

ABSTRACT
Until 2004, many researchers believed that protein methylation in eukaryotic cells was an irreversible reaction. However, the discovery of lysine-specific demethylase 1 in 2004 drastically changed this view and the concept of chromatin regulation. Since then, the enzymes responsible for lysine demethylation and their cellular substrates, biological significance, and selective regulation have become major research topics in epigenetics and chromatin biology. Many cell-permeable inhibitors for lysine demethylases have been developed, including both target-specific and nonspecific inhibitors. Structural understanding of how these inhibitors bind to lysine demethylases is crucial both for validation of the inhibitors as chemical probes and for the rational design of more potent, target-specific inhibitors. This review focuses on published small-molecule inhibitors targeted at the two flavin adenine dinucleotide-dependent lysine demethylases, lysine-specific demethylases 1 and 2, and how the inhibitors interact with the tertiary structures of the enzymes.
KEYWORDS: Chromatin, crystal structure, epigenetics, FAD, histone, LSD1, LSD2, nucleosome, transcription
Abbreviations
- AEL
acute erythroid leukemia
- AMKL
acute megakaryoblastic leukemia
- AML
acute myeloid leukemia
- AOD
amine oxidase domain
- AOF
amine oxidase flavin-containing domain
- BHC
BRAF–histone deacetylase complex
- CoREST
corepressor for RE-1 silencing transcription factor
- FAD
flavin adenine dinucleotide
- HDAC
histone deacetylase
- KDM
lysine (K) demethylase
- LSD1
lysine-specific demethylase 1
- LSD2
lysine-specific demethylase 2
- MAO
monoamine oxidase
- MDS
myelodysplastic syndrome
- NHL
non-Hodgkin's lymphoma
- NPAC
no preservation of anterior capsule
- 2-PCPA
( ± )-trans-2-phenylcyclopropylamine
- PDB
Protein Data Bank
- SCLC
small cell lung cancer
- SWIRM
Swi3/Rsc8/Moira
- THF
tetrahydrofolate
Introduction
In a eukaryotic cell, the activities of chromatin and its associated proteins are often modulated by posttranslational modifications.1 For example, acetylation and methylation of lysine residues are typical modifications observed in the N-terminal flanking tails of chromatinized histones. Possible evidence for the active erasure of histone acetylation in a cell was revealed as early as 1978 by a series of chemical biology studies using nonspecific histone deacetylase (HDAC) inhibitors (e.g., sodium butyrate) or specific HDAC inhibitors (e.g., trichostatin A).2-5 Identification of histone deacetylases Rpd3 and Hda1 in 1996 conclusively confirmed that histone acetylation is dynamically regulated in eukaryotic cells.6,7 In contrast, the reversibility of protein methylation at lysine or arginine residues was not unambiguously confirmed until 2004.8 Because methylated lysines in histones are generally more static than other known chromatin modifications (as exemplified by heritable histone H3K9 methylation, which is associated with heterochromatin),9-11 protein methylation had been assumed to be irreversible. However, identification of nuclear histone demethylase lysine-specific demethylase 1 (LSD1) by Shi and colleagues in 2004 completely changed this view.8
LSD1, also known as AOF2 or BHC110, uses flavin adenine dinucleotide (FAD) as a coenzyme for the removal of one or two methyl groups from a specific mono- or di-methylated lysine side chain of several nuclear proteins, including histone H3. Mammalian LSD1 has only one homolog, designated LSD2 or AOF1, which also uses FAD as a coenzyme and demethylates histone H3 at K4.12 In addition to these two FAD-containing lysine demethylases, another type of lysine demethylases was reported in 2006.13,14 These enzymes, which contain a Jumonji C domain, belong to an Fe(II)-dependent and α-ketoglutarate-dependent dioxygenase family, and they catalyze the removal of one to three methyl groups from mono-, di-, or tri-methylated lysine side chains. Currently, there are thought to be at least 20 different human α-ketoglutarate-dependent lysine demethylases.15 In the nomenclature of histone demethylase families,16 the FAD-dependent enzymes, LSD1/AOF2/BHC110 and LSD2/AOF1, are designated as lysine (K) demethylases 1A and 1B (KDM1A and KDM1B), respectively. They belong to the class I demethylase category. Other α-ketoglutarate-dependent demethylases belong to the class II demethylase category, which is composed of six families, designated KDM2, KDM3, KDM4, KDM5, KDM6, and KDM7.
Because the KDMs in each class share the same catalytic domain, the development of a small-molecule compound or a peptidic compound that inhibits at least some of these enzymes may be possible. Structural information about the KDMs is particularly important not only for designing target-specific inhibitors but also for understanding their mechanism of action and target selectivity at atomic resolution. In particular, the development of specific inhibitors for LSD1 is highly desirable because this enzyme has emerged as a key epigenetic modifier in the self-renewal of several cancer stem cells, such as leukemic stem cells in acute myeloid leukemia (AML) and glioblastoma stem-like tumor-propagating cells.17-19 In fact, treatment of cells with LSD1 inhibitors can result in induction of differentiation and/or loss of cancer stemness,17-20 and some irreversible LSD1 inhibitors have entered clinical trials for the treatment of AML and other diseases,21,22 as summarized in Table 1 and described below. In addition, no LSD2-specific inhibitors have yet been developed, which hinders the study of the chemical biology of LSD2. This review focuses on published small-molecule inhibitors of the FAD-dependent lysine demethylases, LSD1/KDM1A and LSD2/KDM1B, with emphasis on their tertiary structures.
Table 1.
Summary of LSD1/KDM1A inhibitors in clinical trials and clinical use.a
| Drug designation | Organization | Phase | Disease |
|---|---|---|---|
| ORY-1001b | Oryzon (Originator); Roche | I | Small cell lung cancer (SCLC) |
| I/II | Acute myeloid leukemia (AML) | ||
| ORY-2001 | Oryzon (Originator) | I | Alzheimer's disease |
| I | Parkinson's disease | ||
| I | Huntington's disease | ||
| GSK-2879552 | GlaxoSmithKline (Originator) | I | SCLC |
| I | AML | ||
| INCB-059872 | Incyte (Originator) | I/II | Cancer |
| IMG-7289 | Imago Biosciences (Originator) | I/II | AML |
| I/II | Myelodysplastic syndrome (MDS) | ||
| CC-90011 | Celgene (Originator) | I | Solid tumors |
| I | Non-Hodgkin's lymphoma (NHL) | ||
| Tranylcyprominec,d | GlaxoSmithKline (Originator); | Launched | Depression |
As of January 20, 2017.
Other designations: RG-6016, RO-7051790.
Other designation: Parnate.
Monoamine oxidase (MAO) inhibitor.
Structures and functions of LSD1/KDM1A and its isoforms
LSD1 orthologues are found in organisms ranging from fission yeast to humans.8 The domain architecture of human LSD1 is shown in Fig. 1A. The tertiary structure of LSD1 except for approximately 170 residues of the N-terminal disordered region has been elucidated23,24 (Fig. 1B). The enzyme consists of a SWI3/Rsc8/Moira (SWIRM) domain, a Tower domain, and the catalytic amine oxidase domain (AOD). The SWIRM domain is a compact helical domain composed of approximately 85 residues and is found in several chromatin-modulating protein complexes.25,26 It packs against the catalytic domain, and this arrangement presumably contributes to the stability of the protein and the formation of a substrate-binding groove23,24 (Fig. 1B). The AOD possesses sequence similarity to the catalytic domain of small-molecule amine oxidases, such as monoamine oxidases A and B (MAO-A and MAO-B). The catalytic cavity of LSD1 is larger and more open than the cavities of MAOs. The SWIRM domain and the AOD reportedly interact with a G-quadruplex RNA and a long noncoding RNA, respectively.27 Unlike MAOs, LSD1 has the Tower domain inserted in the middle of the AOD. The Tower domain is a slender, 90-Å-long coiled-coil structure protruding from the AOD. It provides the binding site for CoREST, a protein that presumably helps LSD1 tether a nucleosome.28-30 Accordingly, in the absence of the Tower domain, LSD1 cannot associate with CoREST and thus loses its ability to demethylate nucleosomes.24,31
Figure 1.
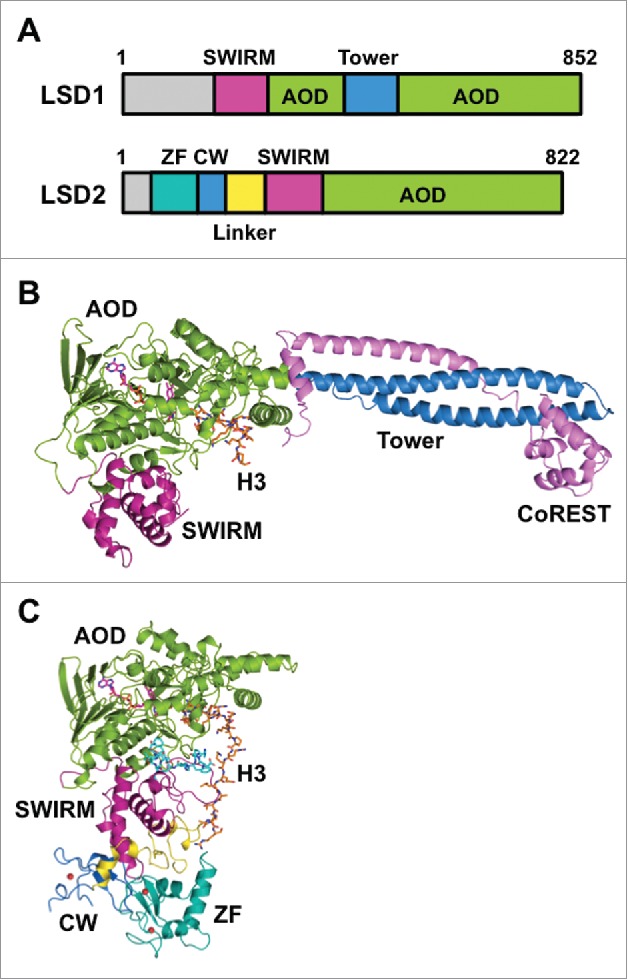
Structures of LSD1/KDM1A and LSD2/KDM1B. (A) Domain architectures of human LSD1 and LSD2. (B) Tertiary structure of LSD1 in complex with CoREST and an H3-like peptide inhibitor (PDB ID: 2V1D). Domains are colored as in panel (A), CoREST is depicted in pink, and the H3-like peptide is depicted in orange. (C) Tertiary structure of LSD2 (PDB ID: 4HSU) in complex with an H3 peptide (residues 1–26) (orange) and the NPAC linker region (cyan).
