

Abstract
Development of reliable methods and site-specific detection of free radicals is an active area of research. Here, we describe the synthesis and radical-trapping properties of new derivatives of DEPMPO and DIPMPO, bearing mitochondria-targeting triphenylphosphonium cationic moiety or guanidinium cationic group. All the spin traps prepared have been observed to efficiently trap superoxide radical anion in cell-free system. The superoxide spin adducts exhibited similar spectral properties indicating no significant differences in the geometry of the cyclic nitroxide moieties of the spin adducts. The superoxide adduct stability was measured and observed to be highest (t1/2 = 73 min) for DIPPMPO nitrone linked to triphenylphosphonium moiety via a short carbon chain (Mito-DIPPMPO). The experimental results and DFT quantum chemical calculations indicate that the cationic property of the triphenylphosphonium group may be responsible for increased superoxide trapping efficiency and adduct stability of Mito-DIPPMPO, as compared to DIPPMPO spin trap. The studies of uptake of the synthesized traps into isolated mitochondria indicated the importance of both cationic and lipophilic properties, with the DEPMPO nitrone linked to triphenylphosphonium moiety via a long carbon chain (Mito10-DEPMPO) exhibiting the highest mitochondrial uptake. We conclude that of the synthesized traps, Mito-DIPPMPO and Mito10-DEPMPO are the best candidates as the potential mitochondria-specific spin traps for use in biologically-relevant systems.
Keywords: Superoxide free radical, Electron paramagnetic resonance (EPR), Spin-trapping, Mitochondria targeting, DEPMPO, ROS
Graphical Abstract
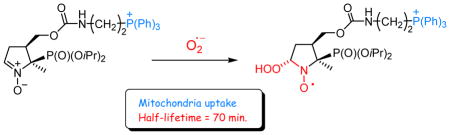
Introduction
There is increased evidence for the involvement of superoxide radical anion (O2•−) in cell damage and disease via direct effects or through formation of secondary reactive oxygen and nitrogen species (ROS, RNS).1–3 The role of these species in the pathogenesis and progression of diseases such as atherosclerosis,4 diabetes,5 cancer,6 neurodegenerative diseases,7,8 is however supported by indirect evidence due to the difficulties of direct detection of ROS and RNS in vivo. To gain information on the mechanisms controlling ROS production at the molecular level, the development of new reliable and efficient techniques for their detection9,10 is needed. Mechanistic studies on the role of O2•− in oxidative stress processes is made particularly difficult by its low steady state concentration and the lack of highly specific probes that allow its unequivocal identification and quantification. Electron paramagnetic resonance (EPR) in combination with the spin trapping technique is the method of choice to detect and characterize radicals such as O2•−.11–15 In the spin trapping technique, a spin trap, typically a nitrone or nitroso probe, is introduced into the system under investigation to scavenge radicals which are too short-lived to be directly detected by EPR. The EPR spectra of the resulting persistent nitroxide spin adducts can usually be recorded and the spectral analysis can bring valuable information on the chemical identity and dynamics of the radical species produced in the investigated system. However, different drawbacks still limit the use and reliability of the spin trapping for in cell and in vivo studies, and its application to the characterization of O2•− and other oxygen-centered radicals still needs to be improved. Important progress has been made over the last years, notably with the development of new spin traps that form superoxide spin adducts exhibiting half-life times significantly longer (17 to 45 min)16–20 than that observed for the most widely used nitrone spin trap, DMPO (~1 min).21–23 Recent research in the oxidative stress field has focused on the development of targeted probes for detecting reactive species in cells.24,25 Oxygen can be partially reduced by mitochondrial electron transfer protein complexes I and III to O2•− leading to cell dysfunction.26,27 Therefore, targeting spin traps to mitochondria might help to characterize the contribution of mitochondrial O2•−. Lipophilic cations such as triphenylphosphonium (TPP+) and N-alkylpyridinium ions have been shown to be effective mitochondria targeting agents. Their uptake across the mitochondrial inner membrane is enhanced by the mitochondrial membrane potential (ΔΨ) according to Nernst equation, with a predicted ~10-fold accumulation of the cation within mitochondria for every ~60 mV increase in ΔΨ.26,28,29 In the recent years, TPP+-conjugated probes have been used in numerous studies focused on mitochondria-associated responses. For instance, Murphy et al.30 have determined that MitoQ, a TPP+-conjugated ubiquinone antioxidant, accumulates up to several hundred-fold in mitochondria matrix and selectively protected mitochondria, both in vitro and in vivo from the oxidative damage.31 Mitochondria-targeted cyclic- and linear nitrones have been studied but usually these reagents have limited spin trapping properties.29,32,33 Recently, we reported the synthesis and the spin trapping properties of the TPP+-conjugated DEPMPO spin trap (Mito-DEPMPO).16,34 Using this new reagent, we demonstrated that the detection of superoxide radical anion generated from intact isolated mitochondria is feasible. This result can be explained by the accumulation of Mito-DEPMPO in mitochondria, but we have also shown that compared with DEPMPO, the rate of trapping of superoxide with Mito-DEPMPO is about two times higher, and that the half-life of the O2•− adduct is about 2.5 times longer. However, the reasons for these differences between DEPMPO and Mito-DEPMPO are not clear and we speculated that electrostatic interactions between the TPP+ cation and the O2•− or / and the presence of stabilizing H-bonds in the O2•− adduct might contribute to the observed improvement in superoxide-trapping properties of Mito-DEPMPO.
To better understand the spin trapping properties of mitochondria-targeted nitrones and to optimize the influence of the TPP+ cation on the spin trapping of O2•−, we have synthesized a series of Mito-DEPMPO analogues 6–12 (Scheme 1). In the series Mito-DIPPMPO 6, Mito5-DIPPMPO 7 and Mito10-DEPMPO 9, the length of the linker (C4-CH2OC(O)NH(CH2)n-TPP+) between the C4 and the TPP+ moiety increases (n = 2, 5 and 10 respectively). Two TPP+ moieties are present in Mito-bis-DIPPMPO 8 (n = 2) and a neutral -N(H)CPh3 group mimicking the lipophilicity of TPP+ was introduced in TritA-DEPMPO 10 (n = 2), finally the TPP+ cation was replaced by a guanidium group in Gua-DIPPMPO 11 and Agm-DIPPMPO 12 (n = 2 and 4 respectively).
Scheme 1.

Chemical structure of spin traps 6–12.
NHS-DIPPMPO and NHS-DEPMPO were used as precursors for the synthesis of the series of derivatives, illustrating the versatility of the post-functionalization step (Scheme 2). For synthetic convenience, NHS-DIPPMPO was mostly used.
Scheme 2.

Reagents and conditions: i, PBu3, C6H12/CH2Cl2, rt; ii, DIBAL-H, CH2Cl2, −78°C; iii, Zn/NH4Cl, H2O/THF, rt; iv, DSC, Et3N, CH3CN, rt.
Hereafter we describe the synthesis of compounds 6–12, their spin trapping properties and the binding/uptake properties of compounds 6–10 to energized mitochondria.
Results and Discussion
Synthesis
NHS-DIPPMPO 5 was prepared (32% overall yield) in a four-step synthetic sequence (Scheme 2) following the procedure described by Hardy et al.16 (SI).
Nitrofuranone 2 was obtained in 74% yield by reacting nitrophosphonate 1 with 2(5H)-furanone in the presence of tributylphosphine as catalyst. Reduction of 2 by DIBAL-H at -78°C led to hemiacetal 3 in good yield (75%). Reductive cyclization of nitroaldehyde 3 in the presence of zinc and ammonium chloride afforded nitrones 4 and 4′ as a mixture of cis / trans diastereoisomers, which were separated on silicagel column chromatography (60% yield for 4). Compound 4 was recrystallized in Et2O / Pentane (8:2) and the geometry obtained by X-ray diffraction confirmed the cis position of the di-isopropylphosphonyl group relative to the hydroxymethylene moiety (Figure 1).
Figure 1.

Geometry of compound 4 from X-ray diffraction analysis (Pov-Ray view)
Reaction of diastereoisomer 4 with N,N′-disuccinimidylcarbonate (DSC) in the presence of triethylamine afforded NHS-DIPPMPO 5 in 95% yield.
Compounds 6–12 were obtained by reacting NHS-DIPPMPO or NHS-DEPMPO with the amino function of the appropriate side chain, with yields ranging from 50 to 91% (Scheme 3 and SI).
Scheme 3.

Synthesis of compounds 6–12.
Except TritA-DIPPMPO 10 and Mito10-DEPMPO 9, all compounds were soluble up to 50 mM concentration in water and in phosphate buffer solutions.
