

ABSTRACT
Malignancies are characterized by the reprogramming of epigenetic patterns. This reprogramming includes gains or losses in DNA methylation and disruption of normal patterns of covalent histone modifications, which are associated with changes in chromatin remodeling processes. This review will focus on the mechanisms underlying this reprogramming and, specifically, on the role of histone modification in chromatin machinery and the modifications in epigenetic processes occurring in brain cancer, with a specific focus on epigenetic therapies for pediatric brain tumors.
KEYWORDS: Acetylation, brain tumor, demethylase, DIPG, epigenetic, histone, methylation, methyltransferase, pediatric
Role of epigenetics in chromatin machinery
Epigenetic background
DNA is packaged in chromatin, whose basic repeating unit, the nucleosome, consists of ∼146 nucleotides wrapped around an octamer of specialized proteins called histones. Each of the 8 histone proteins has amino acid “tails” that stick out from the nucleosome. The modulation of the wrapping of DNA around this octamer constitutes the physical basis for regulation of transcription through nucleosomal DNA.1,2 Epigenetic processes include covalent modifications of DNA (e.g., DNA cytosine methylation and hydroxymethylation) and of histone tails (such as histone lysine acetylation, lysine and arginine methylation, serine and threonine phosphorylation, and lysine ubiquitination or sumoylation).3-5 Enzymes bringing these modifications to DNA or to the histone tail are termed “writers” or “erasers,” while chromatin-associated “readers” are proteins involved in the recruitment of other proteins that contain additional chromatin-modifying activities (including writers and erasers that add or remove, respectively, specific histone posttranslational modifications) (reviewed in6). All together, these modifications determine active and repressive chromatin states of genes and of chromosomal regions, and thus operate as switches to turn gene expression either “on” or “off,” or to modulate gene expression levels (such as by enhancer methylation).7,8
Histone acetylation
Histone acetylation leads to an increased negative charge, which loosens the interaction between the histone and the negatively charged DNA. In addition, acetylated histones recruit specific chromatin-associated proteins that contain bromodomains as the dominant mode of recognition of acetylated lysine residues, present in 47 human proteins (reviewed in6). Histone acetylation also plays an important role in the regulation of RNA Polymerase II (RNAP II) activation by enhancing the search kinetics of transcriptional activators and, later, accelerating the transition of RNAP II from initiation to elongation.9 For example, studies in living cells revealed that H3K27ac associated with active promoters and enhancers can modify downstream transcription kinetics by as much as 50%.9
Histone methylation
Histone methylation, by contrast, does not alter histone charge but, instead, creates a docking site for chromatin-associated proteins that contain specific methyl histone-binding domains. Histone lysine residues can be mono-, di-, or tri-methylated, whereas arginines can be monomethylated or symmetrically/asymmetrically dimethylated, with each modification having a specific biologic effect. Several lysine residues in histone H3 and H4, namely, H3K4, K9, K27, K36, K79, and H4K20, have been found to be methylated and have been extensively studied10 (Fig. 1). Methylation of H3K4, H3K36, and H3K79 is often associated with transcriptionally active euchromatin. By contrast, methylation of H3K9, H3K27, and H4K20 helps specify transcriptionally repressed heterochromatin. Specifically, among the most studied modifications, H3K4me3 is primarily associated with active promoters; H3K4me1 is associated with active chromatin outside of promoters (e.g., enhancers),10,11 while H3K27me3 is associated with silencing by the Polycomb Repressive Complex 2 (PRC2), specifically enhancer of zeste homolog 2 (EZH2 or KMT6) methyltransferase, the enzymatic subunit of PRC2.12,13 Recent discoveries highlight the importance of modifications of these histone methyl marks and/or histone mutations occurring in pediatric brain cancer.
Figure 1.
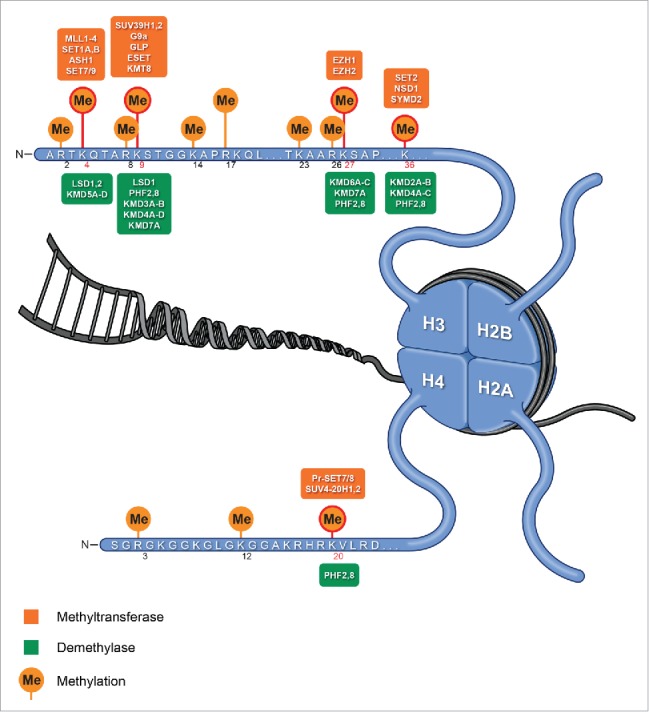
Histone methylases and demethylases. Histones with known lysine methylations: H3K4, K9, K27, K36, K79, and H4K20, and enzymes involved in regulation the methylation status (KMTs and KDMs) of these residues (residues extensively studied in cancer are shown in red circle).
Role of histone modifications in the regulation of transcription
The distribution of histone modifications throughout the genome has been determined by genome-wide comprehensive analyses (e.g., chromatin immunoprecipitation sequencing, ChIP-seq).14,15 These analyses helped to develop predictive models to explore the interaction between histone modifications and measures of transcription at promoters, distinguishing between modifications known to be added as a consequence of transcription (such as H3K36me3 and H3K79me2) and other categories of histone marks.14 These models showed that activating acetylation marks (H3K27ac and H3K9ac) are roughly as informative as activating methylation marks (H3K4me3 and H3K4me2).14 Moreover, repressive histone marks (H3K27me3 or H3K9me3) must be used to accurately predict expression. Studies also show that H3K79me2 occurs preferentially at the 5′ ends of gene bodies and H3K36me3 occurs more often at the 3′ end, supporting the previous model in which the H3K79me2 to H3K36me3 transition occurs at the first 3′ splice site.14 Although these studies describe some relationships between histone modifications and transcription, it has remained unclear how genomic regions modified by histone methylation are specified and formed. For example, recent molecular studies showed that changes in H3K27me3 level, especially in the body of a subset of genes, are triggered by changes in transcriptional activity itself: indeed, deleting the transcription start site increases H3K27me3 level in the gene body.16,17
Role of histone modifications in other biologic processes
The full biologic significance of these histone modifications still needs further investigation, as these modifications are involved in a wide variety of processes. The intensities of some of these signals at enhancers can regulate tissue-specific expression patterns during development.18 Some of these marks have been identified as a chromatin signature linked to transcriptional consistency and cell identity, and highlighting that breadth is a key component of chromatin states.19 The regulation and distribution of histone methylation and of related enzymes can also change during the cell cycle, affecting replication timing (reviewed in20). Of note, the description of these histone posttranslational modifications above is not exhaustive as, for example, phosphorylation also occurs and impacts transcription.21,22
DNA methylation
In addition to histone modifications, DNA methylation plays a key role in coordinating gene expression and chromatin remodeling in brain tumors. DNA methylation, a process occurring at the cytosine of the CpG dinucleotide, is usually associated with gene silencing.23 CpG is underrepresented in the genome, but clusters or “islands” are often found at the 5′ end of a gene. Surveys of DNA methylation in human tissues have established a complex landscape, including both tissue-specific and invariant methylation patterns.24 Current models suggest that DNA methylation helps counteract chromatin disruption, as found in nucleosome displacement during RNAP elongation, for example, while CG-poor regulatory regions generally acquire a low methylation state when occupied by transcription factors (reviewed in23).
Epigenetic changes in brain tumors
Altered epigenetics can play an important part in the development of pediatric brain tumors. The identification of these changes has been important for prognosis and to predict the response to treatment of brain tumors. The possibility to revert epigenetic changes has been valuable to develop and further improve therapeutics, leading to the design of an appropriate therapy for these tumors. Moreover, consortia such as the Roadmap Epigenomics Consortium have extensively advanced our understanding of enhancer/gene regulation across a comprehensive spectrum of cell lines and tissues, establishing the complex cartography of the human regulatory landscape, using diseased and healthy biopsies to link these epigenomic data to the corresponding genetic information.25,26 Comprehensive molecular profiling studies have greatly broadened our knowledge of the underlying genomic and epigenomic aberrations that are associated with the initiation and progression of these brain cancers.27
High-grade glioma
Pediatric high-grade gliomas are clinically and biologically distinct from adult gliomas. Approximately 80% of pediatric high-grade gliomas are restricted to the ventral pons, and are thus named diffuse intrinsic pontine gliomas (DIPGs). DIPGs primarily affect very young children, with peak incidence at 6 y of age, and have the highest mortality of all childhood solid tumors.28 One of the obstacles to improving therapies is that the spectrum of molecular alterations is quite different in tumors from children and adults. Historically, pediatric high-grade glioma was considered similar to secondary glioblastoma multiform (GBM), an adult high-grade glioma that evolves from a less malignant precursor due to changes in gene expression, such as OLIG1 and OLIG2, important neuro-developmental genes.29
K27M mutations in histone H3.1 or H3.3
In contrast to adult tumors, pediatric high-grade gliomas commonly have somatic oncogenic gene mutations (H3F3A and HIST1H3B), resulting in replacement of lysine 27 by methionine (K27M) in the encoded histone H3 proteins (Table 1).27,30 Because it is subject to posttranslational histone modifications, K27 is a key residue in histone H3 variants and can be methylated or acetylated. Substitution of K27 with methionine is thought to contribute to tumorigenesis via defects in chromatin remodeling.27,30 In a recent study aiming to further understand the impact of K27M-histone H3.3 (H3.3K27M) on tumorigenesis in DIPG, immortalized human astrocytes have been used and were transfected with N-terminally flag-tagged expression vectors containing H3.3WT, H3.3K27M, or empty vector control.31 In this study, alterations in both expression and methylation profiles of the H3.3K27M expressing cells vs. both wild type (WT) and controls were observed. The top pathways perturbed by H3.3K27M expression corresponded to molecular and cellular functions, especially an increase in cell-to-cell signaling and a decrease in cell cycle progression. The top molecular and cellular functions affected by methylation changes in H3.3K27M cells were embryonic development, decreased cell growth and proliferation, and increased cell-to-cell signaling. H3.3K27M cells showed reduced global H3K27me3 levels compared with controls. Similarly, immunohistochemical staining of patient-derived DIPGs showed a decrease in global H3K27me3 levels in H3.3K27M-positive tumors compared with WT tumors. Global levels of H3K4me3 and H3K9ac were not altered in a mutation-dependent manner.31
Table 1.