The crystal structure of LSD1 in complex with an H3(1–21)-derived peptide in which the K4 residue has been replaced by methionine (H3K4M(1–21)) reveals that the N-terminal 1–3 residues of H3 adopt a helical conformation that is accommodated in a negatively charged pocket in the catalytic center of LSD1.32 In the pocket, H3R2 forms hydrogen bonds with the D553 and D556 residues of LSD1. The side chain of H3K4 to be demethylated is directed toward FAD along the side chain of Y761 of LSD1. The H3 peptide is compactly folded and interacts extensively with LSD1 residues on the rim of the catalytic cavity. The C-terminal residues after H3P16 are not visible in the electron density map, presumably owing to lack of structural restraints.32 The structure of LSD1 in complex with another H3(1–21)-derived peptide, in which K4 is replaced by a covalent-bond-forming N-methylpropargyl lysine, shows similar enzyme–substrate interactions, although the conformation of the H3 peptide seems to be more constrained owing to the covalent bond to FAD.33 These structures demonstrate that there is little room for extension of the N-terminus of the H3 peptide, which explains the specificity of LSD1 for its major substrate residue, that is, H3K4.32,33 There may be an additional H3-binding site, as indicated by the fact that full-length H3 can tightly bind to LSD1.34
Although LSD1 was first identified as an H3K4 demethylase, it also reportedly demethylates H3K9 when interacting with the androgen receptor.35 The mechanism of demethylation at H3K9 remains unclear, but recent studies have indicated that splicing isoforms of LSD1 exhibit altered substrate specificity with a preference for H3K9 or H4K20.36-39 Thus, there may be another binding mode in which the H3K9 or H4K20 side chain is in close proximity to FAD in the catalytic center of LSD1. The neuron-specific LSD1 isoform has an in-frame insertion of four amino acid residues (DTVK) between A369 and D370. This tetrapeptide forms a short antiparallel β-turn that sticks out from the body of the protein without changing the overall conformation of the LSD1•CoREST complex.36 In addition, phosphorylation of the threonine residue in the insertion sequence triggers a local structural change in LSD1, which may in turn disrupt interactions of LSD1 with CoREST, HDAC1, and HDAC2.40
In addition to histone H3, nonhistone nuclear proteins, including p53, Dnmt1, E2F1, and MYPT1, are also substrates for LSD1.41-44 The functions of these nonhistone proteins are regulated by residue-specific lysine demethylation by LSD1. There is also an LSD1-binding protein that inhibits the demethylation activity of LSD1. Specifically, the N-terminal SNAG domain of the transcription factor SNAIL1, which mediates E-cadherin repression during the epithelial–mesenchymal transition, binds to the AOD of LSD1, thereby recruiting LSD1 to the targeted gene promoter of SNAIL1 for transcriptional repression.45 The first nine residues of SNAG, which are well conserved among species and resemble the first nine residues of H3, tightly bind to the catalytic cavity of LSD1 by mimicking the LSD1-binding mode of H3, resulting in competitive inhibition of LSD1 demethylase activity.46 Therefore, the structure of the SNAG domain has been explored to develop peptidic LSD1 inhibitors (described below).
Structures and functions of LSD2/KDM1B
When LSD1 was discovered as the first lysine demethylase,8 LSD2 was identified as the only mammalian homolog of LSD1. The domain architecture of human LSD2 is shown in Fig. 1A. Like LSD1, LSD2 demethylates mono- and di-methylated H3K4 in an FAD-dependent manner.12,47-49 Mouse LSD2 reportedly demethylates H3K9 at a comparatively higher concentration.50 Unlike the catalytic activity of LSD1, the catalytic activity of LSD2 still seems to be the subject of discussion. The domain architectures of both enzymes (Fig. 1) share the AOD as the catalytic center in the C-terminal region, and they also have the SWIRM domain on the N-terminal side of the AOD. However, the AOD of LSD2 lacks the Tower domain that is present in the AOD of LSD1. LSD2 demethylates nucleosomal histone H3 in vivo.48,49,51 Because LSD2 does not have the nucleosome-binding Tower•CoREST architecture (Fig. 1B), the demethylation of nucleosomal histone H3 by LSD2 must occur in a structurally distinct manner from demethylation by LSD1.
LSD2 has two types of zinc fingers in the N-terminal region (Fig. 1A): a C4H2C2-type zinc finger followed by a CW-type zinc finger. Some of the CW-type zinc fingers function as a reader for K4-methylated histone H3.52-54 The CW domain typically forms an aromatic cage with a preference for hosting the methylated lysine, and the cage-forming residues and four Zn(II)-coordinating cysteine residues are conserved in LSD2. Nevertheless, the CW domain of LSD2 does not bind to the H3 peptide, even though the domain is required for the demethylase activity of LSD2.48,55 Structural comparison of the CW domain of LSD2 with other K4-methylated H3-binding CW domains reveals that L340 and I343 of the neighboring SWIRM domain occupy the binding site of the methylated lysine, and a helix-loop-helix moiety of the SWIRM domain and a helix within the CW domain seem to hinder H3 from binding to LSD2.54-56
As expected from the distinct domain architectures of LSD1 and LSD2, they have distinct binding partners. Specifically, LSD1 associates with HDAC1 and HDAC2,29 whereas LSD2 does not associate with HDACs.48,57 In addition, only LSD1 associates with CoREST, and this association may tether LSD1 to a nucleosome.28-30 Furthermore, NPAC/GLYR1 is an LSD2-specific cofactor that enhances the catalytic activity of LSD2 toward histone H3.56 NPAC binds to LSD2 through the NPAC linker region between the N-terminal putative H3K36me3-binding PWWP domain and the C-terminal dehydrogenase domain. The structure of LSD2 in complex with an NPAC linker region and the H3(1–21) peptide revealed that residues 214–225 of NPAC interact with LSD2 and that this dodecapeptide is sufficient for enhancement of the catalytic activity of LSD2.56 Specifically, F217 of NPAC and Y273, E277, and R285 of LSD2 form a hydrophobic pocket that interacts with L20 of H356 (Fig. 1C). In contrast to the crystal structure of the LSD1•H3(1–21) complex in which only H3(1–16) was visible in the electron density map,32 H3(1–20) was visible in the crystal structures of LSD2•H3(1–21)•NPAC, presumably owing to the interactions between Q19 and L20 of H3 and LSD2•NPAC.56 The LSD2•H3(1–26)•NPAC complex further demonstrated that H3 residues 19–26 bind to a second site on LSD2, which is formed by the linker region between the CW domain and the SWIRM domain. In this complex, NPAC seems to facilitate the interactions between LSD2 and the H3 peptide58 (Fig. 1C). There have been fewer studies of LSD2 than of LSD1, but the studies to date implicate LSD2 in several biological contexts that differ from those of LSD1.47,59
Categories of LSD1/KDM1A inhibitors
Small-molecule inhibitors are useful tools for identifying the functional roles of a target protein. However, the potency and specificity of an inhibitor for the protein of interest are important. When a promiscuous inhibitor is used, drawing valid conclusions from the results can be difficult.60 In early chemical biology studies of LSD1, researchers often used the MAO inhibitor tranylcypromine (( ± )-trans-2-phenylcyclopropylamine; abbreviated as 2-PCPA in this review), which exhibited better LSD1-inhibitory activity than the other MAO inhibitors known at the time.61,62 However, 2-PCPA is now regarded as a promiscuous FAD-inactivating inhibitor, and studies in which 2-PCPA was used as an LSD1 inhibitor must be evaluated carefully. Many research groups have pursued the development of LSD1 inhibitors more specific and potent than 2-PCPA, as described below. In this review, we categorize these new LSD1 inhibitors as irreversible covalent inhibitors and reversible noncovalent inhibitors, and we group the irreversible inhibitors into three subcategories: 2-PCPA derivatives, N-alkylated 2-PCPA derivatives, and inhibitors other than 2-PCPA derivatives.
2-PCPA derivatives that inhibit LSD1/KDM1A
Some typical irreversible LSD1 inhibitors are shown in Fig. 2, and kinetic parameters and structural information (where available) are summarized in Table 2. Most of the irreversible LSD1 inhibitors developed to date are based on the 2-PCPA scaffold (1, Fig. 2).63 Researchers initially focused on chemical modification of the phenyl ring of 1. For example, Ueda et al. attached a lysine-mimicking group at the meta-position (NCL-1, 2, Fig. 2) or the para-position (NCL-2) of the ring, and the resulting compounds show enhanced affinity for LSD1 and are selective for LSD1 over MAOs.64 Mimasu et al. designed a meta-fluoro compound with a benzyloxy group at the ortho-position (designated S1201; the complex with LSD1 is assigned PDB ID 3ABU).65 First, they determined the structure of LSD1 in complex with pentafluorinated 2-PCPA (PDB ID: 3ABT), and then they synthesized various ortho-substituted 2-PCPA derivatives based on the structures of LSD1•2-PCPA complexes66,67 and MAO-B.68 Eventually, they developed a compound, designated S2101 (3, Fig. 2), with one more fluorine atom than S1201. This difluoro compound exhibits submicromolar potency and better selectivity for LSD1 over MAOs. S2101 is also selective for LSD1 over LSD2 (unpublished). S2101 and NCL-1 were shown to be cell active as LSD1-selective inhibitors. In particular, compact cell-permeable compound S2101 has been used in many studies to assess the biological roles of LSD1.19,69-76
Figure 2.
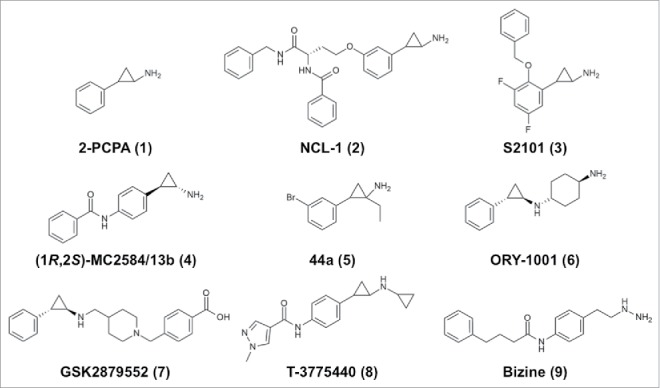
Chemical structures of representative covalent LSD1/KDM1A inhibitors.
Table 2.
Summary of covalent-binding LSD1/KDM1A inhibitors.
| Compound name | LSD1 inhibition (nM) | PDB ID | Resolution (Å) | Reference |
|---|---|---|---|---|
| 2-PCPA derivatives | ||||
| 2-PCPA (1) | 500,000a | 2UXX | 2.75 | 66 |
| 100,000a | 2Z5U, 2Z3Y | 2.25 | 65,67 | |
| 2-PCPA stereoisomers | 284,000a, 168,000a | 2XAH, 2XAJ | 3.1, 3,3 | 77 |
| Br-2-PCPA stereoisomers | 28,000a, 23,000a | 2XAG, 2XAF | 3.1, 3.2 | 77 |
| NCL-1 (2) | 2,500b | 64 | ||
| (1S,2R)-NCL-1 | 1,600b | 84 | ||
| S1201 | 2,400a | 3ABU | 3.1 | 65 |
| S2101 (3) | 610a | 65 | ||
| MC2584/13b | 1,100a | 2XAQ | 3.2 | 77 |
| (1R,2S)-MC2584/13b (4) | 13b | 85 | ||
| MC2584/13b derivative | 84b | 87 | ||
| MC2580/14e | 1,300a | 2XAS | 3.2 | 77 |
| (S,1S,2R)-MC2580/14e | 30b | 86 | ||
| MC2580/14e derivative | 61b | 4UXN | 2.85 | 88 |
| OG-L002 | 20b | 78 | ||
| 1-substituted derivatives | 131b | 4UVCd | 3.1 | 90 |
| 1-substituted “44a” (5) | 31b | 91 | ||
| N-alkylated 2-PCPA derivatives | ||||
| RN-7 | 31b | 92 | ||
| NCL-1 derivative | 380b | 95 | ||
| ORY-1001 (6) | 18b | 21 | ||
| GSK2879552 (7) | 1,700c | 94 | ||
| T-3775440 (8) | 2.1b, 20b | 20 | ||
| Other inhibitors | ||||
| Bizine (9) | 59a | 103 | ||
| Propagyl-K4-H3 (1–21) | 120a | 2UXN | 2.72 | 33 |
Ki;
IC50;
Kiapp;
Related structures: 4UVA, 4UVB, 4UV8, 4UV9.
