

Abstract
Etoposide (VP16) combined with cisplatin (DDP), as the first-line chemotherapy for small cell lung cancer (SCLC), regularly confers drug resistance. The present study applied complementary (c)DNA and micro (mi)RNA microarray to identify gene and miRNA expression profiles associated with multidrug resistance (MDR) in SCLC. The VP16/DDP (VP16 combined with DDP) resistant SCLC H446/EP cell line was derived from the parental H446 cell line by continuous exposure to increasing concentrations of etoposide and cisplatin. The mRNA and miRNA expression profiles between the resistant and parental SCLC cells were analyzed by Phalanx OneArray™ mRNA and miRNA microarray, and the results were confirmed by quantitative polymerase chain reaction. The expression levels of 75 genes were downregulated whilst 40 genes were upregulated in the H446/EP cell line compared with the H446 cell line. The expression levels of 16 miRNAs were upregulated whilst 15 were downregulated in the H446/EP cell line compared with the H446 cell line. Expression profile studies indicate that the particular mRNA and miRNA alteration demonstrated in MDR of SCLC may provide potential biomolecular targets for MDR reversion.
Keywords: small cell lung cancer, gene chips, VP-16, platinol, multidrug resistance
Introduction
Lung cancer is the most common type of malignancy worldwide (1). Small cell lung cancer (SCLC), characterized by a short cell doubling time, rapid progression and early occurrence of blood-bone and lymph metastasis, accounts for 10–15% of all patients with lung cancer with the highest malignancy (2). The combination of etoposide and cisplatin (VP16/DDP) is widely used as a first-line treatment for SCLC, which exhibits a good initial response (3). However, the relatively rapid emergence of multidrug resistance (MDR), resulting in relapse or disease progression, limits the therapeutic benefit and represents a substantial obstacle for SCLC chemotherapy (4,5). It is necessary to identify and understand the aberrant mechanisms underlying drug resistance so as to ameliorate the strategies for SCLC treatment.
Micro (mi)RNAs, a class of non-coding RNA of 19–25 nucleotides, negatively modulate gene expression at the post-transcriptional level (6). As previously demonstrated, miRNAs participate in a number of fundamental biological processes, including proliferation, differentiation and apoptosis (7–9). In addition, it has been demonstrated that miRNA exhibits diverse functions in oncogenesis, as oncogenes or anti-oncogenes (10,11). Aberrant microRNA expression is correlated with tumorigenesis, metastasis, invasiveness and drug resistance (12–14). However, the involvement of miRNAs in drug resistance in SCLC is not clearly defined, and the underlying molecular mechanisms of SCLC-associated miRNAs remain uncharacterized.
A variety of drug resistance mechanisms have been identified in oncogenesis, involving genetic and non-genetic mechanisms (15). However, the exact mechanism of MDR remains unclear, and a small number of miRNAs involved in MDR in SCLC have been identified. In addition, abnormal changes in single genes are not representative of this complex process, and are unable to determine the individualized treatment required for the elimination of MDR. The screening and identification of the molecular targets for a solution to the drug resistance of SCLC are required.
The present study aimed to determine whether alteration of the crosstalk between miRNAs and mRNAs, upon drug treatment, contributes to drug resistance. The VP16/DDP-induced multiple drug resistant SCLC H446/EP cell line was established by continuous exposure to VP16 and DDP. Microarray techniques and bioinformatics analysis were performed to identify the differences in mRNA and miRNA expression between the MDR and the parental cell lines. The successful establishment and characterization of the biological properties of the drug-resistant cell line may facilitate the investigation into the drug resistance mechanisms of SCLC.
Materials and methods
Cell lines and cell culture
The human SCLC NCI-H446 cell line was purchased from Tumor Cell Bank of the Chinese Academy of Medical Science (Shanghai, China). The cell line was maintained in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% calf serum (Hangzhou Sijiqing Bioengineering Material Co., Ltd., Zhejiang, China) with 100 µg/ml penicillin/streptomycin at 37°C with 5% CO2, and was subcultured every two or three days. The cell subline H446/EP was developed from H446 with exposure to pulse and increasing concentrations of etoposide (50, 100, 200, 400, 800 and 1,000 ng/ml) combined with cisplatin (100, 200, 400, 800 and 1,000 ng/ml). H446/EP was preserved in a final concentration of 1,000 ng/ml VP16 and 1,000 ng/ml DDP in the laboratory of Department of Medical Oncology, Jinling Hospital (Nanjing, China). The H446/EP cells were incubated in VP16/DDP-free medium for 1 month prior to additional experiments. The present study was approved by the ethics committee of Jinling Hospital of Nanjing University (Nanjing, China).