Somatic mutations in epigenetic regulator genes or histone genes in brain cancers.
| Gene mutated | Tumor | Reference |
|---|---|---|
| H3F3A (encoding H3.3 with K27M) | High-grade glioma | 29, 30, 41, 42, 46 |
| HIST1H3B (encoding H3.1 with K27M) | High-grade glioma | 30, 41, 46 |
| H3F3A (encoding H3.3 with G34R or G34V) | High-grade glioma | 29, 41, 42, 46 |
| NF1 | GBM | 46, 78 |
| SET2 (KMT3A) | High-grade glioma | 36 |
| GBM | 46 | |
| MLL2 (MLL4, KMT2C) | Medulloblastoma | 50, 95, 96, 97, 98, 99 |
| GBM | 46 | |
| MLL3 (KMT2D) | Medulloblastoma | 95 |
| GBM | 46 | |
| EZH2 (KMT6A) | GBM | 46 |
| LSD1 (KDM1A) | Medulloblastoma | 99 |
| JMJD1A (KDM3A) | Medulloblastoma | 99 |
| JMJD2C (KDM4C) | Medulloblastoma | 99 |
| JARID1A (KDM5A) | Medulloblastoma | 99 |
| JARID1B (KDM5B) | Medulloblastoma | 99 |
| JARID1C (KDM5C) | High-grade glioma | 36 |
| UTX (KDM6A) | Medulloblastoma | 50, 99 |
| GBM | 46 | |
| JMJD1C (KDM7A) | Medulloblastoma | 99 |
| HDAC2 | GBM | 46 |
| HDAC9 | Medulloblastoma | 99 |
To have a better understanding of the role of the H3.3K27M mutation in a specific cell type, another study, from Funato et al., created a model of DIPG by differentiating human embryonic stem cells into neural progenitor cells, and then transducing them with a viral vector carrying the gene encoding H3.3K27M.32 Remarkably, the mutation was only oncogenic in a specific cell type, neural progenitors derived from embryonic stem cells, and not in undifferentiated embryonic stem cells or astrocytes derived from these cells. The neoplastic transformation occurred due to a synergy between H3.3K27M expression with p53 loss and PDGFRA activation in these particular cells. The expression of the stem cell-associated genes (e.g., LIN28B, PLAG1, and PLAGL1) was also upregulated by H3.3K27M, and reducing expression of these genes inhibited tumor cell growth.32 All together, these studies demonstrate the role of the K27M mutations on gene expression, methylation patterns, and transformative capacity. Of potential significance, in addition to the reduction in H3K27 methylation levels, overexpressing H3.3K27M in Drosophila melanogaster caused a strong increase in H3K27 acetylation levels, associated with an increased in bromodomain-containing protein 1 (BRD1) and bromodomain-containing protein 4 (BRD4) in H3.3K27M–containing nucleosomes.33 The same observations have been made when the mutant was overexpressed in mammalian cells. Consistently, the Drosophila melanogaster K27M mutant models resemble PRC2 loss-of-function phenotypes, causing reduction of H3K27 methylation and derepression of PRC2 target genes. This alteration may indicate a similar molecular pathogenesis for K27M pediatric glioma models. Interestingly, these experiments in flies have been extended to other histone lysine-to-methionine mutations (i.e., H3K9M), consistently showing a depletion of methylation levels and a possible role in heterochromatic silencing.33
G34V/R mutation in Histone H3.3
Other pediatric high-grade gliomas involve mutations occurring at glycine at position 34 and result in replacement of glycine 34 by valine or arginine (G34V/R), which are exclusive to H3F3A (Table 1) (for review see34). G34R-mutant and G34V-mutant H3.3 are associated with global DNA hypomethylation, which is particularly pronounced in telomeric regions. These mutations can also interfere with the regulatory H3K36me3 modification, especially at the MYCN locus, associated with increased transcription of this gene, which drives glioma formation in neural stem cells in vivo.35 H3K36me3 can be further disrupted by mutations in the H3K36 trimethyl transferase Suppressor of variegation, Enhancer of zeste, and Trithorax 2 (SET2 or KMT3A).36 Besides its important role in transcription,37 H3K36me3 is involved in recruiting DNA mismatch repair proteins, ensuring replication fidelity by correcting mismatches generated during DNA replication, thereby reducing spontaneous mutation frequency.38 Of note, besides the impact of somatic missense mutations in histone H3 genes in pediatric brain malignancies, mutations are also found in bone malignancies. For example, glycine 34 to tryptophan/leucine (G34W/L) mutations occur in giant cell tumors of the bone, lysine 36 to methionine (K36M) mutations have been reported in ∼95% of chondroblastomas, and H3K36 mutations promote sarcomagenesis.39-42
Associated changes in DNA methylation patterns
DNA methylation patterns have been extensively studied in adult GBM and have helped guide clinical trials, predicting the recurrence of tumors and resistance to radiotherapy. Indeed, CpG island methylator phenotype (G–CIMP) helped subdivide GBMs into glioma, G–CIMP-positive and G–CIMP-negative GBM subsets, partially predicting response to treatment. Several studies have reported promoter-associated hypermethylation of specific loci in GBM, which frequently affects the expression of genes that have known tumor suppressor functions and/or are involved in cell proliferation or radiation sensitivity (reviewed in27). A famous example of a clinical biomarker to predict response to temozolomide in gliomas is the inactivation by promoter hypermethylation of the O6-methylguanine-DNA methyl-transferase–encoding (MGMT) gene in samples from patients with GBM.43-46 Importantly, part of The Cancer Genome Atlas (TCGA) studies focused on examining the genomic and epigenomic modifications occurring in GBM. These studies, applying genome-wide DNA methylation profiling in an adult patient cohort, led to the identification of a G–CIMP positive group associated with isocitrate dehydrogenase 1 (IDH1) mutations, and hypermethylation at a large number of loci was linked to a less severe outcome.47,48 Applied genome-wide DNA methylation profiling in cohorts of pediatric and adult patients also described recurrent age-specific mutations in H3F3A, while tumors enriched for PDGFRA alterations occur in patients from a wider age range.29 These techniques also led to subgrouping DIPG patients based on CpG island methylation, identifying a subgroup with high-level amplification of MYCN and high-grade histology, in which targeting histones would be irrelevant.31 Observations of DNA methylation profiles across all tumor sites in DIPG tissues was strongly associated with alterations in a specific histone 3 variant mark.49 K27M variants recently have been shown to inhibit SET-domain-containing histone methyltransferases (Table 1),37 possibly accounting for the specific DNA methylation pattern observed in H3.3K27M non-brainstem tumors.29 The finding that DNA methylation profiles are associated with the K27M mutation regardless of tumor location supports its role in driving the epigenetic phenotype. Interestingly, gain-of-function mutations in ACVR1, FGFR1, and PDGFRA, encoding 3 growth factor receptors, were also associated with H3K27M variants.49
Medulloblastoma
Medulloblastoma, an aggressively growing pediatric brain tumor that arises in the cerebellum or medulla/brainstem, is also characterized by a reprogramming of DNA methylation patterns. Medulloblastoma is the most common malignant brain tumor in children, and shows tremendous biologic and clinical heterogeneity. Approximately 40% of children experience tumor recurrence, 30% will die from their disease, while the survivors have a significantly reduced quality of life. The genetic and epigenomic characterizations of medulloblastoma have progressed dramatically in recent years.50 For example, H3K27ac- and BRD4-DNA interactions have been found to strongly correlate at active medulloblastoma enhancer loci.51 Interestingly, BRD4 inhibition has been reported to decrease cell viability in medulloblastoma cell lines and xenografts.52,53 In addition to these epigenetic changes occurring in medulloblastoma, reprogramming of DNA methylation patterns in these tumors shows focal regions of low methylation linked to transcription-factor-binding sites, shedding light on differential transcriptional networks between subgroups. At the same time, increased methylation due to re-normalization of repressed chromatin in DNA methylation valleys was positively correlated with gene expression.51,54 Importantly, genetic analysis showed a lack of overlap between the profile of primary tumors and that of matched samples of local or metastatic recurrence,55 bringing another layer of complexity for the treatment of medulloblastomas. Regarding the genetic plasticity occurring in these tumors and the related resistance to treatments, the investigators proposed that a biopsy after recurrence should be mandatory for all future clinical trials for medulloblastoma to determine whether therapeutic targets are present in the dominant clones at recurrence.55
Ependymoma
Ependymoma is also among the most common pediatric brain tumor and remains a leading cause of cancer death in children. Over 90% of pediatric ependymomas are intracranial, with 2 thirds occurring in the posterior fossa, while many adult cases occur in the spinal cord (reviewed in56). Genomic abnormalities have often been observed in ependymoma, including genomic gains and losses or translocations within the ependymoma genome, but these genomic aberrations are more frequent in adult ependymomas. Methylated genes and gene expression profiles are associated with tumor location, patient age at disease onset, grade, and retrospective risk for relapse57 (also reviewed in56). For example, deregulation of genes involved in neural differentiation and maintenance, particularly ion transport and synaptogenesis, highlights the importance of these events in the formation of this supratentorial ependymoma subgroup.58 Importantly, recent studies described that poor-prognosis hindbrain ependymomas exhibit hyperactivity of PRC2 and that associated increased trimethylation of H3K27 leads to tumor suppressor gene silencing, with subsequent gene silencing by DNA CpG hypermethylation/CIMP-positive ependymomas of infancy.59
ATRT
Besides these tumors, atypical teratoid/rhabdoid tumor (ATRT) is a rare, high-grade embryonal brain tumor that occurs most commonly in young children and carries a very poor prognosis.60 ATRTs are characterized by absence of the chromatin remodeling protein SNF5 (SMARCB1).61 The SNF5 gene encodes a subunit of ATP-dependent SWI/SNF chromatin remodeling complexes,62 and complexes oppose epigenetic silencing by PRC2.63 Loss of SNF5 tumor suppressor activity leads to elevated expression of the Polycomb gene EZH2, and polycomb targets are broadly methylated at H3K27 and repressed in ATRT.63 In addition, the SNF5 protein has a role in histone acetylation.64,65 Inhibition of histone deacetylases (HDACs) has been found to specifically restore normal expression of proteins involved in cell cycle, originally altered in ATRT cells, through promoter histone H3 and H4 acetylation, recapitulating the effect of SNF5 restoration in ATRT cells.65
Neuroblastoma
Other extra-cranial solid tumors in children include neuroblastoma, a tumor arising from primitive neural crest cells in the sympathetic nervous system.66 Many of these children will succumb to neuroblastoma despite intensive chemotherapy with autologous stem cell transplantation, surgery and radiation. The pathologic activation of MYCN plays a central role in high-risk neuroblastoma, while bromodomain-mediated inhibition of MYCN impaired growth and induced apoptosis in in vivo neuroblastoma models.67
Histone modifying enzymes: Role in brain cancer therapy
The identification of the functional involvement of chromatin machinery and the precise roles of the different epigenetic modifying enzymes is a necessary step to understand cancer biology, and to develop effective therapeutic strategies in human cancer. Drugs targeting epigenetic modifiers include the inhibitors of enzymatic “writers,” such as DNA and histone methyltransferases, and inhibitors of enzymatic “erasers,” such as histone demethylase and HDACs. Modulation of the epigenetic regulators, known as “readers,” structurally diverse proteins that recognize and bind to covalent modifications of chromatin, has recently emerged as a therapeutic strategy in the treatment of brain cancer and also of other cancers (reviewed in.6,20,68-74).