Several para-substituted 2-PCPA derivatives have been developed, including MC2584/13b and MC2580/14e (structures of their respective complexes with LSD1•CoREST are assigned PDB IDs 2XAQ and 2XAS), which exhibit selectivity for LSD1 over MAO-B and approximately 20-fold enhanced potency against LSD1 relative to that against LSD277 (Table 2). In the LSD1•MC2580/14e structure, one phenylalanine in the linear extension occupies the position of T3 in the LSD1•H3 structure, and the other branched phenyl ring occupies the same position as the ortho-benzyloxy group of S1201. Other para-substituted inhibitors include the following: OG-L002 (Table 2), which inhibits viral infection;78 pan-histone demethylase inhibitors that are hybrids of 2-PCPA and Jumonji C-containing KDM inhibitors;79 compounds coupled with hydroxycinnamic acid;80 and biphenyl derivatives.81
Although 2-PCPA inactivates both LSD1 and MAOs by covalent bonding of the cyclopropylamine group to the FAD, the structures of the resulting adducts are different. In the MAO-B•2-PCPA complex, the covalent bond forms at the C4a atom of FAD.82 In contrast, in the LSD1•2-PCPA complex, the adduct has been modeled as a 5-membered ring in which both the C4a atom and the N5 atom are covalently bonded to the inhibitor66 (PDB ID: 2UXX). There may be an N5 adduct in the LSD1•2-PCPA complex as well67 (see PDB IDs 2Z5U and 2Z3Y for a 5-membered-ring adduct model and an N5 adduct, respectively). The adduct structure is affected by differences in the steric environment near the flavin ring, such as structural differences between the LSD1 and MAO-B cavities and structural differences between the various inhibitors (e.g., 2-PCPA substituents and the configuration of the cyclopropyl carbons; described below).
Because 2-PCPA has two chiral centers on the cyclopropylamine ring, there are four possible stereoisomers. Racemic trans-2-PCPA is in clinical use as a MAO-inhibiting antidepressant (brand name Parnate; Table 1), but investigation of the effects of its chirality is important in the pursuit of more potent and selective inhibitors.83 Binda et al. examined the potencies of the four stereoisomers of 2-PCPA and found that trans-(1R,2S)-PCPA is a more potent inhibitor of LSD1 than trans-(1S,2R)-PCPA and that the two trans-enantiomers are more potent than the two cis-enantiomers.77 These investigators also found that the structures of the FAD•2-PCPA adducts of the two trans-enantiomers differ from each other. trans-(1R,2S)-PCPA forms an N5 adduct in which the phenyl ring extends “vertically” (PDB ID: 2XAJ), whereas trans-(1S,2R)-PCPA forms an N5 adduct in which the phenyl ring extends “laterally” (PDB ID: 2XAH). The structure of the FAD adduct with racemic trans-2-PCPA in the LSD1 complex66,67 corresponds to the “vertical” N5 model of trans-(1R,2S)-PCPA, which is consistent with the higher potency of trans-(1R,2S)-PCPA relative to the other trans-isomer.77
Specific stereoisomers of other compounds that were initially tested as racemic mixtures have also been prepared, including (1S,2R)-NCL-184, (1R,2S)-MC2584/13b (4, Fig. 2),85 and (S,1S,2R)-MC2580/14e86 (Table 2). A specific stereoisomer of an MC2584/13b derivative with a 4-methylpiperazine substituent was prepared and found to be orally active, with an IC50 of 84 nM against LSD1.87 In addition, the stereochemistry of MC2580/14e was simplified by preparation of derivatives in which the chiral amino acid moiety was replaced with an N-carbobenzyloxy-proline residue (PDB ID: 4UXN) and with pyrrole or indole derivatives. The resulting compounds showed higher LSD1-inhibitory activity compared with MC2580/14e; specifically, a pyrrole derivative showed an IC50 of 32 nM against LSD1.88
The atoms on the cyclopropyl core moiety of 2-PCPA can also be modified.89,90 A structure–activity relationship study of α-substituted cyclopropylamines identified a compound (5, Fig. 2) with an IC50 of 31 nM.91 It is noteworthy that the structure of the FAD adducts depends on the substituents on the cyclopropyl ring carbon atoms and on their chirality, as indicated by crystal structures.90
N-alkylated 2-PCPA derivatives that inhibit LSD1/KDM1A
Oryzon Genomics reported that LSD1 inhibition could be substantially improved by using N-alkylated 2-PCPA derivatives, which exhibit nanomolar LSD1-inhibitory potency (WO 2010/043721). ORY-1001 (6, Fig. 2) has an IC50 of 18 nM and greater than 1000-fold selectivity for LSD1 over LSD2 and MAOs.21 Two Oryzon inhibitors have been proven to be efficacious for the treatment of MLL-AF9 AML cells.18 Other N-alkylated compounds, RN-1 and RN-7, have been demonstrated to be brain penetrant, and inhibition of LSD1 by high concentrations of these compounds reportedly impairs long-term memory formation.92 Selectivity for LSD1 may be critical to this result, as indicated by the fact that MAOs are highly expressed in the brain. RN-1 is also selective for LSD1 over LSD2 and reportedly suppresses the growth of AML cells.93
GSK2879552 (7, Fig. 2) and two of its analogues, GSK-LSD1 and GSK2699537, are LSD1-inhibiting N-alkylated compounds that were developed through biochemical screening of a library of 2.5 million compounds by researchers at GlaxoSmithKline.94 A kinetics study showed that the mechanism involves reversible binding followed by time-dependent irreversible inactivation. GSK2879552 is selective for LSD1 over LSD2 and MAOs and is orally bioavailable. The crystal structure of the LSD1•GSK2699537 complex shows the formation of a C4a-type adduct similar to that of the MAO-B•2-PCPA complex.82 AML cell lines and small cell lung cancer (SCLC) cell lines are sensitive to LSD1 inhibition by these compounds.94 Several N-alkylated inhibitors, including ORY-1001, ORY-2001, GSK2879552, INCB-059872, IMG-7289, and CC-90011, are currently in clinical trials for treatments of cancers, such as AML, SCLC, myelodysplastic syndrome (MDS), and non-Hodgkin's lymphoma (NHL), and neurodegenerative diseases (Table 1).21,22
Recently, Takeda Pharmaceuticals reported another N-alkylated 2-PCPA derivative as an LSD1 inhibitor.20 T-3775440 (8, Fig. 2) inhibits LSD1 irreversibly in vitro with an IC50 of 2.1 or 20 nM in the presence or absence of CoREST, respectively (Table 2), and exhibits rapid antiproliferative and pro-apoptotic activities against acute erythroid leukemia (AEL) and acute megakaryoblastic leukemia (AMKL) cells.20
The reason for the enhanced potency of N-alkylated 2-PCPA analogues is not clearly understood. N-alkyl substitution without modification of the aryl substituents enhances inhibitory activity and selectivity for LSD1 over MAOs (WO 2010/043721).92,95,96 The adduct produced by the reaction between FAD and the N-alkylated 2-PCPA analogues is lacking the amine group,94,95,97 which is in accordance with the proposed reaction mechanism.98 Further studies will be necessary to shed light on the role of the N-substituent in conferring high potency against LSD1.
Covalent LSD1/KDM1A inhibitors other than 2-PCPA derivatives
In the early stages of the research on LSD1 inhibitors, the MAO inhibitor pargyline was found to also exhibit LSD1-inhibitory potency,35 and hence it has been used as an LSD1 inhibitor. However, pargyline is a nonselective inhibitor of LSD1, and in most of the early research, pargyline was used at high doses, which makes interpretation of the results problematic.99 An H3 peptide-based inhibitor bearing a propargylated K4 side chain is an effective irreversible inhibitor (Ki = 120 nM).33,100 Using the propargyl group, Schmitt et al. pursued the development of cell-active LSD1-specific inhibitors.101 Another MAO inhibitor that also inhibits LSD1 is phenelzine, which has a hydrazine that covalently binds to FAD.61,102 Structure–activity relationship studies on phenelzine analogues resulted in the development of bizine (9, Fig. 2), which inactivates LSD1 with a Ki of 59 nM and selectivity for LSD1 over LSD2 and MAOs.103
H3 peptide-based inhibitors have specificity for LSD1 over MAOs. Peptide-based irreversible inhibitors have been developed using propargylamine, hydrazine, or 2-PCPA as the moiety that reacts with FAD.100,102,104 Because the SNAG domain of SNAIL1 can inhibit the binding of the H3 N-terminal peptide,46 SNAIL1 peptide-based inhibitors have been explored. These peptidic inhibitors exhibit specificity for LSD1 over not only MAOs but also LSD2,105 which is consistent with the fact that SNAIL1 binds to LSD2 with lower affinity than to LSD1.46
Noncovalent inhibitors of LSD1/KDM1A
In terms of clinical utility, reversible inhibitors would be preferred because they are considered to have a safer metabolic profile than irreversible inhibitors. However, only a limited number of noncovalent LSD1 inhibitors have been developed thus far. Typical examples of reversible LSD1 inhibitors are shown in Fig. 3, and kinetic parameters and structural information (where available) are summarized in Table 3.
Figure 3.
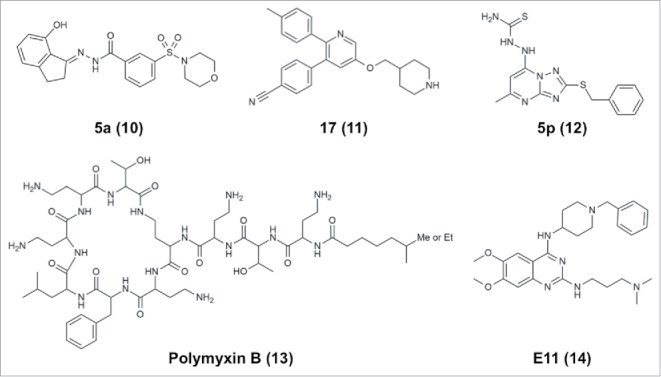
Chemical structures of representative noncovalent LSD1/KDM1A inhibitors.
Table 3.
