

ABSTRACT
Small molecule drugs and probes are important tools in drug discovery, pharmacology, and cell biology. This is of course also true for epigenetic inhibitors. Important examples for the use of established epigenetic inhibitors are the study of the mechanistic role of a certain target in a cellular setting or the modulation of a certain phenotype in an approach that aims toward therapeutic application. Alternatively, cellular testing may aim at the validation of a new epigenetic inhibitor in drug discovery approaches. Cellular and eventually animal models provide powerful tools for these different approaches but certain caveats have to be recognized and taken into account. This involves both the selectivity of the pharmacological tool as well as the specificity and the robustness of the cellular system. In this article, we present an overview of different methods that are used to profile and screen for epigenetic agents and comment on their limitations. We describe not only diverse successful case studies of screening approaches using different assay formats, but also some problematic cases, critically discussing selected applications of these systems.
KEYWORDS: Epigenetics, cellular models, screening, chemical probes, epigenetic drugs, Chip-seq, Chem-seq
Introduction
Cellular models are important tools for the characterization of new small molecule drugs and chemical probes. Vice versa, well-defined chemical tools can be used to probe the mechanistic role of a defined target in a physiologic or pathophysiological pathway. It is very important to know the abilities and limitations of both the cellular system and the small molecule. The small molecule may be designed as a biologic tool or a therapeutic agent, functions that are overlapping but also have complementary requirements. To be successful, a drug needs to achieve a desired phenotype in patients, with a side effect profile that is tolerable. In the rational optimization of drug candidates, a strong mechanistic insight involving knowledge of target potency and selectivity is highly desirable but not absolutely necessary. Thus, a significant number of drugs are successfully used without a clear understanding of their mechanism of action while many others work through interactions with multiple targets. On the other hand, a chemical probe or tool compound needs to produce the expected biologic effects in cellular models, ideally through a high affinity interaction with a single target. The review article of Arrowsmith et al.1 provides a detailed discussion on the differences between drugs and probes. For a high quality probe or pharmacological tool, for example, the Structural Genomics Consortium (SGC) has defined rigid requirements regarding selectivity and potency.9
One important consideration is the probe concentration to be used. The concentration required to demonstrate in vitro activity of the probe in mechanism-based assays for the desired target gives some guidance on the relevant doses for cellular studies. However, due to differences in uptake, stability, and efflux, the necessary concentrations for cellular studies may differ greatly between 2 compounds with similar in vitro potencies. Thus, ideally, a dose titration should be performed and both the target engagement and selectivity should be analyzed before selecting the optimum concentration relevant for the phenotypic response, keeping in mind that only a narrow window might allow the observation of specific effects in a cellular setting.
In this article, we provide an overview of different approaches for the cellular characterization of drugs and pharmacological probes, summarized in Fig. 1. Generally, these approaches are also used for testing the effect of genetic manipulation, e.g., by siRNA. The same restrictions of the cellular models will apply with regard to the interpretation of nucleic acid based interventions. In addition, the effect of chemical inhibition and knockdown or knockout will not necessarily be the same.2 With chemical inhibition, the protein is still present in the cellular network and able to interact with its partners, while a knockout removes it completely and the resulting phenotype may also stem from the alteration of scaffolding functions. For example, it was shown that histone deacetylase 6 (HDAC6) protein knockdown is not functionally equivalent to catalytic inhibition of its deacetylase activity in SKBR3 cells.3 Another example of the separation of catalytic activity and scaffolding functions of an epigenetic modifier is the action of lysine specific demethylase 1 (LSD1) in myeloid leukemia cells. Here, it was proposed that the block of differentiation induced by LSD1 is independent from its demethylase activity. Surprisingly, inhibitors targeted at its demethylase activity were still able to relieve the block of differentiation. It is not clear, therefore, whether the inhibitor action is still dependent on intact amine oxidase activity of LSD1.4 Thus, to analyze the validity of a hypothesis regarding the function of an epigenetic protein, a rescue experiment with a dominant negative function, that is lacking the critical catalytic or recognition function, should be performed. A positive example is a study on the role of the methyl lysine binding protein Spin1 (Spindlin1) in liposarcoma, where re-expression of Spin1 rescued the knockout phenotype but expression of a mutant that cannot bind H3K4me3 failed to do so.5 This proves the dependency of the phenotype on an intact trimethyl lysine recognition and provided significant rationale for drug discovery approaches targeting Spindlin1.6,7
Figure 1.
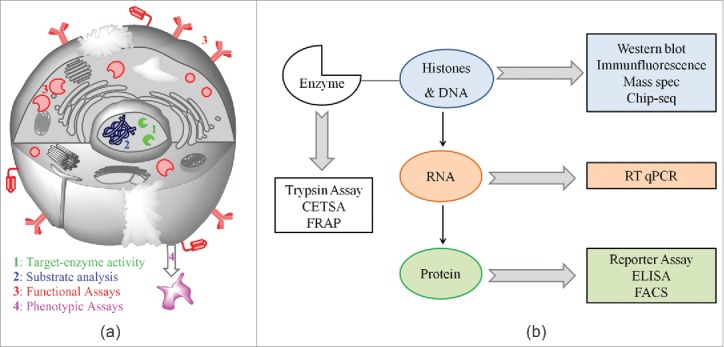
Overview of different approaches for cellular characterization of potential epigenetic modulators.
For pharmacological experiments, reference compounds and negative controls are very important. Ideally, the reference molecules should be derived from a different chemical scaffold to rule out structure specific off-target effects. For the same reason, it is ideal to have a negative control with a similar scaffold to the newly tested probe. For example, among the BRD4 inhibitors that are available from different chemical scaffolds8 the prototypic inhibitor JQ-1 and its inactive enantiomer (−)-JQ-1 can be tested side by side.9 The SGC10 is providing many probes for epigenetic targets with detailed selectivity profiles and also negative controls.
Here, we will present different methods that were useful for the cellular characterization of epigenetic probes and provide selected examples of their use, without the intention of being exhaustive in the coverage of the literature. This article is intended as a commentary and guideline for cellular studies using epigenetic inhibitors with representative examples.
Biochemical assays
Measurement of target-enzyme activity in cells or lysates
Many of the biochemical assays that are used for in vitro assays can also be applied to cellular lysates or may even work in living cells. Generally, interactions with cellular proteins might disrupt the assay; therefore, the selectivity of the assay is of great importance. If an enzyme assay involves a substrate that can be converted by a range of similar enzymes, inhibition results have to be interpreted accordingly.
Trypsin assay
The most widely used HDAC enzyme assays follow the histone deacetylase assay homogenous (HDASH) procedure.11,12 In this protocol, a derivative of acetyl lysine is deacetylated by the enzyme(s) and the product formation is measured by a subsequent detection step. This involves coupling with a protease, usually trypsin, which leads to liberation of the fluorophore AMC with a concomitant shift in fluorescence wavelength. Bonfils et al.13 discovered the cell permeability of the synthetic HDAC substrate Boc-Lys(ε-Ac)-AMC (also termed MAL14) and developed a cellular version of the HDAC assay.15 Only after the substrate got deacetylated by active deacetylases in cells or lysates, the proteinase trypsin accepts the substrate and cleaves it to form the free fluorophore AMC that can be quantitated by fluorescence spectroscopy.12 When cells have been incubated with an inhibitor and, hence, HDACs are not active in the assay, the added synthetic substrate stays acetylated and no free fluorophore is liberated by trypsin cleavage, as illustrated in Fig. 2. Thus, the trypsin assay offers a simple method to measure HDAC activity directly in cells. As most zinc-dependent HDACs accept MAL as substrate, the assay detects global HDAC activity with a moderate subtype selectivity toward some isoforms, such as HDAC6.12 Meanwhile NAD+-dependent deacetylases (sirtuins) can be excluded, as MAL is a poor substrate for these enzymes. The derivative ZMAL15 is converted by both HDACs and sirtuins, while analogs are available with some subtype preference in vitro,16 but this has not been applied widely to the cellular setting.17
Figure 2.
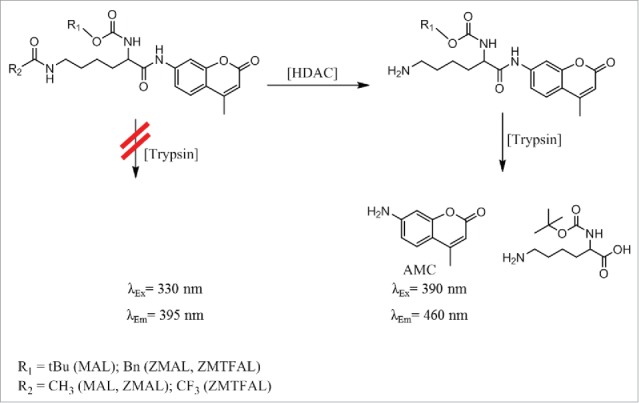
Trypsin-coupled HDAC Assay18.
The coupled trypsin assay offers the determination of IC50-values for deacetylase inhibitors in cells. Since the assay is homogenous and can be performed in 96-well plates, high-throughput testing can easily be performed. However, since deacetylase inhibition reduces cell proliferation,19 inhibitors have to be tested in a proliferation assay for the same incubation time and concentration, to ensure that effects do not arise due to reduced cell numbers. To avoid anti-proliferative effects in trypsin assays, incubation times are usually kept at shorter time scales (a few hours) as compared with antiproliferative tests (2–4 days).
A similar assay, the HADC-Glo I/II, has recently been established for a 1536-well cell-based assay.20 In the assay, a cell permeable HDAC substrate is used that can be converted by a protease into a luciferase substrate upon deacetylation by endogenous HDAC I or II.
Cellular thermal shift assay
Most cellular assays do not measure target engagement directly, but involve readouts of downstream processes. For successful drug discovery efforts, direct target engagement of lead inhibitors should be examined early in the project. Nordlund et al.21 developed an assay to directly monitor target engagement of a drug in cells, the cellular thermal shift assay (CETSA). The underlying principle is the same principle as in thermal shift assays with recombinant isolated proteins.22 The target protein is put under thermal stress in a cellular setting and specific inhibitors may lead to thermal stabilization. This can be performed both with lysates and intact cells. Generally, the temperature-dependent Gibbs free energy of unfolding ΔGu determines protein stability. When proteins are exposed to increasing temperatures their ΔGu usually decreases until it becomes zero at equilibrium, where the concentrations of unfolded and folded proteins are equal. The temperature at the equilibrium is referred to as the melting temperature of the specific protein. If a compound engages its target, it can induce a stabilization of the protein in the free energy of binding, which can result in increased ΔGu and higher melting temperature of the protein will be measured.22 A schematic illustration of this effect is shown in Fig. 3.
Figure 3.
