

Abstract
Bioluminescence imaging with luciferase-luciferin pairs is widely used in biomedical research. Several luciferases have been identified in nature, and many have been adapted for tracking cells in whole animals. Unfortunately, the optimal luciferases for imaging in vivo utilize the same substrate, and therefore cannot easily differentiate multiple cell types in a single subject. To develop a broader set of distinguishable probes, we crafted custom luciferins that can be selectively processed by engineered luciferases. Libraries of mutant enzymes were iteratively screened with sterically modified luciferins, and orthogonal enzyme-substrate “hits” were identified. These tools produced light when complementary enzyme-substrate partners interacted both in vitro and in cultured cell models. Based on their selectivity, these designer pairs will bolster multi-component imaging and enable the direct interrogation of cell networks not currently possible with existing tools. Our screening platform is also general and will expedite the identification of more unique luciferases and luciferins, further expanding the bioluminescence toolkit.
Graphical Abstract
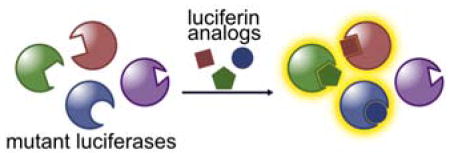
Introduction
Bioluminescence imaging is a popular method for visualizing cells and other biological features in vivo.1 This technology relies on enzymes (luciferases) that catalyze the oxidation of small molecule substrates (luciferins). The oxidation process is accompanied by the release of light (Figure 1A). Since mammalian cells and tissues do not emit substantial numbers of photons, bioluminescent light can facilitate sensitive imaging in these environments.2 Luciferase-labeled cells can also be imaged repeatedly and noninvasively in a variety of preclinical models. This broad dynamic range has enabled numerous studies of fundamental biological processes, including cell homing and differentiation, proliferation, and cell-to-cell communication, in physiologically relevant environments.3
Figure 1. Expanding the bioluminescence toolkit with unique enzyme-substrate pairs.

(A) Luciferase-mediated light production proceeds via an adenylation–oxidation sequence. (B) Strategy to develop orthogonal luciferase-luciferin pairs via substrate resolution. Genetically engineered luciferases and chemically modified luciferins were screened to identify novel partners. Only complementary enzyme-substrate pairs interact to produce light. (C) Model of D-luciferin bound to firefly luciferase (Fluc). (D) Synthesis of C7′ (left) and C4′ (right) sterically modified luciferins.
While versatile, bioluminescence to date has been largely limited to monitoring one cell type or biological feature at a time. This is due, in part, to a lack of distinguishable luciferase-luciferin pairs for in vivo use. The optimal luciferases (from the insect family) use the same substrate, D-luciferin.1,4 Thus, they cannot easily discriminate multiple cell types in a single subject. Additionally, unlike fluorescent protein technologies, a diverse suite of accessible bioluminescent probes does not yet exist. To address this void, D-luciferin analogs have been engineered to emit different colors of light.5–7 However, these substrates are still utilized by the same luciferases, precluding the distinct genetic tagging of individual cell types. Insect luciferases have also been engineered to emit different colors of light with D-luciferin.8–10 The observed emission spectra are not sufficiently resolved, though, for routine use in complex tissues or animals. Discriminating among different wavelengths in bioluminescence (and whole body optical imaging, in general) is exceedingly difficult.
Contrasting with these attempts to achieve spectral resolution, we aimed to obtain distinguishable bioluminescent probes via substrate resolution. Substrate-resolved bioluminescence is well precedented in nature, as structurally distinct luciferase-luciferin pairs have been identified across diverse phyla.11–13 Some of these pairs, including those from the firefly and Renilla reniformis have been used extensively.1,3,14–15 Firefly (Fluc) and Renilla luciferase employ chemically unique substrates (D-luciferin and coelenterazine, respectively), enabling their tandem application in vivo.16–17 Coelenterazine is less ideal for use in these environments, though, owing to its suboptimal bioavailability and stability.1,18 Other naturally occurring luciferases and luciferins can be used in combination with Fluc/D-luciferin or other bioluminescent systems.17,19 However, most of these native pairs remain poorly characterized or ill-suited for routine use.
Artificial (i.e., mutant) luciferases can exhibit altered bioluminescent properties, including tolerance for chemically modified substrates. Fluc itself has been manipulated to process analogs of D-luciferin.20 In elegant work along these lines, Miller and coworkers prepared a class of non-natural aminoluciferins that were found to be robust light emitters with Fluc, but the products inhibited the enzymatic reaction.21 Product inhibition was relieved using mutated versions of the enzyme.22 These same mutations also resulted in sharply reduced emission with D-luciferin, providing key precedent for the development and utilization of orthogonal pairs.23 The mutant enzymes from these studies, though, were less selective for one analog over another perhaps due to the structural similarities between the luciferin scaffolds. Simultaneous enzyme-substrate manipulation has also been applied to aequorin (a marine photoprotein) and the luciferase from the deep-sea shrimp Oplophorus gracilirostris.24–25 In both cases, altered bioluminescent outputs (e.g., colors and stabilities) were achieved, but orthogonal substrate usage was not realized.