Summary of noncovalent-binding LSD1/KDM1A inhibitors.
| Compound name | LSD1 inhibition (nM) | PDB ID | Resolution (Å) | Reference |
|---|---|---|---|---|
| Polyamine analogues | ||||
| Verlindamycin | 13,000b | 106,107 | ||
| Bisthiourea “6d" | 4,800b | 107 | ||
| Small-molecule inhibitors | ||||
| CBB1007 | 5,270b | 108 | ||
| Namoline | 51,000b | 109 | ||
| Amidoxin “22" | 16,800b | 110 | ||
| Phenyl oxazole “9a" | 9,500b | 111 | ||
| Aminothiazole “16k" | 7,500b | 112 | ||
| SP2509/HCI2509 | 13b | 120 | ||
| Benzohydrazide “5a” (10) | 1.4b | 123 | ||
| GSK354/GSK690 | 90b | 119d | ||
| Pyridine containing “17” (11) | 29a | 96 | ||
| XZ09 | 2,410b | 116 | ||
| Pyrimidine-thiourea “6b" | 650b | 117 | ||
| Triazole-based “26" | 2,100b | 113 | ||
| Triazole-based “8k" | 390b | 114 | ||
| Triazole-based “6" | 1,192b | 115 | ||
| Triazole-based “5p” (12) | 154b | 118 | ||
| E11 (14) | 440a | 5L3E | 2.8 | 126 |
| MC3767 | not determineda | 5LBQ | 3.3 | 126 |
| Natural products | ||||
| Resveratrol | 15,000b | 128 | ||
| Geranylgeranoic acid | 22,220b | 129 | ||
| Polymixin B (13) | 157a | 5L3F | 3.5 | 126 |
| Polymixin E | 193a | 5L3G | 3.1 | 126 |
| Baicalin | 3,010b | 130 | ||
| Peptidic inhibitors | ||||
| H3K4M (1–21) | 40a, 50a | 2V1D | 3.1 | 32 |
| SNAIL peptide (1–20) | 210a | 2Y48 | 3.0 | 46 |
| SNAIL peptide PRSFLV | 28,400a | 3ZMTc | 3.1 | 132 |
Ki;
IC50;
Related structures: 3ZMS, 3ZMU, 3ZMV, 3ZMZ, 3ZN0, 3ZN1;
Review.
Polyamine analogues such as bisguanidine and biguanide have been reported to noncovalently inhibit LSD1.106 Their bisurea and bisthiourea derivatives also reportedly inhibit LSD1, with an IC50 of approximately 5 µM for the most potent compound.107 Guanidinium-based compounds such as CBB1007 have been designed to mimic R2 and R8 of H3 in the LSD1•H3(1–21) structure. CBB1007 reportedly inhibits LSD1 with an IC50 of approximately 5 µM but does not inhibit LSD2.108 Apart from the polyamine analogues and guanidium compounds, Namoline is a small-molecule compound reported to inhibit LSD1 selectively over MAOs, although its potency is not high (IC50 = 51 μM).109 Several research groups have developed other noncovalent LSD1 inhibitors.110-118 The IC50 values of some of these compounds are only in the micromolar range, however, inhibitors with submicromolar inhibitory potency have recently started to appear (Table 3).119
Noncovalent LSD1 inhibitors with submicromolar potency include a compound designated SP2509/HCl2509, which contains a benzohydrazide scaffold, inhibits LSD1 at an IC50 of 13 nM and has specificity for LSD1 over MAOs.120 This compound has been shown to impede the growth of Ewing sarcoma tumor cells121 and AML cells.122 Optimization of the structure by means of the introduction of conformational restraints resulted in a benzohydrazide derivative 5a (10, Fig. 3) with an IC50 of 1.4 nM against LSD1 and antiproliferation effects against LSD1-overexpressing cancer cells, including A2780 ovarian cancer cells.123 GSK354/GSK690 (referred to in ref. 119), which inhibits LSD1 with an IC50 of 80 nM and shows selectivity for LSD1 over LSD2 and MAOs, has recently been demonstrated to be effective against AML.93 Wu et al. independently developed an almost identical pyridine-containing molecule (11, Fig. 3) with a piperidine ring at one end instead of the pyrrolidine ring of GSK354/GSK690, and this new molecule inhibits LSD1 with a Ki of 29 nM and shows antitumor activity against four tumor cell lines.96 Finally, Wang et al. recently reported that a [1,2,4]triazolo[1,5-a]pyrimidine-based derivative 5p (12, Fig. 3) inhibited LSD1 with an IC50 of 154 nM.118
The structure of LSD1 in a noncovalent complex with tetrahydrofolate (THF) is interesting. THF binds to LSD1 with moderate affinity (KD = 19.5 µM for (6S)-THF), and when THF is polyglutamated, KD increases to 2.77 µM. However, THF does not exhibit inhibitory activity against LSD1.124 The crystal structure of LSD1•CoREST complexed with THF (PDB ID: 4KUM)125 shows that the THF molecule is located deep in the substrate-binding cavity, occupying the place of the H3K4 side chain. Luka et al. suggest that THF may scavenge formaldehyde molecules generated during demethylation of LSD1, because these investigators found other THF-bound enzymes. They also argue that the natural substrate peptide such as the N-terminal tail of histone H3 may bind to LSD1 in a different conformation from that observed in the crystal structure of LSD1 with an H3(1–21)-derived peptide in which the K4 residue has been replaced by methionine (H3K4M(1–21)).125
Recently, Sperazini et al. reported two groups of LSD1 inhibitors and their binding modes.126 The first group is composed of polymyxin antibiotics, which have a circular peptide moiety with a branched linear aliphatic chain. Polymyxin B (13, Fig. 3) and another similar compound, polymyxin E, inhibit H3K4M binding to LSD1 with Ki values of 157 and 193 nM, respectively. Crystallographic analysis (PDB IDs: 5L3F and 5L3G) demonstrated that these antibiotics bind at the entrance of the LSD1 substrate-binding cavity, possibly interacting with negatively charged regions comprising D555, D556, and E379. The molecules may be able to bind to the site in multiple orientations, as indicated by the lack of electron density for the linear branch of the molecules. The second group reported by Sperazini et al. is composed of quinazoline derivatives. One of the quinazoline derivatives E11 (14, Fig. 3) inhibits methyltransferase G9a and G9a-like protein with an IC50 of 778 nM against G9a-like protein.127 E11 binds to LSD1 with a KD of 243 nM and inhibits association of the H3K4M peptide with a Ki of 440 nM.126 Crystallographic analysis demonstrated that a stack of E11 molecules binds at the entrance of the cavity. In the crystal structure with E11 (PDB ID: 5L3E), five layers of the molecule are modeled. MC3767, a truncated version of E11 with much lower affinity for LSD1 (KD = 7.78 µM), still exhibits stacked binding (PDB ID: 5LBQ). Therefore, E11 may bind to LSD1 as a stack of oligomers, whereas it binds to G9a as a monomer. In addition to the above-mentioned antibiotics, other LSD1-inhibiting natural products, such as polyphenols,128 geranylgeranoic acids,129 and baicalin,130 may be useful as scaffolds for developing potent reversible LSD1 inhibitors; however, polyphenols may affect the peroxidase assay used to detect LSD1 catalytic activity, so the results should be interpreted with caution.
Peptides can also inhibit LSD1, although their poor cell permeability is of concern. For example, an H3K4M(1–21) peptide increased LSD1-binding affinity relative to the native H3 peptide (Ki = 40–50 nM in vitro).32 A modified H3K4M(1–21) peptide in which K5 and E10 are macrocyclized inhibits LSD1 with an IC50 of 2.1 µM and is more stable than a linear peptide.131 As mentioned above, the SNAG domain of SNAIL1 (residues 1–20) binds to the substrate-binding site of LSD1 with a Ki of 210 nM, and the binding mode has been elucidated46 (PDB ID: 2Y48). In addition, the SNAG domain of another SNAG-containing protein, INSM1, also binds to LSD1 with similar affinity (Ki = 240 nM; PDB ID: 3ZMS). The N-terminal hexapeptide of SNAIL1 (PRSFLV) is sufficient to bind to LSD1 (Ki = 28.4 µM), and P1 and R2 are crucial for binding (PDB ID: 3ZMT). Mutation of F4 to methionine increases the affinity approximately 10-fold (Ki = 2.6 µM in vitro).132
Inhibitors of LSD2/KDM1B
Although many LSD1 inhibitors have been developed, the number of reported LSD2 inhibitors is limited. Because LSD2 is the only other known FAD-dependent demethylase, compounds have been assayed against it during the development of LSD1 inhibitors for the purpose of assessing inhibitor specificity. Inhibitors that potently and specifically inhibit LSD2 over LSD1 have not yet been reported.
Early studies demonstrated that the Ki of 2-PCPA against LSD2 (180 µM) in vitro is on the same order of magnitude as the Ki against LSD1 (242 µM).12 Pargyline, deprenyl (Selegiline), and rasagiline do not inhibit LSD2,12 but there is one report indicating that LSD2 may be the target of pargyline inhibition.50 Investigation of inhibition by stereoisomers of 2-PCPA and para-brominated 2-PCPA77 demonstrated that these compounds have similar inhibitory potencies against LSD1 and LSD2, and that para-brominated PCPA isomers have higher potency than 2-PCPA isomers against both LSD1 and LSD2. However, there are subtle differences in the inhibitory behaviors of these compounds against LSD1 and LSD2. Against LSD1, the cis-2-PCPA isomers are generally weaker inhibitors than the trans-isomers, whereas against LSD2, the inhibitory ranges of the cis- and trans-isomers are the same. One enantiomer, (1R,2R)-(−)-cis-2-PCPA, shows higher inhibitory activity against LSD2 (Ki = 68 µM) than against LSD1 (Ki = 506 µM), and its para-brominated analogue remains more active against LSD2 (Ki = 21 µM) than against LSD1 (Ki = 44 µM).77
When bulky substituents are attached to the para-position of the phenyl ring of trans-2-PCPA,77 the resulting compounds are selective for LSD1 over MAO-B. Their inhibitory potencies against LSD1 are higher by one order of magnitude than those of the para-brominated derivatives, whereas no improvement in potency against LSD2 is observed. An interesting exception is MC2581/14l, in which a tryptophanyl group replaces the phenylalanyl group of MC2580/14e. MC2581/14l shows enhanced potency against LSD2 but not against LSD1; the Ki value for LSD2 (12 µM) is slightly better than that for LSD1 (40 µM).77 Meta-substituted 2-PCPA analogue NCL-1 (3, Fig. 2) inhibits LSD2 with a Ki of 26 µM and LSD1 with a Ki of 2.5 µM.133,134 A series of MC2584/13b benzamide-cyclopropylamine derivatives was also tested in vitro against LSD1, LSD2, MAO-A, and MAO-B.87 Whereas all the tested derivatives exhibited submicromolar IC50 values for LSD1 (20–300 nM), their potencies for LSD2 ranged from 4.3 to 75 µM; that is, they were specific for LSD1 over LSD2. However, some derivatives with bulky substituents show >10-fold enhanced potency against LSD2 compared with that of the parent inhibitor, resulting in IC50 values of approximately 5 µM. Thus, bulky substituents on the 2-PCPA phenyl ring generally confer selectivity for LSD1. However, this approach may permit enhancement of LSD2-inhibitory activity and thus facilitate the development of pan-KDM1 inhibitors with selectivity over MAOs, or even the development of inhibitors with selectivity for LSD2.
Kakizawa et al. showed that substitution of 2-PCPA derivatives with lysine-mimicking bulky groups on the nitrogen atom has a detrimental effect on LSD2 inhibition, making the resulting inhibitors more specific for LSD1 over LSD2.134 This result indicates that substituents on the amine group also affect LSD2 specificity. It is noteworthy that when these investigators initially tried to conduct a mass spectrometry-based in vitro enzymatic assay using recombinant LSD2, the demethylase activity was so weak that the LSD2-inhibitory activities of the 2-PCPA derivatives could not be accurately evaluated.134 When Kakizawa et al. used the demethylation activity-enhancing NPAC dodecapeptide in their assay, they were able to evaluate the LSD2-inhibitory activities; the IC50 value of 2-PCPA was 2.3 mM.134 This result indicates that evaluation of the inhibitory activity of 2-PCPA against LSD2 may be hampered by the low catalytic activity of LSD2 as well as the low potency of the inhibitor.
