann-Whitney U test for nonparametric data). (c) Representative images from autoradiography of 64Cu-DOTA-ECL1i binding to lung tissues sections on glass slides. (d) Quantification of autoradiography of lung sections from subjects with COPD. Counts in each blocked sample were compared with those from nonblocked tissues that were set as 1.0 (n = 6). Bars indicate median values, which were significantly different between groups. * = P = .002 (Mann-Whitney U test for nonparametric data).
Figure 6b:

64Cu-DOTA-ECL1i binding in human lung samples from subjects with severe COPD. (a) Photomicrographs of CCR2 immunostaining in lung tissue from non-COPD lung donor and subject with severe COPD (original magnification, ×100) demonstrate a high number of CCR2-expressing cells. Bar = 100 μm. CCR2 is red and DAPI is blue. (b) Quantification of CCR2 staining area relative to DAPI in lung sections from donors (n = 11) and subjects with COPD (n = 16). Data were obtained from photomicrographs acquired at magnification of ×200. Bars indicate median values, which were significantly different between groups. * = P = .002 (Mann-Whitney U test for nonparametric data). (c) Representative images from autoradiography of 64Cu-DOTA-ECL1i binding to lung tissues sections on glass slides. (d) Quantification of autoradiography of lung sections from subjects with COPD. Counts in each blocked sample were compared with those from nonblocked tissues that were set as 1.0 (n = 6). Bars indicate median values, which were significantly different between groups. * = P = .002 (Mann-Whitney U test for nonparametric data).
Accordingly, we next tested the binding of 64Cu-DOTA-ECL1i in the lung tissue sections from COPD and donor subjects by using autoradiography of slides. To enhance the differences in CCR2 detection, we studied samples with high versus low numbers of CCR2-positive cells detected with immunostaining. This comparison showed that 64Cu-DOTA-ECL1i binding was increased in representative samples with high numbers of CCR2-positive cells compared to those with low numbers of CCR2-positive cells (Fig 6c). Qualitative assessment of probe binding was determined by blocking the 64Cu-DOTA-ECL1i signal with nonlabeled ECL1i in tissue samples from subjects with COPD with high levels of CCR2-positive cells. In all samples tested (n = 6), competition with nonlabeled ECL1i diminished the level of 64Cu-DOTA-ECL1i binding on the basis of autoradiographic signal (Fig 6d).
Figure 6c:

64Cu-DOTA-ECL1i binding in human lung samples from subjects with severe COPD. (a) Photomicrographs of CCR2 immunostaining in lung tissue from non-COPD lung donor and subject with severe COPD (original magnification, ×100) demonstrate a high number of CCR2-expressing cells. Bar = 100 μm. CCR2 is red and DAPI is blue. (b) Quantification of CCR2 staining area relative to DAPI in lung sections from donors (n = 11) and subjects with COPD (n = 16). Data were obtained from photomicrographs acquired at magnification of ×200. Bars indicate median values, which were significantly different between groups. * = P = .002 (Mann-Whitney U test for nonparametric data). (c) Representative images from autoradiography of 64Cu-DOTA-ECL1i binding to lung tissues sections on glass slides. (d) Quantification of autoradiography of lung sections from subjects with COPD. Counts in each blocked sample were compared with those from nonblocked tissues that were set as 1.0 (n = 6). Bars indicate median values, which were significantly different between groups. * = P = .002 (Mann-Whitney U test for nonparametric data).
Figure 6d:

64Cu-DOTA-ECL1i binding in human lung samples from subjects with severe COPD. (a) Photomicrographs of CCR2 immunostaining in lung tissue from non-COPD lung donor and subject with severe COPD (original magnification, ×100) demonstrate a high number of CCR2-expressing cells. Bar = 100 μm. CCR2 is red and DAPI is blue. (b) Quantification of CCR2 staining area relative to DAPI in lung sections from donors (n = 11) and subjects with COPD (n = 16). Data were obtained from photomicrographs acquired at magnification of ×200. Bars indicate median values, which were significantly different between groups. * = P = .002 (Mann-Whitney U test for nonparametric data). (c) Representative images from autoradiography of 64Cu-DOTA-ECL1i binding to lung tissues sections on glass slides. (d) Quantification of autoradiography of lung sections from subjects with COPD. Counts in each blocked sample were compared with those from nonblocked tissues that were set as 1.0 (n = 6). Bars indicate median values, which were significantly different between groups. * = P = .002 (Mann-Whitney U test for nonparametric data).