Here we report a strategy for the de novo production of orthogonal luciferase-luciferin pairs. We synthesized a series of sterically modified luciferins that were poor emitters with Fluc, but intrinsically capable of robust light production. We then iteratively screened these analogs with libraries of mutant luciferases and identified substrate-selective enzymes. The “hits” were also biochemically characterized. Importantly, when the mutants and analogs were combined, robust light production was observed when complementary enzyme-substrate partners interacted. Sequential administration of substrates enabled unique luciferases to be illuminated (and thus resolved) within cultured cell models. These tools promise to enable a variety of multi-cellular imaging applications. Importantly, our approach to identifying orthogonal bioluminescence pairs is also general and should enable rapid diversification of the bioluminescence toolkit.
Results and Discussion
Designing and constructing sterically modified luciferins
To expediently identify orthogonal bioluminescence tools, we aimed to screen sterically perturbed luciferins against libraries of mutant luciferases (Figure 1B). We used the Fluc/D-luciferin pair as a starting point for several reasons. First, this duo is the most widely used in biomedical imaging applications owing to the non-toxicity of the reagents and bioavailability of the substrate.26–27 Second, the Fluc/D-luciferin reaction releases the highest percentage of tissue-penetrating light among known bioluminescent families.28 Thus, new enzymes and substrates based on the firefly pair would be more applicable to in vivo studies. Third, a wealth of structural and biochemical information on Fluc could guide our engineering efforts.13,29–32 Finally, D-luciferin derivatives are arguably the most synthetically tractable luciferin architectures.33–34
Generating an expanded set of bioluminescent tools required access to diverse luciferin scaffolds. A variety of D-luciferin analogs have been synthesized over the past four decades,5,7,35–39 and those capable of robust emission with Fluc harbor common features: an electron-donating group at the 6′ position, a carboxylate appendage (for adenylation), and an abstractable proton alpha to the carboxylate.40–41 Beyond these requirements, Fluc can tolerate a surprisingly large variety of modified luciferins,34,36,42 including 6′-amino substituents,20–21,36 alkylated43–45 and acylated46 scaffolds, and even luciferins with non-natural chromophores.6,47 Crystallographic analyses have also corroborated these experimental results, indicating flexibility within the luciferase active site and “space” to accommodate luciferins with appendages at or near the 6′-position.31–32
Unlike most efforts to produce luciferin analogs reported to date, we were attracted to the 4′ and 7′ positions of the luciferin core. These positions lie in close proximity to the Fluc backbone (Figure 1C). Substrates with additional steric bulk at these sites would likely be occluded from the Fluc active site and thus good targets for orthogonal probe development: while poor emitters with the native enzyme, the molecules could potentially give off light with designer mutants. Indeed, preliminary docking studies suggested that only analogs with small (e.g., 2–3 atoms) substituents at C4′ and C7′ could effectively access the active site (Figure S1).
Generating 4′- and 7′-modified luciferins presented an early challenge. These positions have been rarely exploited for analog development, and no prior syntheses were amenable to preparing libraries or large quantities of these probes. Rapid, high-yielding syntheses were essential, as large quantities of luciferins are required for light emission assays. Fortunately, the core benzothiazole unit (1a–c) of the desired analogs could be accessed from a common route (Figure 1D) and in multi-gram quantities.33,38 From this single intermediate, we envisioned installing functional handles at C4′ and C7′, to rapidly assemble a variety of luciferins. We were initially drawn to an aldehyde group, owing to its ease of diversification under mild conditions (e.g., reductive amination) and broad compatibility. Aldehyde installation on 1a was problematic, though, due to formation of a hydrated hemiacetal (Scheme S1).48 To circumvent this issue, we turned to more reactive iminium ions. These electrophiles can be readily trapped by electron-rich aromatics in a Mannich-type reaction.49 Toward this end, benzothiazole 1a was modified with a series of tertiary benzyl amines via in situ iminium formation and coupling (Figure 1D and Schemes S2–S3). The amino appendages were selected to enhance the water solubility of the luciferin core. Importantly, this synthetic approach was modular and amenable to large scale (1–10 g) syntheses. “Matched” probes with steric modifications at C4′ were also prepared (3a–b). A different synthetic approach was necessary, though, as the 4′ position cannot be selectively targeted with electrophiles (Figure 1D and Scheme S4).