Cell viability
Cell viability was evaluated by MTT colorimetric assay. The cells were seeded at 4×103 cells per well in 96-well plates in the presence or absence of the indicated drugs for 48 h. A 0.02 ml MTT solution (5%) in PBS was added to each well followed by incubation at 37°C for 4 h. The medium was removed and the purple formazan product in the cells was measured at a wavelength of 490 nm and the number of viable cells was calculated. Dose-response curves were plotted using data derived from the MTT assay and the half maximal inhibitory concentration (IC50) of each anticancer drug was calculated from this standard curve.
Apoptosis assay
Cell apoptosis was measured using an Annexin V/FITC-PI apoptosis detection kit (Invitrogen; Thermo Fisher Scientific, Inc.) that quantitatively measures the percentages of early apoptotic cells via flow cytometry, according to the manufacturer's protocol.
Whole Genome OneArray®
Total RNA was extracted from cells using the RNeasy Mini kit (Qiagen, Inc., Valencia, CA, USA). Fluorescent antisense RNA (CyDye-aRNA) targets were prepared from 1 or 2.5 µg total RNA samples using OneArray® Amino Allyl aRNA Amplification kit (Phalanx Biotech Group, San Diego, CA, USA) and Cy5 dyes (Amersham Pharmacia; GE Healthcare Life Sciences, Chicago, IL, USA). Aminoallyl-aRNA was produced by adding aminoallyl-uridine-5′-triphosphate, prior to the addition of NHS-CyeDye, which could react with amino allyl. Fluorescent targets were hybridized to the Human Whole Genome OneArray® with Phalanx hybridization buffer using Phalanx Hybridization System. Subsequent to 16 h hybridization at 50°C, non-specific binding targets were washed away by three different washing steps (Wash I 42°C for 5 min; Wash II, 42°C for 5 min, 25°C for 5 min; Wash III, rinse 20 times), and the slides were dried by centrifugation at 671 × g and room temperature for 1 min and scanned by Axon 4000B scanner (Molecular Devices, LLC, Sunnyvale, CA, USA). The intensities of each probe were obtained by GenePix 4.1 software (Molecular Devices, LLC).
The raw intensity of each spot was loaded into Rosetta Resolver System® 7.0 (Rosetta Biosoftware; Merck KGaA, Darmstadt, Germany) to process the data analysis. The error model of Rosetta Resolver System® removed systematic and random errors from the data. Those probes with background signals were filtered out. Probes that passed the criteria were normalized by a 50% median scaling normalization method. The technical repeat data were tested by Pearson correlation coefficient calculation to check the reproducibility (R value>0.975). Normalized spot intensities were transformed to gene expression log2 ratios between the control and treatment groups. The probes with log2 ratio ≥1 or log2 ratio ≤-1 and P<0.05 were defined as differential genes for additional pathway enrichment analysis.
miRNA OneArray
Total RNA was extracted from cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Small RNA was pre-enriched by Nanoseplook (Pall Corporation, Port Washington, NY, USA) from 2.5 µg total RNA samples and labeled with miRNA ULS™ Labeling Kit (Kreatech Diagnostics; Leica Biosystems St. Louis, GmbH, Wetzlar, Germany). Labeled miRNA targets were hybridized to the Human miRNA OneArray® v3 with OneArray® Hybridization System. Subsequent to 16 h hybridization at 37°C, non-specific binding targets were washed away by three different washing steps: Wash I, 37°C for 5 min; Wash II, 37°C for 5 min, then 25°C for 5 min; Wash III, rinse 20 times), and the slides were dried by centrifugation at 671 × g and room temperature for 1 min and scanned by an Axon 4000B scanner (Molecular Devices, LLC). The Cy5 fluorescent intensities of each probe were analyzed by GenePix 4.1 software (Molecular Devices).