EPR / spin trapping
EPR / spin trapping of superoxide
The O2•− trapping properties (Scheme 4) of compounds 6–12 were evaluated using two different O2•− generating systems: hypoxanthine/xanthine oxidase (HX/XO) and KO2/18-crown-6-ether/DMSO (KO2/CE/DMSO) in phosphate buffer. Figure 2 shows the EPR spectra obtained after 10 min incubation of a mixture containing HX (0.4 mM), XO (0.04 U mL−1), DTPA (1 mM) and the spin trap (20 mM of Mito-DIPPMPO; 8 Mito-bis-DIPPMPO; 7 Mito5-DIPPMPO and 9 Mito10-DEPMPO) in oxygen-bubbled phosphate buffer (0.1 M, pH 7.3). The computer calculated EPR spectra, obtained using the parameters reported in Table 1, are shown as gray lines (Figure 2).
Scheme 4.

Spin trapping experiments
Figure 2. Spin trapping of superoxide radical using Mito-DIPPMPO (6), Mito-bis-DIPPMPO (7), Mito5-DIPPMPO (8), Mito10-DEPMPO (9).

(a) EPR spectrum obtained after 10 min incubation of a mixture containing hypoxanthine (HX) (0.4 mM), xanthine oxidase (XO) (0.04 U mL−1), DTPA (1 mM) and 6 (20 mM) in oxygen-bubbled phosphate buffer (0.1 M, pH 7.3); (b) As in (a) but in the presence of SOD (600 U mL−1). (c) As in (a) but containing 7 (20 mM). (d) As in (c) but in the presence of SOD (600 U mL−1). (e) As in (a) but containing 8 (20 mM). (f) As in (e) but in the presence of SOD (600 U mL−1). (g) As in (a) but containing 9 (20 mM) and 20% DMSO. (h) As in (g) but in the presence of SOD (600 U mL−1). Grey lines: calculated spectra (Table 1). Spectrometer settings: microwave power, 10 mW (a–h); modulation amplitude, 0.7 G (a–h); smooth point, 1 (a–h); gain 75 dB (a–h); sweep time, 41.94 s (a–h); conversion time, 40.96 ms (a–h).
Table 1.
Calculated EPR parameters for the superoxide spin adduct of Mito-DEPMPO and newly synthesized nitrones 6–12.
| Spin adduct | Diastereoisomer | Site | k (s−1) * | <aP>(G) | <aN>(G) | <aHβ>(G) |
|---|---|---|---|---|---|---|
| Mito-DEPMPO-OOH | trans (100%) | T1 (70%) T2 (30%) |
0.1×108 | 53.27 52.01 |
12.79 12.97 |
12.37 10.13 |
| Mito-DIPPMPO-OOH (6-OOH) | „ | T1 (88%) T2 (12%) |
4.2×108 | 53.41 48.05 |
12.86 12.76 |
12.13 7.38 |
| Mito-bis-DIPPMPO-OOH (8-OOH) | „ | T1 (54%) T2 (46%) |
2.5×108 | 53.07 45.01 |
12.83 12.69 |
12.17 6.06 |
| Mito5-DIPPMPO-OOH (7-OOH) | „ | T1 (60%) T2 (40%) |
5.1×108 | 54.03 51.86 |
12.80 12.82 |
12.78 10.57 |
| Mito10-DEPMPO-OOH (9-OOH) | „ | T1 (46%) T2 (54%) |
0.67×108 | 54.54 51.48 |
12.72 12.80 |
14.66 9.00 |
| Agm-DIPPMPO-OOH (11-OOH) | „ | T1 (79%) T2 (21%) |
0.8×107 | 53.7 51.8 |
12.8 13.3 |
11.9 9.5 |
| Gnd-DIPPMPO-OOH (12-OOH) | „ | T1 (74%) T2 (26%) |
0.35×107 | 53.2 51.5 |
12.8 13.1 |
12.4 9.4 |
Exchange rate constants in s−1. For DMPO-OOH spin adduct a theoretical study in an explicit water solution based on a combined QM/MM/MD protocol showed that the EPR spectrum can be explained in the light of two sites in chemical exchange. Moreover it was demonstrated that each site consists of an equilibrium between the two main 5-membered ring conformations of DMPO (3T4 and 4T3). For all spin adducts, the g values were very close and measured to be 2.006(4).
In the presence of superoxide dismutase (SOD), no EPR signal was detected (Figure 2, b, d, f, h). Furthermore, for compounds 6–9, identical EPR signals were observed using either the HX/XO or the (KO2/CE/DMSO) system, the signals being particularly long-lasting with the former. After 30 minutes, a weak additional signal (< 10%) is observable and was assigned to the HO-adduct. These results establish that the EPR signals shown in Figure 2 can be unambiguously assigned to the corresponding O2•− spin adducts.
All spectra appear as doublet (31P coupling) of significantly distorted quartets resulting from close couplings with the 14N and 1Hβ, they were calculated (Table 1) using the EPR/ROKI program.35 For all the series the best fit was obtained assuming both that the spin trapping reaction yields only the trans-diastereoisomer (-OOH and -P(O)(OR)2 groups in a trans geometry) and secondly the existence of a chemical exchange between two conformational sites T1 and T2 composed of rapidly exchanging conformers. In the case of the DEPMPO-OOH and DIPPMPO-OOH spin adducts, the same assumptions were made to account for the dramatic alternate line-width observed on the spectra of the trans-diastereoisomers.34–38
Using guanidine salts as inhibitors of XO enzyme,39 and Agm-DIPPMPO 11 and Gua-DIPPMPO 12 no signals were observed in incubations with HX/XO system. Incubations with KO2/CE/DMSO led to detection of very similar EPR signals to Mito-DIPPMPO-OOH (Figure 3), confirming the ability of the probes to trap O2•− and consistent with the inhibitory effects of spin traps 11 and 12 on the XO activity.
Figure 3. Spin trapping of superoxide radical using Gua-DIPPMPO 11 and Agm-DIPPMPO 12.

(i) EPR spectrum obtained after 2 min incubation of a mixture containing the KO2/CE/DMSO (5 mM/5 mM/5% respectively) system and 12 (25 mM) in a phosphate buffer (0.1 M, pH 7.3). (ii) As in (i) but with 11 (50 mM). Grey lines: calculated spectra (Table 1). Spectrometer settings: microwave power, 10 mW; modulation amplitude, 0.7 G; time constant, 1.28 ms; gain 105; sweep time, 21 s; conversion time, 20.48 ms.
Within the series of superoxide adduct of compounds 6–12, the values of the determined EPR parameters are similar and close to the values obtained previously for Mito-DEPMPO-OOH (Table 1), suggesting that the modification of the C4 side-chain does not change significantly the geometry of the preferred conformers of these spin adducts. However, as it is shown below the C4 side chain has a strong influence on their stability.
Decay kinetics of superoxide adduct
Once the concentration of the O2•− spin adduct has reached a plateau (after ~ 9 min), the further formation of O2•− spin adduct was stopped by the addition of a large amount of SOD and the decay kinetics of the O2•− adduct was monitored by following the changes in EPR signal intensity. The kinetic studies were performed at 23°C in 50 μL capillaries, setting the microwave power of the EPR spectrometer at 20 mW. The signal decay was monitored during 70 min. recording successive spectra every 42 s (Figure 4). All the recorded spectra were simulated using the Rocky program.35 The decay curves for the superoxide spin adducts of 6–9 are shown in SI and the apparent half-life times are listed in Table 2. It is worth noting that the half-lifetime values depend strongly on the experimental conditions and various parameters such as temperature, microwave power, EPR cell (capillaries, AquaX cell) must be carefully controlled in order to get reproducible results.
Figure 4.

Kinetics of decay of Mito-bis-DIPPMPO-OOH (8-OOH, a) and Mito-DIPPMPO-OOH (6-OOH, b)
Table 2.
Apparent half-lifetime values for superoxide adducts of DIPPMPO and 6–9, and the ratio values of t1/2 X-OOH / t1/2 DIPPMPO-OOH.
| Spin adducts | t1/2 (min.) | Ratio |
|---|---|---|
| DIPPMPO-OOH | 28 | 1 |
| Mito-DIPPMPO-OOH (6-OOH) | 73 | 2.61 |
| Mito5-DIPPMPO-OOH (7-OOH) | 36.6 | 1.31 |
| Mito-bis-DIPPMPO-OOH (8-OOH) | 29.3 | 1.05 |
| DIPPMPO-OOHi | 25.5 | 0.91 |
| Mito10-DEPMPO-OOHi (9-OOH) | 22 | 0.78 |
in 0.1 M phosphate buffer / DMSO mixture (80:20)
The values of apparent half-lifetime (t1/2) for the superoxide-adducts are reported in Table 2.