KMTs & PRMTs
In addition to the lysine and arginine side chains of a protein, methyltransferases (MTs) can methylate DNA, RNA, and even small molecules, such as a catecholamine. The methyl acceptors for these enzymes can be N (e.g., –NH2 of a lysine), C (e.g., C5-cytosine in DNA), or O (e.g., –OH of a catecholamine) atom69 All methyltransferases use SAM as the enzyme cofactor, with its methyl group (activated by the sulfonium) being the donor. A large family of ≥ 60 histone methyltransferases, including histone lysine methyltransferases (KMTs) and protein/histone arginine methyltransferases (PRMTs), were identified in humans, and the biochemical and biologic functions of many methyltransferases have been characterized.69,75 (Fig. 1). Besides the effects on histones, some of these enzymes have other substrates69,71 Among the non-histone proteins are the cell cycle regulator p53 and the immune response regulator NF-kB. Most KMTs contain the conserved SET domain, excepted DOLT1.69,71 Most of these enzymes have a high degree of specificity for particular residues and the degree of methylation.
Polycomb repressive complex 2
H3K27 methylation is determined by the activity of, the enzymatic subunit of PRC2 (Fig. 1). Despite the importance of EZH2, H3K27 methylation requires other PRC2 components, including EZH1 (functional homolog of EZH2), histone core accessory proteins (EED, SUZ12, and RbAP48), and the PRC2-associated factors JARID2 and ASXL1 (for review see76).
EZH2 is overexpressed in a wide range of cancers, including GBM (but also advanced-stage and high-grade prostate, breast, and lung tumors) (Table 1).70 The PRC2 subunits SUZ12 and EED are frequently deleted in malignant peripheral nerve sheath tumors, GBM, and melanomas, and data suggest that loss of PRC2 function promotes oncogenic Ras signaling.77 Indeed, SUZ12 functions as tumor suppressor in high-grade gliomas by cooperating with mutations in NF1 (which encodes a Ras GTPase-activating protein), and its loss drives cancer by activating Ras.78 Importantly, however, SUZ12 inactivation also triggers an epigenetic switch that sensitizes these cancers to bromodomain inhibitors.77 Moreover, somatic mutations and deletions of EZH2 were also identified in malignancies.79-82 In particular, T cell acute lymphoblastic leukemia (T-ALL) is a hematological malignancy in which H3K27 methylation is reduced by a loss-of-function EZH2 mutation.81,82 Mutations resulting in the replacement of a single tyrosine in the SET domain of the EZH2 protein (tyrosine Y641) can occur in diffuse large B-cell lymphomas and follicular lymphomas and reduce enzyme activity.83 EZH2 has been described as an essential regulator for B cell activation and is involved in germinal center-derived neoplasms.84 In myeloid neoplasia, EZH2 is most often affected by mutations leading to direct abrogation of histone methyltransferase activity, suggesting that EZH2 acts as a tumor suppressor for myeloid malignancies.79
There is evidence for both increased and decreased activity of enzymes controlling H3K27 methylation in other cancers,76,85 supporting the idea that a precise balance of this methylation is critical for normal cell growth and for a context-dependent role of polycomb proteins in oncogenesis. Besides alterations of enzymes controlling H3K27 methylation in cancer, H3K27M gain-of-function mutations often occur in highly aggressive pediatric gliomas (Table 1). These mutations are believed to sequester PRC2, which normally represses gene expression through histone methylation. While H3K27me3 is associated with transcriptionally silenced chromatin, the inactivation of PRC2 through an interaction between EZH2 and the mutant histone results in hypomethylation at H3K27, with consequent transcriptional derepression at these loci.37,86,87 By performing ChIP-seq and whole-genome bisulfite sequencing in primary pediatric high-grade gliomas, Bender and colleagues elegantly demonstrated that reduced H3K27me3 levels and DNA hypomethylation act in concert to activate gene expression in K27M mutant pediatric high-grade gliomas.86
PRC2 dysfunctions have also been found in other brain tumors. As described above, disparate PRC2 H3K27me3 signatures are also found in ependymoma, a phenomenon associated with increased DNA methylation of specific genes, as well as silencing of their expression, reinforcing the rational strategy for the use of drugs targeting PRC2/EZH2, and/or HDAC inhibitors for therapy of this untreatable disease.59 Finally, recent evidence suggests that EZH2 may also have a role in rhabdoid tumors.63,88 Inactivation of the chromatin remodeler SWI/SNF complex component SNF5 is highly prevalent in this disease. Targeted disruption of EZH2 also strongly impairs ATRT cell growth, suppresses tumor cell self-renewal, induces apoptosis, and potently sensitizes these cells to radiation.64
Inhibitors of the catalytic activity of EZH2 have been developed, and suppressing global H3K27 methylation converges upon a common structural feature, a pyridone group, which is required for high affinity target binding (reviewed in71). These inhibitors achieve dose-dependent induction of gene expression, confirming the role of the PRC2 complex in gene silencing, although a prolonged inhibition of EZH2 catalytic activity is required to reduce H3K27me3 to levels that are sufficient to alter patterns of gene expression. As the administered dose of compound correlates with reduction in H3K27me3 levels in tumor tissue and of tumor growth, the clinical use of such inhibitors for cancers in which EZH2 is genetically altered is relevant.88,89 Interestingly, Kim and colleagues discovered that EZH2 enhances STAT3 activation by trimethylating lysine180 in STAT3, and it does so preferentially in glioma stem-like cells.90 The use of the EZH2 inhibitor 3-deazaneplanocin A (DZNep)91,92 and a highly selective EZH2 inhibitor GSK12693 decreases STAT3 activation in glioma stem-like cells. This inhibition reverses the silencing of Polycomb target genes and diminishes STAT3 activity, suggesting therapeutic strategies.90
The therapeutic effects of EZH2 inhibitors are also under evaluation in ATRT. Indeed, DZNep potently suppresses ATRT cell growth, leads to cell cycle alterations, increases apoptosis, potentiates the effect of radiation, decreases key cellular signal transduction pathways, and inhibits tumor sphere formation.64 The first generation EZH2 inhibitors have recently entered phases I and II clinical trials. Tazemetostat (EPZ-6438) is a selective small molecule inhibitor of EZH2 currently being tested in clinical trials for patients having malignant tumors, including ATRT (NCT02875548, NCT02601950, including in pediatric patients NCT02601937) (Table 2).
Table 2.