The raw intensity of each probe was processed by R program (version v2.12.1; https://www.r-project.org). Probes that passed the criteria were normalized by 75% median scaling normalization method. Normalized spot intensities were transformed to gene expression log2 ratios between the control and treatment groups. The spots with log2 ratio ≥1 or log2 ratio ≤-1 and P<0.05 were tested for additional analysis.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the cultured cells with TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and reverse transcribed to produce cDNA using the RT-PCR kit (catalog no., CTB101; Chutian Biosciences, Changzhou, China) on the ABI 9700 thermocycler (Applied Biosystems; Thermo Fisher Scientific, Inc.), following the manufacturer's protocol. EZ gene™ Gel/PCR Ex Kit (Biomiga, San Diego, CA, USA) was used to detect mRNA. qPCR amplifications were performed on Roche LightCycler480 II (Roche Diagnostics, Basel, Switzerland), in 20 µl volumes containing 10 µl 2X Power Taq PCR MasterMix (catalog no., CTB101; Chutian Biosciences). The thermal profile for RT-PCR was: Initial denaturation at 95°C for 5 min; followed by 40 cycles of denaturation at 95°C for 15 sec; annealing at 60°C for 15 sec; and extension at 72°C for 20 sec. Relative mRNA expression was normalized to GAPDH using the comparative ΔΔCq method; values are expressed as 2−ΔΔCq (16). All primers used were provided with the kit (catalog no., CTB000; Chutian Biosciences).
Western blot analysis
The cells were lysed in radioimmunoprecipitation assay buffer for 30 min on ice. Cell lysis supernatant liquid was obtained by centrifugation at 13,765 × g and 4°C for 15 min. Protein concentrations were determined using the bicinchoninic acid assay kit (Pierce; Thermo Fisher Scientific, Inc.). Equal amounts (15 µl) of cell lysate were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Subsequent to blocking nonspecific binding with 5% skim milk in TBS with Tween-20 (TBST) for 2 h at room temperature, membranes were incubated overnight at 4°C with primary antibodies: B-cell lymphoma-2 (BCL-2; 1:1,000; catalog no., ab59348; Abcam, Cambridge, MA, USA); BCL-2-like protein 4 (1:1,000; catalog no., ab32503; Abcam); p-gp (1:500; catalog no., ab103477; Abcam); and β-actin (1:1,000; catalog no., ab8227; Abcam). Membranes were washed with TBST (three washes, 7 min), followed by incubation with secondary antibody (anti-rabbit; catalog no., 074–1506; KPL, Gaithersburg, MD, USA) at room temperature for 2 h. Membranes were then washed with TBST (four times, 10 min) and visualized with a chemiluminescence kit (Invitrogen; Thermo Fisher Scientific, Inc.). A total of three repeats were performed for each antibody.
Statistical analysis
All data were expressed as the mean ± standard deviation. Statistical analyses were performed using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Multiple group comparisons were analyzed with one-way analysis of variance; 2-group comparisons were performed with Student's t test. P<0.05 was considered to indicate a statistically significant difference.
Results
Establishment of the VP16/DDP-resistant cell line H446/EP
Subsequent to continuous exposure to increasing concentrations of VP16 and DDP in vitro for 10 months, the VP16/DDP-resistant H446/EP cell line was established. Analysis suggested that the phenotypic diversity of the two cell lines was significant. The parental cells (Fig. 1A) appeared to be small, and mainly round and spindle shaped. In contrast, the H446/EP cells (Fig. 1B) were characterized as irregular polygons, with increased cell sizes and more intracellular metastasis. The H446/EP cells exhibited more slender pseudopodia prior to cell-fusion, which indicated an aptitude for metastasis. Subsequent to treatment with VP16 and DDP, H446 cells (Fig. 1C) appeared almost completely dead compared with H446/EP cells (Fig. 1D), which were stabilized with normal cell morphology.
Figure 1.

Phenotypic diversity of (A) H446 and (B) H446/EP cell lines was observed under inverted-microscope at magnification, ×400. Subsequent to incubation with VP16 and DDP in the dose of 1 µg/ml. (C) H446 cells and (D) H446/EP cells were observed under inverted-microscope, magnification ×100. (E) Apoptosis was determined by flow cytometric analysis of Annexin-V/PI staining. (F) Western blot analysis demonstrated lower apoptotic rates in the H446/EP cells compared with the H446 cells subsequent to treatment with VP-16 for 72 h. (G) Flow cytometric analysis demonstrated different cell cycle distribution between the H446 and H446/EP cell lines. *P<0.05, **P<0.01. (H) Western blot analysis demonstrated the excretion of P-gP was increased in the H446/EP cell line compared with the H446 cell line. VP-16, Etoposide; BCL-2, B-cell lymphoma 2; BAX, BCL-2-like protein 4; P-gp, P-glycoprotein. G1, cell cycle phase G1; S cell cycle phase S; G2/M, cell cycle G2/metaphase.
As illustrated in Table I, The IC50, or the drug concentration at which cell growth is inhibited by 50%, values for VP16 in the H446 and H446/EP cells were 0.35±0.15 and 19.25±1.49 µg/ml, respectively. The resistance indices of H446/EP cells to diverse anticancer drugs, VP16, DDP, epirubicin, paclitexal, vinorelbine and CPT-11 were significantly higher compared with those in H446. The result indicated that VP16/DDP-resistant cells also exhibited cross-resistance to other drugs.