As one might expected given the results obtained with Mito-DEPMPO, 16 a ratio of 2.6 was found between the apparent half-life time of Mito-DIPPMPO-OOH (n = 2) and that observed with the parent nitrone DIPPMPO-OOH (this ratio amounts to 2.4 – 2.5, in the case of Mito-DEPMPO-OOH and DEPMPO-OOH). However, when the spacer arm linking the TPP+ group and the pyrroline ring was made longer (7, Mito5-DIPPMPO and 9, Mito10-DEPMPO) and when two triphenyphosphonium groups are appended (8, Mito-bis-DIPPMPO), the stability of the corresponding superoxide adducts is very close to that observed for the parent nitrone spin adducts. For Agm-DIPPMPO and Gua-DIPPMPO, attempts to perform a kinetic study using the KO2/CE/DMSO system were frustrated due to the poor reproducibility of the procedure, in addition to decreased persistency of the O2•− adducts. Measures of TritA-DEPMPO-OOH (10) half-lifetime was not possible because of low solubility of TritA-DEPMPO.
EPR characterization of hydroxyl and 1-hydroxyethyl radicals
Hydroxyl radical
When a Fenton system was used in phosphate buffer to generate HO• in the presence of compounds 6–9, complex signals of low intensity corresponding to the superimposition of the spectra of different spin adducts were observed (data not shown). These signals are diminished in intensity in the presence of catalase, indicating that these signals depend on hydrogen peroxide breakdown. A twelve line EPR spectra corresponding to the HO• adducts of 6, 8 and 9 (Figure 5) were obtained by reduction of the corresponding superoxide adducts with glutathione peroxidase / glutathione (GPx/GSH), the hyperfine coupling constants are listed in Table 3.
Figure 5. EPR spectra of radical adducts of Mito-bis-DIPPMPO (8) and Mito10-DEPMPO (9).

(a) EPR spectrum obtained 10 min after reduction of the 8-OOH (a) and 9-OOH (b) adducts (Figure 2) by adding GPx (10 U mL−1) and GSH (1.2 mM) to the incubation mixture and bubbling argon gas for two min. Grey lines: calculated spectra (Table 3). Spectrometers settings: microwave power 30 mW ; modulation amplitude, 0.7; time constant, 1.28 ms; gain 105; sweep time, 20.4 ms; conversion time, 41.9.
Table 3.
EPR parameters of hydroxyl and carbon-centered spin adducts.
| Spin adduct | Generating system | aP (G) | aN (G) | aHβ (G) |
|---|---|---|---|---|
| Mito-bis-DIPPMPO-OH | HX/XO and then GPx/GSH | 52.4 | 13.6 | 10.5 |
| Mito-bis-DIPPMPO-CH(OH)CH3 | Fe2+, H2O2, EtOH (15%) | 56.8 | 14.3 | 19.8 |
| Mito-DIPPMPO-OH | HX/XO and then GPx/GSH | 53.1 | 13.6 | 10.5 |
| Mito-DIPPMPO-CH(OH)CH3 | Fe2+, H2O2, EtOH (15%) | 57.5 | 14.3 | 19.7 |
| Mito10-DEPMPO-OH | HX/XO and then GPx/GSH | 52.9 | 13.4 | 10.2 |
| Mito10-DEPMPO-CH(OH)CH3 | Fe2+, H2O2, EtOH (5%) | 57.6 | 14.2 | 19.4 |
Quantum-Mechanical calculations
Under the experimental conditions we used for our spin trapping experiments, the apparent half life time of Mito-DIPPMPO-OOH is the longest ever observed for the superoxide spin adduct of a spin trap belonging to the pyrroline N-oxide series. In an attempt to rationalize this result, we undertook a Density Functional Theory (DFT) approach to the structure of Mito-DIPPMPO (6) and Mito-DIPPMPO-OOH (6-OOH).
The structures of four conformers (6A to 6D, see SI) have been obtained for 6 and one of lower energy (6A) is shown in Figure 7. For all the calculated conformers, the geometry of the pyrroline ring and its two C5 substituents are almost the same and very close to that determined by X-ray diffraction for compound 4 (Figure 1). This geometry is characterized by an envelope at C4 and a pseudo-axial (iPrO)2P(O)- substituent with its P=O bond directed towards the pyrroline ring. The five calculated conformers differ by the geometry of the C4 substituent. For conformer 6A, the geometry of the (iPrO)2P(O)- and C4 substituents is in agreement with the trans addition of superoxide leading to the observed trans-diastereoisomer spin adduct. Furthermore the kinetic of addition should benefit from the electrostatic interaction of O2−• with the positive charges brought by the TPP+ moiety, and we indeed observed that it increases by a factor 2 compared with DIPPMPO.
Figure 7.

Calculated DFT [B3LYP 6–31G (d,p), PCM (water)] structures of the lowest energy conformers of Mito-DIPPMPO (6A) and Mito-DIPPMPO-OOH (6-OOHA).
The structures of four conformers 6-OOHA to 6-OOHD (Figure 7 and SI) have been obtained for the superoxide adduct of DIPPMPO. The conformers 6-OOHA,B,C correspond to the trans addition of superoxide to 6, and their energies are very close (ΔEmax = 2.93 kJ.mole−1). For these three conformers the positively charged phosphorus atom P8 is close to the hydroperoxyl group (P8-O10 = 4.94 Å for 6-OOHA, Figure 7) and stabilizing interactions can be established between the positive hydrogen atoms (d = + 0.31, Mulliken charge) of one phenyl group and the oxygen lone pairs (H9-O10 = 2.51 Å for 6-OOHA, Figure 7). These interactions of the TPP+ moiety with the hydroperoxyl group likely contribute to the stabilization of the species, resulting in the especially long half-life time observed. For conformer 6-OOHD, the distance P8-O10 is much higher (10.53 Å) and its energy is 6.10 to 9.02 kJ.mole−1 higher compared to conformers 6-OOHA,B,C. For these later conformers the agreement between the calculated and the experimental hyperfine coupling constants is satisfying [for 6-OOHA, the lowest energy conformer, AN = 1.00 mT (1.28)exp ; AP = 5.00 mT (5.27)exp; AHβ = 1.13 mT (1.17)exp].
Mitochondrial uptake studies
The mitochondrial binding/uptake of compounds 6–10 was evaluated from the decrease of the compounds’ concentration after incubation during 30 minutes with respiring mitochondria. No kinetic study was performed and the method was based on the quantification of the compounds remaining in the supernatant (see Experimental Section). The results of mitochondrial uptake experiments are reported in Table 4 represent the percentage of decrease in compounds’ concentration during incubation with energized (with succinate) mitochondria, as compared to mitochondria without succinate. The effects of triphenylphosphonium (TPP+) in improving their affinity for mitochondria is clear from the investigated series, where little effects are seen for DIPPMPO and no membrane potential-dependent uptake is observed for TritA-DEPMPO 10. We conclude that highly delocalized cationic charge on the lipophilic phenyl rings is necessary for the accumulation in mitochondria in membrane potential –dependent manner. Surprisingly, the presence of two TPP+ in Mito-bis-DIPPMPO 8 did not increase the uptake, may be due to the close vicinity of the two moieties.40 The best uptake was obtained with Mito10-DEPMPO 9, probably due its higher hydrophobicity and amphiphilic properties. The mitochondrial uptake of Mito10-DEPMPO 9 and Mito-bis-DIPPMPO 8 has been independently confirmed by LC-MS/MS analysis of the extracts of mitochondrial pellets, indicating significantly higher accumulation of compounds 8 and 9, as compared to non-targeted spin trap, DIPPMPO (data not shown).
Table 4.
Percentage of mitochondria uptake determined by HPLC / UV-Vis detection.
| Spin traps | Mitochondrial uptake (%) |
|---|---|
| DIPPMPO | 2.4 |
| Mito-DIPPMPO (6) | 29 |
| Mito5-DIPPMPO (7) | 21.1 |
| Mito10-DEPMPO (9) | 58 |
| Mito-bis-DIPPMPO (8) | 25.6 |
| TritA-DEPMPO (10) | 0 |
Conclusions
With the aim to develop tools for the study of O2•− formation in mitochondria, we have prepared a series of DIPPMPO and DEPMPO derivatives, with a substituent in the 4-position of the pyrroline ring, having different lengths and bearing a TPP+ cation or a guanidinium group as chain end. We have studied the spin trapping properties of these new spin traps and determined their mitochondria uptake. All the compounds bearing a TPP+ cation efficiently react with O2•− forming stable adducts. The results of quantum-mechanical calculations suggest that the addition of the negatively charged O2•− to Mito-DIPPMPO could be facilitated by the attraction exerted by the TPP+ group. Moreover, in the most stable conformers of Mito-DIPPMPO-OOH, the interactions of the TPP+ moiety with the hydroperoxyl group likely contribute to the stabilization of the species, resulting in the especially long half-life time observed.