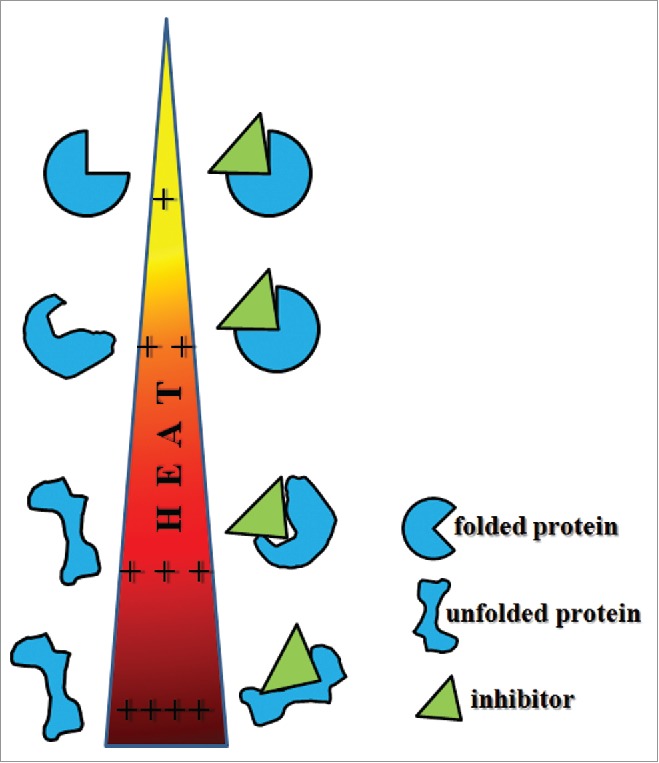
Schematic illustation of protein stability upon heat exposure. Small molecules bound to a protein can result in increased protein stability. CETSA experiments take advantage of this effect.
In a CETSA experiment, whole cells or lysates are aliquoted and heated to different temperatures. The amount of soluble protein in each aliquot can be quantified by different techniques, such as Western blotting, mass spectrometry, or AlphaScreen assays.23 The melting temperature is determined by the inflexion point of a curve plotted as temperature versus quantified soluble protein. The stabilizing effect when compounds bind to a protein is concentration and affinity dependent,22 which enables to monitor drug concentration effects in an isothermal dose-response experiment (ITDRFCETSA). However, half-saturation points of this method are normally reached at higher concentrations than in standard dose-response experiments due to the applied temperatures that go beyond 37°C. Obtained values from ITDRFCETSA experiments would need further modulation for assessment of drug affinity at 37°C. Nevertheless, dose-response effects can be examined with ITDRFCETSA experiments.21 The procedure is time consuming and quite costly and, thus, usually not suitable for screening of larger numbers of compounds. Also, no functional inhibition is monitored and negative results do not exclude target engagement or inhibition of the target enzyme, as not all enzyme/inhibitor engagements increase ΔGu upon binding. Here, a comparison with a reference inhibitor or ligand is very important. Thus, only positive results are meaningful results, at least for new and so far untested target/inhibitor combinations.
When the data of thermal shift assays are interpreted, it is valuable to consider the possible different reasons for stabilization. It can occur because of enthalpy and/or entropy effects and different ligands may exert different impact with respect to these effects. Larger temperature shifts were observed from more entropically-driven binding.22 Thus, it is difficult to quantify the affinity of compounds by their stabilization effect of a target and usually they are best used for ranking within a compound class with similar physicochemical properties.24 Still, models for the quantitative evaluation of thermal shift assays have been established,25,26 and have also been applied to epigenetic targets, at least in vitro. An example is the binding of the ligand A366 to the methyl lysine binding protein Spindlin1.6
Fluorescence recovery after photobleaching
Fluorescence recovery after photobleaching (FRAP) is a method to quantify molecular binding interactions in living cells with fluorescent-labeled proteins (e.g., with GFP). A single fluorescent cell gets exposed to a high-intensity laser pulse, which induces a bleached spot on the cell without alterations of cell function or structure. At frequent intervals after the bleaching, fluorescence images at low laser intensity are taken to determine the rate at which fluorescent molecules relocate into the photobleached spot. Collected data of a FRAP experiment are plotted by fluorescence intensity versus time. These recovery curves give information about diffusion and binding of the fluorescent protein to its targets in a cell. The degree of delaying the fluorescence recovery is dependent to the strength of binding. To examine if binding interactions and not only diffusion are involved in a FRAP recovery curve, the data of an inactive control protein have to be compared with that of active proteins. If recovery time of the curve with active protein is slower than that of the control, binding interactions are involved. Only if diffusion is much faster than binding interactions, it can be neglected. Diffusion-coupled or diffusion-uncoupled FRAP experiments require different mathematical data analysis methods.27
When the fluorescent-labeled enzyme is inhibited by a compound, the binding to its natural target is impaired. This difference between inhibited and natural fluorescence recovery gives information on the engagement of the tested compound with the labeled target protein.
Since epigenetic reader proteins do not have a catalytic activity, or such an activity is located at another domain of the target protein in question, many assays are not applicable. The FRAP technique offers a possibility to study target engagement in these proteins and has been applied, e.g., for potential bromodomain inhibitors. The bromodomain-containing protein of interest is labeled with GFP. When photobleaching has been applied, the rate of diffusion of unbleached proteins into the bleached region is limited by protein binding to chromatin or chromatin-associated complexes. This rate limiting step makes the movement of unbleached proteins in the bleached region slower than freely diffusible molecules. Therefore, the time needed until the bleached spot is recovered is related to protein affinity to the chromatin. If an inhibitor has bound to the bromodomain of interest, the time of recovery should increase.28 Philpott et al.28 showed that FRAP assays are broadly applicable across bromodomains when half recovery times are determined for inhibitors. The method should also be applicable for inhibitor analysis for other reader proteins, like Tudor- or chromodomain-containing proteins that bind to proteins with methylated lysines.29
Substrate analysis
Immunodetection of protein modifications
Posttranslational modifications of histones and other substrate proteins by an epigenetic modifier with enzymatic activity offer the opportunity to examine the cellular activity via immunodetection with specific antibodies. Mostly, Western blots are performed, but also enzyme-linked immunosorbent assays (ELISAs), AlphaLISA®, and immunofluorescence microscopy are possible methods to globally quantify histone modifications. For the final quantitation, normally a secondary antibody labeled with a fluorophore or enzyme (e.g., HRP) is used. In commercially available AlphaLISA® assays, the primary antibody is immobilized to acceptor beads. Donor beads are coated with streptavidin. Via a biotinylated antibody against the histone, acceptor beads and donor beads come in proximity. After laser irradiation of the donor beads, short-lived singlet oxygen gets released and only reaches acceptor beads, which are in proximity via the antibody binding. Singlet oxygen generates a chemiluminescent signal in acceptor beads which can be quantified. Highly specific primary antibodies are very important for reliable results, especially for histone methylation. The acetyl group on a lysine represents a bulky modification with a change in charge, which facilitates the development of specific antibodies for lysine acetylation, also on specific residues. In contrast, mono-, di-, and tri-methylated lysines differ only gradually in size and lipophilicity but not in charge and selective antibodies against one species (e.g., dimethylated lysine) are more difficult to obtain. Engelhofer et al. assessed over 200 antibodies for epigenetic targets and more than 25% failed in specificity tests.30 Due to the lack of uniform standard antibody validation, a website (http://compbio.med.harvard.edu/antibodies/) to share experimental results on antibody specificity has been established by Engelhofer et al.30 Dot blot experiments to exclude cross-reactivity of the antibody with other histone modifications or unmodified histones should always be performed before immunodetection experiments.
Western blot
Target engagement of histone deacetylase inhibitors has been supported in numerous publications via Western blots. Robust histone acetylation can be observed in cell lysates or histone extraction by SDS-PAGE and Western blot with specific antibodies.31 In contrast, changes in global histone methylation, after treatment with methyltransferase or demethylase inhibitors, leads to conflicting results. Several publications show hypermethylation after incubation of cells with potential demethylase inhibitors. However, the protein bands in these blots are not always convincing in their intensity differences. In other cases, after LSD1 knockout significant changes in histone methylation of H3K4me could not be shown via Western blots.32,33 Wang et al.34 recently reported that local changes in histone methylation could be observed after LSD1 inhibition, but global changes were not detected. Also Kruger et al.35 reported that they did not detect significant global changes in H3K4me1/2 levels after LSD1 inhibition. The reason for this effect is not clear. A potential factor could be that other proteins or molecules mask the modification, or demethylation is more locus-specific. Observed global histone hypermethylation in Western blots after the treatment of cells with a (potential) demethylase inhibitor might appear due to off-target effects of the compound, especially when high concentrations are applied. However, local changes in the methylation level of histones near target genes could be revealed via immunoprecipitation in several publications.35,36,4
In summary, target verification via the detection of posttranslational modifications, like histone acetylation or methylation is typically done by Western blot, but changes in global histone methylations have to be interpreted cautiously. Furthermore, Western blotting is a time consuming method and thus only allows a low throughput. ELISA type assays and AlphaLISA assays that also rely on the antibody-mediated quantitation of posttranslational effects are possible and allow a much higher throughput. One example of a successful screening campaign with ELISA assays led to the discovery of the HDAC6 inhibitor tubacin.37
Immunofluorescence, based on specific antibodies against a certain posttranslational histone modification, allows the quantitation of the modification level in single cells and can be applied to a large number of cells. After the measurement, the overall signal is corrected with regard to cell number, which enables the comparison of samples with different cell numbers.
Since detection of global hypermethylation via Western blot might prove difficult, especially in this case, immunofluorescence is a viable alternative. King et al.38 developed an immunofluorescence assay with JumonjiD2-demethylase transfected cells. In addition, immunofluorescence assays without prior transfection of the cells have been published including EC50-values.39,40
Chromatin immunoprecipitation (and variations of it)
Chromatin is a complex of macromolecules found in the nucleus of a cell and consists of DNA, protein, and RNA. The primary structure of chromatin is the nucleosome, which is formed by an octamer of 4 different histone proteins, each present twice, and wrapped around by 146 base pairs of DNA. Gene expression regulation can be achieved through direct modification of the DNA with modified nucleobases, including 5-methylcytosine and 5-hydroxymethylcytosine or through histone posttranslational modifications (PTMs), including methylation, acetylation, phosphorylation, and ubiquitylation.41 Histone PTMs are key components of the epigenome and they can affect chromatin transcriptional output, as they can modulate interactions between histones and DNA and directly affect chromatin condensation. Moreover, histone PTMs also act as docking sites for proteins that can read the mark and recruit complexes to activate or repress transcription. Histone PTMs can show altered profiles in cancer cells or after drug treatment.42
Chromatin immunoprecipitation (ChIP) is an invaluable method for studying interactions between specific proteins or modified forms of proteins and a genomic DNA region. A great advantage is that local chromatin changes can be followed. ChIP can be used to determine whether a transcription factor interacts with a candidate target gene or recognizes a certain sequence motif in a genome-wide fashion.43 The method is used with equal frequency to monitor the presence of histones with posttranslational modifications at specific genomic locations.