Table I.
IC50 and RI values of H446 and H446/EP cell lines.
| IC50 (mean ± SD) | |||
|---|---|---|---|
| Drug | H446 | H446/EP | RI |
| VP16 | 0.35±0.15 | 19.25±1.49a | 55 |
| DDP | 0.52±0.29 | 14.56±1.35a | 28 |
| EPI | 0.65±0.34 | 5.32±0.27 | 8.18 |
| TAX | 0.11±0.43 | 0.57±0.60 | 5.18 |
| NVB | 3.39±1.35 | 7.32±1.84 | 2.16 |
| CPT-11 | 21.43±2.45 | 98.87±6.95 | 4.61 |
VP16, etoposide; DDP, cisplatin; EPI, epirubicin; TAX, paclitexal; NVB, vinorelbine; CPT-11, irinotecan. Data were represented as mean ± SD of three independent experiments.
P<0.01 vs. H446 cell lines. SD, standard deviation; RI, resistance indices.
The apoptotic rates of H446 and H446/EP increased in a dose-dependent manner subsequent to treatment with VP-16 for 72 h (Table II; Fig. 1E). The H446/EP cell line exhibited a slightly increased apoptotic rate (P<0.05) compared with the H446 cell line, although the two cell lines exhibited almost the same apoptotic rate without administration of VP-16 (Fig. 1F).
Table II.
Apoptotic rates of the H446 and H446/EP cells.
| Group | Apoptotic rate (mean ± standard deviation %) |
|---|---|
| H446 | 5.44±0.45 |
| H446 (VP16 500 ng/ml) | 11.71±0.54a |
| H446(VP16 1,000 ng/ml) | 18.42±0.51a,b |
| H446/EP | 5.72±0.55 |
| H446/EP (VP16 500 ng/ml) | 6.97±1.67 |
| H446/EP (VP16 1,000 ng/ml) | 8.6±0.47c |
P<0.01 vs. the same cell line without drug treatment
P<0.01 vs. the same cell line treated with VP16 of 500 ng/ml
P<0.05 vs. the same cell line without drug treatment.
The cell cycle distribution demonstrated a marked change (Fig. 1G). The H446/EP cell line was observed to gradually increase the S phase and G2/M population (P<0.05). This increase was accompanied by a concomitant decrease of the cell number in the G1 phase (P<0.01).
The expression level of the P-glycoprotein (P-gP) was significantly higher in the H446/EP cell line compared with the H446 cell line, indicating the increase of P-gP excretion in the drug-resistant cells (Fig. 1H). The ratios of accumulation of rhodamine fluorescence were 28.94±1.32% in H446 and 6.97±0.56% in H446/EP. The H446/EP cell line demonstrated a lower accumulation rate (P<0.05), compared with H446 (Fig. 2A).
Figure 2.

(A) Detecting accumulation of rhodamine via fluorescence demonstrated a lower accumulation of rhodamine in the H446/EP cell line compared with the H446 cell line. (B) Flow cytometry was used to quantitate the CD44 and CD133 expression. (C and D) Fluorescent exchange experiment: Green point represents Cy3 fluorescein, red dots represents Cy5 fluorescein. (E) Volcano diagram was depicted with the abscissa of log2 (fold change) and the ordinate of -log10 (P-value), using the filter conditions (>1 times fold change and P<0.05). CD, cluster of differentiation; Cy, cyanine.
As illustrated in Fig. 2B, the H446/EP cell line demonstrated a higher CD44+/CD133+ expression compared with the H446 cell line (P<0.01), and the CD44-/CD133- expression was lower (P<0.01).
mRNA expression profiles in the H446 and 446/EP cell lines
As illustrated in Fig. 2C and D, the signal of microarray hybridization was clear, and the results of local fluorescence hybridization were selected to distinguish the differences in gene expression. Subsequent to t-test analysis, significant differential expressed genes were presented in blue in the scatterplot (Fig. 2E).
Compared with H446/EP, 115 genes were expressed differently (log2, |fold change |≥3), of which 75 genes were upregulated and 40 were downregulated. Based on an analysis of the literature and significant sequences of fold change, the differences in the mRNA expression of 42 genes (Table III) were verified via RT-qPCR. PCR amplification and melting curves were as illustrated in Fig. 3A. The RT-qPCR results were 95.2% consistent with the high throughput microarray analysis results. The top 10 downregulated and 10 upregulated genes are demonstrated in Fig. 3B.