Results from the mitochondria uptake studies showed the key role of the TPP+ moiety in the targeting properties. It should be noted, however, that other factors such as the accessibility of the TPP+ group or the amphiphilic properties of the molecule can modulate the mitochondria uptake, which cannot readily be predicted and experimental determination is required. Spin traps that can be compartmentalized or localized at particular biological sites are part of the toolbox required for maximizing the amount of useful information, as it is now clear that data obtained from multiple techniques is often required to obtain a definitive picture on the formation and role of free radicals in biological systems. The apparent half-life time of the O2•− adduct and the mitochondrial uptake are 73 min and 30 % for Mito-DIPPMPO (6), and 22 min. and 60 % for Mito10-DEPMPO (9), making these new spin traps suitable candidates for mitochondrial superoxide trapping.
Experimental Section
Materials
CH2Cl2 was distilled under dry argon atmosphere in the presence of P2O5. All reagents were used as received without further purification. The reactions were monitored by TLC on silica gel and by 31P NMR. Crude materials were purified by flash chromatography on Silica gel 60 (0.040–0.063 mm). 31P NMR, 1H NMR and 13C NMR spectra were recorded with a 300 or 400 spectrometers at 121.49, 300.13 and 75.54 MHz respectively. 31P NMR was taken in CDCl3 using 85% H3PO4 as an external standard with broad-band 1H decoupling. 1H NMR and 13C NMR were taken in CDCl3 using TMS or CDCl3 as internal reference respectively. Chemical shifts (δ) are reported in ppm and coupling constant J values in Hertz. The assignments of NMR signals were facilitated by use of the DEPT 135 sequence for all the nitrones. High resolution MS experiments (HRMS) were performed with a mass spectrometer equipped with an electrospray ionization source operated in the positive ion mode. In this hybrid instrument, ions were measured using an orthogonal acceleration time-of-flight (oa-TOF) mass analyzer.
Synthesis of the nitrone NHS-DIPPMPO 5
The synthesis was performed adapting the procedure described by Hardy et al.41 The nitrophosphonate 1 has already been described.42
4-(1-Diisopropyloxyphosphoryl-1-nitroethyl)-tetrahydrofuran-2-one 2
The product 2 was obtained as a yellow oil (25 g, 74 %) corresponding to a mixture of two diastereoisomers. 31P NMR (121.49 MHz) δ 12.84 (60%), 12.95 (40%); 1H NMR (300.13 MHz) δ 1.40–1.30 (12H, m), 1.75 (3H, d, J = 14.4), 2.74–2.36 (2H, m), 3.82–3.63 (1H, m), 4.54–4.07 (2H, m), 4.86–4.66 (2H, m).13C NMR (75.47 MHz) δ 174.7 (CIV, s), 174.6 (CIV, s), 90.5 (CIV, d, J = 148.0), 90.2 (d, J = 147.1), 74.4 (s), 74.3 (s), 74.2 (d, J = 1.8), 74.0 (d, J = 1.4), 68.4 (d, J = 2.8), 68.2 (s), 40.2 (s), 39.7 (s), 30.3 (d, J = 2.8), 29.9 (d, J = 8.3), 24.1 (d, J = 3.2), 23.9 (d, J = 1.8), 23.8 (d, J = 1.8), 23.5 (d, J = 1.4), 23.4 (s), 23.3 (d, J = 1.4), 16.2 (d, J = 1.4), 16.1 (d, J = 1.4). HMRS calcd. for C12H22NO7P; [C12H22NO7P+NH4]+; 341.1472, found: 341.1474.
4-(1-Diisopropyloxyphosphoryl-1-nitroethyl)-2-hydroxytetrahydrofurane 3
The product 3 was obtained as a yellow oil (3 g, 75%) corresponding to a mixture of four diastereoisomers. 31P NMR (121.49 MHz) δ14.58 (44%), 14.75 (22%), 14.95 (34%); 1H NMR (300.13 MHz) δ 1.40–1.25 (12H, m), 1.66&1.78&1.83 (3H, 3d, J = 14.4, 14.5, 14,0), 1.98–1.87 (1H, m), 3.57–3.32 (1H, m), 3.94–3.60 (2H, m), 4.21–4.01 (1H, m), 4.85–4.62 (2H, m), 5.51 (1H, t, J = 3.2); 13C NMR (75.47 MHz) δ 97.8 (s), 97.7 (s), 91.4 (CIV, d, J = 148.4), 73.8 (d, J = 6.7), 73.7 (d, J = 6.7), 73.5 (d, J = 7.3), 67.1 (d, J = 0.9), 66.9 (d, J = 0.9), 43.0 (s), 42.3 (s), 35.2, (s), 34.9 (s), 34.8 (s), 24.2 (d, J = 7.8), 24.5 (d, J = 5.0), 24.4 (d, J = 5.9), 15.2 (s), 14.9 (s). HMRS calcd. for C12H24NO7P; [C12H22NO7P+H]+; 326.1363, found: 326.1363.
5-Diisopropyloxyphosphoryl-5-methyl-4-hydroxymethyl-1-pyrroline N-Oxide 4 and 4′
The nitrones 4 and 4′ were obtained in 60% yield (7 g) corresponding to a mixture of 2 diastereoisomers. (4R*, 5R*)-4-HMDIPPMPO 4; 31P NMR (121.49 MHz) δ 21.19; 1H NMR (300.13 MHz) δ 1.31 (3H, d, J = 6.2), 1.32 (3H, d, J = 6.0), 1.37 (3H, d, J = 6.2), 1.41 (3H, d, J = 6.2), 1.70 (3H, d, J = 14.5), 2.75–2.37 (3H, m), 3.92–3.82 (2H, m), 4.18 (1H, m), 4.95–4.71 (2H, m), 6.88 (1H, dt, J = 2.4, 2.4); 13C NMR (75.47 MHz) δ 134.1 (1C, d, J = 7.7), 77.0 (1C, d, J = 152.0), 73.3 (1C, d, J = 7.1), 72.8 (1C, d, J = 7.7), 62.4 (1C, d, J = 5.5), 49.3 (1C, d, J = 5.7), 29.2 (1C, s), 24.2 (1C, d, J = 2.7), 24.0 (1C, d, J = 2.7), 24.8 (1C, d, J = 6.0), 23.6 (1C, d, J = 5.5), 21.4 (1C, d, J = 1.6); ESI-MS : m/z : 293 [M + H]+. HMRS calcd. for C12H24NO5P; [C12H25NO5P+H]+; 294.1465, found: 294.1465.
(4S*,5R*)-4-HMDIPPMPO 4′ 31P NMR (121.49 MHz) δ 21.97; 1H NMR (300.13 MHz) δ 1.31&1.32 (12H, 2d, J = 6.2, 6.2), 1.57 (3H, d, J = 16.0), 2.60–2.34 (2H, m), 3.14–2.72 (2H, m), 3.79–3.69 (1H, m), 4.2–3.90 (1H, m), 4.90–4.67 (2H, m), 6.82 (1H, dt, J = 2.7, 2.6); 13C NMR (75.47 MHz) δ 134.2 (1C, d, J = 8.8), 76.7 (1C, d, J = 159.7), 73.0 (1C, d, J = 7.1), 72.1 (1C, d, J = 7.7), 61.4 (1C, d, J = 6.0), 40.9 (1C, s), 30.0 (1C, d, J = 4.9), 24.2 (1C, d, J = 2.7), 23.9 (1C, d, J = 4.4), 23.8 (1C, d, J = 4.9), 23.5 (1C, d, J = 6.6), 14.2 (1C, s).
(4R*,5R*)-5-diisopropyloxyphosphoryl-5-methyl-4-(succinimidyloxycarbonyloxymethyl)-1-pyrroline N-Oxide 5
The nitrone NHS-DIPPMPO 5 was obtained as a white crystal (1.4 g, 100%); 31P NMR (81.01 MHz):δ 18.0; 1H NMR (300.13 MHz) δ 1.41–1.26 (12H, m), 1.66 (3H, d, J = 14.0), 2.90–2.63 (3H, m), 2.8 (4H, s), 4.63–4.48 (1H, m), 4.86–4.67 (3H, m), 6.92 (1H, m); 13C NMR (50.32 MHz) δ 168.5 (2C, s), 151.3 (1C, s), 133.5 (1C, d, J = 7.3), 75.9 (1C, d, J = 149.8), 73.9 (1C, d, J = 6.4), 72.1 (1C, d, J = 7.8), 70.5 (1C, d, J = 3.2), 45.6 (1C, d, J = 2.3), 30.0 (1C, d, J = 0.9), 25.4 (2C, s), 24.5 (1C, d, J = 1.4), 23.8 (1C, d, J = 1.4), 23.7 (1C, d, J = 1.8), 23.4 (1C, d, J = 7.3), 20.3 (1C, s). HMRS calcd. for C17H27N2O9P; [C17H27N2O9P +H]+; 435.1527, found: 435.1527.