The first ChIP assay was developed by Gilmour and Lis44 as a technique to monitor the association of RNA polymerase II with transcribed and poised genes in Drosophila. In those early ChIP studies, UV light from a transilluminator was used to crosslink proteins to DNA irreversibly. The crosslinked chromatin was then either sonicated or cleaved with restriction enzymes to generate smaller DNA fragments, followed by immunoprecipitation with the desired antibodies. The precipitated protein-DNA fragments were then purified, treated with a protease, and analyzed by dot blot or Southern blot using a radiolabeled probe derived from the cloned DNA fragment of interest. These classical ChIP assays required very high amounts of material. The use of formaldehyde as a reversible protein-DNA and protein-protein crosslinking agent for ChIP and the use of polymerase chain reaction (PCR) to detect precipitated DNA fragments were later added as components of the modern ChIP procedure. Recently, the ChIP procedure is coupled to massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins or PTMs genome wide. Variations of classical ChIP include native ChIP that is performed in the absence of crosslinking agents and is used to examine proteins that remain stably associated with DNA during chromatin processing and immunoprecipitation. Native ChIPs, when performed to detect histone PTMs, can be performed with as little as 200 cells as starting material.45
Drug substances can also affect the transcriptional output of chromatin.46 They can alter chromatin by either directly targeting the DNA backbone or by targeting the proteins associated with it. For example, small molecules can interfere or block an epigenetic modifier protein interacting with the chromatin and therefore cause ensuing transcriptional changes. Local changes in the methylation level of histones near target genes could be revealed via immunoprecipitation after treatment of cells with LSD1 inhibitors.35,36,4 Although many drugs against epigenetic modifiers have been developed, it still remains unknown to what extent a given drug prevents or enhances protein-chromatin interactions. Therefore, an unbiased genome-wide approach has been recently established to identify functional targets of small molecules in cells and in animals to unravel drug effects across the genome; this technique is called Chem-seq.47
Briefly, cultured cells or animals are treated or fed with a biotinylated version of a given small molecule. Subsequently, the cells or the tissue from the treated animals are fixed with formaldehyde to crosslink DNA, proteins, and the small biotinylated molecules. As in regular ChIP, the DNA is extracted, sonicated, and enriched for regions containing the biotinylated molecule of interest by incubation with streptavidin magnetic beads. The enriched DNA fraction is then purified, eluted from the beads, and subjected to next generation sequencing. In contrast to a regular ChIP, where specific antibodies are used, Chem-seq makes use of a synthetic compound to identify the genome-wide binding profile of a target molecule.47 Both methods, illustrated in Fig. 4, can complement each other. For a successful Chem-seq, the biotinylated version of the synthetic compound should have a similar strength in binding the target as the unmodified drug.
Figure 4.
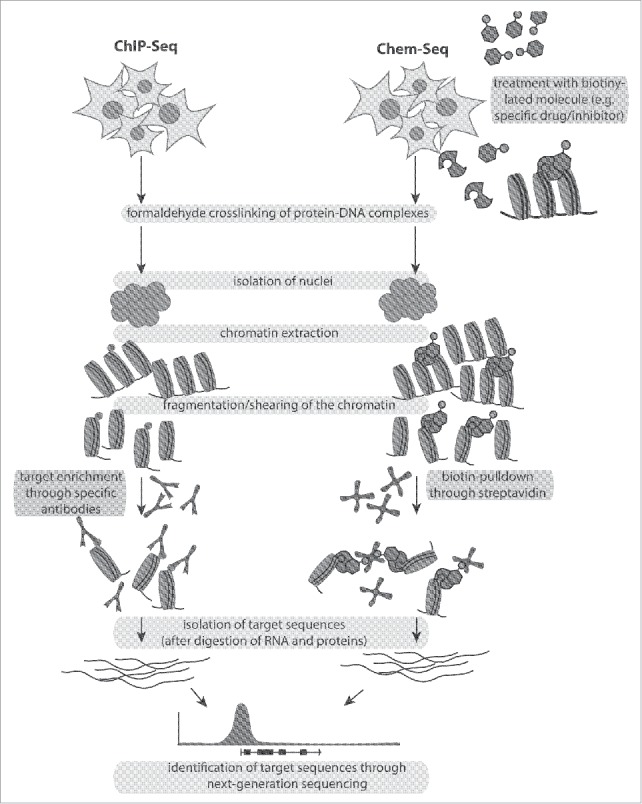
Comparative illustration of the methods ChIP-seq and Chem-seq.
JQ-1, a well-studied bromodomain inhibitor, binds to the bromodomain family members BRD2, BRD3, and BRD4 in MM1.S multiple myeloma cells. Chem-seq profiling of both the inhibitor and proteins revealed that BRD4 and JQ-1 co-occupancy occurs at >99% of the target genes. In addition, molecules that intercalate with DNA can also be mapped using Chem-Seq, e.g., psoralen could be mapped to the transcription start site of active genes.47 An in vitro variation of the protocol was successfully used for a non-membrane permeable compound. For this, the small biotinylated molecule was added after fragmentation of the chromatin.
Fluorescence resonance energy transfer imaging probes
The method, adapted by Yoshida et al.48 to analyze histone acetylation is based on conformational changes in a fusion protein of the substrate histone H4 and via a flexible linker a bromodomain-containing protein which is further fused with a donor and acceptor fluorescent protein. These 2 fluorescent proteins (CFP and Venus) serve as a FRET system. Upon conformational changes in the protein probe, the intramolecular FRET from CFP and Venus changes, resulting in a change in signal. The conformational changes occur when the bromodomain binds to acetylated substrate histone H4Kac, thereby changes in the acetylation level result in a change in signal. Similar FRET imaging probes using different bromodomains can be used to visualize different acetylation sites. Yoshida et al. have developed 2 FRET imaging probes, Histac48 with the bromodomain BRDT for acetylation of H4K5/K8 and Histac-K1249 with BRD2 for acetylation of H4K12. They have shown that both FRET probes incorporate into chromatin without causing apparent abnormalities in the nucleosome. Similar acetylation kinetics of the FRET probes and endogenous histone H4 approved the FRET probes as reversible indicators of H4 acetylation in living cells and target engagement has been visualized of various HDAC inhibitors.49 As the method has been shown to be more sensitive than immunoblot analysis of histone acetylation, it represents a powerful tool for HDAC inhibitor target engagement. But not only the cellular response on HDAC- or HAT- inhibitors can be analyzed, the technique also serves as a tool to evaluate target engagement of small molecules that interact with the bromodomains of FRET probes.
Histone mass spectrometry
The development of high sensitivity methods and their broad application has made mass spectrometry a key tool in histone tail modification analysis. However, it is a complex method and requires high expertise and modern MS/MS technical equipment with high-resolution detector systems. Also, demanding methods for the enrichment of peptides before mass spectrometry, such as capillary electrophoresis or reversed phase chromatography, are required for reliable results.50 Hung et al.51 recently published a review about the “Quantitative Proteomic Analysis of Histone Modifications” to which we want to refer here.
Chemo-proteomics by mass spectrometry has become a widely used method for the determination of selectivity and duration of target engagement. The investigated inhibitor is fused to an affinity tag to form a probe. After incubation in a cellular environment, this probe enables the isolation of inhibitor-complexes via specific trapping of the affinity tag. The methods for quantitative probe-protein complex isolation differ between chemo-proteomic methods and depict one of the biggest challenges of the method. Isolated probe-protein complexes are analyzed via quantitative mass spectrometry techniques. Thus, ideally, all targeted proteins in a cellular proteome of the inhibitor can be identified. Different methods have been established for chemo-proteomics approaches, e.g., affinity pull-down, capture compound mass spectrometry (CCMS), or activity-based protein profiling. The affinity pull-down approach uses small molecules like biotin as affinity tags combined with affinity chromatography or the inhibitor is immobilized to a solid phase directly. In the capture compound mass spectrometry method a third moiety for crosslinking is attached to an inhibitor-probe. The covalent crosslinking of probe and targets prevents equilibrium-based losses of proteins during affinity purification, resulting in higher selectivity and sensitivity of the method. The affinity-based protein profiling method adds a functional reporter tag to the inhibitor enabling visualization and enrichment of targets.52 Recently, mass spectrometry probes for epigenetic inhibitors of deacetylases, bromodomains, demethylases, and methyltransferases have been reviewed by Weigt et al.53
Generally, inhibitor derived probes should not change the functionality, specificity, or potency drastically. Different attachment sites of the sorting moiety on the inhibitor might be explored to define structure activity relationships of inhibitor and probes. A phenotypic cell response upon probe incubation versus inhibitor ought to be analyzed before proceeding with chemo-proteomic experiments.52 If structural information is available on the target-inhibitor interaction, the probe can be designed rationally.54
Functional assays
These assays cover downstream events in transcription and translation that can be functionally tied to the inhibition of epigenetic agents.
Protein analysis, invasion assay, reporter assay
Several cancer types show upregulated epigenetic enzymes, which are responsible for abnormal gene expression. This phenomenon can be used in inhibitor discovery as treatment of cells with the compound leads to changes in downstream gene transcription.55,56 Van Lint et al.57 found changes of 2% in cellular gene expression upon treatment with the HDAC inhibitor trichostatin A (TSA) in a lymphoid cell line. This unique gene change can be used as readout for target engagement of an inhibitor. Obviously, the gene change depends highly on the cell line and target enzyme subtype. Glaser et al.58 even suggests, that gene expression depends on the mechanism of inhibition rather than the chemical structure of the inhibitor, since they observed different results with the known HDAC inhibitors TSA and vorinostat (SAHA) versus entinostat (MS-275). Changes in expression patterns can be observed via Western blot, ELISA, q-PCR, microarrays, reporter gene assays, or flow cytometry (FACS), and dependent on the method, results may differ. Also, concentration and time of incubation influence results. However, as protein expression is controlled by multifaceted processes, a marker as signal of enzyme inhibition has to be chosen carefully and confirmed by several positive and negative controls before screening of new compounds can be examined. Two examples are Chou et al.,59 who reported increased cellular prostatic acid phosphatase (cPAcP) levels in prostate cancer upon treatment with the HDAC inhibitor valproic acid, and Ward et al.,60 who proposed phosphocholine to be a biomarker for HDAC inhibition in breast cancer cells. Scoumanne et al.61 identified various genes, such as DEK, S100A8, PLCL1, and ADAMTS1 regulated by LSD1. As HDACs and LSD1 have been reported to be involved in cell cycle progression,62,61,63 FACS offers a suitable method for their analysis.64 The method is highly applied in clinical studies, since only small cell numbers are required. Yu et al.65 observed, upon LSD1 inhibition with pargyline in MCF-7 breast cancer cells, significantly reduced E2-mediated S100A7 expression. In addition, differentiation markers can be epigenetically regulated and be examined via FACS. For example, Lynch et al.66 ascertained the differentiation marker CD86 as a biomarker for LSD1 inhibition in human acute myeloid leukemia (THP1 cells). They applied flow cytometry and developed an ELISA. This exemplifies the strategy for the development of a cell-based assay with a biomarker as surrogate for enzyme inhibition. However, HDAC inhibition has also been associated with increased CD86 expression.67 Differentiation of cancer and/or stem cells can show the process of epithelial-mesenchymal transition (EMT or MET). This process has been found to be controlled by epigenetic regulators68 and can be observed via cell invasion assays. Invasion is defined as cell movement through a 3D matrix. The capability of invasion requires modification of cell shape, adhesion, migration, and proteolysis of extracellular matrix components. Multiple epigenetic processes have been described in the EMT process, such as the interaction of LSD1 with Snail and Slug on the CDH1 promoter;69 also, HDAC1 and HDAC2 are involved in the process.70 However, more extracellular matrix-related genes are regulated by epigenetic enzymes, as reported for HDAC by Whetstine et al.71 This makes a cell-based invasion assay a useful tool for epigenetic inhibitor target engagement experiments and screening.