Table III.
Differential expression of genes of H446 cells vs. H446/EP cells.
| Gene | Fold change | P-value |
|---|---|---|
| ABCC6 | 1.23a | 0.810 |
| CDKN2B | 17.03 | 0.050 |
| IFI27 | 652.58 | 0.000 |
| MMP7 | 25.11 | 0.055 |
| ABCB1 | 28.05 | 0.040 |
| HSD17B2 | 6.96a | 0.140 |
| RTN1 | 133.44a | 0.010 |
| TNFSF10 | 32.45 | 0.020 |
| SOCS2 | 22.47a | 0.040 |
| NFE2 | 6.68a | 0.150 |
| FIBIN | 2.79 | 0.210 |
| FZD10 | 30.91 | 0.036 |
| PYCARD | 32.00 | 0.035 |
| CNRIP1 | 4.96a | 0.205 |
| KCTD12 | 6.50a | 0.158 |
| GNG4 | 9.38a | 0.116 |
| TRPC7 | 15.35a | 0.077 |
| TCEA3 | 2.89 | 0.238 |
| FLI1 | 35.51a | 0.030 |
| MTA1 | 15.35a | 0.050 |
| ITGB2 | 7.46a | 0.138 |
| ANPEP | 9.38a | 0.118 |
| PLAC8 | 5.98 | 0.484 |
| LUM | 15.25a | 0.068 |
| MMP1 | 155.42a | 0.012 |
| ROR2 | 4.69 | 0.115 |
| NTSR1 | 11.16a | 0.090 |
| SERPINB2 | 50.91a | 0.021 |
| PDE1A | 168.90 | 0.011 |
| DUSP10 | 3.63 | 0.235 |
| SERPINB10 | 52.71a | 0.024 |
| PLS1 | 11.96 | 0.157 |
| SDR16C5 | 1.35a | 0.747 |
| PRKAR2B | 61.39a | 0.020 |
| KCTD12 | 3.12a | 0.326 |
| PAGE1 | 15.35a | 0.050 |
| ABCG1 | 16.22 | 0.077 |
| CYGB | 2.20a | 0.450 |
| CXCR7 | 265.03 | 0.013 |
| STEAP4 | 91.14 | 0.021 |
| RERG | 74.54 | 0.025 |
| KRT81 | 1.88a | 0.530 |
Downregulated.
Figure 3.

[(A) Polymerase chain reaction amplification and melting curves. (i, ii) amplification curves and melting curves of H446 cells. (iii, iv) amplification curves and melting curves of H446/EP cells]. (B) Fold change expressions of mRNAs between H446/EP and H446 cells. The top 10 increased and 10 decreased genes were illustrated. (C) Histogram of fold change. (D) The volcano plot of H446 vs. H446/EP cells. Standard selection criteria to identify differentially expressed genes are established at a fold change ≥0.585 and P<0.05. (E) Heat map of 31 microRNAs that increased or decreased in expression at least 2-fold in the H446/EP cells compared with their expression in the H446 cells (columns: Cell lines; rows: Probe sets). Heat map indicates high or low expression relative to mean, as demonstrated in the scale. (C) blue dots, differentially expressed genes; (E) Red, high expression level; green, low expression level.
miRNA expression profiles in the H446 and H446/EP cell lines
The H446 and H446/EP cell lines demonstrated significant differences in the expression of miRNA (Fig. 3C and D). Compared with H446/EP, 31 miRNA were expressed differently (log2 |fold change| ≥1), of which 15 were upregulated and 16 were downregulated. In addition, a hierarchical cluster analysis based on the expression patterns of these miRNAs accurately separated the H446/EP cells from the H446 cells (Fig. 3E).
Bioinformatic analysis
As described above, there were 115 genes which exhibited marked differences in gene expression. The pathway enrichment analysis revealed that the main changes were concentrated in the tumor protein (p) 53 hypoxia pathway, apoptosis, the transforming growth factor β signaling pathway, colorectal cancer, the mitogen activated protein kinase signaling pathway, the peroxisome proliferator-activated receptor signaling pathway, pathways in cancer (including oncogenesis, proliferation, differentiation and apoptosis), basal cell carcinoma and the Wnt signaling pathway (Fig. 4).
Figure 4.