Synthesis of the nitrone Mito-DIPPMPO 6
Mito-DIPPMPO 6
To a mixture of NHS-DIPPMPO (0.2 g, 0.46 mmol) and (2-aminoethyl) triphenylphosphonium Bromide (0.18 g, 0.46 mmol) in CH2Cl2 (15 mL) was added at room temperature under inert atmosphere triethylamine (141 μL, 1.06 mmol). The reaction mixture was stirred for 3 h. The solution was washed with 8 ml of distilled water, and extracted 3 times with CHCl3. The organic layers were dried over Na2SO4 and the solvent distilled under reduced pressure. Purification of the crude product by flash chromatography on silicagel (CH2Cl2/EtOH 80:20) afforded a white powder (0.28 g, 86 %), corresponding to Mito-DIPPMPO 6. 31P NMR (121.49 MHz) δ 17.8, 20.9; 1H NMR (300.13 MHz) δ 1.38–1.27 (12H, m), 1.62 (3H, d, J = 14.1), 2.68–2.52 (3H, broad band), 3.57–3.68 (2H, m), 3.95–3.33 (2H, m), 4.17–4.07 (1H, m), 4.36–4.29 (1H, m), 4.79–4.67 (2H, m), 6.91 (1H, m), 7.52 (1H, t, J = 6.0), 7.82–7.63 (15H, broad band); 13C NMR (75.47 MHz) δ 156.5 (1C, s), 135.2 (3C, d, J = 2.9), 134.6 (1C, d, J = 7.3), 133.6 (6C, d, J = 10.3), 130.5 (6C, d, J = 12.4), 117.5 (3C, d, J = 86.5), 75.1 (1C, d, J = 149.6), 73.4 (1C, d, J = 6.6), 71.8 (1C, d, J = 8.0), 64.5 (1C, s), 46.4 (1C, d, J = 2.2), 35.2 (1C, s), 30.7 (1C, s), 24.5 (1C, d, J = 1.5), 24.0 (1C, d, J = 3.7), 25.8 (1C, d, J = 5.1), 23.6 (1C, d, J = 7.3), 23.3 (1C d, J = 48.4), 20.3 (1C, s). HMRS calcd. for [C33H43N2O6P2]+, Br−; [C33H43N2O6P2]+ 625.2591, found: 625.2587.
Synthesis of the nitrone Mito-bis-DIPPMPO 8
Bis-[2-(triphenylphosphonium bromide)-ethyl]-amine A
A mixture containing Bis-(2-bromo-ethyl)-amine43 (7 g, 0.03 mol) and triphenylphosphane (16 g, 0.061 mol) in acetonitrile (50 mL) was refluxed for 48 hours. The solvent distilled under reduced pressure. Purification of the crude product by flash chromatography on a silicagel (CH2Cl2/EtOH 80:20) afforded a brown solid A (12 g, 52%).31P NMR (121.49 MHz) 23.56, 1H NMR (300.13 MHz) δ 3.06–3.12 (4H, m), 3.72–3.80 (4H, m), 7.60–7.82 (30H, m); 13C NMR (75.47 MHz) δ 134.6 (6C, d, J = 2.9), 133.8 (12C, d, J = 10.3), 130.4 (12C, d, J = 12.5), 118.5 (6C, d, J = 86.6), 41.7 (2C, s), 23.7 (2C, d, J = 50.6). ESI-MS : m/z : 297.6 [M + H]++.
Mito-bis-DIPPMPO 8
To a mixture of NHS-DIPPMPO (0.2 g, 0.46 mmol) and Bis-[2-(triphenylphosphonium bromide)-ethyl]-amine A (0.37 g, 0.46 mmol) in CH2Cl2 (5 mL) was added at room temperature under inert atmosphere triethylamine (141 μL, 1.06 mmol). The reaction mixture was stirred for 3 h. The solution was washed with 8 ml of distilled water, and extracted 3 times with CHCl3. The organic layers were combined and dried over Na2SO4 and the solvent removed under reduce pressure. Purification of the crude product by flash chromatography on silicagel (CH2Cl2/EtOH 80:20) afforded a white powder (0.25 g, 50 %), corresponding to Mito-bis-DIPPMPO 8. 31P NMR (121.49 MHz) 17.61, 21.89; 1H NMR (300.13 MHz) δ 1.38–1.26 (12H, m), 1.53 (3H, d, J = 13.8), 2.82–2.62 (3H, m), 4.10–3.90 (4H, m), 4.35–4.12 (4H, m), 4.60–4.39 (2H, m), 4.79–4.62 (2H, m), 6.83 (1H, m), 7.60–7.98 (30H, m); 13C NMR (75.47 MHz) δ 154.9 (1C, s), 134.8 (3C, d, J = 2.9), 134.7 (3C, d, J = 2.9), 134.6 (1C, d, J = 8.8), 134.2 (6C, d, J = 7.3), 134.1 (6C, d, J = 7.3), 130.4 (6C, d, J = 12.5), 130.3 (6C, d, J = 12.5), 117.9 (3C, d, J = 86.6), 117.8 (3C, d, J = 86.8), 75.8 (1C, d, J = 149.6), 73.5 (1C, d, J = 6.6), 71.7 (1C, d, J = 7.3), 64.3 (1C, d, J = 2.2), 45.8 (1C, d, J = 2.2), 42.9 (1C, s), 42.6 (1C, s), 29.2 (1C, s), 24.5 (1C, s), 24.0 (1C, d, J = 4.4), 23.9 (1C, d, J = 4.4), 23.4 (1C, d, J = 7.3), 23.1 (1C, d, J = 46.2), 22.1 (1C, d, J = 47.7), 20.3 (1C, s). HMRS calcd. for [C53H61N2O6P3]2+, 2Br−; [C53H61N2O6P3]2+ 457.1866, found: 457.1868.
Synthesis of the nitrone Mito5-DIPPMPO 7
Mito5-DIPPMPO 7
To a mixture of NHS-DIPPMPO (0.200 g, 0.46 mmol) and (2-Aminopentyl) triphenylphosphonium bromide44 (0.190 g, 0.46 mmol) in CH2Cl2 (20 mL) was added at room temperature under inert atmosphere triethylamine (147 μL, 6 mmol). The reaction mixture was stirred for 3 h. The solution was washed with 8 ml of distilled water, and extracted 3 times with CHCl3. The organic layers were combined and dried over Na2SO4 and the solvent removed under reduce pressure. Purification of the crude product by flash chromatography on silicagel (CH2Cl2/EtOH 80:20) afforded a white powder (0.171 g, 50 %), corresponding to Mito5-DIPPMPO 7. 31P NMR (121.49 MHz) δ 18.0, 24.3; 1H NMR (300.13 MHz) δ 1.38–1.26 (12H, m), 1.62 (3H, d, J = 14.0), 1.71–1.57 (6H, m), 2.72–2.61 (3H, broad band), 3.13–3.09 (2H, m), 3.73–3.66 (2H, m), 4.21–4.16 (1H, m), 4.45–4.40 (1H, m), 4.77–4.69 (2H, m), 6.29 (1H, t, J = 5.5), 6.91 (1H, m), 7.82–7.63 (15 H, broad band); 13C NMR (75.47 MHz) δ 156.6 (1C, s), 135.0 (3C, d, J = 2.9), 135.5 (6C, d, J = 10.3), 130.4 (6C, d, J = 12.5), 118.2 (3C, d, J = 85.8), 75.9 (1C, d, J = 150.4), 73.2 (1C, d, J = 6.6), 71.6 (1C, d, J = 7.3), 64.0 (1C, s), 46.5 (1C, d, J = 2.2), 39.9 (1C, s), 30.7 (1C, s), 29.5 (1C, s), 28.3 (1C, s), 26.9 (1C, d, J = 16.7), 24.5 (1C, d, J = 1.5), 23.9 (1C, d, J = 3.7), 23.7 (1C, d, J = 5.4), 23.4 (1C, d, J = 6.6), 22.5 (1C, d, J = 49.9), 21.8 (1C, d, J = 4.4), 20.3 (1C, s). HMRS calcd. for [C36H49N2O6P2]+, Br−; [C36H49N2O6P2]+ 667.3060, found: 667.3060.