Future directions for the development of LSD2/KDM1B-specific inhibitors
In the Protein Data Bank, there is an entry for LSD2 in complex with an NPAC peptide and 2-PCPA (PDB ID: 4GUU),56 although to our knowledge the structure of this complex has not been described in the literature. The structure shows that LSD2•2-PCPA contains a 5-membered-ring adduct. However, superimposition of the LSD2•2-PCPA complex with the LSD1•2-PCPA complex (PDB ID: 2Z5U)67 shows that the phenyl ring of the FAD•2-PCPA adduct in the former complex extends in a slightly different direction than that in the latter complex. This difference appears to be due to steric hindrance between the phenyl ring of 2-PCPA and N440 in LSD2, whereas the corresponding residue in LSD1 is T335, which has a smaller side chain than N440 (Fig. 4). The superimposition also shows amino acid differences between LSD1 and LSD2 in the vicinity of the isoalloxazine ring of FAD, which may also affect inhibitor binding. These residues include N440 in LSD2 and T335 in LSD1 (hereafter, the corresponding residues in LSD1 are shown in parentheses), I438 (V333), Y545 (F538), and Q803 (A809). It is noteworthy that in all cases, the residue in LSD2 has a bulkier side chain than the corresponding residue in LSD1, making the catalytic cavity of LSD2 smaller than that of LSD1. This structural feature is consistent with the assay results showing that 2-PCPA derivatives with bulky substituents generally have reduced inhibitory activities against LSD2 but maintain their LSD1-inhibitory activities.77,87,134
Figure 4.
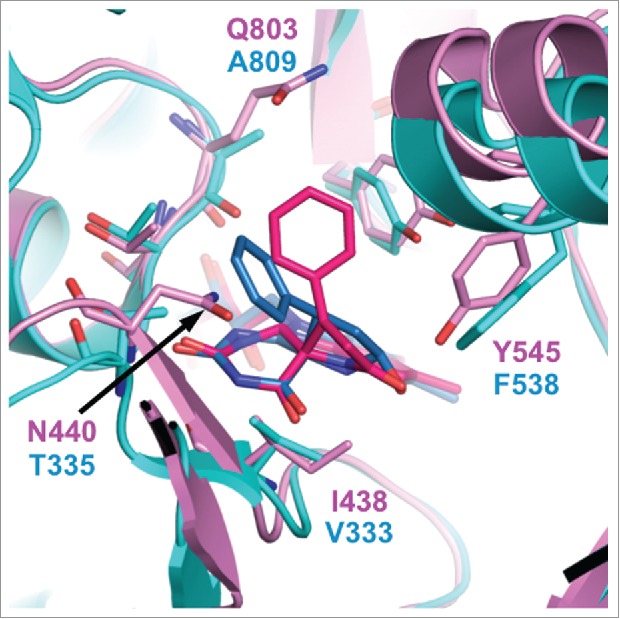
Superimposition of the catalytic cavities of LSD1/KDM1A (cyan) and LSD2/KDM1B (magenta) in complex with 2-PCPA. The FAD•2-PCPA adduct in the LSD1•2-PCPA complex (PDB ID: 2Z5U) is depicted in blue, and that in the LSD2•2-PCPA complex (PDB ID: 4GUU) is depicted in red.
Overall, the amino acid sequences and tertiary structures of the catalytic domains of LSD1 and LSD2 are similar. However, a few of the amino acid differences mentioned above may confer selectivity in the binding of inhibitors. Structural analyses of LSD2 in complexes with 2-PCPA derivatives with various shapes can be expected to facilitate the development of LSD2-specific inhibitors. The surface structures of the AOD in LSD1 and LSD2 differ considerably.55 Furthermore, only LSD2 has the NPAC-binding site56 and the second H3-binding site between the CW domain and the SWIRM domain.58 These features may be useful for the development of LSD2-specific inhibitors.
Given that the crystal structures of LSD1 complexes with reversible inhibitors have been elucidated,126 structural information on their binding modes and possible additional druggable sites will be useful in developing additional LSD1-specific inhibitors or in developing LSD2-specific inhibitors that are useful for studying chemical biology of LSD2. Pan-KDM1 inhibitors with potency and selectivity for both LSD1 and LSD2 over other FAD-containing enzymes may also be developed and can be expected to be useful for drug discovery.
Conclusions
The discovery of FAD-dependent demethylases and α-ketoglutarate-dependent demethylases enabled researchers to modulate epigenetic histone methylation by controlling the reverse (i.e., demethylation) process. Herein, we have reviewed published small-molecule inhibitors for LSD1/KDM1A and pan-KDM1 inhibitors. The inhibitors can be categorized as either irreversible or reversible, and the irreversible inhibitors can be classified as 2-PCPA derivatives, N-alkylated 2-PCPA derivatives, or non-2-PCPA-type derivatives, based on the chemical structures of their FAD-inactivating functional groups. Some of the irreversible inhibitors show high selectivity and potency toward LSD1, and several of them are currently in clinical trials for the treatments of cancers and neurodegenerative diseases. That is, the development of LSD1-specific inhibitors has generally been successful, and the prospects for drug discovery are promising. In contrast, few LSD2-specific inhibitors have been discovered, although some compounds have shown selectivity for inhibition of LSD2, as described herein. Additional information about LSD1 and LSD2 in complexes with their natural ligands or synthetic compounds can be expected to aid the development of more potent, ligand-efficient, and target-specific inhibitors for these FAD-dependent lysine demethylases.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Prof. Naoki Umezawa (Nagoya City University) for critical reading of the manuscript, and Dr. Takehiro Fukami and Ms. Mayuko Yasuda for the investigation of clinical studies. This study was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (no. JP16H05089) and by the Japan Science and Technology Agency PRESTO program.
References
- 1.Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41-5; PMID: 10638745; https://doi.org/ 10.1038/47412 [DOI] [PubMed] [Google Scholar]
- 2.Simpson RT. Structure of chromatin containing extensively acetylated H3 and H4. Cell 1978; 13:691-9; PMID: 657272; https://doi.org/ 10.1016/0092-8674(78)90219-2 [DOI] [PubMed] [Google Scholar]
- 3.Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 1978; 14:105-13; PMID: 667927; https://doi.org/ 10.1016/0092-8674(78)90305-7 [DOI] [PubMed] [Google Scholar]
- 4.Sealy L, Chalkley R. The effect of sodium butyrate on histone modification. Cell 1978; 14:115-21; PMID: 667928; https://doi.org/ 10.1016/0092-8674(78)90306-9 [DOI] [PubMed] [Google Scholar]
- 5.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. The Journal of biological chemistry 1990; 265:17174-9; PMID: 2211619 [PubMed] [Google Scholar]
- 6.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science (New York, NY) 1996; 272:408-11; https://doi.org/ 10.1126/science.272.5260.408 [DOI] [PubMed] [Google Scholar]
- 7.Carmen AA, Rundlett SE, Grunstein M. HDA1 and HDA3 are components of a yeast histone deacetylase (HDA) complex. The Journal of biological chemistry 1996; 271:15837-44. PMID: 8663039; https://doi.org/ 10.1074/jbc.271.26.15837 [DOI] [PubMed] [Google Scholar]
- 8.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119:941-53; PMID: 15620353; https://doi.org/ 10.1016/j.cell.2004.12.012 [DOI] [PubMed] [Google Scholar]
- 9.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science (New York, NY) 2001; 292:110-3; https://doi.org/ 10.1126/science.1060118 [DOI] [PubMed] [Google Scholar]
- 10.Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell 2002; 109:801-6; PMID: 12110177; https://doi.org/ 10.1016/S0092-8674(02)00798-5 [DOI] [PubMed] [Google Scholar]
- 11.Pedersen MT, Helin K. Histone demethylases in development and disease. Trends in cell biology 2010; 20:662-71; PMID: 20863703; https://doi.org/ 10.1016/j.tcb.2010.08.011 [DOI] [PubMed] [Google Scholar]
- 12.Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. A novel mammalian flavin-dependent histone demethylase. The Journal of biological chemistry 2009; 284:17775-82; PMID: 19407342; https://doi.org/ 10.1074/jbc.M109.003087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006; 439:811-6; PMID: 16362057; https://doi.org/ 10.1038/nature04433 [DOI] [PubMed] [Google Scholar]
- 14.Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006; 125:483-95; PMID: 16603237; https://doi.org/ 10.1016/j.cell.2006.03.027 [DOI] [PubMed] [Google Scholar]
- 15.Markolovic S, Leissing TM, Chowdhury R, Wilkins SE, Lu X, Schofield CJ. Structure-function relationships of human JmjC oxygenases-demethylases versus hydroxylases. Current opinion in structural biology 2016; 41:62-72; PMID: 27309310; https://doi.org/ 10.1016/j.sbi.2016.05.013 [DOI] [PubMed] [Google Scholar]
- 16.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, et al.. New nomenclature for chromatin-modifying enzymes. Cell 2007; 131:633-6; PMID: 18022353; https://doi.org/ 10.1016/j.cell.2007.10.039 [DOI] [PubMed] [Google Scholar]
- 17.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et al.. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nature medicine 2012; 18:605-11; PMID: 22406747; https://doi.org/ 10.1038/nm.2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al.. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012; 21:473-87; PMID: 22464800; https://doi.org/ 10.1016/j.ccr.2012.03.014 [DOI] [PubMed] [Google Scholar]
- 19.Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al.. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014; 157:580-94; PMID: 24726434; https://doi.org/ 10.1016/j.cell.2014.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishikawa Y, Gamo K, Yabuki M, Takagi S, Toyoshima K, Nakayama K, Nakayama A, Morimoto M, Miyashita H, Dairiki R, et al.. A novel LSD1 inhibitor T-3775440 disrupts GFI1B-containing complex leading to transdifferentiation and impaired growth of AML cells. Molecular cancer therapeutics 2016; PMID: 27903753 [DOI] [PubMed] [Google Scholar]
- 21.Maes T, Carceller E, Salas J, Ortega A, Buesa C. Advances in the development of histone lysine demethylase inhibitors. Current opinion in pharmacology 2015; 23:52-60; PMID: 26057211; https://doi.org/ 10.1016/j.coph.2015.05.009 [DOI] [PubMed] [Google Scholar]
- 22.Morera L, Lubbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics 2016; 8:57; PMID: 27222667; https://doi.org/ 10.1186/s13148-016-0223-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stavropoulos P, Blobel G, Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nature structural & molecular biology 2006; 13:626-32; https://doi.org/ 10.1038/nsmb1113 [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proceedings of the National Academy of Sciences of the United States of America 2006; 103:13956-61; PMID: 16956976; https://doi.org/ 10.1073/pnas.0606381103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aravind L, Iyer LM. The SWIRM domain: a conserved module found in chromosomal proteins points to novel chromatin-modifying activities. Genome biology 2002; 3:Research0039; https://doi.org/ 10.1186/gb-2002-3-8-research0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tochio N, Umehara T, Koshiba S, Inoue M, Yabuki T, Aoki M, Seki E, Watanabe S, Tomo Y, Hanada M, et al.. Solution structure of the SWIRM domain of human histone demethylase LSD1. Structure (London, England : 1993) 2006; 14:457-68; PMID: 16531230; https://doi.org/ 10.1016/j.str.2005.12.004 [DOI] [PubMed] [Google Scholar]