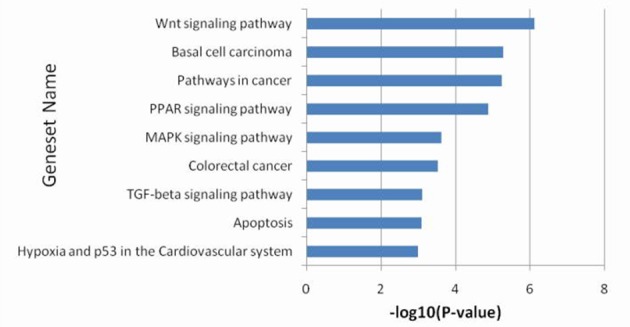
Gene set enrichment analysis of pathway was performed according to the top 10 differentially expressed genes. PPAR, peroxisome proliferator-activated receptor; MAPK, mitogen activated protein kinase; TGF, transforming growth factor.
Discussion
Although the number of clinical experiences of SCLC have increased, additional insight into the bionomics and effective treatment of this disease are required (17). The establishment of viable cell lines is essential for the study of SCLC oncogenesis, development, invasion, metastasis, and drug-resistance. Obtaining drug-resistant cells by interval or continuous exposure to chemotherapy drugs in vitro, resembling a clinical scenario, has use in investigating the mechanisms of clinical drug resistance. Previous studies have usually developed resistant cells through administrating one single drug, which is inconsistent with clinical scenarios. The H446/EP cell line is a reliable multidrug-resistant cell subline of human SCLC developed through continuous exposure to increasing concentrations of VP16/DDP. The H446/EP cells exhibited alterations in morphology compared with the parental cells, which inferred increases in invasion and metastasis capabilities.
Apoptosis is programmed cell death, and apoptosis induced by chemotherapeutic agents serves an important role in the anticancer activity of the cell (18). The specific mechanism of inhibited cell proliferation and induced cell death caused by anticancer drugs is complicated, amongst which accelerated apoptosis is one of the important mechanisms. In the present study, flow cytometry demonstrated a minimal change in the rate of apoptosis for the H446/EP cells compared with the H446 cells subsequent to incubation with VP16 for 72 h. The western blot of P-gP illustrated that the increase of P-gP excretion in H446/EP may be a potential mechanism of drug resistance (19). These data indicate that the drug-resistant cell line does not possess the biological apoptosis mechanisms that are induced by chemotherapy drugs, and it may be one of the potential mechanisms of drug resistance of SCLC.
CD44 and CD133 are important markers of lung cancer stem cells, and cancer stem cells serve an important role in the early diagnosis, survival, proliferation, metastasis and recurrence of lung cancer (20). Cancer stem cells maintain the vitality of cancer cells by self-renewal and unlimited proliferation. The results demonstrated that the proportion of CD44+/CD133+ cells increased significantly, indicating the activation, growth and proliferation of the lung cancer stem cells, and may partly explain the drug resistance in SCLC.
A previous study considered nine gene mutations to be the key drivers of SCLC, including the inactivation of antigen p53 and retinoblastoma-associated protein (RB1), recombinant mutant of histone modification gene CREB binding protein, E1A binding protein P300 and myeloid/lymphoid mixed-lineage leukemia 1, together with phosphatase and tensin homolog (PTEN), Slit guidance ligand 2, Cordon bleu WH2 repeat protein and EPHA receptor 7. Additionally, amplification of fibroblast growth factor 1, deletion of chromosome 3p, 13q, RB1, and 17p, p53, and acquisition of chromosome 3q, SRY-Box 2 (SOX2) and 5p were also significant (21). Rudin et al (22) confirmed the significance of 22 special gene mutations including p53, RB1, phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit α, cyclin-dependent kinase inhibitor 2A, PTEN, SOX2 and rearranged L-myc fusion-v-myc avian myelocytomatosis viral oncogene lung carcinoma derived homolog in the pathogenesis of SCLC. The expression of suppressor of cytokine signalling 2 was positively proportional to the malignance of cancer cells, but the exact drug-resistant mechanism requires additional examination (23). Reversing the silence of apoptosis-associated speck-like protein promotes apoptosis whilst treating cancer cells with DNA damaging agent (24). Phosphodiestera-1A (PDE1A), which may induce the growth inhibition and cycle capture of Jurkat cell (25), was predicted to be one cause of MDR in the H446 cell line, as the expression of PDE1A in the H446 cells was 168.9-fold higher compared with that in H446/EP. Reticulon-1 (RTN1) associated with intracellular transport, cell division, migration and apoptosis, were expressed in the majority of neuroendocrine tumor cells including SCLC and neuroblastoma (26). The expression of RTN1 in H446/EP cells was 133.44-fold higher compared with that in H446 cells, which indicated that neuroendocrine may be relevant to MDR of SCLC.