Synthesis of the nitrone Mito10-DEPMPO 9
(10-phtalimidodecyl) triphenylphosphonium bromide B
A mixture containing Bromophtalimide (7 g, 0.019 mol) and triphenylphosphane (5 g, 0.019 mol) in acetonitrile (60 mL) was refluxed for 15 hours. The solvent distilled under reduced pressure. Purification of the crude product by flash chromatography on a silicagel (CH2Cl2/EtOH 80:20) afforded a white solid B (9 g, 73%). MS calcd for [C36H39NO2P]+, Br−; [C36H39NO2P]+, 548.3, found: 548.3.
(10-Aminodecyl) triphenylphosphonium Bromide C
To a solution of B (7 g, 0.011 mol) in EtOH (70 mL) was added hydrazine (0.54 mL, 0.011 mol). The mixture was refluxed for 15 hours. The solvent is distilled and the impurity was recrystallized using a mixture Et2O/EtOH (2/1). The product was purified by flash chromatography on silicagel (CH2Cl2/EtOH 80:20) afforded a yellow solid C (4 g, 73%). 31P NMR (121.49 MHz) δ 24.61; 1H NMR (300.13 MHz) δ 7.95–7.73 (15H, m), 3.70–3.55 (2H, m), 2.80–2.70 (2H, m), 1.60–1.40 (6H, m), 1.35–1.10 (10H, m). MS calcd for [C28H37NP]+, Br−; [C28H37NP]+, 418.2, found: 418.2.
Mito10-DEPMPO 9
To a mixture of NHS-DEPMPO (0.25 g, 0.61 mmol) and (10-Aminodecyl) triphenylphosphonium bromide C (0.32 g, 0.62 mmol) in CH2Cl2 (20 mL) was added at room temperature under inert atmosphere triethylamine (0.23 mL, 1.61 mmol). The reaction mixture was stirred for 3 h, then washed with water (15 mL). The organic layer was dried over Na2SO4 and the solvent distilled under reduce pressure. Purification of the crude product by flash chromatography on silicagel (CH2Cl2/EtOH 70:30) afforded a white powder (0.31 g, 64 %), corresponding to Mito10-DEPMPO 9. 31P NMR (121.49 MHz) δ 20.58, 25.48; 1H NMR (300.13 MHz) δ 1.39–1.16 (17H, m), 1.50–1.40 (2H, m), 1.65–1.57 (3H, m), 1.68 (3H, d, J = 14.0), 2.80–2.55 (3H, m), 3.19–3.06 (2H, m), 3.90–3.60 (3H, m), 4.30–4.10 (5H, m), 4.56–4.45 (1H, m), 6.97 (1H, dt, J = 2.4, 2.4), 7.92–7.62 (15H, m); 13C NMR (75.47 MHz) δ 156.1 (1CIV, s), 134.8 (1C, d, J = 8.2), 134.9 (3C, d, J = 3), 133.7 (6C, d, J = 9.7), 130.4 (6C, d, J = 12.6), 118.5 (3CIV, d, J = 85.5), 76.2 (1C, d, J = 155.7), 64.3 (1C, d, J = 6.3), 63.9 (1C, s), 62.5 (1C, d, J = 8.0), 46.7 (1C, d, J = 2.3), 41.0 (1C, s), 30.4 (1C, s), 30.2 (1C, s), 29.7 (1C, s), 29.0 (1C, s), 28.9 (2C, s), 26.52 (1C, s), 23.0 (1C, d, J = 49.3), 22.6 (1C, d, J = 4.0), 22.7 (1C, d, J = 49.3), 20.3 (1C, s), 16.4 (1C, d, J = 5.7), 16.3 (1C, d, J = 5.7); HRMS calcd for [C39H55N2O6P2]+, Br−; [C39H55N2O6P2]+ 709.3530, found: 709.3529.
Synthesis of the nitrone TritA-DEPMPO 10
TritA-DEPMPO 10
To a mixture of NHS-DEPMPO (0.15 g, 3.6 mmol) and N-tritylethylenediamine hydrobromide (0.14 g, 3.6 mmol) in CH2Cl2 (10 mL) was added triethylamine (103 μL, 1.61 mmol) at room temperature under argon. The reaction mixture was stirred for 5 h and then washed with water (15 mL). The organic layer was dried over Na2SO4 and the solvent distilled under reduced pressure. Purification of the crude product by flash chromatography on a silicagel (CH2Cl2/EtOH 97:03) afforded a white powder (0.2 g, 91%), corresponding to TritA-DEPMPO 10. 31P NMR (121.49 MHz) δ 19.75. 1H NMR (300.13 MHz) δ 7.49–7.43 (5H, m), 7.33–7.28 (4H, m), 7.27–7.20 (6H, m), 7.18 (1H, t, J = 1.2), 7.01 (1H, m), 5.08 (1H, t, J = 4.2), 4.62–4.53 (1H, m), 4.38–4.12 (5H, m), 3.37–3.21 (2H, m), 2.84–2.57 (3H, m), 2.30 (2H, t, J = 6.0), 1.73 (3H, d, J = 14.0), 1.36 (6H, dt, J = 7.0). 13C NMR (75.47 MHz) δ 156.1 (1CIV, s), 145.7 (3CIV, s), 134.5 (1C, d, J = 8.0), 128.5 (6C, s), 127.9 (6C, s), 126.4 (3C, s), 76.1 (1C, d, J = 160.6), 70.6 (1CIV, s), 64.3 (1C, d, J = 6.3), 64.1 (1C, s), 62.4 (1C, d, J = 7.4) 46.7 (1C, s), 43.6 (1C, s), 41.7 (1C, s), 30.3 (1C, s), 20.3 (1C, s), 16.3 (1C, d, J = 5.7), 16.2 (1C, d, J = 5.7). HRMS calcd for [C32H40N3O6P] ; [C32H40N2O6P]+ 594.2728, found: 594.2735.
Synthesis of the nitrone Gua-DIPPMPO 11
Gua-DIPPMPO 11
To a mixture of (2-aminoethyl)-guanidine (0.13 g, 1,1 mmol) in acetonitrile (5 mL) was added NHS-DIPPMPO 5 (0.4 g, 0.92 mmol) in 10 mL of anhydrous acetonitrile following by the addition of N-ethyldiisopropylamine (0,24 mL, 1,38 mmol). The reaction mixture was stirred overnight. The solvent was removed under reduce pressure. Purification of the crude product by flash chromatography on basic alumina (CH2Cl2/EtOH 85:15) afforded a pale yellow powder (0.23 g, 60%). 31P NMR (121.49 MHz, D2O) δ 17.81.1H NMR (300.13 MHz) δ 6.91 (1H, d, J = 3.02), 4.78–4.67 (2H, m), 4.59–4.53 (1H, m), 4.41–4.35 (1H, m), 3.87–3.69 (2H, m), 3.65–3.57 (2H, m), 2.78–2.52 (3H, m), 1.64 (3H, d, J = 14), 1.35–1.27 (12H, m). 13C NMR (75.47 MHz) δ 161.5 (1CIV, s), 158.5 (1C, s), 144.7 (1C, d, J = 8.0), 76.6 (1CIV, d, J = 153.74Hz), 75.7 (1C, d, J = 8.0), 75.58 (1C, d, J = 8.0), 61.8 (1C, d, J = 3.5), 49.1 (1C, d, J = 2.3), 43.4 (2C, s), 31.7 (1C, s), 24.1 (1C, d, J = 3.4), 23.9 (1C, d, J = 3.4), 23.7 (1C, d, J = 5.2), 23.5 (1C, d, J = 5.2), 19.89 (1C, d, J = 1.7). ESI-MS/MS : [M+H+]: 422.22.