Reporter gene assays
In standard reporter assays, the target enzyme regulates the activity of an expression vector via promoter modifications. The promoter is cloned to a reporter gene, which suits as selection marker either directly, like GFP, or via an enzyme that catalyzes the formation of a quantifiable signal. As bioluminescence of reporter enzyme reactions is more sensitive than fluorescent reporters themselves, reporter enzymes became popular and widely used. The most commonly applied reporter protein in research is luciferase, which catalyzes the conversion of the substrate luciferin in an ATP-dependent manner to the luminescent oxyluciferin. However, high-throughput assays usually use the β-lactamase reporter due to several advantages, reviewed by Zlokarnik.72 For the application in epigenetic drug discovery, expression of the reporter genes is correlated with epigenetically regulated promoter activity. Martinez et al.73 described the establishment of a GFP-reporter assay for HDAC and DNMT activity with a CMV promoter. The lack of promoter activity is related to repressive chromatin structure, regulated via HDAC activity. Recently, this GFP-reporter assay has been used for screening and identification of new HDAC inhibitors.77 Another example is the promoter of p21, which is associated with HDAC binding sites.74 The HDAC inhibitor program of Novartis that led to the approval of panobinostat for the treatment of multiple myeloma, originated from a campaign that was looking for compounds that induce the overexpression of p21.75 Several promoters have also been found to be regulated by LSD1, e.g., BAT gene promoters76 or the mouse PGC-1α promoter.78 The challenge with histone modifying enzyme is the incorporation of the transgene into the nucleosome. Only stable transfection enables the cloned gene construct to engage with histones, which can be verified in Chip experiments. However, this challenge can be avoided when the function of a target enzyme as a mediator in a signal pathway is used for the regulation of the promoter of a reporter gene, e.g., reporter constructs of the TGFβ signaling pathway for HDAC inhibitor screenings.79 As LSD1 is involved in estrogen- and androgen-receptor regulation, ERα-80 and androgen-receptor81-dependent luciferase assays have been developed.
Once a reporter assay has been established, screening can be done easily and high-throughput assays are possible, which was implemented by Ashburner et al.82 with a NF-κB-dependent reporter gene. Before reporter assays are performed for inhibitor testing, compound effects on cell growth are usually assessed. Strong anti-proliferative action would show a downregulation of the reporter, leading to either false positive or false negative results. This problem can be avoided when dual reporter assays are established, where a second reporter is used to normalize gene expression and, thus, cytotoxic effects are noticed. One important complication can be direct inhibitors of luciferase, which lead to an increased luciferase activity. This is counterintuitive but can be explained by stabilization of the enzyme in the workup.83 Thus, the hits from an enzymatic reporter gene assay always need to be counter-screened for an influence on the reporter's activity.
Phenotypic assays
Phenotypic assays are methods where the effect of compounds is measured with a relatively simple global readout in a cellular setting. Phenotypic assays allow the capture of information about the reaction of a complex system to a compound. These assay systems can screen for proliferation or viability of cells, cytotoxicity, apoptosis, differentiation, and control of cell cycle. Since epigenetic modifications regulate gene expression, all of these global processes can be affected by inhibitors of epigenetic targets.
Proliferation, viability, and cytotoxicity assays
MTS assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay)
The MTS assay is a standard colorimetric method to quantify the viability of cells after they have been incubated with a compound at specific concentrations. Viability is measured via the conversion of the substrate MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] with an electron coupling reagent (phenazine ethosulfate, PES, or phenazine methosulfate, PMS) by NAD(P)H-dependent dehydrogenases in metabolically active cells, as portrayed in Fig. 5. The formazan product is quantified by measuring its absorbance of light with a wavelength of 490 nm. Cell viability is directly proportional to the amount of absorbance at 490 nm.84 Compounds have to be removed before the measurement to exclude false results due to the possibility of their absorbance at 490 nm. Through incubating cells at different concentrations of the same compound, determinations of GI50-values are possible.
Figure 5.
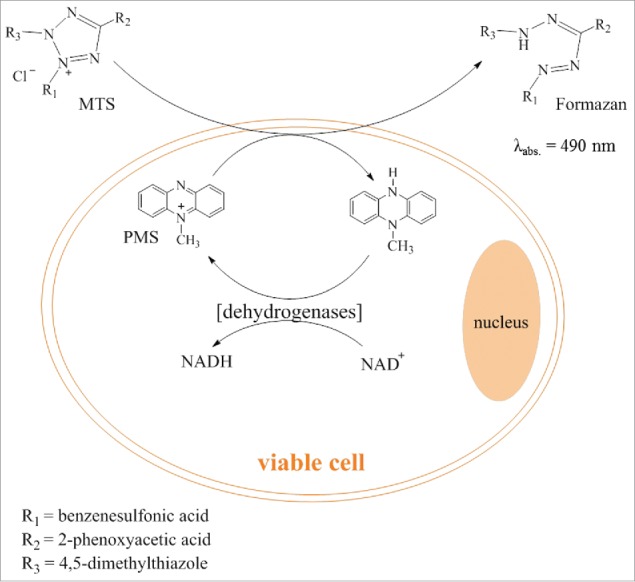
Illustration of the MTS Assay for determination of cytotoxicity.
Other assay systems, such as the CellTiter-Blue®Cell Viability Assay (resazurin) or CellTiter-Glo® Luminescent Cell Viability Assay, exist to determine cytotoxicity or viability of cells.84 The MTS assay procedure is easy and fast to perform, which makes it a widely applied assay system, also for high-throughput screening. Many publications show growth arrest of cancer cells after HDAC inhibition due to diverse reasons, such as apoptosis, autophagy, or senescence.19,56 However, LSD1 inhibition has been reported to be insensitive to cell proliferation or viability.85 Mohammad et al.86 monitored proliferation of more than 150 cancer cell lines during incubation with an LSD1 inhibitor and found only acute monocytic leukemia (AML) and some small cell lung cancer (SCLC) cell lines to be sensitive. Nevertheless, reduced cell numbers have been observed upon LSD1 knockout in breast cancer cells after extended incubation times comparable to colony formation assays.87 Since apoptosis and cell arrest are not specific readouts for a specific enzyme, off-target effects cannot be discriminated from target engagement effect in MTS assays, especially when high compound concentrations are applied. It is not clear yet if all types of Jumonji-demethylase inhibitors reduce proliferation of all cell types. Many publication show the effect; however, Itoh et al.,88 for example, only observed reduced cell growth when cells were incubated with their inhibitors in combination with the HDAC inhibitor vorinostat and, thus, off-target effects might also be the reason for cell death. Still, a phenotypic screen for the reversal of ras-transformed cells by Fujisawa was the basis for the discovery of the HDAC inhibitor romidepsin, which is approved for the treatment of cutaneous and peripheral T-cell lymphoma (CTCL and PTCL).89
Colony formation assays
Clonogenic assays utilize the ability of single cells to undergo unlimited division and proliferate into a clonogenic colony of at least 50 cells.90 Changes in the chromatin structure can result in the loss of this ability. Thus, this method offers a platform to screen epigenetic compounds. In the assay, cells are usually incubated for 10–20 days. During this prolonged incubation, all forms of cell death and the ability to proliferate have an impact on the readout. LSD1 inhibition results in gene modulation, which affects the clonogenic potential of cancer cells. Therefore, colony formation assays can be applied to test potential LSD1 inhibitors on their effect on cells. Cells can be treated before or after plating single cells on a petri dish91 or special semi solid matrix for suspension cells.92
The incorporation of several mechanisms in a single readout indicates colony formation assays results are closer to the in vivo situation than viability assays. However, the conditions of the isolated cells in a petri dish with growth media and normalized oxygen concentrations still vastly differ from the environment of a cancer cell in vivo.90
Issues with chemical probes
Characterization of chemical probes
In using epigenetic chemical probes described in the literature, a major issue is the lack of detail provided. Of course, this is often unavoidable when a new chemical probe is published and one has to wait for independent verification from other groups. Until then, the validity of the probe primarily comes from mechanism-based in vitro assays that measure its affinity for the desired target. Nowadays, kits are commercially available for many epigenetic targets and batch to batch variation can compromise assay results. Furthermore, all assays are prone to artifacts and interfering agents and the published literature should be carefully read to understand if such possibilities were considered for the new probe. Should the data for target affinity appear robust, then the next question is the probe's selectivity against related targets and the evidence for target engagement in cells or in vivo. A poorly characterized probe is not necessarily a bad one and many that were originally not described in much detail have gone on to be highly successful and widely used. However, should there be an absence of extensive literature regarding the probe, one should proceed with caution and the fact that it may be sold by catalog companies should not be taken generally as an endorsement of its validity.
Santacruzamate A (1) is a marine natural product that was recently isolated from a Symploca sp. marine cyanobacterium and bears a superficial resemblance to the clinically approved HDAC inhibitor vorinostat (2) (structures are shown in Fig. 6). Based on assay kits, santacruzamate A has a reported IC50 of 0.11 nM against HDAC2, 433 nM against HDAC4, and >1 µM against HDAC6.93 The high level of activity against HDAC2 and the selectivity are both surprising, given the absence of an obvious zinc-binding warhead typical of HDAC inhibitors and the simplicity of the structure. Although no evidence for cellular target engagement was provided, the compound was patented and sold by catalog companies. Ganesan and Wen synthesized santacruzamate A and a set of analogs, which all proved to be inactive in HDAC assays.94 The original report was clearly mistaken although the compound continues to be available as an isoform-selective HDAC inhibitor.
Figure 6.
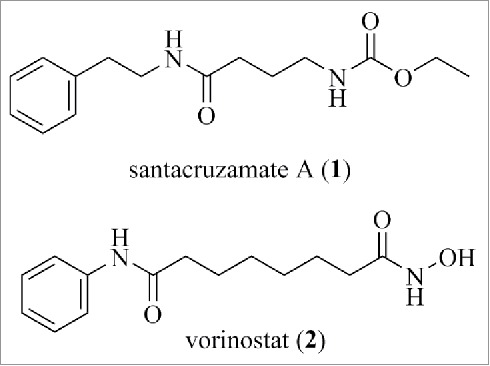
Structures of santacruzamate A (1) and vorinostat (2).