Analyzing bioluminescent light emission with modified luciferins
With the modified luciferins in hand, we first evaluated their optical properties with Fluc. All analogs were competent light emitters and could be processed by the enzyme (Figures 2A and S2). However, the emission intensities were much weaker than those observed with D-luciferin, the native substrate. Interestingly, the largest analog (2d) was not the weakest emitter, suggesting that steric modification alone does not dictate luciferin utilization. Similar trends in light emission were observed across a range of physiological pH values (Scheme S3). Consistent with the observed light outputs, the measured kinetic constants for all analogs showed reduced performance relative to D-luciferin (Table S10). For example, the measured Km values were ~100-fold larger than the native substrate, with the largest analogs (2c and 2d) exhibiting the lowest relative binding affinities. Despite their large Km values, 2b–d exhibited emission spectra similar to D-luciferin (Figures S4–S5). Only the C4′-modifed analog 3b emitted noticeably red-shifted bioluminescent light, likely due to poor Fluc binding in the excited state32 or the luminophore being forced into a more polar environment.50–51
Figure 2. Measuring luciferin light emission.

(A) Bioluminescence from luciferin analogs (100 μM) incubated with 1 μg of Fluc. Emission intensities are plotted as total photon flux values on a log scale. Error bars represent the standard deviation of the mean for n ≥ 3 experiments. (B) Chemiluminescence with luciferin analogs. Emission intensities are plotted as counts per molar luciferin on a log scale. Error bars represent the standard error of the mean for n ≥ 3 experiments.
Measuring the light-emitting potential of luciferin analogs
We attributed the weak bioluminescence of the analogs to poor utilization by Fluc. It was possible, though, that the luciferins were simply not capable of photon production upon activation and oxidation in the active site. For productive bioluminescence, an analog must be able to reach an electronic excited state (S1) and relax back to the ground state with concomitant photon release.52–53 If an analog cannot reach S1 or emit efficiently from that state, reduced photon outputs would be expected. Such molecules would also be poor candidates for orthogonal probe development. To ensure that our lead analogs were intrinsically capable of light emission, we utilized a previously described chemiluminescence assay.54 This process mimics the enzymatic reaction itself via formation of an activated ester intermediate, followed by proton abstraction and subsequent reaction with molecular oxygen.41,53,55 When analogs 2a–d and 3a–b were subjected to the assay, robust light emission was observed (Figure 2B). In fact, photon outputs for some of the weakest bioluminescent emitters (including 2c and 3b) were on par with D-luciferin. A control compound (6′-deoxylucifeirn) lacking an electron-dense residue on the aromatic ring (a key feature of luciferins) exhibited only weak levels of emission. These results provided assurance that while luciferin scaffolds may be poor substrates for Fluc, they are still capable of photon production and thus good candidates for orthogonal tool development.
Evolving substrate-specific luciferases
Having prepared candidate orthogonal luciferins, we set out to identify mutant luciferases that could selectively process the molecules. Predicting enzyme mutations that confer substrate selectivity or otherwise beneficial properties is challenging. Fluc is a highly dynamic enzyme,31,56 complicating the selection of residues from static structural or sequence data. Moreover, amino acids known to play key roles in enzyme function have been identified far from the luciferin binding site;23 such critical residues are often revealed only by random mutagenesis approaches.57–58 Screening libraries of completely random mutants was impractical in our case, though, owing to the large library sizes needed to achieve adequate enzyme coverage.59 Screening in bulk is also difficult as bioluminescent light emission is too weak to detect on conventional cell sorters or other high-throughput instruments. Thus, each enzyme-substrate combination must be physically segregated (to a certain extent) and interrogated for light emission with a sensitive camera.
Recognizing that manual screening necessitated the use of smaller libraries, we developed focused, semi-rational libraries where the mutations were confined to regions known to modulate substrate binding.60 “Hits” from these smaller, individual libraries could then be easily combined and assayed in subsequent library generations for improved function. We initially targeted residues 218, 249–251, and 314–316 for mutagenesis (Figure 3A). These selections were partially based on phylogenetic data gathered from across the insect luciferase family,13,61 along with previous biochemical assays: Arg218 is known to interact with D-luciferin and influence the local structure of the binding pocket;29 F250 lies in close proximity (~3 Å) to the benzothiazole ring of D-luciferin; T251 has been shown to potentiate substrate binding;30 residues 314–316 line a critical edge near the luciferin phenolate and C7′ position. Mutations at all of these target sites have been shown to perturb D-luciferin binding (and thus light emission), while preserving the overall structural integrity of the enzyme.9,30,62
Figure 3. Generating mutant luciferase libraries and screening for orthogonal pairs.

(A) Amino acids targeted for mutagenesis. These residues were selected based on their proximity to the 4′ and 7′ positions of luciferin. (B) Library screening strategy. An initial on-plate screen identified functional mutants. These “hits” were subjected to a secondary screen for orthogonality with other mutants and luciferin analogs.