Discussion
We selected a CCR2 binding peptide as a PET imaging target on the basis of several observations. First, the CCR2–chemokine ligand type 2 axis recruits inflammatory monocytes and other types of immune cells into the lung (1). Second, CCR2 is elevated in lung cells in acute respiratory distress syndrome, COPD, experimental asthma, and pulmonary fibrosis as well as in common nonpulmonary diseases, including atherosclerosis and malignancy (6,8–10,17,25–28). Third, there is substantial evidence that genetic deletion or pharmacologic inhibition of CCR2 ameliorates disease in animal models, leading to industry efforts to develop and test CCR2 antagonists for respiratory disorders and other disease (2,18,19).
Current tools are limited for the noninvasive assessment of inflammation in lung disease. To test 64Cu-DOTA-ECL1i as an agent for detecting inflammation, we chose LPS injury, a well-established model that has been characterized relative to CCR2-mediated inflammation (13,24,29). Previous reports demonstrated that LPS directs accumulation of CCR2-expressing cells in the lung in wild-type but not CCR2−/− mice or after anti-CCR2 antibody blockade (13,24,30). Consistent with these findings, 64Cu-DOTA-ECL1i activity in the lung was increased only during the acute phase of injury and was not present in CCR2−/− mice. The specificity of 64Cu-DOTA-ECL1i activity was further shown by the ability to block detection of LPS-induced activity by using nonradioactive ECL1i.
Because CCR2 deletion in mice alters immune responses, including decreased neutrophil influx and other inflammatory cells in the lung (13–15), we sought alternative approaches to study the in vivo performance of ECL1i. To determine the cell types binding ECL1i, we created a fluorescently tagged ECL1i imaging agent for in vivo labeling of immune cells in wild-type mice. Injection of ECL1i–Dylight 550 and analysis of whole lung cell preparations with flow cytometry revealed ECL1i signal in lung monocytes and macrophages as well as in small numbers of dendritic cells, which are also known to express CCR2 (12,26,29). Thus, future in vivo studies with ECL1i–Dylight 550 and CCR2 reporter mice may provide additional information as to cell targets. Ultimately, the characterization of the complete identity of cell types that bind 64Cu-DOTA-ECL1i in vivo will be difficult owing to inherent differences in the sensitivity of detection methods for fluorescent and radionuclide labels.
64Cu-DOTA-ECL1i joins a small number of agents developed for PET of nonmalignant lung disease. The strength of the ECL1i radiotracer is its ability to enable imaging of CCR2-related inflammation, as we have shown in the lung by using the mouse endotoxin and a mouse ischemia-reperfusion model of lung transplantation (21). 64Cu-DOTA-ECL1i activity in the bone marrow and extrapulmonary organs suggests that it may also be possible to follow CCR2 cell trafficking. Moving forward, it will be important to compare the utility of 64Cu-DOTA-ECL1i with radiotracers that are less specific for a defined immune cell population, such as fluorine 18 fluorodeoxyglucose PET (5,31), and using single photon emission CT or planar imaging with technetium 99m hexamethylpropylene amine oxime (32) as well as targeted approaches including the translocator protein (33) and folate receptor β (34) to identify CCR2-dependent inflammation.
We identified that the percentage of CCR2-expressing cells detected with immunostaining was elevated in lung tissue from subjects with severe COPD compared with that from the lung donor group. This finding was consistent with previous reports of elevated levels of CCR2-positive cells in other types of samples obtained from individuals with COPD (6,7). There was some overlap in levels between the COPD and donor groups, possibly owing to underlying phenotypic differences in COPD and inflammation that may occur in the donor lungs before harvest for transplantation. Thus, further study will be needed to define precise differences for CCR2-positive cells in COPD; however, the present observations identify a set of clinical samples and a strategy that can be used to validate whether the level of CCR2-positive cells in tissue correlates with the level detected by using our probe for noninvasive imaging.