Synthesis of the nitrone Agm-DIPPMPO 12
Agm-DIPPMPO 12
To a mixture of agmantine (0,21 g, 0,84 mmol) in acetonitrile (5 mL) was added NHS-DIPPMPO (0.28 g, 0.64 mmol) in 8 mL of anhydrous acetonitrile followed by the addition of N-ethyldiisopropylamine (0.33 mL, 1.94 mmol). The reaction mixture was stirred overnight. The solvent was removed under reduce pressure. Purification of the crude product by flash chromatography on basic alumina (CH2Cl2/EtOH 75:25) afforded a pale yellow powder (0.277 g, 95%). 31P NMR (121.49 MHz, D2O) δ 18.88. 1H NMR (300.13 MHz) δ 7,36 (1H, d, J = 2.8), 4.85–4.75 (2H, m), 4.36–4.29 (1H, m), 4.20–4.14 (1H, m), 3.14–3.05 (4H, m), 2.98–2.60 (3H, m), 1.67 (3H, d, J = 14.9), 1.60–1.50 (4H, m), 1.31–1.26 (12H, m). 13C NMR (75.47 MHz) δ 162.9 (1CIV, s), 158.9 (1C, s), 144.3 (1C,d, J = 7.5), 77.3 (1CIV, d, J = 153.7), 75.6 (1C, d, J = 7.5), 75.39 (1C, d, J = 8.0), 64.6 (1C, d, J = 2.8), 46.2 (1C, d, J = 2.3), 41.51 (1C, s), 40.6 (1C, s), 30.7 (1C, s), 26.9 (1C, s), 26.0 (1C, s), 24.1 (1C, d, J = 2.9), 24.0 (1C, d, J = 4.0), 23.7 (1C, d, J = 6.9), 23.6 (1C, d, J = 4.6), 19.99 (1C, s). ESI-MS/MS : [M+H+]: 450.25.
Mitochondrial uptake studies
Mitochondria were isolated from rat heart as described by Sethumadhavan S. et al.45 Briefly, freshly isolated heart tissue was homogenized in modified Chappell Perry medium: 10 mM HEPES, 100 mM KCl, 1 mM EGTA, 5 mM MgSO4, 1 mM ATP, and 0.2% BSA, pH 7.4. Homogenates were centrifuged at 700 × g for 15 min at 4°C. The supernatant was transferred to a cold clean tube and subjected to a high speed centrifugation (10,000 × g, 15 min, 4°C). The final supernatant was discarded, and the mitochondrial pellet was washed twice. Finally, the pellets were resuspended in storage buffer: 10 mM HEPES, 100 mM KCl, and 1 mM EGTA, pH 7.4. Mitochondrial protein was quantified by bicinchoninic acid method and used immediately for uptake assays. Incubations were performed in 10 mM HEPES buffer (pH = 7.2) containing KCl (120 mM), EGTA (1 mM), succinate (5 mM) with 10μM of the spin trap solution. Aliquots of the supernatant were collected at 0 and 30 min of incubation for HPLC analysis. Aliquots of the initial solutions and samples at time 0 and 30 min were analyzed, and the initial reagent solution after 30 min was also re-injected to confirm the stability of the compounds tested over the course of experiment.
After incubation, the mixtures were centrifugated (10 min x 1,000g, 4°C) and supernatant was analyzed by HPLC with UV-Vis absorption detection.
Supplementary Material
Figure 6. Spin trapping of carbon-centered radical by Mito-bis-DIPPMPO (8) and Mito10-DEPMPO (9).

(a) Signal obtained after 15 min incubation of a mixture containing Mito-bis-DIPPMPO (20 mM), H2O2 (2 mM), FeSO4 (2 mM), EtOH (15%), DTPA (1 mM) in phosphate buffer (0.1 M, pH 7.3); (b) Signal obtained after 1 min incubation of a mixture containing Mito10-DEPMPO (20 mM), H2O2 (2 mM), FeSO4 (2 mM), EtOH (15%), DTPA (1 mM) in phosphate buffer (0.1 M, pH 7.3). Grey lines: calculated spectra (Table 3). Spectrometers settings: microwave power 30 mW (a–b); modulation amplitude, 0.7 (a–b); time constant, 1.28 ms (a–b); gain 105 (a–b); sweep time, 20.4 ms (a–b); conversion time, 41.9 (a–b).
Acknowledgments
A. R. thanks the Hungarian Science Fund for partial funding of this work (grant OTKA T-046953). The authors thank Patrick Bernasconi (Aix-Marseille University) for EPR technical support and F. Peyrot, J.-L. Boucher and Y. M. Frapart for discussions.
Funding Sources. This work was supported by Aix-Marseille University, CNRS and by “Agence Nationale de la Recherche” (ANR-09-BLAN-0193-02, SPIN BioRad).
Abbreviations
- DEPMPO
5-(diethoxyphosphoryl)-5-methyl-pyrroline N-oxide
- DIPPMPO
5-(diisopropyloxyphosphoryl)-5-methyl-pyrroline N-oxide
- DMPO
5,5-dimethyl-pyrroline N-oxide
- NHS-DIPPMPO
(N-hydroxysuccinimidyl-DIPPMPO)
- EMPO
5-ethoxycarbonyl-5-methyl-pyrroline N-oxide
- HX
hypoxanthine
- Mito-DIPPMPO
(4R*, 5R*) 5-(diisopropyloxyphosphoryl)-5-methyl-4-[({[2-(triphenylphosphonio)-ethyl]carbamoyl}oxy)methyl]pyrroline N-oxide bromide
- Mito10-DEPMPO
(4R*, 5R*) 5-( diethoxyphosphoryl)-5-methyl-4-[({[2-(triphenylphosphonio)-decyl]carbamoyl}oxy)methyl]pyrroline N-oxide bromide
- Mito5-DIPPMPO
(4R*, 5R*) 5-(diisopropyloxyphosphoryl)-5-methyl-4-[({[2-(triphenylphosphonio)-pentyl]carbamoyl}oxy)methyl]pyrroline N-oxide bromide
- Mito-bis-DIPPMPO
(4R*, 5R*) 5-(diisopropyloxyphosphoryl)-5-methyl-4-[({bis [2-(triphenylphosphonio)-ethyl]carbamoyl}oxy)methyl]pyrroline N-oxide bromide
- TrA-DEPMPO
5-(diethoxyphosphoryl)-5-methyl-4-[({trityl-2-aza-ethyl}-carbamoyl)-oxy-methyl]pyrroline N-oxide
- Gua-DIPPMPO
5-(diisopropyloxyphosphoryl)-5-methyl-4-[({[2-(guanidino)-ethyl] carbamoyl}oxy)methyl]-5-methyl-1-pyrroline N-oxide
- Agm-DIPPMPO
5-( diisopropyloxyphosphoryl)-5-methyl-4-[({[2-(guanidino)-buthyl] carbamoyl}oxy)methyl]-5-methyl-1-pyrroline N-oxide
- SOD
superoxide dismutase
- XO
xanthine oxidase
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
Footnotes
The authors declare no competing financial interest.