Saturation mutagenesis was used to prepare the desired libraries. The degree of mutation applied at each residue was based on the following considerations: sequence conservation among the insect luciferase family, the identity of the native residue, and the location of the residue. For example, non-conserved residues were mutated to a higher degree compared to conserved residues in the active site. Codon compression methods were further used to eliminate redundancies and reduce the number of transformants (Tables S1–S2).63 The final libraries ranged from 19–4800 members in size, and were constructed using synthetic gene assembly64 in combination with circular polymerase extension cloning (CPEC) (Tables S3–S9).65
The libraries were screened for orthogonal substrate usage using a two-tiered approach. Library DNA was first introduced into bacteria, and the transformants were arrayed across agar plates containing embedded luciferins (Figure 3B). Light-emitting colonies were easily identified (Figure S6) and, in some cases, the light emission values were on par with native Fluc and D-luciferin (Figure S7). A handful of the corresponding mutants were sequenced. Some mutations were observed for multiple analogs, suggesting that they might be selective for bulky luciferins (Figure S8). Other mutations were unique to each compound, which is notable, given the subtle structural differences between some of the analogs. The number of colonies screened was ~3X the calculated diversity for each library (Table S1).
While initial screens revealed functional mutants (and quickly culled non-functional enzymes), they did not report on selective substrate usage (i.e., orthogonality). The on-plate screens also did not control for overall expression levels and differences in compound transport. To address these parameters, we performed a secondary screen. Colonies emitting detectable levels of light on-plate were selected and expanded overnight. These cultures were then lysed and imaged with analogs. Mutants that provided light emission on par with native Fluc were identified as bona fide “hits” and used to create next-generation sequences. This iterative process was performed to evolve large pools of diverse, but functional enzymes. “Hits” from these subsequent generations were ultimately tested with all luciferin analogs in secondary screens.
To mine the entire collection of imaging data for substrate-selective pairs, we first developed a measure of orthogonality (equation shown in Figure 4A). Favorable values are obtained when two mutants (e.g., A and B) react robustly with unique substrates (e.g., cmpd 1 and 2, respectively), in a mutually exclusive manner. Thus, the more selective a pair of enzymes for their cognate substrates, the larger the orthogonality rating. Since the number of potential pairings exceeded 3000 in our dataset, we wrote a computer script to rapidly examine all pairs in an unbiased fashion. The program iterated through each possible combination, calculating the corresponding orthogonality rating. The script ultimately returned a list of pairs ranked by their potential for orthogonality (and thus utility for multi-component imaging).
Figure 4. Analyzing orthogonal enzyme-substrate pairs.

(A) Representative emission of luciferase mutants screened against a panel of luciferin analogs. These data were analyzed with a computer algorithm to determine lead mutants with the strongest orthogonality. (B) Purified mutants exhibit orthogonality. Enzyme (1 μg) was incubated with 100 μM of luciferin analogs and emission intensities were used to determine the orthogonality quotient (the ratio of the total flux for the C4/C7 or C7/C4 pairings). The geometric mean is plotted and the error bars represent the 95% confidence intervals for n > 4 experiments. (C) Total flux for lead mutants B and C highlights substrate selectivity between C4′ and C7′ sterically modified luciferins. Error bars represent the standard error of the mean for n > 4 experiments.
The top pairs identified by the script exhibited selectivity for analogs 3b (mutant A and B) and 2b–d (mutant C). The magnitude of each mutant’s preference—defined as the orthogonality quotient—was analyzed. As shown in Figures 4B–C, mutants A and B exhibited nearly a 100-fold preference for 3b over other analogs, while mutant C strongly favored C7′ modified analogs. Similar trends in orthogonal substrate usage were observed using bacterial lysates (Figures S9) and across a range of luciferin concentrations (Figure S10–S11). Biochemical analyses further indicated that the “brightest” mutant enzymes were those capable of most efficient substrate turnover (Table 1).
Table 1.
Biochemical analyses of orthogonal enzyme-substrate pairs.
| Enzyme | % WT light emissiona | Normalized kcat/KMb | λmax (nm) | Compound |
|---|---|---|---|---|
|
| ||||
| A | 1.2 ± 0.35 | 0.041±0.016 | 612 |
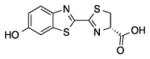 D-luc |
| B | 0.92 ±0.17 | 0.013 ± 0.004 | 616 | |
| C | 94 ± 8.4 | 5.22 ± 0.58 | 570 | |
|
| ||||
| A | 0.19±0.02 | 0.034 ± 0.008 | 614 |
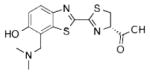 2b |
| B | 0.33 ± 0.09 | 0.050 ± 0.020 | 614 | |
| C | 17 ±5.2 | 5.0 ±1.3 | 574 | |
|
| ||||
| A | 0.16 ±0.02 | 0.253 ±0.065 | 614 |
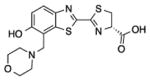 2c |
| B | 3.7 ± 0.76 | 1.09 ± 0.36 | 618 | |
| C | 16 ± 2.3 | 8.2 ±2.2 | 600 | |
|
| ||||
| A | 0.47 ±0.01 | 0.121 ±0.025 | _c |
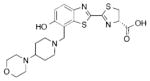 2d |
| B | 0.81 ± 0.09 | 0.155 ±0.061 | 604 | |
| C | 22 ±2.3 | 6.0 ±1.7 | 570 | |
|
| ||||
| A | 38 ±13 | 17.1 ± 6.4 | 622 |
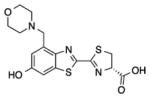 3b |
| B | 200 ± 41 | 83 ±37 | 628 | |
| C | 13 ± 2 | 13.1 ± 5.7 | 626 | |
Values normalized to each compound’s corresponding emission with WT Fluc. Errors represent standard error of the mean for n = 3 measurements.