This study has several limitations. We assessed radiotracer activity in a limited number of mice and 64Cu-DOTA-ECL1i has a very rapid blood clearance (<1% ID per gram of tissue at 1 hour after injection), so that the sensitivity of detection may be limited. Chemical modification of the radiotracer could improve the pharmacokinetics to increase the blood retention time. Although we have shown that ECL1i can bind human tissues from subjects with COPD, sampling issues inherent in this heterogeneous disease prevent generalization to the whole lung. The assessment of mice is also limited because relatively small numbers have been studied with PET/CT after 64Cu-DOTA-ECL1i injection and models of chronic lung disease such as COPD are lacking; cigarette smoke in mice does not induce inflammation similar to that in humans (35). Interpretation of results is also limited by a lack of current approval for clinical testing. Moving to human trials may be the only way to test our proposal that a 64Cu-DOTA-ECL1i signal is increased in the lungs of subjects with COPD. Although the 64Cu decay half-life of approximately 13 hours may be less desirable for imaging, the high specific activity and the longer physical half-life will permit production and distribution of intact radiotracer for nationwide trials. For expanded clinical use of ECL1i, radiochemistry could be developed for fluoride-18 labeling. Moreover, future human studies of PET with use of radiolabeled ECL1i in combination with CT may provide synergistic information regarding the localization of inflammation and the relationship to specific lung structures and pathologic changes.
Practical applications: A CCR2 binding peptide adapted as a PET probe can help detect lung inflammation in a mouse model and human tissues and may serve as a tool for the management of human lung disease. Our study of lungs of subjects with clinically similar, very severe COPD showed that expression of CCR2 varied markedly among subjects, supporting the concept that the molecular mechanisms underlying chronic inflammatory lung abnormalities are not necessarily alike but could be differentiated by means of noninvasive detection of certain biomarkers. There are few available treatments for COPD, and CCR2 detection could be an important step for developing therapies, personalizing treatment, and monitoring treatment response.
Advances in Knowledge
■ Chemokine receptor type 2 (CCR2) binding peptide extrcellular loop 1 inverso (ECL1i), adapted as a PET radiotracer, can be used to detect 3.5-fold greater activity in the lungs of mice injured after intratracheal delivery of lipopolysaccharide compared with a control group receiving saline (P < .001).
■ Levels of CCR2-positive cells varied in human lung tissue among subjects with chronic obstructive pulmonary disease (COPD) (range, 1.62%–24.11% cells per tissue sample).
■ The radiotracer copper 64–tetraazacyclododecane tetraacetic acid–ECL1i can be used to detect inflammation in human lung tissue from subjects with COPD by using autoradiography (specificity determined with nonradioactive blockade, 54.5%; P = .002), which suggests that there may be a possible clinical application if approved for in vivo human use.
APPENDIX
SUPPLEMENTAL FIGURES
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Acknowledgments
The authors appreciate assistance provided by the Small Animal Imaging Core and Cyclotron Facilities.
Received June 19, 2016; revision requested June 1; revision received August 12; accepted September 27; final version accepted October 25.
Supported by National Heart, Lung, and Blood Institute (HHN268201000046C, NHLBI-HV-10-08, R01 HL131908, U19 AI070489, and R01 HL121791). S.L.B. is the Hubert S. and Dorothy R. Moog Professor of Pulmonary Medicine and is supported by the Foundation for Barnes-Jewish Hospital.