A series of synthetic aurones was reported as HDAC inhibitors, exemplified by (3) (structure is shown in Fig. 7) with an IC50 of 8 µM against HDACs from HeLa nuclear extracts, 11 µM against HDAC1, 5 µM against HDAC2, and 27 µM against HDAC6.95 Evidence for target engagement came from measurements of histone H3 acetylation in living cells using a bioluminescent resonance energy transfer technology (BRET)-based assay. Subsequently, Suzuki has shown that the aurone is a strong quencher of fluorescence in the HDAC assays and found no evidence for increased H3K9 or tubulin acetylation by Western blotting.96 Recently, the same authors who reported the aurones have identified hyrazide (4) (structure is shown in Fig. 7) as a non-hydroxamic acid selective HDAC inhibitor with an IC50 of 13 µM against HDAC6 in the fluorescent assay.97 The only evidence for target engagement was increased tubulin acetylation levels and further investigation is necessary to substantiate these claims given the unprecedented nature of hydrazides as zinc binding warheads in HDAC inhibitors.
Figure 7.
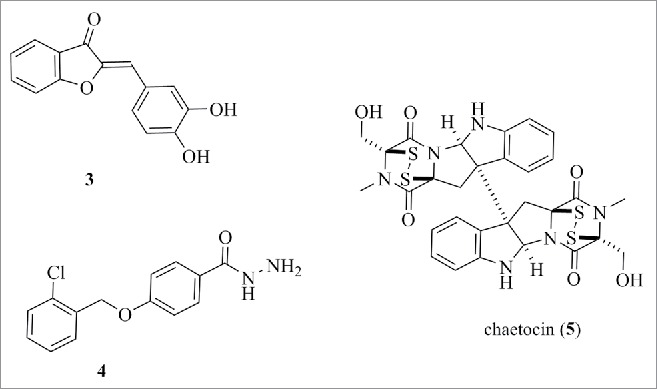
Structure of 3, 4 and chaetocin (5).
Through screening of a compound library, the natural product chaetocin (5) (structure is shown in Fig. 7) was identified as a lysine methyltransferase inhibitor. The initial profiling suggested the compound was a specific inhibitor of SU(VAR)3–9 methyltransferase, competitive with the enzyme cofactor SAM, and the activity was unrelated to the presence of the disulfide bridge.98 Later studies by Fuchter with chaetocin and synthetic analogs suggest these compounds react with the enzyme in a time-dependent and nonspecific manner and are not competitive with SAM.99,100 There is data that chaetocin is a selective inhibitor within a certain concentration range,101 but, given the chemical reactivity of disulfides and their propensity for nonselective reaction with protein thiol residues, it—and other members of this class—should not be the first choice of cellular probes for methyltransferases.
Selectivity and target affinity of chemical probes
Besides probes that are poorly characterized, another major issue arises from probes that are poor in selectivity. Some of these compounds may have robust and reproducible evidence from multiple laboratories to support engagement with an epigenetic target and even advanced to clinical development as therapeutic agents on this basis. Nevertheless, the fact remains that their target affinity is relatively modest (micromolar to almost millimolar) and the data are based on experiments using high concentrations of the probe. At such levels, there are likely to be off-target effects and, indeed, it is a common feature of such molecules to be associated with multiple unrelated targets. While some might consider such promiscuity beneficial, it will confound the interpretation of cellular assays and there needs to be convincing evidence that the phenotypic effects can be attributed to the epigenetic target.
Butyric acid (6), as the sodium salt, was the first HDAC inhibitor to be identified and this led to studies with other short chain fatty acids. Valproic acid (7), for example, is an approved drug for the treatment of epilepsy and it works primarily as a blocker of voltage-dependent sodium channels for this indication. In addition, the compound is a HDAC inhibitor with a reported IC50 of 400 µM against HDAC1.102 There are numerous reports investigating valproic acid, butyric acid and phenylbutyric acid (8) (structures are shown in Fig. 8) as HDAC inhibitors and, on this basis, the compounds are undergoing clinical trials as anticancer agents. Nevertheless, all these compounds have an IC50 > 100 µM and are likely to exhibit polypharmacology, which needs to be taken into account in the interpretation of biologic experiments. Certainly, it is difficult to understand why these fatty acids continue to be widely used as chemical probes of HDAC function, while a plethora of inhibitors, such as hydroxamic acids, benzamides, or the disulfide-containing romidepsin, have IC50 values that are 1,000-fold higher in potency. Depudecin (9) (structure is shown in Fig. 8) is another probe sold as a HDAC inhibitor although the relatively high IC50 of 5 µM in the original report103 and the presence of reactive epoxide functionality do not promote confidence in its application as a selective chemical probe.
Figure 8.
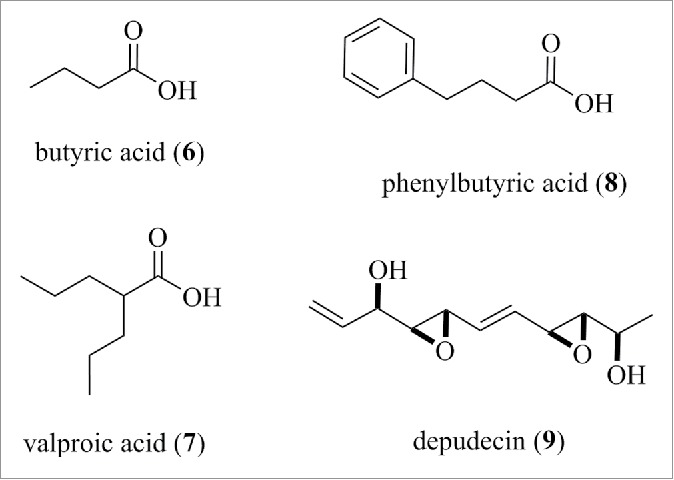
Structure 6, 7, 8, and 9.
Extensive polypharmacology is a common feature of dietary chemicals. Rather than potently inhibiting a single protein, these natural products tend to interact with large numbers of targets. While this might be useful nutritionally and help provide a therapeutic benefit when taken as dietary mixtures, such compounds in their pure state are poor choices as selective cellular probes and more likely to result in pain.104 Curcumin (10) from turmeric, epigallocatechin gallate (11) from green tea, and quercetin (12) (structures are shown in Fig. 9) in fruits and vegetables, for example, are reported to modulate multiple epigenetic pathways albeit at high concentrations.105
Figure 9.
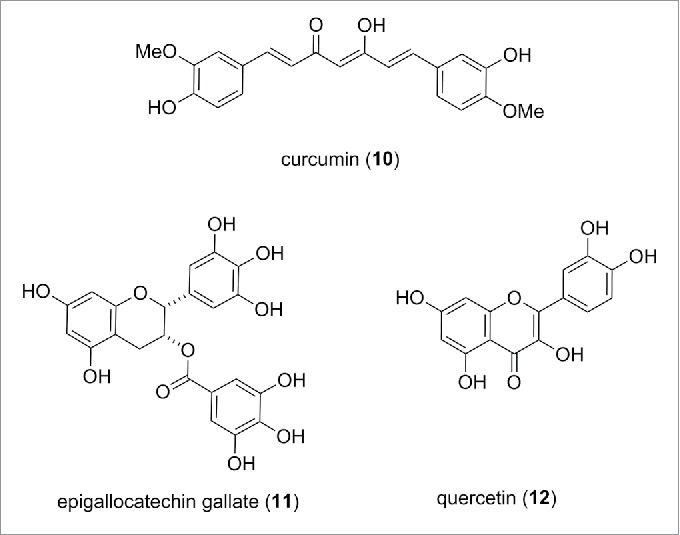
Structure of curcumin, epigallocatechin gallate and quercetin.
Recently, the flavone chrysin (13) (structure is shown in Fig. 10), which is even simpler in structure than quercetin, was screened against HDAC isoforms and found by 2 groups to selectively inhibit HDAC8,106,107 with one reporting an IC50 of 40 µM with HDAC8 and 120 µM with HDAC2. This level of selectivity for a simple flavone that does not contain a high-affinity zinc-binding warhead is unexpected. In support of target engagement, both groups showed p21 induction, although changes in histone H3 acetylation levels were contradictory. The compound displayed antitumor activity in vivo in a xenograft model, although further work is necessary to prove if this is primarily due to HDAC inhibition and if it is indeed isoform-selective. Resveratrol (14) (structure is shown in Fig. 10) was initially reported as a sirtuin activator, although this is now controversial.108 At a high concentration of 100 µM, it appears to inhibit all HDAC isoforms by 15–50%, although the physiologic relevance at such high levels is moot.109 The saturated anacardic acid (15) (structure is shown in Fig. 10), a byproduct from cashew nut shell oil, was reported as a micromolar inhibitor of the p300 histone acetyltransferase.110 The molecule's high lipophilicity, its ability to act as a surfactant, and extensive literature on other targets111 suggest its biologic effects are at best complex in nature.
Figure 10.
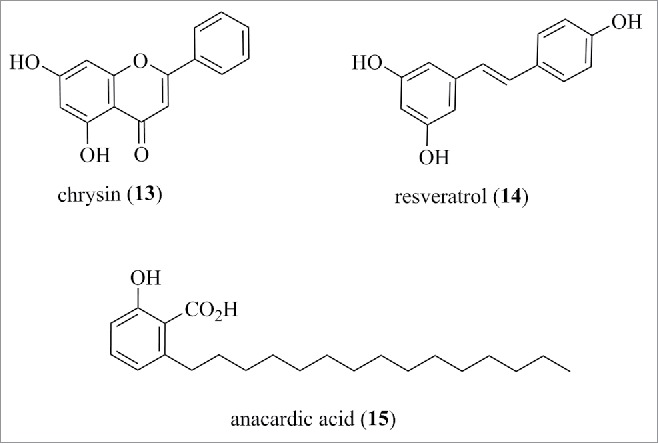
Structure of chrysin, resveratrol, anacardic acid.
Strategy for avoiding issues with chemical probes
When selecting an epigenetic chemical probe, it is helpful to go through a checklist (Table 1) before making a decision.
-
Does the probe have a high affinity (< 1 µM) for the target in vitro?
For some epigenetic targets, there is no choice as the best probes do not reach this level of affinity. However, if you are working on a target for which higher affinity probes are available, there must be a good reason for selecting one with lower affinity.
-
Is there evidence of target engagement in cellular or in vivo models?
This is critical in the choice of probe. Ideally, there should be supporting evidence of target engagement from multiple experiments and multiple laboratories.
-
Is the probe selective for a particular member of the epigenetic target family?
The importance of this question lies in the biology that is being investigated. For example, if you are interested in bromodomains generally, a nonspecific ligand like bromosporine would be a good choice. Conversely, if selectivity for the BET subfamily of bromodomains is required, then a ligand like JQ1 would be preferable.
-
Are compounds available for control experiments?
If a biologic effect is observed with the probe, it should be reproducible by a chemically unrelated probe for the same epigenetic target. On the other hand, a probe analog that is inactive is a highly useful negative control. This may, for example, involve switching a hydroxamic acid HDAC inhibitor for the corresponding amide or ester that would have much weaker affinity for the enzyme. Stereochemical switches are another option, as the enantiomers of JQ1 or i-BET ligands are equivalent in chemical connectivity and functional groups but are inactive in binding to the bromodomain.
-
Is the probe well documented in the literature?