Epigenetic inhibitors for brain cancer therapy.
| Enzyme/protein | Inhibitor | Mechanism | Pre-clinical studies | Clinical trials (ongoing or recently completed or terminated) |
|---|---|---|---|---|
| EZH2 | Tazemetostat | EPZ-6438 (N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[1,1′-biphenyl]-3-carboxamide) is a selective small molecule inhibitor of the histone-lysine methyltransferase EZH2 (competition with S-adenosyl methionine (SAM)) | Reduces intratumoral trimethylation levels of lysine 27 on histone H3 (88) | A Phase II, Multicenter Study of the EZH2 Inhibitor Tazemetostat in Adult Subjects With INI1-Negative Tumors or Relapsed/Refractory Synovial Sarcoma (NCT02601950) |
| Induces the apoptosis of ATRT cell-like (88) | Tazemetostat Rollover Study (TRuST): An Open-Label Rollover Study (NCT02875548) | |||
| A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma (NCT02601937) | ||||
| EZH2 | DZNep | 3-deazaneplanocin A (DZNep) is a potent S-adenosylhomocysteine hydrolase inhibitors (92) | Suppresses self-renewal and induces radiation sensitivity in ATRT cells (64) | |
| It depletes EZH2 indirectly by increasing levels of adenosylhomocysteine, which in turn, inhibits EZH2 and its methyltransferase activity | Decreases STAT3 activation in glioma stem-like cells, which reverses the silencing of polycomb target genes (90) | |||
| Has been found to suppress EZH2-mediated H3K27 methylation (91) | ||||
| EZH2 | GSK126 | Potent, highly selective, S-adenosyl-methionine-competitive, small-molecule inhibitor of EZH2 methyltransferase activity (93) | Decreases STAT3 activation in glioma stem-like cells, which reverses the silencing of polycomb target genes (90) | |
| Decreases global H3K27me3 levels and reactivates silenced PRC2 target genes (93) | ||||
| MEN1 | MI-2–2, MI-3 | MI-2–2 and −3 belong to the thienopyrimidine class, and can block menin-MLL interaction | Inhibits tumor cell growth in vitro in H3K27M mutant cells and in xenografts in mice (32) | |
| Inhibits self-renewal of adult GBM stem-like cells, in combination with KDM inhibitors (101) | ||||
| UTX /JMJD3 | GSK-J1/J4 | Small-molecule catalytic site inhibitor, selective for the H3K27me3-specific JMJ subfamily (107) | Increases K27me2 and K27me3 and a dose-dependent reduction in cell viability and proliferation of H3K27M mutant cells (108) | |
| Chelation of the Fe(II) active site and binding of a propanoic acid side-chain to the 2OG binding site | Increases animal survival and reduced tumor growth in H3K27M human brainstem tumor xenografts (108) | |||
| Reduces the lipopolysaccharide-induced proinflammatory response in human macrophages (107) | ||||
| HDAC | Panobinostat | Hydroxamic acid, non-specific HDAC inhibitor. HDACs 1–4, 7 and 9 but less so against HDAC6 and, especially, HDAC8 (reviewed in 125) | Despite a promotion of growth arrest and apoptotic cells death in glioma models (mostly due to anti-angiogenic effects), the sample sizes in the first high-grade glioma trials were too small to understand its effects in human (120) | Panobinostat and Stereotactic Radiation Therapy in Treating Patients With Brain Tumors (NCT01324635) |
| Recent work shows that panobinostat is active against diffuse intrinsic pontine glioma (119) | Panobinostat in Treating Younger Patients With Progressive Diffuse Intrinsic Pontine Glioma (NCT02899715) | |||
| Increases in global H3 acetylation and H3K27 trimethylation (dose-dependant fashion) in H3K27M cells (117) | Trial of Panobinostat in Children With Diffuse Intrinsic Pontine Glioma (NCT02717455) | |||
| Panobinostat synergized with KDM inhibitor GSK-J4 to significantly decrease cell viability in H3K27M mutant (117) | Study of LBH589 (Panobinostat) to Treat Malignant Brain Tumors (NCT00848523) | |||
| Panobinostat caN-terminally differentiate ATRT tumor cells and reduce their ability to self-renew (118) | ||||
| HDAC | Vorinostat | Suberoylanilide hydroxamic acid - SAHA, non-specific HDAC inhibitor (reviewed in 125) | Despite a significant synergistic cytotoxicity between HDAC inhibitors and proteasome inhibitors in GBM cell lines, a similar combination did not provide a significant survival benefit in previous trials (121) | Study of the Combination of Vorinostat and Radiation Therapy for the Treatment of Patients With Brain Metastases (NCT00838929) |
| Study of Suberoylanilide Hydroxamic Acid (SAHA) With Temsirolimus in Children With Diffuse Intrinsic Pontine Glioma (DIPG) (NCT02420613) | ||||
| Vorinostat and Temozolomide in Treating Patients With Malignant Gliomas (NCT00268385) | ||||
| N2007–03: Vorinostat and 131-I MIBG in Treating Patients With Resistant or Relapsed Neuroblastoma (NCT01019850) | ||||
| Vorinostat and Radiation Therapy Followed by Maintenance Therapy With Vorinostat in Treating Younger Patients With Newly Diagnosed Diffuse Intrinsic Pontine Glioma (NCT01189266) | ||||
| Vorinostat, Temozolomide, and Radiation Therapy in Treating Patients With Newly Diagnosed Glioblastoma Multiforme (NCT00731731) | ||||
| Vorinostat, Temozolomide, or Bevacizumab in Combination With Radiation Therapy Followed by Bevacizumab and Temozolomide in Young Patients With Newly Diagnosed High-grade Glioma (NCT01236560) | ||||
| Vorinostat and Isotretinoin in Treating Patients With High-Risk Refractory or Recurrent Neuroblastoma (NCT01208454) | ||||
| Vorinostat, Isotretinoin and Temozolomide in Adults With Recurrent Glioblastoma Multiforme (GBM) (NCT00555399) | ||||
| Magnetic Resonance Spectroscopy Imaging in Predicting Response to Vorinostat and Temozolomide in Patients With Recurrent or Progressive Glioblastoma (NCT01342757) | ||||
| phase I/II Adaptive Randomized Trial of Bevacizumab Versus Bevacizumab Plus Vorinostat in Adults With Recurrent Glioblastoma (NCT01266031) | ||||
| High-Dose Vorinostat and Fractionated Stereotactic Body Radiation Therapy in Treating Patients With Recurrent Glioma (NCT01378481) | ||||
| Vorinostat and Bortezomib in Treating Young Patients With Refractory or Recurrent Solid Tumors, Including Central Nervous System Tumors and Lymphoma (NCT00994500) | ||||
| HDAC | Belinostat | Hydroxamic acid, non-specific HDAC inhibitor. Belinostat is quite active against HDACs 1–10 (reviewed in 125) | MRSI to Predict Response to RT/TMZ ± Belinostat in GBM (NCT02137759) | |
| HDAC | Romidepsin | FK228, Cyclic peptide, once converted to its active form (redFK) by cellular reducing activity, can inhibit HDACs 1, 2, 4 and 6 (reviewed in 125) | Induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo (122) | FR901228 in Treating Patients With Recurrent High-grade Gliomas (NCT00085540) |
| HDAC | Valproate | Short-chain fatty acids. Valproate is quite active against HDACs 1–5, 7 and 9 but less so against HDACs 6 and 10 (reviewed in 125) | Combined with radiotherapy, valproate treatement was associated with a better outcome than other HDAC inhibitors in patients with GBM (123; 124) | Valproate and Etoposide for Patients With Neuronal Tumors and Brain Metastases (NCT00513162) |
| Stereotactic Radiosurgery With Nivolumab and Valproate in Patients With Recurrent Glioblastoma (NCT02648633) | ||||
| Sorafenib Tosylate, Valproic Acid, and Sildenafil Citrate in Treating Patients With Recurrent High-grade Glioma (NCT01817751) | ||||
| An International Clinical Program for the Diagnosis and Treatment of Children With Ependymoma (NCT02265770) | ||||
| phase I Study of Temozolomide, Valproic Acid and Radiation Therapy in Patients With Brain Metastases (NCT00437957) | ||||
| Valproic Acid and Radiation Followed by Maintenance Valproic Acid and Bevacizumab in Children With High Grade Gliomas or Diffuse Intrinsic Pontine Glioma (NCT00879437) | ||||
| Valproic Acid in Treating Young Patients With Recurrent or Refractory Solid Tumors or CNS Tumors (NCT00107458) | ||||
| BET | OTX015 | OTX015 (MK-8628) specifically targets the recognition of acetylated lysine residues of BETs (133) | Displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models (133) | A Trial With Dose Optimization of OTX015 in Recurrent Glioblastoma Multiforme (GBM) Patients (NCT02296476) |
| BET | INCB057643 | INCB057643 specifically targets the recognition of acetylated lysine residues of BETs | A Phase 1/2, Open-Label Safety and Tolerability Study of INCB057643 in Subjects With Advanced Malignancies (NCT02711137) | |
| BET | INCB054329 | INCB054329 inhibited binding of BRD2, BRD3 and BRD4 to an acetylated histone H4 | An Open-Label, Dose-Escalation Study of INCB054329 in Patients With Advanced Malignancies (solid tumors of all types, including brain tumors) (NCT02431260) | |
| BET | JQ1 | JQ1 specifically targets the recognition of acetylated lysine residues of BETs (127; 129) | Represses growth of orthotopic glioblastoma tumors (136) | JQ1 is not being tested in clinical trials due to its short half life. |
| BET | I-BET151 | I-BET151 specifically targets the recognition of acetylated lysine residues of BETs (131) | Inhibits GBM cell proliferation (135) |
Mixed-lineage leukemia
One of the first mutations affecting histone modification patterns was described in mixed-lineage leukemia (MLL) KMT2A, which encodes an H3K4me3 MT (Table 1). Indeed, more than 50 different translocations or partial tandem duplications involving MLL on chromosome 11q23 have been described and are found to be associated with poor prognosis in acute lymphoblastic leukemia or normal-karyotype acute myeloid leukemia (AML) (reviewed in94). Of interest, on the basis of genome-wide sequencing data, mutations of other MLLs, MLL2 (also called MLL4, KMT2D) and MLL3 (KMT2C), have been described in both GBM and medulloblastoma,46,50,95,96,97,98,99 further increasing the interest for these MTs in cancer. Importantly, UTX (KDM6A) is part of the MLL3/4 complexes, while JMJD3 (KDM6B) associates with the common MLL complex proteins WDR5, ASH2L, and RBBP5. Interestingly, some studies suggest a role for MLL in contributing to the epigenetic heterogeneity between tumor-initiating and non-tumor-initiating cells in GBM.100 More recently, Gallo et al. showed that MLL5 induces reorganization of chromatin structure and decreases expression of H3.3. Reduced H3.3 expression favors self-renewal properties in adult GBM cells and phenocopies pediatric GBM with H3.3 mutations, indicating potential therapeutic strategies for adult GBM.101 The authors observe that MLL5 and H3.3 have antagonistic roles on glioma stem-like cell self-renewal and that these mechanisms are reversible. Because there are no available MLL5 inhibitors, the authors use the TCGA database to search for alterations in other chromatin targets that may co-occur with alterations in MLL5 and H3.3 and identify the histone demethylases JMJD3/KDM6B and UTX/KDM6A and the trithorax-complex subunit Multiple Endocrine Neoplasia 1 (MEN1, menin)101 (Table 2). Treatment of adult GBM stem-like cells with histone demethylase and MEN1 inhibitors (such as MI-2, which blocks menin-MLL interaction), recently was shown to be efficacious in preclinical models of K27M mutant gliomas (described below in the section on lysine demethylases), inhibiting the self-renewal properties of GBM cells.101 Treatment with MEN1 inhibitors was previously found to decrease tumor cell growth in vitro and in mice using human embryonic stem cells to model pediatric gliomas with the H3.3K27M histone mutation.32 Although the role of MEN1 in DIPG is unclear, these studies suggest that it may be an important therapeutic target. Further details of UTX (KDM6A) and JMJD3 (KDM6B) are described below in the section “UTX (KDM6A) and JMJD3 (KDM6B).”