Disclosures of Conflicts of Interest: Y.L. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: disclosed no relevant relationships. Other relationships: has a patent pending. S.P.G. disclosed no relevant relationships. D.H.S. disclosed no relevant relationships. H.P.L. disclosed no relevant relationships. Y.Z. disclosed no relevant relationships. T.S.B. disclosed no relevant relationships. Z.B. disclosed no relevant relationships. J.P. disclosed no relevant relationships. D.E.B. disclosed no relevant relationships. J.J.A. disclosed no relevant relationships. D.K. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: disclosed no relevant relationships. Other relationships: has a patent pending. M.J.H. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: is a consultant for Boehringer-Ingelheim; is a DSMB member for Astra-Zeneca Quintiles. Other relationships: has a patent. R.J.G. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: is a consultant for Biomedical Systems and GE; is on the advisory panel for Sanofi; received a grant from Astellas, Bayer, and GE. Other relationships: has a patent pending. C.C. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: disclosed no relevant relationships. Other relationships: has a patent pending. S.L.B. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: disclosed no relevant relationships. Other relationships: has a patent pending.
Abbreviations:
- CCR2
- chemokine receptor type 2
- COPD
- chronic obstructive pulmonary disease
- DAPI
- 49,6-diamidino-2-phenylindole
- DOTA
- tetraazacyclododecane tetraacetic acid
- ECL1i
- extracellular loop 1 inverso
- ID
- injected dose
- LPS
- lipopolysaccharide
- PBS
- phosphate-buffered saline
References
- 1.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 2006;354(6):610–621. [DOI] [PubMed] [Google Scholar]
- 2.Tomankova T, Kriegova E, Liu M. Chemokine receptors and their therapeutic opportunities in diseased lung: far beyond leukocyte trafficking. Am J Physiol Lung Cell Mol Physiol 2015;308(7):L603–L618. [DOI] [PubMed] [Google Scholar]
- 3.Byers DE, Holtzman MJ. Alternatively activated macrophages and airway disease. Chest 2011;140(3):768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deshane JS, Redden DT, Zeng M, et al. Subsets of airway myeloid-derived regulatory cells distinguish mild asthma from chronic obstructive pulmonary disease. J Allergy Clin Immunol 2015;135(2):413–424.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen DL, Schuster DP. Imaging pulmonary inflammation with positron emission tomography: a biomarker for drug development. Mol Pharm 2006;3(5):488–495. [DOI] [PubMed] [Google Scholar]
- 6.de Boer WI, Sont JK, van Schadewijk A, Stolk J, van Krieken JH, Hiemstra PS. Monocyte chemoattractant protein 1, interleukin 8, and chronic airways inflammation in COPD. J Pathol 2000;190(5):619–626. [DOI] [PubMed] [Google Scholar]
- 7.Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax 2002;57(7):590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartl D, Griese M, Nicolai T, et al. A role for MCP-1/CCR2 in interstitial lung disease in children. Respir Res 2005;6:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kallinich T, Schmidt S, Hamelmann E, et al. Chemokine-receptor expression on T cells in lung compartments of challenged asthmatic patients. Clin Exp Allergy 2005;35(1):26–33. [DOI] [PubMed] [Google Scholar]
- 10.An JL, Ishida Y, Kimura A, Tsokos M, Kondo T. Immunohistochemical detection of CCR2 and CX3CR1 in sepsis-induced lung injury. Forensic Sci Int 2009;192(1-3):e21–e25. [DOI] [PubMed] [Google Scholar]
- 11.Tsou CL, Peters W, Si Y, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 2007;117(4):902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol 2008;180(4):2562–2572. [DOI] [PubMed] [Google Scholar]
- 13.Maus U, von Grote K, Kuziel WA, et al. The role of CC chemokine receptor 2 in alveolar monocyte and neutrophil immigration in intact mice. Am J Respir Crit Care Med 2002;166(3):268–273. [DOI] [PubMed] [Google Scholar]
- 14.Maus UA, Waelsch K, Kuziel WA, et al. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol 2003;170(6):3273–3278. [DOI] [PubMed] [Google Scholar]