Kinetic constants are apparent values, determined via measurements of initial rates of light emission over a range of 2 μM to 10 mM. Errors represent standard error of the mean for n≥3 measurements. kcat values are relative to each compound’s corresponding value with WT Fluc. Errors represent standard error of the mean for n ≥ 3 measurements.
λmax value could not be determined due to low level of light emission.
Analyzing the origins of orthogonality
The identities of the mutant “hits” provided some insights into the origins of substrate orthogonality. Mutant A had a single arginine to alanine mutation at amino acid 218. Mutant B comprised the same R218A mutation, but harbored additional mutations at residue 250 (Phe to Met), 314 (Ser to Thr), and 316 (Gly to Thr). These residues are known to play a role in modulating binding and interaction with the luciferin substrate. The R218A mutant is especially interesting, as it is known to greatly reduce light production and red shift emission with D-luciferin.29 It has been hypothesized that the smaller Ala group allows more water molecules to access the active site, potentially quenching light emission.29 The bulky morpholino substituent of 3b could fill this active site void to retain photon production. The third mutant (mutant C) was more selective for the C7′-modified luciferins compared to the C4′-modified compound. Mutant C harbored a single mutation, R218K. R218K may slightly enlarge the active site of the luciferase. This mutation has also been shown to boost activity with bulkier cyclic aminoluciferin analogs.22 The improved selectivity with 2b–d could be the result of active site positioning. The C7′-subsitituents could potentially place the luminophore in a more advantageous spot for light emission.
To delve into the origins of selectivity, we prepared a small library of additional mutants based on enzyme B (R218A, F250M, S314T, G316T). R218A seemed critical for discriminating the regioisomeric compounds, so this residue was held constant across the series. All possible combinations of the remaining mutations (F250M, S314T, G316T, or native Fluc residues) were then allowed. Imaging analyses of these combinatorial mutants indicated that R218A and F250M were critical for luciferin discrimination (Figures S12–13). Both mutations should result in a larger active site, but why they preferentially accommodate 3b over other analogs remains unknown. It is possible that the mutations disrupt critical binding interactions with the luciferin core, but that steric appendages (e.g., on the C4′ side) retain sufficient contacts for subsequent oxidation. Indeed, when 3b was incubated with R218A/F250M, light emission was maintained (as compared to Fluc, Figure S13C). When D-luciferin and the C7′ modified analogs were incubated with this same mutant, though, light emission was drastically reduced. Interestingly, the R218A/S314T mutant exhibited an opposite trend in analog selectivity: 2b was preferred to 3b (Figure S13C). Collectively, these results suggest that mutant luciferases can be tuned to respond to unique substrates. It is also possible that enzyme orthogonality is most readily achieved not by improving the utilization of one substrate, but by diminishing reactivity with all other substrates.
Cellular imaging with orthogonal pairs
As a step toward multi-component imaging applications, we evaluated the orthogonal enzymes and probes in cultured cell models. Mammalian cell lines (HEK293 and DB7) were engineered to express orthogonal mutants A–C. Equivalent expression levels were confirmed using flow cytometry (Figure S14). Cells were then incubated with analogs 2b–d and 3b, and photon outputs were measured. As shown in Figure 5, the substrates were able to cross cell membranes and access the relevant luciferases, resulting in sustained emission. Photon production was also confined to cells expressing the complementary luciferase for each orthogonal luciferin: cells with mutant B were only visible upon treatment with analog 3b, while cells with mutant C were only visible upon treatment with analog 2b–d (Figure S15–18). Importantly, the orthogonal pairs could also distinguish unique cell types in a single imaging session. For example, DB7 cells stably expressing mutants B or C could be readily detected via sequential administration of the requisite substrates (Figure 5). Similar trends were observed upon imaging HEK293 cells (Figure S19) and co-cultures (Figure S20). These data suggest that cross-reactivity between mutants B and C and their non-orthogonal substrates is minimal. The orthogonal pairs also exhibit unique emission spectra (Figure S21) that can further enhance some multi-component imaging applications (Figure S22).
Figure 5. Imaging cells with orthogonal luciferase-luciferin pairs.

(A) Mutant luciferase-expressing DB7 cells were plated (1.5 x 105 cells/well) in 96-well black plates and sequentially incubated with C4′ and C7′ sterically modified luciferins (750 μM). Representative bioluminescence images are shown. (B) Quantification of the images from (A) after initial substrate addition. Error bars represent the standard error of the mean for experiments performed in triplicate.