As time goes by, a good probe will receive multiple citations and these should be examined for further information regarding its affinity and selectivity. Inevitably, the more the probe gets used, the more will be reported about its off-target effects. Having this knowledge allows one to decide whether these are relevant or not to the planned experiments and is preferable to using a new probe which appears clean simply because it has not been thoroughly investigated. Natural products may have an additional concern due to minor impurities that lead to false activity and this may be resolved only when the compound is independently made synthetically.
Table 1.
Five questions for epigenetic probe selection.
| 1. Does the probe have a high affinity (<1 µM) for the target? |
| 2. Is there evidence of target engagement in cellular or in vivo models? |
| 3. Is the probe selective for a particular member of the epigenetic target family? |
| 4. Are additional compounds available for control experiments? |
| 5. Is the probe well documented in the literature? |
Conclusions
Many methods are available to characterize the cellular effects of epigenetic inhibitors.112 These methods are applied to cultured cells, possibly also to animal samples as well as patient derived material. Some directly measure target engagement while others monitor downstream events. For an enzyme the next closest readout is the change of a level in substrate abundance, then effects on gene transcription and protein biosynthesis can be monitored subsequently, which ultimately culminate in changes of phenotypic properties, such as cell differentiation, apoptosis, or viability. The further away the assay is from the direct target of course the higher the chance that other effects lead to the same downstream response. For example, standard cytotoxic agents such as ethanol may also lead to tubulin hyperacetylation just like specific HDAC6 or Sirt2 inhibitors do.113
Thus, it is very important to be critical regarding the level of information that a cellular test provides and alternative explanations for a given readout should be considered. Still, cellular assays can be very useful for the discovery and characterization of new epigenetic drugs. Ideally, the whole chain of assays from in vitro biochemistry, target engagement, and functional and phenotypic assays is performed and good positive and negative controls are used to support the implications drawn from such assays.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the Deutsche Forschungsgemeinschaft (DFG) for funding within CRC992 (Medical Epigenetics, Project A04 to MJ, Project B06 to NI). We also thank the COST Action CM1406 Epigenetic Chemical Biology for support.
References
- 1.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, et al.. The promise and peril of chemical probes. Nat Chem Biol 2015; 11(8):536-41; PMID: 26196764; https://doi.org/ 10.1038/nchembio.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Connor CJ, Laraia L, Spring DR. Chemical genetics. Chem Soc Rev 2011; 40(8):4332-45; PMID: 21562678; https://doi.org/ 10.1039/c1cs15053g [DOI] [PubMed] [Google Scholar]
- 3.Scott GK, Marx C, Berger CE, Saunders LR, Verdin E, Schäfer S, Jung M, Benz CC. Destabilization of ERBB2 transcripts by targeting 3´ UTR mRNA associated HuR and histone deacetylase-6 (HDAC6). Mol Can Res 2008; 6(7):1250-8; PMID:18644987; https://doi.org/26893353 10.1158/1541-7786.MCR-07-2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lynch JT, Spencer GJ, Harris WJ, Maiques-Díaz A, Ciceri F, Huang X, Somervaille TCP. Pharmacological inhibitors of LSD1 promote differentiation of myeloid leukemia cells through a mechanism Independent of Histone Demethylation. Blood 2014; 124(21):267. [Google Scholar]
- 5.Franz H, Greschik H, Willmann D, Ozreti L, Jilg CA, Wardelmann E, Jung M, Buettner R, Schüle R. The histone code reader SPIN1 controls RET signaling in liposarcoma. Oncotarget 2014; 6(7):4773-89; PMID:25749382; https://doi.org/26893353 10.18632/oncotarget.3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner T, Greschik H, Burgahn T, Schmidtkunz K, Schott A-K, McMillan J, Baranauskiene L, Xiong Y, Fedorov O, Jin J, et al.. Identification of a small-molecule ligand of the epigenetic reader protein Spindlin1 via a versatile screening platform. Nucleic Acids Res 2016; 44(9):e88; PMID: 26893353; https://doi.org/ 10.1093/nar/gkw089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robaa D, Wagner T, Luise C, Carlino L, McMillan J, Flaig R, Schüle R, Jung M, Sippl W. Identification and structure-activity relationship studies of small-molecule inhibitors of the methyllysine reader Protein Spindlin1. ChemMedChem 2016; 11:1-13; PMID:27634332; https://doi.org/27382528 10.1002/cmdc.201600362 [DOI] [PubMed] [Google Scholar]
- 8.Wadhwa E, Nicolaides T, Muacevic A, Adler JR. Bromodomain inhibitor review: Bromodomain and extra-terminal family protein inhibitors as a potential New Therapy in Central Nervous System Tumors. Cureus 2016; 8(5):e620; PMID: 27382528; https://doi.org/ 10.7759/cureus.620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al.. Selective inhibition of BET bromodomains. Nature 2010; 468(7327):1067-73; PMID: 20871596; https://doi.org/ 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards AM. Structural Genomics Consortium. Toronto: SGC Toronto; 2003. [accessed 2016October17]. http://www.thesgc.org/ [Google Scholar]
- 11.Heltweg B, Jung M. A homogeneous nonisotopic histone deacetylase activity assay. J Biomol Screening 2003; 8(1):89-95; PMID:12855002; https://doi.org/16087348 10.1177/1087057102239644 [DOI] [PubMed] [Google Scholar]
- 12.Heltweg B, Trapp J, Jung M. In vitro assays for the determination of histone deacetylase activity. Methods 2005; 36(4):332-7; PMID: 16087348; https://doi.org/ 10.1016/j.ymeth.2005.03.003 [DOI] [PubMed] [Google Scholar]
- 13.Bonfils C, Kalita A, Dubay M, Siu LL, Carducci MA, Reid G, Martell RE, Besterman JM, Li Z. Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human, using a novel HDAC enzyme assay. Clin Cancer Res 2008; 11(14):3441-9; PMID:18519775; https://doi.org/10198441 10.1158/1078-0432.CCR-07-4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann K, Jung M, Brosch G, Loidl P. A non-isotopic assay for histone deacetylase activity. Nucleic Acids Res 1999; 27(9):2057-8; PMID: 10198441; https://doi.org/ 10.1093/nar/27.9.2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heltweg B, Dequiedt F, Verdin E, Jung M. Nonisotopic substrate for assaying both human zinc and NAD+dependent histone deacetylases. Anal Biochem 2003; 319(1):42-8; PMID: 12842105; https://doi.org/ 10.1016/S0003-2697(03)00276-8 [DOI] [PubMed] [Google Scholar]
- 16.Heltweg B, Dequiedt F, Marshall BL, Brauch C, Yoshida M, Nishino N, Verdin E, Jung M. Subtype selective substrates for histone deacetylases. J Med Chem 2004; 47(21):5235-43; PMID: 15456267; https://doi.org/ 10.1021/jm0497592 [DOI] [PubMed] [Google Scholar]
- 17.Gajer (née Wagner) J. Zelluläre Charakterisierung von Hemmstoffen der Histon-Acetyltransferasen und -Desacetylasen [dissertation]. Freiburg: Albert-Ludwigs-Universität Freiburg, Fakultät für Chemie, Pharmazie und Geowissenschaften; 2011. [Google Scholar]
- 18.Hauser A-T, Gajer (née Wagner) J, Jung M. Nonradioactive in vitro assays for histone deacetylases. Methods Mol Biol 2013; 981:211-27; PMID: 23381865; https://doi.org/ 10.1007/978-1-62703-305-3_17 [DOI] [PubMed] [Google Scholar]
- 19.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 37(26):5541-52; PMID:17694093; https://doi.org/26858181 10.1038/sj.onc.1210620 [DOI] [PubMed] [Google Scholar]
- 20.Hsu C-W, Shou D, Huang R, Khuc T, Dai S, Zheng W, Klumpp-Thomas C, Xia M. Identification of HDAC Inhibitors Using a Cell-Based HDAC I/II Assay. J Biomol Screen 2016; 21(6):643-52; PMID: 26858181; https://doi.org/ 10.1177/1087057116629381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013; 341(6141):84-7; PMID: 23828940; https://doi.org/ 10.1126/science.1233606 [DOI] [PubMed] [Google Scholar]
- 22.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2007; 2(9):2212-21; PMID: 17853878; https://doi.org/ 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
- 23.Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundbäck T, Nordlund P, Molina DM. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014; 9(9):2100-22; PMID: 25101824; https://doi.org/ 10.1038/nprot.2014.138 [DOI] [PubMed] [Google Scholar]
- 24.Holdgate GA, Ward WH. Measurements of binding thermodynamics in drug discovery. Drug Discovery Today 2005; 10(22):1543-50; PMID: 16257377; https://doi.org/ 10.1016/S1359-6446(05)03610-X [DOI] [PubMed] [Google Scholar]
- 25.Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry Using ThermoFluor. Biochem 2005; 44(13):5258-66; PMID:15794662; https://doi.org/18599640 10.1021/bi048135v [DOI] [PubMed] [Google Scholar]
- 26.Cimmperman P, Baranauskienė L, Jachimovič; iū tė S, Jachno J, Torresan J, Michailovienė V, Matulienė J, Sereikaitė J, Bumelis V, Matulis D. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys J 2008; 95(7):3222-31; PMID: 18599640; https://doi.org/ 10.1529/biophysj.108.134973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sprague BL, McNally JG. FRAP analysis of binding: proper and fitting. Trends Cell Biol 2005; 15(2):84-91; PMID: 15695095; https://doi.org/ 10.1016/j.tcb.2004.12.001 [DOI] [PubMed] [Google Scholar]
- 28.Philpott M, Rogers CM, Yapp C, Wells C, Lambert J-P, Strain-Damerell C, Burgess-Brown NA, Gingras A-C, Knapp S, Müller S. Assessing cellular efficacy of bromodomain inhibitors using fluorescence recovery after photobleaching. Epigenetics Chromatin 2014; 7(1):1-12; PMID: 24393457; https://doi.org/ 10.1186/1756-8935-7-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner T, Robaa D, Sippl W, Jung M. Mind the methyl: Methyllysine binding proteins in epigenetic regulation. ChemMedChem 2014; 9(3):466-83; PMID: 24449612; https://doi.org/ 10.1002/cmdc.201300422 [DOI] [PubMed] [Google Scholar]
- 30.Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung M-S, Day DS, Gadel S, Gorchakov AA, et al.. An assessment of histone-modification antibody quality. Nat Struct Mol Biol 2011; 18(1):91-3; PMID: 21131980; https://doi.org/ 10.1038/nsmb.1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc 2007; 2(6):1445-57; PMID: 17545981; https://doi.org/ 10.1038/nprot.2007.202 [DOI] [PubMed] [Google Scholar]