- 27.Hirschi A, Martin WJ, Luka Z, Loukachevitch LV, Reiter NJ. G-quadruplex RNA binding and recognition by the lysine-specific histone demethylase-1 enzyme. RNA 2016; 22:1250-60; PMID: 27277658; https://doi.org/ 10.1261/rna.057265.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005; 437:432-5; PMID: 16079794 [DOI] [PubMed] [Google Scholar]
- 29.Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Molecular cell 2005; 19:857-64; PMID: 16140033; https://doi.org/ 10.1016/j.molcel.2005.08.027 [DOI] [PubMed] [Google Scholar]
- 30.Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Molecular cell 2006; 23:377-87; PMID: 16885027; https://doi.org/ 10.1016/j.molcel.2006.07.012 [DOI] [PubMed] [Google Scholar]
- 31.Burg JM, Makhoul AT, CWt Pemble, Link JE, Heller FJ, McCafferty DG. A rationally-designed chimeric KDM1A/KDM1B histone demethylase tower domain deletion mutant retaining enzymatic activity. FEBS Lett 2015; 589:2340-6; PMID: 26226427; https://doi.org/ 10.1016/j.febslet.2015.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. The Journal of biological chemistry 2007; 282:20070-4; PMID: 17537733; https://doi.org/ 10.1074/jbc.C700100200 [DOI] [PubMed] [Google Scholar]
- 33.Yang M, Culhane JC, Szewczuk LM, Gocke CB, Brautigam CA, Tomchick DR, Machius M, Cole PA, Yu H. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nature structural & molecular biology 2007; 14:535-9; https://doi.org/ 10.1038/nsmb1255 [DOI] [PubMed] [Google Scholar]
- 34.Burg JM, Gonzalez JJ, Maksimchuk KR, McCafferty DG. Lysine-Specific Demethylase 1A (KDM1A/LSD1): Product Recognition and Kinetic Analysis of Full-Length Histones. Biochemistry 2016; 55:1652-62; PMID: 26673564; https://doi.org/ 10.1021/acs.biochem.5b01135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005; 437:436-9; PMID: 16079795 [DOI] [PubMed] [Google Scholar]
- 36.Zibetti C, Adamo A, Binda C, Forneris F, Toffolo E, Verpelli C, Ginelli E, Mattevi A, Sala C, Battaglioli E. Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J Neurosci 2010; 30:2521-32; PMID: 20164337; https://doi.org/ 10.1523/JNEUROSCI.5500-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wada T, Koyama D, Kikuchi J, Honda H, Furukawa Y. Overexpression of the shortest isoform of histone demethylase LSD1 primes hematopoietic stem cells for malignant transformation. Blood 2015; 125:3731-46; PMID: 25904247; https://doi.org/ 10.1182/blood-2014-11-610907 [DOI] [PubMed] [Google Scholar]
- 38.Laurent B, Ruitu L, Murn J, Hempel K, Ferrao R, Xiang Y, Liu S, Garcia BA, Wu H, Wu F, et al.. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Molecular cell 2015; 57:957-70; PMID: 25684206; https://doi.org/ 10.1016/j.molcel.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Telese F, Tan Y, Li W, Jin C, He X, Basnet H, Ma Q, Merkurjev D, Zhu X, et al.. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat Neurosci 2015; 18:1256-64; PMID: 26214369; https://doi.org/ 10.1038/nn.4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toffolo E, Rusconi F, Paganini L, Tortorici M, Pilotto S, Heise C, Verpelli C, Tedeschi G, Maffioli E, Sala C, et al.. Phosphorylation of neuronal Lysine-Specific Demethylase 1LSD1/KDM1A impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2. Journal of neurochemistry 2014; 128:603-16; PMID: 24111946; https://doi.org/ 10.1111/jnc.12457 [DOI] [PubMed] [Google Scholar]
- 41.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, et al.. p53 is regulated by the lysine demethylase LSD1. Nature 2007; 449:105-8; PMID: 17805299; https://doi.org/ 10.1038/nature06092 [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, et al.. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nature genetics 2009; 41:125-9; PMID: 19098913; https://doi.org/ 10.1038/ng.268 [DOI] [PubMed] [Google Scholar]
- 43.Nicholson TB, Chen T. LSD1 demethylates histone and non-histone proteins. Epigenetics 2014; 4:129-32; https://doi.org/ 10.4161/epi.4.3.8443 [DOI] [PubMed] [Google Scholar]
- 44.Hamamoto R, Saloura V, Nakamura Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nature reviews Cancer 2015; 15:110-24; PMID: 25614009; https://doi.org/ 10.1038/nrc3884 [DOI] [PubMed] [Google Scholar]
- 45.Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J 2010; 29:1803-16; PMID: 20389281; https://doi.org/ 10.1038/emboj.2010.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baron R, Binda C, Tortorici M, McCammon JA, Mattevi A. Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure (London, England : 1993) 2011; 19:212-20; PMID: 21300290; https://doi.org/ 10.1016/j.str.2011.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009; 461:415-8; PMID: 19727073; https://doi.org/ 10.1038/nature08315 [DOI] [PubMed] [Google Scholar]
- 48.Yang Z, Jiang J, Stewart MD, Qi S, Yamane K, Li J, Zhang Y, Wong J. AOF1 is a histone H3K4 demethylase possessing demethylase activity-independent repression function. Cell Res 2010; 20:276-87; PMID: 20101264; https://doi.org/ 10.1038/cr.2010.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, Lan F, Mei P, Yuan GC, Lian C, et al.. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Molecular cell 2010; 39:222-33; PMID: 20670891; https://doi.org/ 10.1016/j.molcel.2010.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Essen D, Zhu Y, Saccani S. A feed-forward circuit controlling inducible NF-kappaB target gene activation by promoter histone demethylation. Molecular cell 2010; 39:750-60; PMID: 20832726; https://doi.org/ 10.1016/j.molcel.2010.08.010 [DOI] [PubMed] [Google Scholar]
- 51.Nagaoka K, Hino S, Sakamoto A, Anan K, Takase R, Umehara T, Yokoyama S, Sasaki Y, Nakao M. Lysine-specific demethylase 2 suppresses lipid influx and metabolism in hepatic cells. Molecular and cellular biology 2015; 35:1068-80; PMID: 25624347; https://doi.org/ 10.1128/MCB.01404-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He F, Umehara T, Saito K, Harada T, Watanabe S, Yabuki T, Kigawa T, Takahashi M, Kuwasako K, Tsuda K, et al.. Structural insight into the zinc finger CW domain as a histone modification reader. Structure (London, England : 1993) 2010; 18:1127-39; PMID: 20826339; https://doi.org/ 10.1016/j.str.2010.06.012 [DOI] [PubMed] [Google Scholar]
- 53.Hoppmann V, Thorstensen T, Kristiansen PE, Veiseth SV, Rahman MA, Finne K, Aalen RB, Aasland R. The CW domain, a new histone recognition module in chromatin proteins. Embo j 2011; 30:1939-52; PMID: 21522130; https://doi.org/ 10.1038/emboj.2011.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y, Tempel W, Zhang Q, Liang X, Loppnau P, Qin S, Min J. Family-wide Characterization of Histone Binding Abilities of Human CW Domain-containing Proteins. The Journal of biological chemistry 2016; 291:9000-13; PMID: 26933034; https://doi.org/ 10.1074/jbc.M116.718973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Q, Qi S, Xu M, Yu L, Tao Y, Deng Z, Wu W, Li J, Chen Z, Wong J. Structure-function analysis reveals a novel mechanism for regulation of histone demethylase LSD2/AOF1/KDM1b. Cell Res 2013; 23:225-41; PMID: 23266887; https://doi.org/ 10.1038/cr.2012.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fang R, Chen F, Dong Z, Hu D, Barbera AJ, Clark EA, Fang J, Yang Y, Mei P, Rutenberg M, et al.. LSD2/KDM1B and its cofactor NPAC/GLYR1 endow a structural and molecular model for regulation of H3K4 demethylation. Molecular cell 2013; 49:558-70; PMID: 23260659; https://doi.org/ 10.1016/j.molcel.2012.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang Y, Vasilatos SN, Boric L, Shaw PG, Davidson NE. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res Treat 2012; 131:777-89; PMID: 21452019; https://doi.org/ 10.1007/s10549-011-1480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen F, Yang H, Dong Z, Fang J, Wang P, Zhu T, Gong W, Fang R, Shi YG, Li Z, et al.. Structural insight into substrate recognition by histone demethylase LSD2/KDM1b. Cell Res 2013; 23:306-9; PMID: 23357850; https://doi.org/ 10.1038/cr.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burg JM, Link JE, Morgan BS, Heller FJ, Hargrove AE, McCafferty DG. KDM1 class flavin-dependent protein lysine demethylases. Biopolymers 2015; 104:213-46; PMID: 25787087; https://doi.org/ 10.1002/bip.22643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, et al.. The promise and peril of chemical probes. Nature chemical biology 2015; 11:536-41; PMID: 26196764; https://doi.org/ 10.1038/nchembio.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol 2006; 13:563-7; PMID: 16793513; https://doi.org/ 10.1016/j.chembiol.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 62.Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007; 46:4408-16; PMID: 17367163; https://doi.org/ 10.1021/bi0618621 [DOI] [PubMed] [Google Scholar]
- 63.Zheng YC, Yu B, Chen ZS, Liu Y, Liu HM. TCPs: privileged scaffolds for identifying potent LSD1 inhibitors for cancer therapy. Epigenomics 2016; 8:651-66; PMID: 27102879; https://doi.org/ 10.2217/epi-2015-0002 [DOI] [PubMed] [Google Scholar]
- 64.Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. Journal of the American Chemical Society 2009; 131:17536-7; PMID: 19950987; https://doi.org/ 10.1021/ja907055q [DOI] [PubMed] [Google Scholar]
- 65.Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T, Yokoyama S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 2010; 49:6494-503; PMID: 20568732; https://doi.org/ 10.1021/bi100299r [DOI] [PubMed] [Google Scholar]
- 66.Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, Cole PA, Yu H. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 2007; 46:8058-65; PMID: 17569509; https://doi.org/ 10.1021/bi700664y [DOI] [PubMed] [Google Scholar]