Finally, although there is no (or very little) evidence of the involvement of other MT in brain cancer, the impact of some enzymes, including DOT1L (KMT4), NSD proteins, H3K9MT, and PRMT has been extensively studied in several cancers (reviewed in70).
DOT1L (KMT4)
DOT1L methylates lysine 79 on H3K79 within the globular histone domain on which DNA is wrapped, and not on the N-terminal tails. Genome-wide studies show that actively transcribed genes are enriched for H3K79 methylation.10 Several studies highlight the impact of DOT1L in leukemia, and DOT1L inhibitors show enhanced anti-proliferative activity against MLL-rearranged leukemia cells.102
Nuclear receptor SET domain-containing (NSD) proteins 1, 2, and 3
Nuclear receptor SET domain-containing (NSD) MTs methylate H3K36. NSDs have been involved in some cases of AML or breast cancers,70 and NSD2 has been reported as highly overexpressed in GBM.103
H3K9MTs
These enzymes have been found to be upregulated in various human tumors.102
Protein arginine methyltransferases
Methylation of arginines by protein arginine methyltransferases (PRMT1–9) has been associated with both positive and negative regulation of transcription. Arginine methylations have also been involved in cancer, and especially in various MLL and non-MLL. For example, PRMT1 is necessary for leukemic transformation, which requires co-recruitment of KDM4C, an H3K9 demethylase.75,104
Lysine demethylases
Histone methylation is also dynamically controlled by histone/protein lysine demethylases (KDMs), enzymes that remove the methyl group(s) from a methylated lysine side chain (Fig. 1). The opposite functions of MTs and DMs work to maintain balanced histone methylation levels. KDM protein families are based on the organization of their catalytic domains and the type of oxidative mechanism that underlies the demethylation reaction. The Jumonji domain-containing KDM family members utilize 2-oxoglutarate (2-OG; α-ketoglutarate) as a cofactor, while KDM1A (LSD1, BHC110, AOF2) and KDM1B (LSD2) require FAD. Histone methylation patterns can also be altered by KDMs. Mutations have been reported in the genes encoding KDM5A (JARID1A); KDM5C (JARID1C), which affects H3K4 methylation; and KDM6A (UTX), which affects H3K27 methylation.50,81,82,98,105,106
UTX (KDM6A) and JMJD3 (KDM6B)
Besides EZH2 methyltransferase, H3K27 methylation is also determined by the activity of demethylases, including UTX/KDM6A and JMJD3/KDM6B. Somatic loss-of-function mutations of UTX are found in multiple myeloma, esophageal squamous cell carcinomas, renal carcinomas, and occasionally in breast cancer, AML, T-ALL, GBM, and colorectal and bladder cancer (Table 1) (for review see.70,81,82), suggestive of a tumor suppressor role for UTX. Both JMJD3 and UTX function as part of a transcriptional activator complex that includes the MLL3/MLL4 H3K4 methyltransferases, indicating that these enzymes have a dual role involving removal of H3K27 methyl marks and addition of methyl groups to H3K4 (reviewed in76).
GSK-J1/J4 were first discovered as JMJD3 (KMD6B) and UTX (KMD6A) inhibitors, and the treatment of human primary macrophages with these pharmacological substances reduces the lipopolysaccharide-induced pro-inflammatory response107 Remarkably, GSK-J4 recently has been used successfully to treat brainstem gliomas in vitro and in vivo (108 and reviewed in109-112). The mechanism of action of GSK-J4 is chelation of the Fe(II) active site and binding of a propanoic acid side-chain to the 2OG binding site. In this study, a panel of patient-derived cell lines has been examined: 2 lines derived from patients with DIPG that harbor a heterozygous H3.3K27M dominant mutation, 2 pediatric glioblastoma lines wild type for H3, a fifth pediatric glioblastoma line that carries the H3.3 G34V mutation, and isogenic cell lines derived from human astrocytes with and without transgene expression of the H3.3 K27M mutation.108 Both patient-derived cell lines had global hypomethylation of H3K27 compared with the wild type models. Treatment of K27M mutant cells with GSK-J4 resulted in increased K27me2 and K27me3 and a dose-dependent reduction in cell viability and proliferation (Fig. 2) (Table 2). Treatment of 2 H3K27M human brainstem tumor xenografts with GSK-J4 increased animal survival and reduced tumor growth but did not achieve the same effect in animals with xenografts containing wild type H3.3.108 The confirmation of the delivery of GSK-J4 into the pons using HPLC analysis of non-tumor-bearing mice further supports the use of GSK-J4 as a potentially clinically effective targeted therapy.108 Of interest, this compound has a therapeutic potential for T-ALL, which can be driven by oncogenic activation of NOTCH1 signaling, loss-of-function mutations and deletion of the EZH2 and SUZ12 genes, and in which JMJD3 is more highly expressed compared with other types of leukemia.81,113,114 Indeed, treatment of T-ALL cells with GSK-J4 also resulted in reduced cell proliferation and increased K27M methylation.82 In this study, the antitumor activities of the GSK-J4 inhibitor was also specific for T-ALL cells, and the main action of this inhibitor in T-ALL is also thought to be channeled through the inhibition of JMJD3 activity.82
Figure 2.
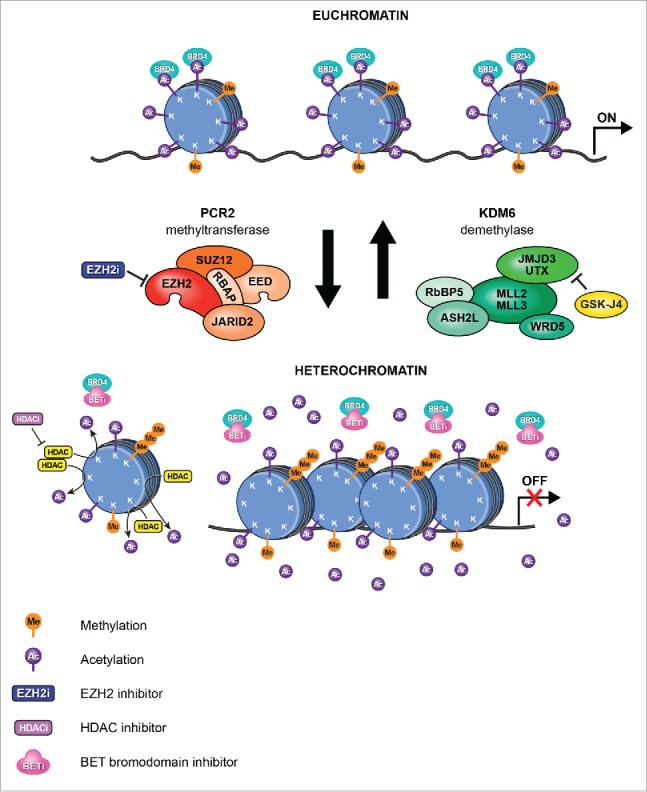
New epigenetic therapies for pediatric brain tumors. In DIPG, H3K27M mutations (histone H3.1 or H3.3) lead to hypomethylation of H3K27, which promotes a more accessible chromatin state characterized by H3K27 acetylation and aberrant gene expression (upper: Euchromatin). H3K27M mutations inhibit the major H3K27 methylase PRC2. Treatment of DIPG with the K27 demethylase inhibitor GSKJ4 results in increased K27me2 and K27me3 and reduced tumor growth (lower: Heterochromatin). Moreover, treatment with the non-specific HDAC inhibitor panobinostat resulted in an increase in global H3 acetylation, increasing/restoring H3K27me3 levels. In addition, competitive binding with BET bromodomain inhibitors prevents the interaction of BRD4 with acetylated histones, leading to repression of BRD4 transcriptional targets and also limiting tumor growth.
Histone acetyltransferases and deacetylases
Histone acetyltransferases (HATs) and HDACs (including the NAD-dependent Sirtuins) function antagonistically to control histone acetylation. Because acetylation is a histone mark for active transcription, HATs have been associated with active and HDACs with inactive genes. A recent description of genome-wide mapping of HAT and HDAC binding on chromatin has been made, finding that both are found at active genes with acetylated histones.115 These data provide evidence that HATs and HDACs are both targeted to transcribed regions of active genes by phosphorylated RNAP II, and the majority of HDACs in the human genome function to reset chromatin by removing acetylation at active genes.115 Besides the finding of genome-wide mapping, the most recent study revealed that HDAC mediates Ras signaling-induced silencing of the studied genes and the subsequent H3K27me3 increase, while H3K27me3 accumulation is reversed by forced activation of transcription.16 Rare mutations have been described in tumor genomes for almost all members of this protein family (Table 1).