Previous studies have demonstrated that miRNAs function as important posttranscriptional regulators in the biological and pathological processes of lung cancer cells (27), but the associations with chemoresistance have not been fully characterized. Multiple miRNAs may target a single mRNA, whilst a single miRNA may regulate a number of mRNA molecules (28). In the present study, 31 differentially expressed miRNAs were identified between the parental and drug resistant SCLC cells, consistent with previous reports. miRNA (miR)-27a is suggested to serve important roles in proliferation and drug resistance of gastric cancer as a useful target for cancer therapy (29,30), and contributes to the chemoresistance of lung adenocarcinoma cells to cisplatin (31). By downregulating RegIV, miR-24 functions as a novel tumor suppressor in gastric cancer (32). Compared with the parental cell SGC-7901, miR-5096 is downregulated in the 5-Fluorouracil-induced drug-resistant cell line (33). The overexpression of miR-103a inhibits growth, invasion and migration of gastric cancer cells by suppressing the transcriptional activator Myb gene (34).
Multiple genes and miRNAs are involved in the resistance to chemotherapy for SCLC. Gene-chip techniques are effective in screening drug resistant genes involved in multidrug resistance of SCLC, which may reveal the mechanism of drug resistance and discover novel therapeutic targets. The data of the present study suggests the role of miRNAs and their molecular targets in drug resistance, and provides a novel data investigating chemo-sensitizing strategies through the manipulation of mRNA and miRNA expressions.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant no. 81402492) and the Natural Science Foundation of Jinling Hospital (grant nos. 2013060 and 2014013).
Glossary
Abbreviations
- SCLC
small-cell lung cancer
- DDP
cisplatin
- VP16
etoposide
- VP16/DDP
VP16 combined with DDP
- MDR
multidrug resistance
- IC50
half maximal inhibitory concentration
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Lara PN, Jr, Chansky K, Shibata T, Fukuda H, Tamura T, Crowley J, Redman MW, Natale R, Saijo N, Gandara DR. Common arm comparative outcomes analysis of phase 3 trials of cisplatin + irinotecan versus cisplatin + etoposide in extensive stage small cell lung cancer: Final patient-level results from Japan Clinical Oncology Group 9511 and Southwest Oncology Group 0124. Cancer. 2010;116:5710–5715. doi: 10.1002/cncr.25532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 5.Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract. 2005;14(Suppl 1):S35–S48. doi: 10.1159/000086183. [DOI] [PubMed] [Google Scholar]
- 6.Chekanova JA, Belostotsky DA. MicroRNAs and messenger RNA turnover. Methods Mol Biol. 2006;342:73–85. doi: 10.1385/1-59745-123-1:73. [DOI] [PubMed] [Google Scholar]
- 7.Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, Enzo E, Guzzardo V, Rondina M, Spruce T, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141:1195–1207. doi: 10.1016/j.cell.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 8.Mo YY. MicroRNA regulatory networks and human disease. Cell Mol Life Sci. 2012;69:3529–3531. doi: 10.1007/s00018-012-1123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death and tumorigenesis. Br J Cancer. 2006;94:776–780. doi: 10.1038/sj.bjc.6603023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. 2012;6:590–610. doi: 10.1016/j.molonc.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, Zucman-Rossi J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–1963. doi: 10.1002/hep.22256. [DOI] [PubMed] [Google Scholar]
- 12.Ge YZ, Xin H, Lu TZ, Xu Z, Yu P, Zhao YC, Li MH, Zhao Y, Zhong B, Xu X, et al. MicroRNA expression profiles predict clinical phenotypes and prognosis in chromophobe renal cell carcinoma. Sci Rep. 2015;5:10328. doi: 10.1038/srep10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H, Li M, Han Y, Hong L, Gong T, Sun L, Zheng X. Down-regulation of miR-27a might reverse multidrug resistance of esophageal squamous cell carcinoma. Dig Dis Sci. 2010;55:2545–2551. doi: 10.1007/s10620-009-1051-6. [DOI] [PubMed] [Google Scholar]