- 32.Jin L, Hanigan CL, Wu Y, Wang W, Park BH, Woster PM, Casero RA. Loss of LSD1 (lysine-specific demethylase 1) suppresses growth and alters gene expression of human colon cancer cells in a p53- and DNMT1(DNA methyltransferase 1)-independent manner. Biochem J 2013; 449(2):459-68; PMID: 23072722; https://doi.org/ 10.1042/BJ20121360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foster CT, Dovey OM, Lezina L, Luo JL, Gant TW, Barlev N, Bradley A, Cowley SM. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol Cell Biol 2010; 30(20):4851-63; PMID: 20713442; https://doi.org/ 10.1128/MCB.00521-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Przespolewski A, Wang ES. Inhibitors of LSD1 as a potential therapy for acute myeloid leukemia. Expert Opin Invest Drugs 2016; 25(7):771-80; PMID:27077938; https://doi.org/26175415 10.1080/13543784.2016.1175432 [DOI] [PubMed] [Google Scholar]
- 35.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al.. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 2015; 28(1):57-69; PMID: 26175415; https://doi.org/ 10.1016/j.ccell.2015.06.002 [DOI] [PubMed] [Google Scholar]
- 36.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein H-U, Popescu AC, Burnett A, Mills K, et al.. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med 2012; 4(18):605-11; PMID:22406747; https://doi.org/12677000 10.1038/nm.2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A 2003; 100(8):4389-94; PMID: 12677000; https://doi.org/ 10.1073/pnas.0430973100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.King ONF, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, et al.. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase Inhibitors. PLoS One 2010; 5(11):e15535; PMID: 21124847; https://doi.org/ 10.1371/journal.pone.0015535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo X, Liu Y, Kubicek S, Myllyharju J, Tumber A, Ng S, Che KH, Podoll J, Heightman TD, Oppermann U, et al.. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. JACS 2011; 133(24):9451-6; PMID:21585201; https://doi.org/27505861 10.1021/ja201597b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morera L, Roatsch M, Furst MCD, Hoffmann I, Senger J, Hau M, Franz H, Schüle R, Heinrich MR, Jung M. 4-Biphenylalanine- and 3-Phenyltyrosine-derived hydroxamic acids as inhibitors of the JumonjiC-domain-containing histone demethylase KDM4A. ChemMedChem 2016; 11(18):2063-83; PMID: 27505861; https://doi.org/ 10.1002/cmdc.201600218 [DOI] [PubMed] [Google Scholar]
- 41.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 2011; 12(1):7-18. ENG; PMID: 21116306; https://doi.org/ 10.1038/nrg2905 [DOI] [PubMed] [Google Scholar]
- 42.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science 2013; 339(6127):1567-70. ENG; PMID: 23539597; https://doi.org/ 10.1126/science.1230184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farnham P. Insights from genomic profiling of transcription factors. Nat Rev Genet 2009; 10(9):605-16; PMID: 19668247; https://doi.org/ 10.1038/nrg2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilmour DS, Lis JT. In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Mol Cell Biol 1985; 5(8):2009-18; PMID: 3018544; https://doi.org/ 10.1128/MCB.5.8.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang B, Zheng H, Huang B, Li W, Xiang Y, Peng X, Ming J, Wu X, Zhang Y, Xu Q, et al.. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 2016; 537(7621):553-7; PMID: 27626382; https://doi.org/ 10.1038/nature19361 [DOI] [PubMed] [Google Scholar]
- 46.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 2012; 11(5):384-400; PMID: 22498752; https://doi.org/ 10.1038/nrd3674 [DOI] [PubMed] [Google Scholar]
- 47.Anders L, Guenther MG, Qi J, Fan ZP, Marineau JJ, Rahl PB, Loven J, Sigova AA, Smith WB, Lee TI, et al.. Genome-wide localization of small molecules. Nat Biotech 2014; 32(1):92-6; PMID:24336317; https://doi.org/19805290 10.1038/nbt.2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasaki K, Ito T, Nishino N, Khochbin S, Yoshida M. Real-time imaging of histone H4 hyperacetylation in living cells. PNAS 2009; 106(38):16257-62; PMID: 19805290; https://doi.org/ 10.1073/pnas.0902150106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ito T, Umehara T, Sasaki K, Nakamura Y, Nishino N, Terada T, Shirouzu M, Padmanabhan B, Yokoyama S, Ito A, et al.. Real-time imaging of histone H4K12–specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem Biol 2011; 18(4):495-507; PMID: 21513886; https://doi.org/ 10.1016/j.chembiol.2011.02.009 [DOI] [PubMed] [Google Scholar]
- 50.Villar-Garea A, Imhof A. Histone modification analysis using mass spectrometry. Curr Protoc Protein Sci 2008; 51(14):87-97; PMID:18429056; https://doi.org/25688442 10.1002/0471140864.ps1410s51 [DOI] [PubMed] [Google Scholar]
- 51.Huang H, Lin S, Garcia BA, Zhao Y. Quantitative proteomic analysis of histone modifications. Chem Rev 2015; 115(6):2376-418; PMID: 25688442; https://doi.org/ 10.1021/cr500491u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urh M. Chemo proteomics, a valuable tool for biomarker and drug discovery. Mol Biol 2014; 3(1):e117; https://doi.org/ 10.4172/2168-9547.1000e117 [DOI] [Google Scholar]
- 53.Weigt D, Hopf C, Medard G. Studying epigenetic complexes and their inhibitors with the proteomics toolbox. Clin Epigenet 2016; 8(76):1-16; PMID:27437033; https://doi.org/26748890 10.1186/s13148-016-0244-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schiedel M, Rumpf T, Karaman B, Lehotzky A, Gerhardt S, Ovadi J, Sippl W, Einsle O, Jung M. Structure-based development of an affinity probe for sirtuin 2. Angewandte Chemie 2016; 55(6):2252-6. ENG; PMID:26748890; https://doi.org/ 10.1002/anie.201509843 [DOI] [PubMed] [Google Scholar]
- 55.Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R, Mita M, Beck J, Burris H, Ryan G, et al.. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression Profiles in Cutaneous T-Cell Lymphoma. Clin Cancer Res 2008; 14(14):4500-10; PMID: 18628465; https://doi.org/ 10.1158/1078-0432.CCR-07-4262 [DOI] [PubMed] [Google Scholar]
- 56.Glaser KB, Li J, Staver MJ, Wei R-Q, Albert DH, Davidsen SK. Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem Biophys Res Commun 2003; 310(2):529-36; PMID: 14521942; https://doi.org/ 10.1016/j.bbrc.2003.09.043 [DOI] [PubMed] [Google Scholar]
- 57.van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr 1996; 5(9):245-53. PMID: 8723390. [PMC free article] [PubMed] [Google Scholar]
- 58.Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: Defining a common gene set produced by HDAC Inhibition in T24 and MDA Carcinoma Cell Lines. Mol Can Ther 2003; 2(2):151-63; PMID:1258903225601894 [PubMed] [Google Scholar]
- 59.Chou Y-W, Lin F-F, Muniyan S, Lin FC, Chen C-S, Wang J, Huang C-C, Lin M-F. Cellular prostatic acid phosphatase (cPAcP) serves as a useful biomarker of histone deacetylase (HDAC) inhibitors in prostate cancer cell growth suppression. Cell Biosci 2015; 5(38):1-9; PMID: 25601894; https://doi.org/ 10.1186/s13578-015-0033-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ward CS, Eriksson P, Izquierdo-Garcia JL, Brandes AH, Ronen SM, Cheriyath V. HDAC inhibition induces increased choline uptake and elevated phosphocholine levels in MCF7 Breast Cancer Cells. PLoS One 2013; 8(4):e62610; PMID: 23626839; https://doi.org/ 10.1371/journal.pone.0062610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scoumanne A, Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. J Biol Chem 2007; 282(21):15471-5; PMID: 17409384; https://doi.org/ 10.1074/jbc.M701023200 [DOI] [PubMed] [Google Scholar]
- 62.Kim BY, Ki WS, Yoshida M, Horinouchi S. Mechanism of cell cycle arrest caused by histone deacetylase inhibitors in human carcinoma cells. J Antibiot 2000; 53(10):1191-200; PMID: 11132966; https://doi.org/ 10.7164/antibiotics.53.1191 [DOI] [PubMed] [Google Scholar]
- 63.Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 2001; 1(3):194-202; PMID: 11902574; https://doi.org/ 10.1038/35106079 [DOI] [PubMed] [Google Scholar]
- 64.Pozarowski P, Darzynkiewicz Z. Analysis of cell cycle by flow cytometry. Methods Mol Biol 2004; 281:301-311; PMID:15220539; 23000163https://doi.org/10.1385/1-59259-811-0:301 [DOI] [PubMed] [Google Scholar]
- 65.Yu SE, Jang YK. The histone demethylase LSD1 is required for estrogen-dependent S100A7 gene expression in human breast cancer cells. Biochem Biophys Res Commun 2012; 427(2):336-42; PMID: 23000163; https://doi.org/ 10.1016/j.bbrc.2012.09.057 [DOI] [PubMed] [Google Scholar]
- 66.Lynch JT, Cockerill MJ, Hitchin JR, Wiseman DH, Somervaille TC. CD86 expression as a surrogate cellular biomarker for pharmacological inhibition of the histone demethylase lysine-specific demethylase 1. Anal Biochem 2013; 442(1):104-6; PMID: 23911524; https://doi.org/ 10.1016/j.ab.2013.07.032 [DOI] [PubMed] [Google Scholar]
- 67.Maeda T, Towatari M, Kosugi H, Saito H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood 2000; 96(12):3847-56. PMID: 11090069 [PubMed] [Google Scholar]
- 68.Kiesslich T, Pichler M, Neureiter D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol Clin Oncol 2012; 1(1):3-11; PMID: 24649114; https://doi.org/ 10.3892/mco.2012.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010; 29(35):4896-904; PMID: 20562920; https://doi.org/ 10.1038/onc.2010.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peinado H, Ballestar E, Manuel Esteller, Amparo Cano. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol 2004; 24(1):306-19; PMID: 14673164; https://doi.org/ 10.1128/MCB.24.1.306-319.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Whetstine JR, Ceron J, Ladd B, Dufourcq P, Reinke V, Shi Y. Regulation of tissue-specific and extracellular matrix-related genes by a class I histone deacetylase. Mol Cell 2005; 18(4):483-90; PMID: 15893731; https://doi.org/ 10.1016/j.molcel.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 72.Zlokarnik G. [15]Fusions to β-lactamase as a reporter for gene expression in live mammalian cells. Methods Enzymol 2000; 326:221-41; PMID: 11036645; https://doi.org/ 10.1016/S0076-6879(00)26057-6 [DOI] [PubMed] [Google Scholar]
- 73.Martinez ED, Dull AB, Beutler JA, Hager GL. High‐Content fluorescence‐based screening for epigenetic modulators. Methods Enzymol 2006; 414:21-36; PMID: 17110184; https://doi.org/ 10.1016/S0076-6879(06)14002-1 [DOI] [PubMed] [Google Scholar]