Conclusions
We developed a general strategy to evolve and identify mutant versions of firefly luciferase that accept distinct, chemically modified luciferins. Bioluminescence has been largely limited to visualizing one biological feature at a time, as the most advantageous luciferases and luciferins for whole animal imaging utilize the same substrate and cannot be distinguished in vivo. To address this void, we generated a family of sterically modified luciferins that were poor substrates for firefly luciferase, but inherently capable of producing light. Using an on-plate screen, mutant versions of luciferase were identified that could also catalyze light emission with other analogs. Pools of these functional mutants were then further mined for orthogonal pairs. Some of the mutants could selectively process individual luciferins both in vitro and in cells, setting the stage for multi-component in vivo imaging.
Future studies will be aimed at generating additional bioluminescent probes with improved brightness and other optical properties. The enzyme-substrate “hits” reported here, while immediately useful, are weaker light emitters than native bioluminescent systems. Improved light outputs can be achieved using additional rounds of mutagenesis and screening. Previous studies have also demonstrated that distant mutations can profoundly influence the architecture of the luciferase active site, and these regions will be incorporated into future libraries. The screening strategy is also broadly applicable to diverse luciferins, including analogs with altered chromophores that could provide drastically different colors of light. Our results suggest that enzymes capable of discriminating even subtle substrate modifications can be readily identified. Such an outcome bodes well for generating additional orthogonal pairs and filling a long-standing void in imaging capabilities. We anticipate that collections of designer luciferins and luciferases will inspire new discoveries in a variety of disciplines, similar to how fluorescent protein technology enabled seminal advancements in numerous fields.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health (R01 GM107630 to J.A.P). W.B.P. was supported by the National Science Foundation via the BEST IGERT program (DGE-1144901) and an Allergan Graduate Fellowship. K.A.J. was supported by an institutional Chemical and Structural Biology Training Grant predoctoral fellowship (T32-GM10856). C.M.R. was supported by the National Science Foundation Graduate Research Fellowship under grant No. DGE-1321846. J.A.P. is also an Alfred P. Sloan Fellow and Dreyfus Scholar. Some experiments reported in this paper were performed at the Laboratory for Fluorescence Dynamics (LFD) at UC Irvine. The LFD is supported jointly by the National Institute of General Medical Sciences of the National Institutes of Health (8P41GM103540) and UC Irvine. Additional thanks to the members of Pedersen, Weiss, and Martin labs for reagents and experimental assistance, along with Prof. Aaron Leconte (Claremont-McKenna) and members of the Prescher lab for helpful discussions.
Footnotes
The authors declare no conflict of interest.
Experimental details, full spectroscopic data for all new compounds, and additional images. The Supporting Information is available free of charge on the ACS Publications website
References
- 1.Paley MA, Prescher JA. Medchemcomm. 2014;5:255. doi: 10.1039/C3MD00288H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prescher JA, Contag CH. Curr Opin Chem Biol. 2010;14:80. doi: 10.1016/j.cbpa.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Badr CE, Tannous BA. Trends Biotechnol. 2011;29:624. doi: 10.1016/j.tibtech.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams ST, Miller SC. Curr Opin Chem Biol. 2014;21:112. doi: 10.1016/j.cbpa.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Branchini BR, Hayward MM, Bamford S, Brennan PM, Lajiness EJ. Photochem Photobiol. 1989;49:689. doi: 10.1111/j.1751-1097.1989.tb08442.x. [DOI] [PubMed] [Google Scholar]