- 67.Mimasu S, Sengoku T, Fukuzawa S, Umehara T, Yokoyama S. Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 A. Biochemical and biophysical research communications 2008; 366:15-22; PMID: 18039463; https://doi.org/ 10.1016/j.bbrc.2007.11.066 [DOI] [PubMed] [Google Scholar]
- 68.Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proceedings of the National Academy of Sciences of the United States of America 2003; 100:9750-5; PMID: 12913124; https://doi.org/ 10.1073/pnas.1633804100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, Umehara T, Yokoyama S, Kosai K, Nakao M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun 2012; 3:758; PMID: 22453831; https://doi.org/ 10.1038/ncomms1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yatim A, Benne C, Sobhian B, Laurent-Chabalier S, Deas O, Judde JG, Lelievre JD, Levy Y, Benkirane M. NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Molecular cell 2012; 48:445-58; PMID: 23022380; https://doi.org/ 10.1016/j.molcel.2012.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Konovalov S, Garcia-Bassets I. Analysis of the levels of lysine-specific demethylase 1 (LSD1) mRNA in human ovarian tumors and the effects of chemical LSD1 inhibitors in ovarian cancer cell lines. Journal of ovarian research 2013; 6:75; https://doi.org/ 10.1186/1757-2215-6-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schooley A, Moreno-Andres D, De Magistris P, Vollmer B, Antonin W. The lysine demethylase LSD1 is required for nuclear envelope formation at the end of mitosis. Journal of cell science 2015; 128:3466-77; https://doi.org/ 10.1242/jcs.173013 [DOI] [PubMed] [Google Scholar]
- 73.Li A, He Y, Sun S, Cai C, Li H. Lysine-specific demethylase 1 inhibitors protect cochlear spiral ganglion neurons against cisplatin-induced damage. Neuroreport 2015; 26:539-47. [DOI] [PubMed] [Google Scholar]
- 74.Hirano K, Namihira M. LSD1 Mediates Neuronal Differentiation of Human Fetal Neural Stem Cells by Controlling the Expression of a Novel Target Gene, HEYL. Stem cells (Dayton, Ohio) 2016; 34:1872-82; PMID: 27018646; https://doi.org/ 10.1002/stem.2362 [DOI] [PubMed] [Google Scholar]
- 75.Di Stefano B, Collombet S, Jakobsen JS, Wierer M, Sardina JL, Lackner A, Stadhouders R, Segura-Morales C, Francesconi M, Limone F, et al.. C/EBPalpha creates elite cells for iPSC reprogramming by upregulating Klf4 and increasing the levels of Lsd1 and Brd4. Nature cell biology 2016; 18:371-81; PMID: 26974661; https://doi.org/ 10.1038/ncb3326 [DOI] [PubMed] [Google Scholar]
- 76.Sato T, Cesaroni M, Chung W, Panjarian S, Tran A, Madzo J, Okamoto Y, Zhang H, Chen X, Jelinek J, et al.. Transcriptional Selectivity of Epigenetic Therapy in Cancer. Cancer Res 2016; 77(2):470-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A, Ciossani G, Botrugno OA, Forneris F, Tardugno M, et al.. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. Journal of the American Chemical Society 2010; 132:6827-33; PMID: 20415477; https://doi.org/ 10.1021/ja101557k [DOI] [PubMed] [Google Scholar]
- 78.Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. MBio 2013; 4:e00558-12; https://doi.org/ 10.1128/mBio.00558-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rotili D, Tomassi S, Conte M, Benedetti R, Tortorici M, Ciossani G, Valente S, Marrocco B, Labella D, Novellino E, et al.. Pan-histone demethylase inhibitors simultaneously targeting Jumonji C and lysine-specific demethylases display high anticancer activities. J Med Chem 2014; 57:42-55. [DOI] [PubMed] [Google Scholar]
- 80.Han Y, Wu C, Lv H, Liu N, Deng H. Novel Tranylcypromine/Hydroxylcinnamic Acid Hybrids as Lysine-Specific Demethylase 1 Inhibitors with Potent Antitumor Activity. Chemical & pharmaceutical bulletin 2015; 63:882-9. [DOI] [PubMed] [Google Scholar]
- 81.Miyamura S, Araki M, Ota Y, Itoh Y, Yasuda S, Masuda M, Taniguchi T, Sowa Y, Sakai T, Suzuki T, et al.. C-H activation enables a rapid structure-activity relationship study of arylcyclopropyl amines for potent and selective LSD1 inhibitors. Org Biomol Chem 2016; 14:8576-85; PMID: 27548471; https://doi.org/ 10.1039/C6OB01483F [DOI] [PubMed] [Google Scholar]
- 82.Bonivento D, Milczek EM, McDonald GR, Binda C, Holt A, Edmondson DE, Mattevi A. Potentiation of ligand binding through cooperative effects in monoamine oxidase B. The Journal of biological chemistry 2010; 285:36849-56; PMID: 20855894; https://doi.org/ 10.1074/jbc.M110.169482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Agranat I, Caner H, Caldwell J. Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov 2002; 1:753-68; PMID: 12360254; https://doi.org/ 10.1038/nrd915 [DOI] [PubMed] [Google Scholar]
- 84.Ogasawara D, Suzuki T, Mino K, Ueda R, Khan MN, Matsubara T, Koseki K, Hasegawa M, Sasaki R, Nakagawa H, et al.. Synthesis and biological activity of optically active NCL-1, a lysine-specific demethylase 1 selective inhibitor. Bioorg Med Chem 2011; 19:3702-8; PMID: 21227703; https://doi.org/ 10.1016/j.bmc.2010.12.024 [DOI] [PubMed] [Google Scholar]
- 85.Valente S, Rodriguez V, Mercurio C, Vianello P, Saponara B, Cirilli R, Ciossani G, Labella D, Marrocco B, Monaldi D, et al.. Pure enantiomers of benzoylamino-tranylcypromine: LSD1 inhibition, gene modulation in human leukemia cells and effects on clonogenic potential of murine promyelocytic blasts. European journal of medicinal chemistry 2015; 94:163-74; PMID: 25768700; https://doi.org/ 10.1016/j.ejmech.2015.02.060 [DOI] [PubMed] [Google Scholar]
- 86.Valente S, Rodriguez V, Mercurio C, Vianello P, Saponara B, Cirilli R, Ciossani G, Labella D, Marrocco B, Ruoppolo G, et al.. Pure Diastereomers of a Tranylcypromine-Based LSD1 Inhibitor: Enzyme Selectivity and In-Cell Studies. ACS Med Chem Lett 2015; 6:173-7; PMID: 25699146; https://doi.org/ 10.1021/ml500424z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vianello P, Botrugno OA, Cappa A, Dal Zuffo R, Dessanti P, Mai A, Marrocco B, Mattevi A, Meroni G, Minucci S, et al.. Discovery of a Novel Inhibitor of Histone Lysine-Specific Demethylase 1A (KDM1A/LSD1) as Orally Active Antitumor Agent. J Med Chem 2016; 59:1501-17; PMID: 26702542; https://doi.org/ 10.1021/acs.jmedchem.5b01209 [DOI] [PubMed] [Google Scholar]
- 88.Rodriguez V, Valente S, Rovida S, Rotili D, Stazi G, Lucidi A, Ciossani G, Mattevi A, Botrugno OA, Dessanti P, et al.. Pyrrole- and indole-containing tranylcypromine derivatives as novel lysine-specific demethylase 1 inhibitors active on cancer cells. Med Chem Commun 2015; 6:665-70. [Google Scholar]
- 89.Gooden DM, Schmidt DM, Pollock JA, Kabadi AM, McCafferty DG. Facile synthesis of substituted trans-2-arylcyclopropylamine inhibitors of the human histone demethylase LSD1 and monoamine oxidases A and B. Bioorg Med Chem Lett 2008; 18:3047-51; PMID: 18242989; https://doi.org/ 10.1016/j.bmcl.2008.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vianello P, Botrugno OA, Cappa A, Ciossani G, Dessanti P, Mai A, Mattevi A, Meroni G, Minucci S, Thaler F, et al.. Synthesis, biological activity and mechanistic insights of 1-substituted cyclopropylamine derivatives: a novel class of irreversible inhibitors of histone demethylase KDM1A. European journal of medicinal chemistry 2014; 86:352-63; PMID: 25173853; https://doi.org/ 10.1016/j.ejmech.2014.08.068 [DOI] [PubMed] [Google Scholar]
- 91.Pieroni M, Annunziato G, Azzali E, Dessanti P, Mercurio C, Meroni G, Trifiro P, Vianello P, Villa M, Beato C, et al.. Further insights into the SAR of alpha-substituted cyclopropylamine derivatives as inhibitors of histone demethylase KDM1A. European journal of medicinal chemistry 2015; 92:377-86; PMID: 25585008; https://doi.org/ 10.1016/j.ejmech.2014.12.032 [DOI] [PubMed] [Google Scholar]
- 92.Neelamegam R, Ricq EL, Malvaez M, Patnaik D, Norton S, Carlin SM, Hill IT, Wood MA, Haggarty SJ, Hooker JM. Brain-penetrant LSD1 inhibitors can block memory consolidation. ACS Chem Neurosci 2012; 3:120-8; PMID: 22754608; https://doi.org/ 10.1021/cn200104y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McGrath JP, Williamson KE, Balasubramanian S, Odate S, Arora S, Hatton C, Edwards TM, O'Brien T, Magnuson S, Stokoe D, et al.. Pharmacological Inhibition of the Histone Lysine Demethylase KDM1A Suppresses the Growth of Multiple Acute Myeloid Leukemia Subtypes. Cancer Res 2016; 76:1975-88; PMID: 26837761; https://doi.org/ 10.1158/0008-5472.CAN-15-2333 [DOI] [PubMed] [Google Scholar]
- 94.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al.. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015; 28:57-69; PMID: 26175415; https://doi.org/ 10.1016/j.ccell.2015.06.002 [DOI] [PubMed] [Google Scholar]
- 95.Ahmed Khan MN, Tsumoto H, Itoh Y, Ota Y, Suzuki M, Ogasawara D, Nakagawa H, Mizukami T, Miyata N, Suzuki T. Design, synthesis, and biological activity of N-alkylated analogue of NCL1, a selective inhibitor of lysine-specific demethylase 1. Med Chem Commun 2015; 6:407-12; https://doi.org/ 10.1039/C4MD00330F [DOI] [Google Scholar]
- 96.Wu F, Zhou C, Yao Y, Wei L, Feng Z, Deng L, Song Y. 3-(Piperidin-4-ylmethoxy)pyridine Containing Compounds Are Potent Inhibitors of Lysine Specific Demethylase 1. J Med Chem 2016; 59:253-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ogasawara D, Itoh Y, Tsumoto H, Kakizawa T, Mino K, Fukuhara K, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, et al.. Lysine-specific demethylase 1-selective inactivators: protein-targeted drug delivery mechanism. Angew Chem Int Ed Engl 2013; 52:8620-4. [DOI] [PubMed] [Google Scholar]
- 98.Culhane JC, Cole PA. LSD1 and the chemistry of histone demethylation. Current opinion in chemical biology 2007; 11:561-8; PMID: 17851108; https://doi.org/ 10.1016/j.cbpa.2007.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pollock JA, Larrea MD, Jasper JS, McDonnell DP, McCafferty DG. Lysine-specific histone demethylase 1 inhibitors control breast cancer proliferation in ERalpha-dependent and -independent manners. ACS Chem Biol 2012; 7:1221-31; PMID: 22533360; https://doi.org/ 10.1021/cb300108c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA. A mechanism-based inactivator for histone demethylase LSD1. Journal of the American Chemical Society 2006; 128:4536-7; PMID: 16594666; https://doi.org/ 10.1021/ja0602748 [DOI] [PubMed] [Google Scholar]