- 74.Lin Y-C, Lin J-H, Chou C-W, Chang Y-F, Yeh S-H, Chen C-C. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res 2008; 68(7):2375-83; PMID: 18381445; https://doi.org/ 10.1158/0008-5472.CAN-07-5807 [DOI] [PubMed] [Google Scholar]
- 75.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009; 280(2):233-41; PMID:19344997; https://doi.org/27264172 10.1016/j.canlet.2009.02.019 [DOI] [PubMed] [Google Scholar]
- 76.Sambeat A, Gulyaeva O, Dempersmier J, Tharp KM, Stahl A, Paul SM, Sul HS. LSD1 interacts with Zfp516 to promote UCP1 transcription and brown fat program. Cell Rep 2016; 15(11):2536-49; PMID: 27264172; https://doi.org/ 10.1016/j.celrep.2016.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Son D, Kim CS, Lee KR, Park H-J. Identification of new quinic acid derivatives as histone deacetylase inhibitors by fluorescence-based cellular assay. Bioorg Med Chem Lett 2016; 26(9):2365-69; PMID: 26996372; https://doi.org/ 10.1016/j.bmcl.2016.03.010 [DOI] [PubMed] [Google Scholar]
- 78.Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, Umehara T, Yokoyama S, K-i Kosai, Nakao M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun 2012; 3(758):1-12; PMID:22453831; https://doi.org/27325688 10.1038/ncomms1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Glaser KB, Li J, Aakre M, Morgan D, Sheppard G, Stewart K, Pollock J, Lee P, O'Connor C, Anderson S, et al.. Transforming growth factor ß mimetics: Discovery of 7-[4-(4-Cyanophenyl)phenoxy]-Heptanohydroxamic acid, a biaryl hydroxamate inhibitor of histone deacetylase. Mol Cancer Ther 2002; 1(10):759-68; PMID:1249210827325688 [PubMed] [Google Scholar]
- 80.Bennesch MA, Segala G, Wider D, Picard D. LSD1 engages a corepressor complex for the activation of the estrogen receptor alpha by estrogen and cAMP. Nucleic Acids Res 2016; 44(18):8655-8670; PMID: 27325688; https://doi.org/ 10.1093/nar/gkw522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters , Antoine HFM, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005; 437(7057):436-9; PMID: 16079795; https://doi.org/ 10.1038/nature04020 [DOI] [PubMed] [Google Scholar]
- 82.Ashburner BP, Westerheide SD, Baldwin AS. The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 To negatively regulate gene expression. Mol Cell Biol 2001; 21(20):7065-77; PMID: 11564889; https://doi.org/ 10.1128/MCB.21.20.7065-7077.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Auld DS, Southall NT, Jadhav A, Johnson RL, Diller DJ, Simeonov A, Austin CP, Inglese J. Characterization of chemical libraries for luciferase inhibitory activity. J. Med. Chem 2008; 51(8):2372-86; PMID: 18363348; https://doi.org/ 10.1021/jm701302v [DOI] [PubMed] [Google Scholar]
- 84.Promega Corporation Protocols and Applications Guide, Cell Viability. [Google Scholar]
- 85.Shi L, Cui S, Engel JD, Tanabe O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med 2013; 3(19):291-294; PMID:23416702; https://doi.org/27308632 10.1038/nm.3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mohammad HP, Kruger RG. Antitumor activity of LSD1 inhibitors in lung cancer. Mol Cell Oncol 2016; 3(2):e1117700; PMID: 27308632; https://doi.org/ 10.1080/23723556.2015.1117700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pollock JA, Larrea MD, Jasper JS, McDonnell DP, McCafferty DG. Lysine-specific histone demethylase 1 inhibitors control breast cancer proliferation in ERα-dependent and independent manners. ACS Chem Biol 2012; 7(7):1221-31; PMID: 22533360; https://doi.org/ 10.1021/cb300108c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Itoh Y, Sawada H, Suzuki M, Tojo T, Sasaki R, Hasegawa M, Mizukami T, Suzuki T. Identification of Jumonji AT-rich interactive domain 1A inhibitors and their effect on cancer cells. ACS Med Chem Lett 2015; 6(6):665-70; PMID: 26101571; https://doi.org/ 10.1021/acsmedchemlett.5b00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.VanderMolen KM, McCulloch W, Pearce CJ, Oberlies NH. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. J. Antibiotics 2011; 64(8):525-531; PMID:21587264; https://doi.org/15738985 10.1038/ja.2011.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown M, Attardi L. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 2005; 5(3):231-7; PMID: 15738985; https://doi.org/ 10.1038/nrc1560 [DOI] [PubMed] [Google Scholar]
- 91.Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006; 1(5):2315-9; PMID: 17406473; https://doi.org/ 10.1038/nprot.2006.339 [DOI] [PubMed] [Google Scholar]
- 92.Wognum B, Yuan N, Lai B, Miller CL. Colony forming cell assays for human hematopoietic progenitor cells. Methods Mol Biol 2013; 946:267-83; PMID: 23179838; https://doi.org/ 10.1007/978-1-62703-128-8_17 [DOI] [PubMed] [Google Scholar]
- 93.Pavlik CM, Wong CYB, Ononye S, Lopez DD, Engene N, McPhail KL, Gerwick WH, Balunas MJ. Santacruzamate A, a potent and selective histone deacetylase (HDAC) inhibitor from the panamanian marine cyanobacterium cf. symploca sp. J Nat Prod 2013; 76(11):2026-33; PMID: 24164245; https://doi.org/ 10.1021/np400198r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu Q, Lu W, Ma M, Liao J, Ganesan A, Hu Y, Wen S, Huang P. Synthesis and biological evaluation of santacruzamate A and analogs as potential anticancer agents. RSC Adv 2015; 5(2):1109-12; https://doi.org/ 10.1039/C4RA13889A [DOI] [Google Scholar]
- 95.Zwick V, Chatzivasileiou A-O, Deschamps N, Roussaki M, Simoes-Pires CA, Nurisso A, Denis I, Blanquart C, Martinet N, Carrupt P-A, et al.. Aurones as histone deacetylase inhibitors: identification of key features. Bioorg Med Chem Lett 2014; 24(23):5497-501; PMID: 25455492; https://doi.org/ 10.1016/j.bmcl.2014.10.019 [DOI] [PubMed] [Google Scholar]
- 96.Itoh Y, Suzuki M, Matsui T, Ota Y, Hui Z, Tsubaki K, Suzuki T. False HDAC Inhibition by Aurone Compound. Chem Pharm Bull 2016; 64(8):1124-8; PMID: 27477650; https://doi.org/ 10.1248/cpb.c16-00123 [DOI] [PubMed] [Google Scholar]
- 97.Goracci L, Deschamps N, Randazzo GM, Petit C, Dos Santos Passos C, Carrupt P-A, Simoes-Pires C, Nurisso A. A Rational Approach for the Identification of Non-Hydroxamate HDAC6-Selective Inhibitors. Sci Rep 2016; 6(12):29086; PMID: 27404291; https://doi.org/ 10.1038/srep29086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat Chem Biol 2005; 1(3):143-45; PMID: 16408017; https://doi.org/ 10.1038/nchembio721 [DOI] [PubMed] [Google Scholar]
- 99.Cherblanc FL, Chapman KL, Brown R, Fuchter MJ. Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases. Nat Chem Biol 2013; 9(3):136-7; PMID: 23416387; https://doi.org/ 10.1038/nchembio.1187 [DOI] [PubMed] [Google Scholar]
- 100.Cherblanc FL, Chapman KL, Reid J, Borg AJ, Sundriyal S, Alcazar-Fuoli L, Bignell E, Demetriades M, Schofield CJ, DiMaggio PA JR, et al.. On the histone lysine methyltransferase activity of fungal metabolite chaetocin. J Med Chem 2013; 56(21):8616-25; PMID: 24099080; https://doi.org/ 10.1021/jm401063r [DOI] [PubMed] [Google Scholar]
- 101.Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Reply to “Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases”. Nat Chem Biol 2013; 9(3):137; PMID: 23416388; https://doi.org/ 10.1038/nchembio.1188 [DOI] [PubMed] [Google Scholar]
- 102.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001; 276(39):36734-41; PMID: 11473107; https://doi.org/ 10.1074/jbc.M101287200 [DOI] [PubMed] [Google Scholar]
- 103.Kwon HJ, Owa T, Hassig CA, Shimada J, Schreiber SL. Depudecin induces morphological reversion of transformed fibroblasts via the inhibition of histone deacetylase. PNAS 1998; 95(7):3356-61; PMID: 9520369; https://doi.org/ 10.1073/pnas.95.7.3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baell JB. Feeling nature's PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J Nat Prod 2016; 79(3):616-28; PMID: 26900761; https://doi.org/ 10.1021/acs.jnatprod.5b00947 [DOI] [PubMed] [Google Scholar]
- 105.Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr 2011; 6(2):93-108; PMID: 21516481; https://doi.org/ 10.1007/s12263-011-0222-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pal-Bhadra M, Ramaiah MJ, Reddy TL, Krishnan A, Pushpavalli SNCVL, Babu KS, Tiwari AK, Rao JM, Yadav JS, Bhadra U. Plant HDAC inhibitor chrysin arrest cell growth and induce p21WAF1 by altering chromatin of STAT response element in A375 cells. BMC Cancer 2012; 12(180); PMID: 23020297; https://doi.org/ 10.1186/1471-2407-12-180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun L-P, Chen A-L, Hung H-C, Chien Y-H, Huang J-S, Huang C-Y, Chen Y-W, Chen C-N. Chrysin: a histone deacetylase 8 inhibitor with anticancer activity and a suitable candidate for the standardization of Chinese propolis. J Agric Food Chem 2012; 60(47):11748-58; PMID: 23134323; https://doi.org/ 10.1021/jf303261r [DOI] [PubMed] [Google Scholar]
- 108.Dang W. The controversial world of sirtuins. Drug Discovery Today 2014; 12:e9-e17; PMID: 25027380; https://doi.org/ 10.1016/j.ddtec.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Venturelli S, Berger A, Bocker A, Busch C, Weiland T, Noor S, Leischner C, Schleicher S, Mayer M, Weiss TS, et al.. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone corrected proteins in human-derived hepatoblastoma cells. PLoS One 2013; 8(8):e73097; PMID: 24023672; https://doi.org/ 10.1371/journal.pone.0073097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem 2003; 278(21):19134-40; PMID: 12624111; https://doi.org/ 10.1074/jbc.M301580200 [DOI] [PubMed] [Google Scholar]
- 111.Hemshekhar M, Sebastin Santhosh M, Kemparaju K, Girish KS. Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin Physiol Pharmacol 2012; 110(2):122-32; PMID:22103711; https://doi.org/19291822 10.1111/j.1742-7843.2011.00833.x [DOI] [PubMed] [Google Scholar]
- 112.Jung M, Yong K-J, Velena A, Lee S. Epigenetic targets in drug discovery: cell-based assays for HDAC inhibitor hit validation In: Epigenetic Targets in Drug Discovery. Vol. 42 Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2010. p. 119-137; https://doi.org/10.1002/9783527627073.ch6 [Google Scholar]
- 113.Shepard BD, Tuma PL. Alcohol-induced protein hyperacetylation: Mechanisms and consequences. World J Gastroenterol 2009; 15(10):1219-30; PMID: 19291822; https://doi.org/ 10.3748/wjg.15.1219 [DOI] [PMC free article] [PubMed] [Google Scholar]