- 6.Jathoul AP, Grounds H, Anderson JC, Pule MA. Angew Chem Int Ed. 2014;53:13059. doi: 10.1002/anie.201405955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mofford DM, Reddy GR, Miller SC. J Am Chem Soc. 2014;136:13277. doi: 10.1021/ja505795s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Branchini BR, Ablamsky DM, Murtiashaw MH, Uzasci L, Fraga H, Southworth TL. Anal Biochem. 2007;361:253. doi: 10.1016/j.ab.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 9.Branchini BR, Ablamsky DM, Rosenman JM, Uzasci L, Southworth TL, Zimmer M. Biochemistry. 2007;46:13847. doi: 10.1021/bi7015052. [DOI] [PubMed] [Google Scholar]
- 10.Mezzanotte L, Que I, Kaijzel E, Branchini B, Roda A, Lowik C. PLoS One. 2011;6:e19277. doi: 10.1371/journal.pone.0019277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haddock SHD, Moline MA, Case JF. Ann Rev Mar Sci. 2010;2:443. doi: 10.1146/annurev-marine-120308-081028. [DOI] [PubMed] [Google Scholar]
- 12.Oba Y, Schultz DT. Adv Biochem Eng Biotechnol. 2014;144:3. doi: 10.1007/978-3-662-43385-0_1. [DOI] [PubMed] [Google Scholar]
- 13.Viviani VR. Cell Mol Life Sci. 2002;59:1833. doi: 10.1007/PL00012509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porterfield WB, Prescher JA. Curr Opin Chem Biol. 2015;24:121. doi: 10.1016/j.cbpa.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Massoud TF, Paulmurugan R, De AJ, Ray P, Gambhir SS. Curr Opin Biotechnol. 2007;18:31. doi: 10.1016/j.copbio.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhaumik S, Gambhir SS. Proc Natl Acad Sci USA. 2002;99:377. doi: 10.1073/pnas.012611099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maguire CA, Bovenberg MS, Crommentuijn MHW, Niers JM, Kerami M, Teng J, Sena-Esteves M, Badr CE, Tannous BA. Mol Ther Nucleic Acids. 2013;2:e99. doi: 10.1038/mtna.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pichler A, Prior JL, Piwnica-Worms D. Proc Natl Acad Sci USA. 2004;101:1702. doi: 10.1073/pnas.0304326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubinnyi MA, Kaskova ZM, Rodionova NS, Baranov MS, Gorokhovatsky AY, Kotlobay A, Solntsev KM, Tsarkova AS, Petushkov VN, Yampolsky IV. Angew Chem Int Ed. 2015;54:7065. doi: 10.1002/anie.201501668. [DOI] [PubMed] [Google Scholar]
- 20.Evans MS, Chaurette JP, Adams ST, Reddy GR, Paley MA, Aronin N, Prescher JA, Miller SC. Nat Methods. 2014;11:393. doi: 10.1038/nmeth.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reddy GR, Thompson WC, Miller SC. J Am Chem Soc. 2010;132:13586. doi: 10.1021/ja104525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harwood KR, Mofford DM, Reddy GR, Miller SC. Chem Biol. 2011;18:1649. doi: 10.1016/j.chembiol.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams ST, Mofford DM, Reddy GSKK, Miller SC. Angew Chem Int Ed. 2016;55:4943. doi: 10.1002/anie.201511350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rowe L, Dikici E, Daunert S. Anal Chem. 2009;81:8662. doi: 10.1021/ac9007286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. ACS Chem Biol. 2012;7:1848. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berger F, Paulmurugan R, Bhaumik S, Gambhir SS. Eur J Nucl Med Mol Imaging. 2008;35:2275. doi: 10.1007/s00259-008-0870-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Contag CH, Spilman SD, Contag PR, Oshiro M, Eames B, Dennery P, Stevenson DK, Benaron DA. Photochem Photobiol. 1997;66:523. doi: 10.1111/j.1751-1097.1997.tb03184.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhao H, Doyle TC, Coquoz O, Kalish F, Rice BW, Contag CH. J Biomed Opt. 2005;10:041210. doi: 10.1117/1.2032388. [DOI] [PubMed] [Google Scholar]
- 29.Branchini BR, Magyar RA, Murtiashaw MH, Portier NC. Biochemistry. 2001;40:2410. doi: 10.1021/bi002246m. [DOI] [PubMed] [Google Scholar]
- 30.Branchini BR, Southworth TL, Murtiashaw MH, Boije H, Fleet SE. Biochemistry. 2003;42:10429. doi: 10.1021/bi030099x. [DOI] [PubMed] [Google Scholar]
- 31.Sundlov JA, Fontaine DM, Southworth TL, Branchini BR, Gulick AM. Biochemistry. 2012;51:6493. doi: 10.1021/bi300934s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakatsu T, Ichiyama S, Hiratake J, Saldanha A, Kobashi N, Sakata K, Kato H. Nature. 2006;440:372. doi: 10.1038/nature04542. [DOI] [PubMed] [Google Scholar]
- 33.McCutcheon DC, Porterfield WB, Prescher JA. Org Biomol Chem. 2015;13:2117. doi: 10.1039/c4ob02529f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meroni G, Rajabi M, Santaniello E. Arkivoc. 2009:265. [Google Scholar]
- 35.White EH, Worther H, Field GF, Mcelroy WD. J Org Chem. 1965;30:2344. [Google Scholar]