- 101.Schmitt ML, Hauser AT, Carlino L, Pippel M, Schulz-Fincke J, Metzger E, Willmann D, Yiu T, Barton M, Schule R, et al.. Nonpeptidic propargylamines as inhibitors of lysine specific demethylase 1 (LSD1) with cellular activity. J Med Chem 2013; 56:7334-42; https://doi.org/ 10.1021/jm400792m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Culhane JC, Wang D, Yen PM, Cole PA. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. Journal of the American Chemical Society 2010; 132:3164-76; PMID: 20148560; https://doi.org/ 10.1021/ja909996p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Prusevich P, Kalin JH, Ming SA, Basso M, Givens J, Li X, Hu J, Taylor MS, Cieniewicz AM, Hsiao PY, et al.. A selective phenelzine analogue inhibitor of histone demethylase LSD1. ACS Chem Biol 2014; 9:1284-93; PMID: 24707965; https://doi.org/ 10.1021/cb500018s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kakizawa T, Ota Y, Itoh Y, Tsumoto H, Suzuki T. Histone H3 peptide based LSD1-selective inhibitors. Bioorg Med Chem Lett 2015; 25:1925-8; PMID: 25827526; https://doi.org/ 10.1016/j.bmcl.2015.03.030 [DOI] [PubMed] [Google Scholar]
- 105.Itoh Y, Aihara K, Mellini P, Tojo T, Ota Y, Tsumoto H, Solomon VR, Zhan P, Suzuki M, Ogasawara D, et al.. Identification of SNAIL1 Peptide-Based Irreversible Lysine-Specific Demethylase 1-Selective Inactivators. J Med Chem 2016; 59:1531-44; https://doi.org/ 10.1021/acs.jmedchem.5b01323 [DOI] [PubMed] [Google Scholar]
- 106.Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA Jr.. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proceedings of the National Academy of Sciences of the United States of America 2007; 104:8023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nowotarski SL, Pachaiyappan B, Holshouser SL, Kutz CJ, Li Y, Huang Y, Sharma SK, Casero RA Jr., Woster PM. Structure-activity study for (bis)ureidopropyl- and (bis)thioureidopropyldiamine LSD1 inhibitors with 3-5-3 and 3-6-3 carbon backbone architectures. Bioorg Med Chem 2015; 23:1601-12; PMID: 25725609; https://doi.org/ 10.1016/j.bmc.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, Liu Y, Ward D, Quan J, Ye T, et al.. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res 2011; 71:7238-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Willmann D, Lim S, Wetzel S, Metzger E, Jandausch A, Wilk W, Jung M, Forne I, Imhof A, Janzer A, et al.. Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Int J Cancer 2012; 131:2704-9; PMID: 22447389; https://doi.org/ 10.1002/ijc.27555 [DOI] [PubMed] [Google Scholar]
- 110.Hazeldine S, Pachaiyappan B, Steinbergs N, Nowotarski S, Hanson AS, Casero RA Jr., Woster PM. Low molecular weight amidoximes that act as potent inhibitors of lysine-specific demethylase 1. J Med Chem 2012; 55:7378-91; PMID: 22876979; https://doi.org/ 10.1021/jm3002845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dulla B, Kirla KT, Rathore V, Deora GS, Kavela S, Maddika S, Chatti K, Reiser O, Iqbal J, Pal M. Synthesis and evaluation of 3-amino/guanidine substituted phenyl oxazoles as a novel class of LSD1 inhibitors with anti-proliferative properties. Org Biomol Chem 2013; 11:3103-7; PMID: 23575971; https://doi.org/ 10.1039/c3ob40217g [DOI] [PubMed] [Google Scholar]
- 112.Hitchin JR, Blagg J, Burke R, Burns S, Cockerill MJ, Fairweather EE, Hutton C, Jordan AM, McAndrew C, Mirza A, et al.. Development and evaluation of selective, reversible LSD1 inhibitors derived from fragments. MedChemComm 2013; 4:1513-22; https://doi.org/ 10.1039/c3md00226h [DOI] [Google Scholar]
- 113.Zheng YC, Duan YC, Ma JL, Xu RM, Zi X, Lv WL, Wang MM, Ye XW, Zhu S, Mobley D, et al.. Triazole-dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J Med Chem 2013; 56:8543-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ye X-W, Zheng Y-C, Duan Y-C, Wang M-M, Yu B, Ren J-L, Ma J-L, Zhang E, Liu H-M. Synthesis and biological evaluation of coumarin-1,2,3-triazole-dithiocarbamate hybrids as potent LSD1 inhibitors. MedChemComm 2014; 5:650-4. [Google Scholar]
- 115.Kutz CJ, Holshouser SL, Marrow EA, Woster PM. 3,5-Diamino-1,2,4-triazoles as a novel scaffold for potent, reversible LSD1 (KDM1A) inhibitors. Medchemcomm 2014; 5:1863-70; PMID: 25580204; https://doi.org/ 10.1039/C4MD00283K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou C, Kang D, Xu Y, Zhang L, Zha X. Identification of Novel Selective Lysine-Specific Demethylase 1 (LSD1) Inhibitors Using a Pharmacophore-Based Virtual Screening Combined with Docking. Chemical biology & drug design 2015; 85:659-71. [DOI] [PubMed] [Google Scholar]
- 117.Ma LY, Zheng YC, Wang SQ, Wang B, Wang ZR, Pang LP, Zhang M, Wang JW, Ding L, Li J, et al.. Design, synthesis, and structure-activity relationship of novel LSD1 inhibitors based on pyrimidine-thiourea hybrids as potent, orally active antitumor agents. J Med Chem 2015; 58:1705-16. [DOI] [PubMed] [Google Scholar]
- 118.Wang S, Zhao LJ, Zheng YC, Shen DD, Miao EF, Qiao XP, Zhao LJ, Liu Y, Huang R, Yu B, et al.. Design, synthesis and biological evaluation of [1,2,4]triazolo[1,5-a]pyrimidines as potent lysine specific demethylase 1 (LSD1/KDM1A) inhibitors. European journal of medicinal chemistry 2017; 125:940-51. [DOI] [PubMed] [Google Scholar]
- 119.Mould DP, McGonagle AE, Wiseman DH, Williams EL, Jordan AM. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date. Medicinal research reviews 2015; 35:586-618; PMID: 25418875; https://doi.org/ 10.1002/med.21334 [DOI] [PubMed] [Google Scholar]
- 120.Sorna V, Theisen ER, Stephens B, Warner SL, Bearss DJ, Vankayalapati H, Sharma S. High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J Med Chem 2013; 56:9496-508; PMID: 24237195; https://doi.org/ 10.1021/jm400870h [DOI] [PubMed] [Google Scholar]
- 121.Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, Sorna V, Beckerle MC, Sharma S, Lessnick SL. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res 2014; 20:4584-97; PMID: 24963049; https://doi.org/ 10.1158/1078-0432.CCR-14-0072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K, Iyer SP, Bearss D, Bhalla KN. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014; 28:2155-64; PMID: 24699304; https://doi.org/ 10.1038/leu.2014.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhou Y, Li Y, Wang WJ, Xiang P, Luo XM, Yang L, Yang SY, Zhao YL. Synthesis and biological evaluation of novel (E)-N′-(2,3-dihydro-1H-inden-1-ylidene) benzohydrazides as potent LSD1 inhibitors. Bioorg Med Chem Lett 2016; 26:4552-7. [DOI] [PubMed] [Google Scholar]
- 124.Luka Z, Moss F, Loukachevitch LV, Bornhop DJ, Wagner C. Histone demethylase LSD1 is a folate-binding protein. Biochemistry 2011; 50:4750-6; PMID: 21510664; https://doi.org/ 10.1021/bi200247b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Luka Z, Pakhomova S, Loukachevitch LV, Calcutt MW, Newcomer ME, Wagner C. Crystal structure of the histone lysine specific demethylase LSD1 complexed with tetrahydrofolate. Protein science : a publication of the Protein Society 2014; 23:993-8; PMID: 24715612; https://doi.org/ 10.1002/pro.2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Speranzini V, Rotili D, Ciossani G, Pilotto S, Marrocco B, Forgione M, Lucidi A, Forneris F, Mehdipour P, Velankar S, et al.. Polymyxins and quinazolines are LSD1/KDM1A inhibitors with unusual structural features. Sci Adv 2016; 2:e1601017; PMID: 27626075; https://doi.org/ 10.1126/sciadv.1601017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chang Y, Ganesh T, Horton JR, Spannhoff A, Liu J, Sun A, Zhang X, Bedford MT, Shinkai Y, Snyder JP, et al.. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. Journal of molecular biology 2010; 400:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Abdulla A, Zhao X, Yang F. Natural Polyphenols Inhibit Lysine-Specific Demethylase-1 in vitro. Journal of biochemical and pharmacological research 2013; 1:56-63; PMID: 23662249 [PMC free article] [PubMed] [Google Scholar]
- 129.Sakane C, Okitsu T, Wada A, Sagami H, Shidoji Y. Inhibition of lysine-specific demethylase 1 by the acyclic diterpenoid geranylgeranoic acid and its derivatives. Biochemical and biophysical research communications 2014; 444:24-9; https://doi.org/ 10.1016/j.bbrc.2013.12.144 [DOI] [PubMed] [Google Scholar]
- 130.Zheng YC, Shen DD, Ren M, Liu XQ, Wang ZR, Liu Y, Zhang QN, Zhao LJ, Zhao LJ, Ma JL, et al.. Baicalin, a natural LSD1 inhibitor. Bioorganic chemistry 2016; 69:129-31; PMID: 27814566; https://doi.org/ 10.1016/j.bioorg.2016.10.004 [DOI] [PubMed] [Google Scholar]
- 131.Kumarasinghe IR, Woster PM. Synthesis and evaluation of novel cyclic Peptide inhibitors of lysine-specific demethylase 1. ACS Med Chem Lett 2014; 5:29-33; PMID: 24883177; https://doi.org/ 10.1021/ml4002997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tortorici M, Borrello MT, Tardugno M, Chiarelli LR, Pilotto S, Ciossani G, Vellore NA, Bailey SG, Cowan J, O'Connell M, et al.. Protein recognition by short peptide reversible inhibitors of the chromatin-modifying LSD1/CoREST lysine demethylase. ACS Chem Biol 2013; 8:1677-82; PMID: 23721412; https://doi.org/ 10.1021/cb4001926 [DOI] [PubMed] [Google Scholar]
- 133.Mino K, Nishimura S, Ninomiya S, Tujii H, Matsumori Y, Tsuchida M, Hosoi M, Koseki K, Wada S, Hasegawa M, et al.. Regulation of tissue factor pathway inhibitor-2 (TFPI-2) expression by lysine-specific demethylase 1 and 2 (LSD1 and LSD2). Bioscience, biotechnology, and biochemistry 2014; 78:1010-7; PMID: 25036127; https://doi.org/ 10.1080/09168451.2014.910104 [DOI] [PubMed] [Google Scholar]
- 134.Kakizawa T, Mizukami T, Itoh Y, Hasegawa M, Sasaki R, Suzuki T. Evaluation of phenylcyclopropylamine compounds by enzymatic assay of lysine-specific demethylase 2 in the presence of NPAC peptide. Bioorg Med Chem Lett 2016; 26:1193-5; PMID: 26794039; https://doi.org/ 10.1016/j.bmcl.2016.01.036 [DOI] [PubMed] [Google Scholar]