- 15.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 2006;7(3):311–317. [DOI] [PubMed] [Google Scholar]
- 16.Hildebrandt GC, Duffner UA, Olkiewicz KM, et al. A critical role for CCR2/MCP-1 interactions in the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood 2004;103(6):2417–2426. [DOI] [PubMed] [Google Scholar]
- 17.Bhatia M, Zemans RL, Jeyaseelan S. Role of chemokines in the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol 2012;46(5):566–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Struthers M, Pasternak A. CCR2 antagonists. Curr Top Med Chem 2010;10(13):1278–1298. [DOI] [PubMed] [Google Scholar]
- 19.Carter PH. Progress in the discovery of CC chemokine receptor 2 antagonists, 2009–2012. Expert Opin Ther Pat 2013;23(5):549–568. [DOI] [PubMed] [Google Scholar]
- 20.Auvynet C, Baudesson de Chanville C, Hermand P, et al. ECL1i, d(LGTFLKC), a novel, small peptide that specifically inhibits CCL2-dependent migration. FASEB J 2016;30(6):2370–2381. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Li W, Luehmann HP, et al. Noninvasive imaging of CCR2(+) cells in ischemia-reperfusion injury after lung transplantation. Am J Transplant 2016;16(10):3016–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Abendschein D, Woodard GE, et al. Molecular imaging of atherosclerotic plaque with (64)Cu-labeled natriuretic peptide and PET. J Nucl Med 2010;51(1):85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Ibricevic A, Cohen JA, et al. Impact of hydrogel nanoparticle size and functionalization on in vivo behavior for lung imaging and therapeutics. Mol Pharm 2009;6(6):1891–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maus UA, Wellmann S, Hampl C, et al. CCR2-positive monocytes recruited to inflamed lungs downregulate local CCL2 chemokine levels. Am J Physiol Lung Cell Mol Physiol 2005;288(2):L350–L358. [DOI] [PubMed] [Google Scholar]
- 25.Lee YG, Jeong JJ, Nyenhuis S, et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol 2015;52(6):772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robays LJ, Maes T, Lebecque S, et al. Chemokine receptor CCR2 but not CCR5 or CCR6 mediates the increase in pulmonary dendritic cells during allergic airway inflammation. J Immunol 2007;178(8):5305–5311. [DOI] [PubMed] [Google Scholar]
- 27.Okuma T, Terasaki Y, Kaikita K, et al. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J Pathol 2004;}204(5):594–604. [DOI] [PubMed] [Google Scholar]
- 28.Charo IF, Peters W. Chemokine receptor 2 (CCR2) in atherosclerosis, infectious diseases, and regulation of T-cell polarization. Microcirculation 2003;10(3-4):259–264. [DOI] [PubMed] [Google Scholar]
- 29.Martinu T, Gowdy KM, Nugent JL, et al. Role of C-C motif ligand 2 and C-C motif receptor 2 in murine pulmonary graft-versus-host disease after lipopolysaccharide inhalations. Am J Respir Cell Mol Biol 2014;51(6):810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang D, Tong L, Wang D, Wang Y, Wang X, Bai C. Roles of CC chemokine receptors (CCRs) on lipopolysaccharide-induced acute lung injury. Respir Physiol Neurobiol 2010;170(3):253–259. [DOI] [PubMed] [Google Scholar]
- 31.de Prost N, Tucci MR, Melo MF. Assessment of lung inflammation with 18F-FDG PET during acute lung injury. AJR Am J Roentgenol 2010;195(2):292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Audi SH, Clough AV, Haworth ST, et al. 99MTc-hexamethylpropyleneamine oxime imaging for early detection of acute lung injury in rats exposed to hyperoxia or lipopolysaccharide treatment. Shock 2016;46(4):420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatori A, Yui J, Yamasaki T, et al. PET imaging of lung inflammation with [18F]FEDAC, a radioligand for translocator protein (18 kDa). PLoS One 2012;7(9):e45065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han W, Zaynagetdinov R, Yull FE, et al. Molecular imaging of folate receptor β-positive macrophages during acute lung inflammation. Am J Respir Cell Mol Biol 2015;53(1):50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Churg A, Sin DD, Wright JL. Everything prevents emphysema: are animal models of cigarette smoke-induced chronic obstructive pulmonary disease any use? Am J Respir Cell Mol Biol 2011;45(6):1111–1115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.