HDAC inhibitors work by preventing histone deacetylation, thereby facilitating an open chromatin structure and resulting in gene activation. A non-selective potent HDAC inhibitor drug, panobinostat, a hydroxamic acid (LBH589, a non-selective FDA-approved potent HDAC inhibitor), has been developed for the treatment of a various cancers.116 A recent study aimed to screen 83 drugs selected by pediatric neuro-oncologists as either promising targeted agents for cancer or traditional chemotherapeutic agents in use for pediatric brain tumor therapy.117 The chemical screen and subsequent validation experiments revealed patient-derived DIPG cell sensitivity to HDAC inhibitors. Indeed, Western blot analyses of cells expressing the H3.3K27M mutation demonstrated a dose-dependent increase in global H3 acetylation and H3K27 trimethylation following panobinostat treatment, suggesting that the drug produced partial rescue of the H3K27M-induced hypotrimethylation phenotype117 (Fig. 2). Increased H3K27 trimethylation showed that polyacetylation of the H3 N-terminal tail can ‘detoxify’ K27M-induced inhibition of PRC2 by rescuing the methylation levels. RNA-seq performed on panobinostat- or vehicle-treated DIPG cells revealed a normalization of the K27M gene expression signature. In addition to clinical trials for brain tumors in adults (NCT01324635, NCT00848523), the side effects and best dose of panobinostat are currently being tested for the treatment of younger patients with DIPG (NCT02899715, NCT02717455). Because panobinostat was shown to reduce the expression of oncogenes in DIPG cells (e.g., MK167, CCND1), and to increase global H3 acetylation and H3K27 trimethylation, this drug has also been tested in combination with GSK-J4. Consistent with previous findings,108 the study confirmed that GSK-J4 decreased cell viability in H3K27M DIPG cell cultures, and further demonstrated that panobinostat synergized with GSK-J4 to significantly decrease cell viability in H3K27M mutant DIPG cell cultures.117
In addition to glioma therapy, the therapeutic potential of HDAC inhibitors has also been investigated in ATRT. The rationale was that SNF5 protein loss leads to direct impairment of chromatin remodeling and changes in histone acetylation. Using mouse xenograft models, sustained low-dose panobinostat treatment caused tumor growth arrest. The use of low-dose HDAC inhibitors as a novel therapeutic approach warrants further investigation.118 Panobinostat,119120 and other HDAC inhibitors, including vorinostat,121 belinostat, romidepsin122 and valproate123,124 (reviewed in125), are currently been used in clinical trials for patients with brain tumors (Table 2).
Bromodomain and extraterminal
The bromodomain and extraterminal (BET) protein BRD4 recently was found possibly to play an important role in the development of brain cancer. The involvement of BRDs has been examined not only in GBM126 but also in medulloblastoma, where enrichment of H3K27ac and BRD4 ChIP-seq peaks strongly correlated at active medulloblastoma enhancer loci.51 BRD4 is a “reader” of lysine acetylation, involved in initial recognition of acetylated histones followed by recruitment of Mediator, the transcription initiation cofactor, on promoter regions, leading to phosphorylation of RNAP II. BRD4 also facilitates transcription elongation. Highly selective BET bromodomain inhibitors, such as JQ-1,127-129 I-BET151,130,131 I-BET762,132 INCB054329,126 and OTX-015,133 have been generated to specifically target the recognition of acetylated lysine residues of BETs. Competitive binding with a BET bromodomain inhibitor blocks the binding between BRD4 and acetylated histones, leading to transcriptional inactivation (Fig. 2) (Table 2).128 These inhibitors seem to be a promising therapeutic strategy in medulloblastoma,53,134 and GBM.126 A number of preclinical studies has been performed in GBM models with the BRD inhibitors JQ1 and I-BET151, showing promising antitumor activity.135,136 Moreover, OTX015 (MK-8628), a novel BET inhibitor, has been tested recently at the clinical level in dose-finding studies in GBM patients (NCT02296476), and displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in GBM models.126 The safety and tolerability of another BET inhibitor, INCB057643, is also currently being tested in subjects with GBM (NCT02711137) (Table 2). Interestingly, a recent study suggests that the anti-proliferative effects of BET inhibitors in GBM might be at least partially mediated by a decrease in the tumor-promoting long noncoding RNA HOX transcript antisense RNA (HOTAIR).137
Safety and feasibility of epigenetic targeted therapy
Toxicological studies should confirm the accessibility of agents targeting epigenetic modifiers or the administration at the appropriate location in the CNS. The compound should be delivered in effective concentrations to inhibit the target (thereby requiring a good blood brain penetration if administered systemically), with a sufficient therapeutic window for such pharmacological inhibition and have an acceptable toxicity profile, taking into account the continuing normal development of pediatric patients. Given the important functions of histone methylation and acetylation in normal physiology, inhibition of related enzymes could cause off-target effects with systemic administration. Moreover, these treatments have an anti-proliferative activity, and the dose used plays a key role. For this purpose, the information obtained during the nonclinical safety studies needs to be used to estimate an initial safe starting dose and dose range of the pharmaceutical epigenetic treatment of the human trials and to identify parameters for clinical monitoring for potential adverse effects. In clinical trials, a wide range of toxic effects has been reported during the systemic use of epigenetic targeted therapies.138 For example, fatigue, thrombocytopenia, nausea, and diarrhea have been reported in GBM patients treated with vorinostat121,139 and similar toxic side effects can be accompanied by hypophosphatemia and hemorrhage with panobinostat.120
In addition to the evaluation of anti-cancer activity based on tumor size, and to the evaluation of molecular effects of therapy through analysis of biopsy tissue obtained before and/or after treatment, it is relevant to have a detailed understanding of the molecular impact of a drug on healthy CNS cells as well as on peripheral organs if the drug is administered systemically. Much progress and improvement are still needed in the treatment of malignant brain tumors, both in the preclinical and clinical settings. The penetration of the various agents in development into the central nervous system remains an important issue and is being tested. As an example, OTX015 crosses the blood–brain barrier and selectively penetrates tumor tissue.133 OTX015 tumor levels were 7–15 times higher than in normal brain tissues in several in vivo models.133 As another example, GSK-J4 brain access has been assessed in murine DIPG models, after being administered to mice by intraperitoneal injection.108 After brain resection, each brainstem was dissected from the surrounding brain. GSK-J1, the hydrolysis product and activated derivative of GSK-J4 and the most selective KDM inhibitor yet disclosed,140 was found in all brainstem samples, indicating that the drug entered the brain and accessed the site of brainstem tumor development.108 Extensive additional experiments confirmed the anti-tumor activity of GSK-J4 as involving inhibition of JMJD3,108 but did not exclude the possibility of other histone demethylase activities being affected. Recent studies described that besides being a potent inhibitor of jumonji proteins, with activity toward H3K27me3/me2 (KDM6), GSK-J1 can also inhibit KDM5 activity on H3K4me3/me2 in U2OS cells in vitro.141 Considering the effect of JMJD3 on several biologic functions (e.g., inflammatory response in macrophages107) and those of KDM5,142 optimizing the dose and frequency of administration of such inhibitors to maximize the therapeutic window and limit toxicity will be a key factor for reaching a successful clinical outcome. The combination of agents with different mechanisms of action, as recently proposed117 may offer a better chance for tumor control with limited toxicity.
Conclusions
Due to the recent links to brain cancer, some KDMs have come under investigation as potential therapeutic targets. The precise impact of the mutations occurring in brain cancer and related changes in chromatin machinery, as well as their complex interactions with other known oncogenes, need to be further explored. It will be also relevant to investigate the many processes that can regulate the activity of these enzymes, including phosphorylation, ubiquitination, and changes in metabolism (reviewed in6,143), as well as fluctuations in the availability of energy substrates, cofactors, and coenzymes144,145 to improve specific therapies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Michael Gallagher for illustration of the figures and Accixx Biomedical Consulting (www.Accixx.com) for editorial assistance.
Funding
R.H. was supported by US National Institutes of Health (NIH) grant NS093079, the Bear Necessities Pediatric Cancer Foundation and Rally Foundation, and the John McNicholas Pediatric Brain Tumor Foundation.