- 14.Farazi TA, Hoell JI, Morozov P, Tuschl T. MicroRNAs in human cancer. Adv Exp Med Biol. 2013;774:1–20. doi: 10.1007/978-94-007-5590-1_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug resistance in cancer: An overview. Cancers (Basel) 2014;6:1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.El-Khoury V, Breuzard G, Fourre N, Dufer J. The histone deacetylase inhibitor trichostatin A downregulates human MDR1 (ABCB1) gene expression by a transcription-dependent mechanism in a drug-resistant small cell lung carcinoma cell line model. Br J Cancer. 2007;97:562–573. doi: 10.1038/sj.bjc.6603914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hodkinson PS, Mackinnon AC, Sethi T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Int J Radiat Biol. 2007;83:733–741. doi: 10.1080/09553000701570204. [DOI] [PubMed] [Google Scholar]
- 19.Yeh JJ, Hsu NY, Hsu WH, Tsai CH, Lin CC, Liang JA. Comparison of chemotherapy response with P-glycoprotein, multidrug resistance-related protein-1, and lung resistance-related protein expression in untreated small cell lung cancer. Lung. 2005;183:177–183. doi: 10.1007/s00408-004-2532-1. [DOI] [PubMed] [Google Scholar]
- 20.Cichy J, Kulig P, Puré E. Regulation of the release and function of tumor cell-derived soluble CD44. Biochim Biophys Acta. 2005;1745:59–64. doi: 10.1016/j.bbamcr.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoefer J, Kern J, Ofer P, Eder IE, Schäfer G, Dietrich D, Kristiansen G, Geley S, Rainer J, Gunsilius E, et al. SOCS2 correlates with malignancy and exerts growth-promoting effects in prostate cancer. Endocr Relat Cancer. 2014;21:175–187. doi: 10.1530/ERC-13-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong S, Hwang I, Lee YS, Park S, Lee WK, Fernandes-Alnemri T, Alnemri ES, Kim YS, Yu JW. Restoration of ASC expression sensitizes colorectal cancer cells to genotoxic stress-induced caspase-independent cell death. Cancer Lett. 2013;331:183–191. doi: 10.1016/j.canlet.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abusnina A, Alhosin M, Keravis T, Muller CD, Fuhrmann G, Bronner C, Lugnier C. Down-regulation of cyclic nucleotide phosphodiesterase PDE1A is the key event of p73 and UHRF1 deregulation in thymoquinone-induced acute lymphoblastic leukemia cell apoptosis. Cell Signal. 2011;23:152–160. doi: 10.1016/j.cellsig.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 26.Yan R, Shi Q, Hu X, Zhou X. Reticulon proteins: emerging players in neurodegenerative diseases. Cell Mol Life Sci. 2006;63:877–889. doi: 10.1007/s00018-005-5338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du L, Pertsemlidis A. microRNA regulation of cell viability and drug sensitivity in lung cancer. Expert Opin Biol Ther. 2012;12:1221–1239. doi: 10.1517/14712598.2012.697149. [DOI] [PubMed] [Google Scholar]
- 28.Cherni I, Weiss GJ. miRNAs in lung cancer: Large roles for small players. Future Oncol. 2011;7:1045–1055. doi: 10.2217/fon.11.74. [DOI] [PubMed] [Google Scholar]
- 29.Zhao X, Yang L, Hu J. Down-regulation of miR-27a might inhibit proliferation and drug resistance of gastric cancer cells. J Exp Clin Cancer Res. 2011;30:55. doi: 10.1186/1756-9966-30-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian Y, Fu S, Qiu GB, Xu ZM, Liu N, Zhang XW, Chen S, Wang Y, Sun KL, Fu WN. MicroRNA-27a promotes proliferation and suppresses apoptosis by targeting PLK2 in laryngeal carcinoma. BMC Cancer. 2014;14:678. doi: 10.1186/1471-2407-14-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Wang Y, Song Y, Fu Z, Yu W. miR-27a regulates cisplatin resistance and metastasis by targeting RKIP in human lung adenocarcinoma cells. Mol Cancer. 2014;13:193. doi: 10.1186/1476-4598-13-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan Y, Hu L, Liu B, Yu B, Li J, Yan M, Yu Y, Li C, Su L, Zhu Z, et al. Tumor suppressor miR-24 restrains gastric cancer progression by downregulating RegIV. Mol Cancer. 2014;13:127. doi: 10.1186/1476-4598-13-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Gu X, Li Z, Xiang J, Jiang J, Chen Z. microRNA expression profiling in multidrug resistance of the 5-Fu-induced SGC7901 human gastric cancer cell line. Mol Med Rep. 2013;7:1506–1510. doi: 10.3892/mmr.2013.1384. [DOI] [PubMed] [Google Scholar]
- 34.Liang J, Liu X, Xue H, Qiu B, Wei B, Sun K. MicroRNA-103a inhibits gastric cancer cell proliferation, migration and invasion by targeting c-Myb. Cell Prolif. 2015;48:78–85. doi: 10.1111/cpr.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]