1H, 31P, 13C NMR and EPR spectra, X-Ray data and Quantum-Mechanical calculations. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, Davies KJA. Free radical biology and medicine: it’s a gas, man! Am J Physiol-Reg I. 2006;291:R491–R511. doi: 10.1152/ajpregu.00614.2005. [DOI] [PubMed] [Google Scholar]
- 2.Floyd RA. Serendipitous findings while researching oxygen free radicals. Free Radic Biol Med. 2009;46:1004–1013. doi: 10.1016/j.freeradbiomed.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halliwell B, Gutteridge JMC. Free Radicals in Biology And Medicine. 4. Cary, North Carolina, U.S.A: Oxford Univ Pr; 2007. [Google Scholar]
- 4.Hadfield KA, Pattison DI, Brown BE, Hou LM, Rye KA, Davies MJ, Hawkins CL. Myeloperoxidase-derived oxidants modify apolipoprotein A-I and generate dysfunctional high-density lipoproteins: comparison of hypothiocyanous acid (HOSCN) with hypochlorous acid (HOCI) Biochem J. 2013;449:531–542. doi: 10.1042/BJ20121210. [DOI] [PubMed] [Google Scholar]
- 5.Vasquez-Vivar J. Tetrahydrobiopterin, superoxide, and vascular dysfunction. Free Radic Biol Med. 2009;47:1108–1119. doi: 10.1016/j.freeradbiomed.2009.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright RM, McManaman JL, Repine JE. Alcohol-induced breast cancer: a proposed mechanism. Free Radic Biol Med. 1999;26:348–354. doi: 10.1016/s0891-5849(98)00204-4. [DOI] [PubMed] [Google Scholar]
- 7.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 8.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 9.Zielonka J, Zielonka M, Sikora A, Adamus J, Joseph J, Hardy M, Ouari O, Dranka BP, Kalyanaraman B. Global profiling of reactive oxygen and nitrogen species in biological systems. J Biol Chem. 2012;287:2984–2995. doi: 10.1074/jbc.M111.309062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 11.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BSS, Karoui H, Tordo P, Pritchard KA. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. P Natl Acad Sci USA. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buettner GR, Mason RP. Spin-trapping methods for detecting superoxide and hydroxyl free-radicals in-vitro and in-vivo. Methods in Enzymology. 1990;186:127–133. doi: 10.1016/0076-6879(90)86101-z. [DOI] [PubMed] [Google Scholar]
- 13.Velayutham M, Hemann C, Zweier JL. Removal of H2O2 and generation of superoxide radical: role of cytochrome c and NADH. Free Radic Biol Med. 2011;51:160–170. doi: 10.1016/j.freeradbiomed.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell DG, Rosen GM, Tseitlin M, Symmes B, Eaton SS, Eaton GR. Use of rapid-scan EPR to improve detection sensitivity for spin-trapped radicals. Biophys J. 2013;105:338–342. doi: 10.1016/j.bpj.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawkins CL, Davies MJ. Detection and characterisation of radicals in biological materials using EPR methodology. Biochim Biophys Acta. 2013;840:708–721. doi: 10.1016/j.bbagen.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 16.Hardy M, Chalier F, Ouari O, Finet JP, Rockenbauer A, Kalyanaraman B, Tordo P. Mito-DEPMPO synthesized from a novel NH2-reactive DEPMPO spin trap: a new and improved trap for the detection of superoxide. Chem Commun. 2007;(10):1083–1085. doi: 10.1039/b616076j. [DOI] [PubMed] [Google Scholar]
- 17.Frejaville C, Karoui H, Tuccio B, Lemoigne F, Culcasi M, Pietri S, Lauricella R, Tordo P. 5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide - a new efficient phosphorylated nitrone for the in-vitro and in-vivo spin-trapping of oxygen-centered radicals. J Med Chem. 1995;38:258–265. doi: 10.1021/jm00002a007. [DOI] [PubMed] [Google Scholar]
- 18.Olive G, Mercier A, Le Moigne F, Rockenbauer A, Tordo P. 2-Ethoxycarbonyl-2-methyl-3,4-dihydro-2H-pyrrole-1-oxide: evaluation of the spin trapping properties. Free Radic Biol Med. 2000;28:403–408. doi: 10.1016/s0891-5849(99)00254-3. [DOI] [PubMed] [Google Scholar]
- 19.Kim SU, Liu Y, Nash KM, Zweier JL, Rockenbauer A, Villamena FA. Fast reactivity of a cyclic nitrone-calix[4]pyrrole conjugate with superoxide radical anion: theoretical and experimental studies. J Am Chem Soc. 2010;132:17157–17173. doi: 10.1021/ja105198c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villamena F, Gallucci J, Velayutham M, Hadad C, Zweier J. Spin trapping by 5-carbamoyl-5-methyl-1-pyrroline N-oxide (AMPO): theoretical and experimental studies. Free Radic Biol Med. 2003;35:S15–S15. doi: 10.1021/jo049244i. [DOI] [PubMed] [Google Scholar]
- 21.Finkelstein E, Rosen GM, Rauckman EJ. Spin trapping - kinetics of the reaction of superoxide and hydroxyl radicals with nitrones. J Am Chem Soc. 1980;102:4994–4999. [Google Scholar]
- 22.Finkelstein E, Rosen GM, Rauckman EJ. Production of hydroxyl radical by decomposition of superoxide spin-trapped adducts. Mol Pharmacol. 1982;21:262–265. [PubMed] [Google Scholar]
- 23.Hardy M, Bardelang D, Karoui H, Rockenbauer A, Finet JP, Jicsinszky L, Rosas R, Ouari O, Tordo P. Improving the trapping of superoxide radical with a beta-cyclodextrin-5-diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide (DEPMPO) conjugate. Chemistry-a European Journal. 2009;15:11114–11118. doi: 10.1002/chem.200901342. [DOI] [PubMed] [Google Scholar]
- 24.Smith RAJ, Hartley RC, Murphy MP. Mitochondria-targeted small molecule therapeutics and probes. Antioxid Redox Sign. 2011;15:3021–3038. doi: 10.1089/ars.2011.3969. [DOI] [PubMed] [Google Scholar]
- 25.Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Targeting mitochondria. Acc Chem Res. 2008;41:87–97. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- 26.Sheu SS, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: a new therapeutic direction. Bba-Mol Basis Dis. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev. 2002;54:101–127. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- 28.Smith RAJ, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 29.Robertson L, Hartley RC. Synthesis of N-arylpyridinium salts bearing a nitrone spin trap as potential mitochondria-targeted antioxidants. Tetrahedron. 2009;65:5284–5292. doi: 10.1016/j.tet.2009.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James AM, Cocheme HM, Smith RAJ, Murphy MP. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species - Implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- 31.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 32.Xu YK, Kalyanaraman B. Synthesis and ESR studies of a novel cyclic nitrone spin trap attached to a phosphonium group-a suitable trap for mitochondria-generated ROS? Free Radical Res. 2007;41:1–7. doi: 10.1080/10715760600911147. [DOI] [PubMed] [Google Scholar]
- 33.Quin C, Trnka J, Hay A, Murphy MP, Hartley RC. Synthesis of a mitochondria-targeted spin trap using a novel Parham-type cyclization. Tetrahedron. 2009;65:8154–8160. doi: 10.1016/j.tet.2009.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardy M, Rockenbauer A, Vasquez-Vivar J, Felix C, Lopez M, Srinivasan S, Avadhani N, Tordo P, Kalyanaraman B. Detection, characterization, and decay kinetics of ROS and thiyl adducts of Mito-DEPMPO spin trap. Chem Res Toxicol. 2007;20:1053–1060. doi: 10.1021/tx700101d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rockenbauer A, Korecz L. Automatic computer simulations of ESR spectra. Appl Magn Reson. 1996;10:29–43. [Google Scholar]
- 36.Clement JL, Ferre N, Siri D, Karoui H, Rockenbauer A, Tordo P. Assignment of the EPR spectrum of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) superoxide spin adduct. J Org Chem. 2005;70:1198–1203. doi: 10.1021/jo048518z. [DOI] [PubMed] [Google Scholar]
- 37.Rockenbauer A, Clement JL, Culcasi M, Mercier A, Tordo P, Pietri S. Combined ESR and thermodynamic studies of the superoxide adduct of 5-(diethoxyphosphoryl)-5-methyl-1-oyrroline N-oxide (DEPMPO): hindered rotation around the O-O bond evidenced by two-dimensional simulation of temperature-dependent spectra. J Phys Chem A. 2007;111:4950–4957. doi: 10.1021/jp070679u. [DOI] [PubMed] [Google Scholar]
- 38.Houriez C, Ferre N, Siri D, Tordo P, Masella M. Structure and spectromagnetic properties of the superoxide radical adduct of DMPO in water: elucidation by theoretical investigations. J Phys Chem B. 2010;114:11793–11803. doi: 10.1021/jp1033307. and references cited herein. [DOI] [PubMed] [Google Scholar]
- 39.Hausladen A, Fridovich I. Competitive-inhibition of xanthine-oxidase by guanidinium - dependence upon monovalent anions and effects on production of superoxide. Arch Biochem Biophys. 1993;304:479–482. doi: 10.1006/abbi.1993.1378. [DOI] [PubMed] [Google Scholar]
- 40.Ross MF, Da Ros T, Blaikie FH, Prime TA, Porteous CM, Severina II, Skulachev VP, Kjaergaard HG, Smith RAJ, Murphy MP. Accumulation of lipophilic dications by mitochondria and cells. Biochem J. 2006;400:199–208. doi: 10.1042/BJ20060919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardy M, Chalier F, Ouari O, Finet JP, Rockenbauer A, Kalyanaraman B, Tordo P. Mito-DEPMPO synthesized from a novel NH2 -reactive DEPMPO spin trap: a new and improved trap for the detection of superoxide. Chem Commun. 2007:1083–1085. doi: 10.1039/b616076j. [DOI] [PubMed] [Google Scholar]
- 42.Zon J. Synthesis of diisopropyl 1-nitroalkanephosphonates from diisopropyl 1-oxoalkanephosphonate. Synthesis. 1984;8:661–663. [Google Scholar]
- 43.Li S, Zhou X, Wang L, Xu C, Ruan C, Lin C, Xiao J, Zheng Z, Liu H, Xie Y, Zhong W, Cui H. Preparation of tyrosine derivs. substituted by alkanoyl as hPPARα and & hPPARγ agonists. WO2005116018 (A1) PCT Int Appl. 2005
- 44.McAllister PR, Dotson MJ, Grim SO, Hillman GR. Effects of phosphonium compounds on schistosoma mansoni. J Med Chem. 1980;23:862–865. doi: 10.1021/jm00182a010. [DOI] [PubMed] [Google Scholar]
- 45.Sethumadhavan S, Vasquez-Vivar J, Migrino RQ, Harmann L, Jacob HJ, Lazar J. Mitochondrial DNA variant for complex I reveals a role in diabetic cardiac remodeling. J Biol Chem. 2012;287:22174–22182. doi: 10.1074/jbc.M111.327866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.