References
- 1.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 2011; 469:368-73; PMID:21248844; https://doi.org/ 10.1038/nature09652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science 2009; 325:626-8; PMID:19644123; https://doi.org/ 10.1126/science.1172926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rivera CM, Ren B. Mapping human epigenomes. Cell 2013; 155:39-55; PMID:24074860; https://doi.org/ 10.1016/j.cell.2013.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bird A. Perceptions of epigenetics. Nature 2007; 447:396-8; PMID:17522671; https://doi.org/ 10.1038/nature05913 [DOI] [PubMed] [Google Scholar]
- 5.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet 2008; 9:15-26; PMID:18059368; https://doi.org/ 10.1038/nrg2206 [DOI] [PubMed] [Google Scholar]
- 6.Kaelin WG Jr., McKnight SL. Influence of metabolism on epigenetics and disease. Cell 2013; 153:56-69; PMID:23540690; https://doi.org/ 10.1016/j.cell.2013.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furey TS, Sethupathy P. Genetics. Genetics driving epigenetics. Science 2013; 342:705-6; PMID:24202168; https://doi.org/10638745 10.1126/science.1246755 [DOI] [PubMed] [Google Scholar]
- 8.Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41-5; PMID:10638745; https://doi.org/ 10.1038/47412 [DOI] [PubMed] [Google Scholar]
- 9.Stasevich TJ, Hayashi-Takanaka Y, Sato Y, Maehara K, Ohkawa Y, Sakata-Sogawa K, Tokunaga M, Nagase T, Nozaki N, McNally JG, et al.. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature 2014; 516:272-5; PMID:25252976; https://doi.org/ 10.1038/nature13714 [DOI] [PubMed] [Google Scholar]
- 10.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823-37; PMID:17512414; https://doi.org/ 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 11.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al.. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108-12; PMID:19295514; https://doi.org/ 10.1038/nature07829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002; 111:197-208; PMID:12408864; https://doi.org/ 10.1016/S0092-8674(02)00976-5 [DOI] [PubMed] [Google Scholar]
- 13.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 2002; 111:185-96; PMID:12408863; https://doi.org/ 10.1016/S0092-8674(02)00975-3 [DOI] [PubMed] [Google Scholar]
- 14.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; https://doi.org/ 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al.. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43-9; PMID:21441907; https://doi.org/ 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosogane M, Funayama R, Shirota M, Nakayama K. Lack of transcription triggers H3K27me3 accumulation in the gene body. Cell Rep 2016; 16:696-706; PMID:27396330; https://doi.org/ 10.1016/j.celrep.2016.06.034 [DOI] [PubMed] [Google Scholar]
- 17.Hosogane M, Funayama R, Nishida Y, Nagashima T, Nakayama K. Ras-induced changes in H3K27me3 occur after those in transcriptional activity. PLoS Genet 2013; 9:e1003698; PMID:24009517; https://doi.org/ 10.1371/journal.pgen.1003698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al.. A map of the cis-regulatory sequences in the mouse genome. Nature 2012; 488:116-20; PMID:22763441; https://doi.org/ 10.1038/nature11243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, et al.. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 2014; 158:673-88; PMID:25083876; https://doi.org/ 10.1016/j.cell.2014.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 2012; 48:491-507; PMID:23200123; https://doi.org/ 10.1016/j.molcel.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell 2009; 138:1122-36; PMID:19766566; https://doi.org/ 10.1016/j.cell.2009.07.031 [DOI] [PubMed] [Google Scholar]
- 22.Basnet H, Su XB, Tan Y, Meisenhelder J, Merkurjev D, Ohgi KA, Hunter T, Pillus L, Rosenfeld MG. Tyrosine phosphorylation of histone H2A by CK2 regulates transcriptional elongation. Nature 2014; 516:267-71; PMID:25252977; https://doi.org/ 10.1038/nature13736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schubeler D. Function and information content of DNA methylation. Nature 2015; 517:321-6; PMID:25592537; https://doi.org/ 10.1038/nature14192 [DOI] [PubMed] [Google Scholar]
- 24.Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, et al.. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015; 523:212-6; PMID:26030523; https://doi.org/ 10.1038/nature14465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al.. Integrative analysis of 111 reference human epigenomes. Nature 2015; 518:317-30; PMID:25693563; https://doi.org/ 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romanoski CE, Glass CK, Stunnenberg HG, Wilson L, Almouzni G. Epigenomics: Roadmap for regulation. Nature 2015; 518:314-6; PMID:25693562; https://doi.org/ 10.1038/518314a [DOI] [PubMed] [Google Scholar]
- 27.Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al.. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 2014; 14:92-107; PMID:24457416; https://doi.org/ 10.1038/nrc3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donaldson SS, Laningham F, Fisher PG. Advances toward an understanding of brainstem gliomas. J Clin Oncol 2006; 24:1266-72; PMID:16525181; https://doi.org/ 10.1200/JCO.2005.04.6599 [DOI] [PubMed] [Google Scholar]
- 29.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al.. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22:425-37; PMID:23079654; https://doi.org/ 10.1016/j.ccr.2012.08.024 [DOI] [PubMed] [Google Scholar]
- 30.Zadeh G, Aldape K. ACVR1 mutations and the genomic landscape of pediatric diffuse glioma. Nat Genet 2014; 46:421-2; PMID:24769718; https://doi.org/ 10.1038/ng.2970 [DOI] [PubMed] [Google Scholar]
- 31.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, et al.. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 2014; 46:451-6; PMID:24705254; https://doi.org/ 10.1038/ng.2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014; 346:1529-33; PMID:25525250; https://doi.org/ 10.1126/science.1253799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herz HM, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, Shilatifard A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 2014; 345:1065-70; PMID:25170156; https://doi.org/ 10.1126/science.1255104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waldmann T, Schneider R. Targeting histone modifications–epigenetics in cancer. Curr Opin Cell Biol 2013; 25:184-9; PMID:23347561; https://doi.org/ 10.1016/j.ceb.2013.01.001 [DOI] [PubMed] [Google Scholar]
- 35.Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, Grimmer MR, Lau J, Chesler L, Perry A, et al.. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012; 21:601-13; PMID:22624711; https://doi.org/ 10.1016/j.ccr.2012.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, Liu XY, Sturm D, Korshunov A, Jones DT, Witt H, Kool M, Albrecht S, et al.. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol 2013; 125:659-69; PMID:23417712; https://doi.org/ 10.1007/s00401-013-1095-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013; 340:857-61; PMID:23539183; https://doi.org/ 10.1126/science.1232245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013; 153:590-600; PMID:23622243; https://doi.org/ 10.1016/j.cell.2013.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, et al.. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016; 352:844-9; PMID:27174990; https://doi.org/ 10.1126/science.aac7272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al.. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 2013; 45:1479-82; PMID:24162739; https://doi.org/ 10.1038/ng.2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al.. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012; 44:251-3; PMID:22286216; https://doi.org/ 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al.. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482:226-31; PMID:22286061; https://doi.org/ 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- 43.Malmstrom A, Gronberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, Abacioglu U, Tavelin B, Lhermitte B, Hegi ME, et al.. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol 2012; 13:916-26; PMID:22877848; https://doi.org/ 10.1016/S1470-2045(12)70265-6 [DOI] [PubMed] [Google Scholar]
- 44.Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M, Nikkhah G, Papsdorf K, Steinbach JP, Sabel M, et al.. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol 2012; 13:707-15; PMID:22578793; https://doi.org/ 10.1016/S1470-2045(12)70164-X [DOI] [PubMed] [Google Scholar]
- 45.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 1999; 59:793-7; PMID:10029064 [PubMed] [Google Scholar]
- 46.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al.. The somatic genomic landscape of glioblastoma. Cell 2013; 155:462-77; PMID:24120142; https://doi.org/ 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al.. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse Glioma. Cell 2016; 164:550-63; PMID:26824661; https://doi.org/ 10.1016/j.cell.2015.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al.. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010; 17:510-22; PMID:20399149; https://doi.org/ 10.1016/j.ccr.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, et al.. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 2014; 46:462-6; PMID:24705250; https://doi.org/ 10.1038/ng.2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM, et al.. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012; 488:100-5; PMID:22832583; https://doi.org/ 10.1038/nature11284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, et al.. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 2016; 530:57-62; PMID:26814967; https://doi.org/ 10.1038/nature16546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay P, Bergthold G, Masoud S, Nguyen B, Vue N, et al.. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med 2014; 20:732-40; PMID:24973920; https://doi.org/ 10.1038/nm.3613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, et al.. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res 2014; 20:912-25; PMID:24297863; https://doi.org/ 10.1158/1078-0432.CCR-13-2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, et al.. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 2014; 510:537-41; PMID:24847876; https://doi.org/ 10.1038/nature13268 [DOI] [PubMed] [Google Scholar]
- 55.Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, Remke M, Cavalli FM, Ramaswamy V, Lindsay PE, et al.. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016; 529:351-7; PMID:26760213; https://doi.org/ 10.1038/nature16478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao Y, Mack SC, Taylor MD. Molecular genetics of ependymoma. Chin J Cancer 2011; 30:669-81; PMID:21959044; https://doi.org/ 10.5732/cjc.011.10129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R, Benner A, Hielscher T, Milde T, Remke M, et al.. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 2011; 20:143-57; PMID:21840481; https://doi.org/ 10.1016/j.ccr.2011.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, Rand V, Leary SE, White E, Eden C, et al.. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 2010; 466:632-6; PMID:20639864; https://doi.org/ 10.1038/nature09173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, et al.. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014; 506:445-50; PMID:24553142; https://doi.org/ 10.1038/nature13108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, Walter AW, Rorke LB, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 2004; 22:2877-84; PMID:15254056; https://doi.org/ 10.1200/JCO.2004.07.073 [DOI] [PubMed] [Google Scholar]
- 61.Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, Klekota J, Tamayo P, Nguyen PT, Tolstorukov M, et al.. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med 2010; 16:1429-33; PMID:21076395; https://doi.org/ 10.1038/nm.2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Versteege I, Medjkane S, Rouillard D, Delattre O. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene 2002; 21:6403-12; PMID:12226744; https://doi.org/ 10.1038/sj.onc.1205841 [DOI] [PubMed] [Google Scholar]