- 36.White EH, Worther H, Seliger HH, Mcelroy WD. J Am Chem Soc. 1966;88:2015. [Google Scholar]
- 37.Woodroofe CC, Meisenheimer PL, Klaubert DH, Kovic Y, Rosenberg JC, Behney CE, Southworth TL, Branchini BR. Biochemistry. 2012;51:9807. doi: 10.1021/bi301411d. [DOI] [PubMed] [Google Scholar]
- 38.McCutcheon DC, Paley MA, Steinhardt RC, Prescher JA. J Am Chem Soc. 2012;134:7604. doi: 10.1021/ja301493d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conley NR, Dragulescu-Andrasi A, Rao JH, Moerner WE. Angew Chem Int Ed. 2012;51:3350. doi: 10.1002/anie.201105653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White EH, Worther H. J Org Chem. 1966;31:1484. doi: 10.1021/jo01343a039. [DOI] [PubMed] [Google Scholar]
- 41.Branchini BR, Behney CE, Southworth TL, Fontaine DM, Gulick AM, Vinyard DJ, Brudvig GW. J Am Chem Soc. 2015;137:7592. doi: 10.1021/jacs.5b03820. [DOI] [PubMed] [Google Scholar]
- 42.Seliger HH, Mcelroy WD, Field GF, White EH. Proc Natl Acad Sci USA. 1961;47:1129. doi: 10.1073/pnas.47.8.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woodroofe CC, Shultz JW, Wood MG, Osterman J, Cali JJ, Daily WJ, Meisenheimer PL, Klaubert DH. Biochemistry. 2008;47:10383. doi: 10.1021/bi800505u. [DOI] [PubMed] [Google Scholar]
- 44.Kojima R, Takakura H, Ozawa T, Tada Y, Nagano T, Urano Y. Angew Chem Int Ed. 2013;52:1175. doi: 10.1002/anie.201205151. [DOI] [PubMed] [Google Scholar]
- 45.Takakura H, Kojima R, Urano Y, Terai T, Hanaoka K, Nagano T. Chem Asian J. 2011;6:1800. doi: 10.1002/asia.201000873. [DOI] [PubMed] [Google Scholar]
- 46.Shinde R, Perkins J, Contag CH. Biochemistry. 2006;45:11103. doi: 10.1021/bi060475o. [DOI] [PubMed] [Google Scholar]
- 47.Kuchimaru T, Iwano S, Kiyama M, Mitsumata S, Kadonosono T, Niwa H, Maki S, Kizaka-Kondoh S. Nat Commun. 2016;7:11856. doi: 10.1038/ncomms11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones PR, Gelinas RM. J Org Chem. 1981;46:194. [Google Scholar]
- 49.Phillips JP, Barrall EM. J Org Chem. 1956;21:692. [Google Scholar]
- 50.Hirano T, Nagai H, Matsuhashi T, Hasumi Y, Iwano S, Ito K, Maki S, Niwa H, Viviani VR. Photochem Photobio Sci. 2012;11:1281. doi: 10.1039/c2pp25106j. [DOI] [PubMed] [Google Scholar]
- 51.Viviani VR, Neves DR, Amaral DT, Prado RA, Matsuhashi T, Hirano T. Biochemistry. 2014;53:5208. doi: 10.1021/bi500160m. [DOI] [PubMed] [Google Scholar]
- 52.da Silva LP, da Silva JCGE. Chemphyschem. 2012;13:2257. [Google Scholar]
- 53.Hopkins TA, Seliger HH, White EH, Cass MW. J Am Chem Soc. 1967;89:7148. doi: 10.1021/ja01002a076. [DOI] [PubMed] [Google Scholar]
- 54.Steinhardt RC, Rathbun CM, Krull BT, Yu JM, Yang Y, Nguyen BD, Kwon J, McCutcheon DC, Jones KA, Furche F, Prescher JA. ChemBioChem. 2016 doi: 10.1002/cbic.201600564. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kato D, Shirakawa D, Polz R, Maenaka M, Takeo M, Negoro S, Niwa K. Photochem Photobio Sci. 2014;13:1640. doi: 10.1039/c4pp00250d. [DOI] [PubMed] [Google Scholar]
- 56.Mao Y. Protein Eng Des Sel. 2011;24:341. doi: 10.1093/protein/gzq109. [DOI] [PubMed] [Google Scholar]
- 57.Chen MMY, Snow CD, Vizcarra CL, Mayo SL, Arnold FH. Protein Eng Des Sel. 2012;25:171. doi: 10.1093/protein/gzs004. [DOI] [PubMed] [Google Scholar]
- 58.Reetz MT, Prasad S, Carballeira JD, Gumulya Y, Bocola M. J Am Chem Soc. 2010;132:9144. doi: 10.1021/ja1030479. [DOI] [PubMed] [Google Scholar]
- 59.Reetz MT, Kahakeaw D, Lohmer R. ChemBioChem. 2008;9:1797. doi: 10.1002/cbic.200800298. [DOI] [PubMed] [Google Scholar]
- 60.Kille S, Acevedo-Rocha CG, Parra LP, Zhang ZG, Opperman DJ, Reetz MT, Acevedo JP. ACS Synth Biol. 2013;2:83. doi: 10.1021/sb300037w. [DOI] [PubMed] [Google Scholar]
- 61.Amaral DT, Arnoldi FGC, Viviani V. Luminescence. 2012;27:96. [Google Scholar]
- 62.Viviani VR, Amaral DT, Neves DR, Simões A, Arnoldi FGC. Biochemistry. 2013;52:19. doi: 10.1021/bi300740y. [DOI] [PubMed] [Google Scholar]
- 63.Pines G, Pines A, Garst AD, Zeitoun RI, Lynch SA, Gill RT. ACS Synth Biol. 2014;4:604. doi: 10.1021/sb500282v. [DOI] [PubMed] [Google Scholar]
- 64.Ness JE, Kim S, Gottman A, Pak R, Krebber A, Borchert TV, Govindarajan S, Mundorff EC, Minshull J. Nat Biotechnol. 2002;20:1251. doi: 10.1038/nbt754. [DOI] [PubMed] [Google Scholar]
- 65.Quan J, Tian J. Nat Protoc. 2011;6:242. doi: 10.1038/nprot.2010.181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.