- 63.Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010; 18:316-28; PMID:20951942; https://doi.org/ 10.1016/j.ccr.2010.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alimova I, Birks DK, Harris PS, Knipstein JA, Venkataraman S, Marquez VE, Foreman NK, Vibhakar R. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol 2013; 15:149-60; PMID:23190500; https://doi.org/ 10.1093/neuonc/nos285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Algar EM, Muscat A, Dagar V, Rickert C, Chow CW, Biegel JA, Ekert PG, Saffery R, Craig J, Johnstone RW, et al.. Imprinted CDKN1C is a tumor suppressor in rhabdoid tumor and activated by restoration of SMARCB1 and histone deacetylase inhibitors. PLoS One 2009; 4:e4482; PMID:19221586; https://doi.org/ 10.1371/journal.pone.0004482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maris JM. Recent advances in neuroblastoma. N Engl J Med 2010; 362:2202-11; PMID:20558371; https://doi.org/ 10.1056/NEJMra0804577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al.. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov 2013; 3:308-23; PMID:23430699; https://doi.org/ 10.1158/2159-8290.CD-12-0418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zahnow CA, Topper M, Stone M, Murray-Stewart T, Li H, Baylin SB, Casero RA Jr.. Inhibitors of DNA methylation, histone deacetylation, and histone demethylation: A perfect combination for cancer therapy. Adv Cancer Res 2016; 130:55-111; PMID:27037751; https://doi.org/ 10.1016/bs.acr.2016.01.007 [DOI] [PubMed] [Google Scholar]
- 69.Song Y, Wu F, Wu J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J Hematol Oncol 2016; 9:49; PMID:27316347; https://doi.org/ 10.1186/s13045-016-0279-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Popovic R, Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov 2012; 2:405-13; PMID:22588878; https://doi.org/ 10.1158/2159-8290.CD-12-0076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McGrath J, Trojer P. Targeting histone lysine methylation in cancer. Pharmacol Ther 2015; 150:1-22; PMID:25578037; https://doi.org/ 10.1016/j.pharmthera.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, Liu K, Qin S, Xu C, Min J. Epigenetic targets and drug discovery: part 1: histone methylation. Pharmacol Ther 2014; 143:275-94; PMID:24704322; https://doi.org/ 10.1016/j.pharmthera.2014.03.007 [DOI] [PubMed] [Google Scholar]
- 73.Liu K, Liu Y, Lau JL, Min J. Epigenetic targets and drug discovery Part 2: Histone demethylation and DNA methylation. Pharmacol Ther 2015; 151:121-40; PMID:25857453; https://doi.org/ 10.1016/j.pharmthera.2015.04.001 [DOI] [PubMed] [Google Scholar]
- 74.Arcipowski KM, Martinez CA, Ntziachristos P. Histone demethylases in physiology and cancer: a tale of two enzymes, JMJD3 and UTX. Curr Opin Genet Dev 2016; 36:59-67; PMID:27151432; https://doi.org/ 10.1016/j.gde.2016.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer 2013; 13:37-50; PMID:23235912; https://doi.org/ 10.1038/nrc3409 [DOI] [PubMed] [Google Scholar]
- 76.Ezponda T, Licht JD. Molecular pathways: deregulation of histone h3 lysine 27 methylation in cancer-different paths, same destination. Clin Cancer Res 2014; 20:5001-8; PMID:24987060; https://doi.org/ 10.1158/1078-0432.CCR-13-2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, et al.. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014; 514:247-51; PMID:25119042; https://doi.org/ 10.1038/nature13561 [DOI] [PubMed] [Google Scholar]
- 78.Cancer Genome Atlas Research N Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061-8; PMID:18772890; https://doi.org/ 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, et al.. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010; 42:722-6; PMID:20601953; https://doi.org/ 10.1038/ng.621 [DOI] [PubMed] [Google Scholar]
- 80.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, et al.. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 2010; 42:665-7; PMID:20601954; https://doi.org/ 10.1038/ng.620 [DOI] [PubMed] [Google Scholar]
- 81.Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, et al.. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 2012; 18:298-301; PMID:22237151; https://doi.org/ 10.1038/nm.2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, Loizou E, Holmfeldt L, Strikoudis A, King B, et al.. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014; 514:513-7; PMID:25132549; https://doi.org/ 10.1038/nature13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al.. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010; 42:181-5; PMID:20081860; https://doi.org/ 10.1038/ng.518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al.. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013; 23:677-92; PMID:23680150; https://doi.org/ 10.1016/j.ccr.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martinez-Garcia E, Licht JD. Deregulation of H3K27 methylation in cancer. Nat Genet 2010; 42:100-1; PMID:20104248; https://doi.org/ 10.1038/ng0210-100 [DOI] [PubMed] [Google Scholar]
- 86.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al.. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013; 24:660-72; PMID:24183680; https://doi.org/ 10.1016/j.ccr.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 87.Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al.. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013; 27:985-90; PMID:23603901; https://doi.org/ 10.1101/gad.217778.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, et al.. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 2013; 110:7922-7; PMID:23620515; https://doi.org/ 10.1073/pnas.1303800110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al.. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 2012; 8:890-6; PMID:23023262; https://doi.org/ 10.1038/nchembio.1084 [DOI] [PubMed] [Google Scholar]
- 90.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al.. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013; 23:839-52; PMID:23684459; https://doi.org/ 10.1016/j.ccr.2013.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 2007; 21:1050-63; PMID:17437993; https://doi.org/ 10.1101/gad.1524107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther 2009; 8:1579-88; PMID:19509260; https://doi.org/ 10.1158/1535-7163.MCT-09-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E, et al.. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492:108-12; PMID:23051747; https://doi.org/ 10.1038/nature11606 [DOI] [PubMed] [Google Scholar]
- 94.Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet 2013; 14:765-80; PMID:24105274; https://doi.org/ 10.1038/nrg3554 [DOI] [PubMed] [Google Scholar]
- 95.Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, et al.. The genetic landscape of the childhood cancer medulloblastoma. Science 2011; 331:435-9; PMID:21163964; https://doi.org/ 10.1126/science.1198056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dahlin AM, Hollegaard MV, Wibom C, Andersson U, Hougaard DM, Deltour I, Hjalmars U, Melin B. CCND2, CTNNB1, DDX3X, GLI2, SMARCA4, MYC, MYCN, PTCH1, TP53, and MLL2 gene variants and risk of childhood medulloblastoma. J Neurooncol 2015; 125:75-8; PMID:26290144; https://doi.org/ 10.1007/s11060-015-1891-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lopez GY, Grant GA, Fuchs HE, Leithe LG, Gururangan S, Bigner DD, Yan H, McLendon RE, He Y. Clinico-pathological description of three paediatric medulloblastoma cases with MLL2/3 gene mutations. Neuropathol Appl Neurobiol 2014; 40:217-20; PMID:23659599; https://doi.org/ 10.1111/nan.12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, et al.. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012; 488:106-10; PMID:22820256; https://doi.org/ 10.1038/nature11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, et al.. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012; 488:43-8; PMID:22722829; https://doi.org/ 10.1038/nature11213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gallo M, Ho J, Coutinho FJ, Vanner R, Lee L, Head R, Ling EK, Clarke ID, Dirks PB. A tumorigenic MLL-homeobox network in human glioblastoma stem cells. Cancer Res 2013; 73:417-27; PMID:23108137; https://doi.org/ 10.1158/0008-5472.CAN-12-1881 [DOI] [PubMed] [Google Scholar]
- 101.Gallo M, Coutinho FJ, Vanner RJ, Gayden T, Mack SC, Murison A, Remke M, Li R, Takayama N, Desai K, et al.. MLL5 orchestrates a cancer self-renewal state by repressing the histone variant H3.3 and globally reorganizing chromatin. Cancer Cell 2015; 28:715-29; PMID:26626085; https://doi.org/ 10.1016/j.ccell.2015.10.005 [DOI] [PubMed] [Google Scholar]
- 102.Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, Doench JG, Xu H, Chu SH, Qi J, et al.. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med 2015; 21:335-43; PMID:25822366; https://doi.org/ 10.1038/nm.3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li J, Yin C, Okamoto H, Mushlin H, Balgley BM, Lee CS, Yuan K, Ikejiri B, Glasker S, Vortmeyer AO, et al.. Identification of a novel proliferation-related protein, WHSC1 4a, in human gliomas. Neuro Oncol 2008; 10:45-51; PMID:18182627; https://doi.org/ 10.1215/15228517-2007-036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, Finn MG, Lenhard B, Chan LC, So CW. Targeting aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute myeloid leukemia. Cancer Cell 2016; 29:32-48; PMID:26766589; https://doi.org/ 10.1016/j.ccell.2015.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD, et al.. Medulloblastomics: the end of the beginning. Nat Rev Cancer 2012; 12:818-34; PMID:23175120; https://doi.org/ 10.1038/nrc3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al.. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012; 150:1107-20; PMID:22980975; https://doi.org/ 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et al.. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012; 488:404-8; PMID:22842901; https://doi.org/ 10.1038/nature11262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, et al.. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 2014; 20:1394-6; PMID:25401693; https://doi.org/ 10.1038/nm.3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ramaswamy V, Remke M, Taylor MD. An epigenetic therapy for diffuse intrinsic pontine gliomas. Nat Med 2014; 20:1378-9; PMID:25473916; https://doi.org/ 10.1038/nm.3769 [DOI] [PubMed] [Google Scholar]
- 110.Morales La Madrid A, Hashizume R, Kieran MW. Future clinical trials in DIPG: Bringing epigenetics to the clinic. Front Oncol 2015; 5:148; PMID:26191506; https://doi.org/ 10.3389/fonc.2015.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lulla RR, Saratsis AM, Hashizume R. Mutations in chromatin machinery and pediatric high-grade glioma. Sci Adv 2016; 2:e1501354; PMID:27034984; https://doi.org/ 10.1126/sciadv.1501354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Becher OJ, Wechsler-Reya RJ. Cancer. For pediatric glioma, leave no histone unturned. Science 2014; 346:1458-9; PMID:25525232; https://doi.org/ 10.1126/science.aaa3814 [DOI] [PubMed] [Google Scholar]
- 113.Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, Tosello V, Della Gatta G, Paietta E, Racevskis J, et al.. ETV6 mutations in early immature human T cell leukemias. J Exp Med 2011; 208:2571-9; PMID:22162831; https://doi.org/ 10.1084/jem.20112239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, et al.. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med 2004; 350:1617-28; PMID:15084694; https://doi.org/ 10.1056/NEJMoa040465 [DOI] [PubMed] [Google Scholar]
- 115.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009; 138:1019-31; PMID:19698979; https://doi.org/ 10.1016/j.cell.2009.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R, Mita M, Beck J, Burris H, Ryan G, et al.. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res 2008; 14:4500-10; PMID:18628465; https://doi.org/ 10.1158/1078-0432.CCR-07-4262 [DOI] [PubMed] [Google Scholar]
- 117.Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, Quist MJ, Davis LE, Huang EC, Woo PJ, et al.. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 2015; 21:555-9; PMID:25939062; https://doi.org/ 10.1038/nm.3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Muscat A, Popovski D, Jayasekara WS, Rossello FJ, Ferguson M, Marini KD, Alamgeer M, Algar EM, Downie P, Watkins DN, et al.. Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of malignant rhabdoid tumors. Clin Cancer Res 2016; 22:3560-70; PMID:26920892; https://doi.org/ 10.1158/1078-0432.CCR-15-2260 [DOI] [PubMed] [Google Scholar]
- 119.Bagcchi S. Panobinostat active against diffuse intrinsic pontine glioma. Lancet Oncol 2015; 16:e267; PMID:25971867; https://doi.org/ 10.1016/S1470-2045(15)70230-5 [DOI] [PubMed] [Google Scholar]
- 120.Drappatz J, Lee EQ, Hammond S, Grimm SA, Norden AD, Beroukhim R, Gerard M, Schiff D, Chi AS, Batchelor TT, et al.. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J Neurooncol 2012; 107:133-8; PMID:21984064; https://doi.org/ 10.1007/s11060-011-0717-z [DOI] [PubMed] [Google Scholar]
- 121.Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, Schwerkoske J, Mazurczak M, Gross H, Pajon E, et al.. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol 2012; 14:215-21; PMID:22090453; https://doi.org/ 10.1093/neuonc/nor198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sawa H, Murakami H, Kumagai M, Nakasato M, Yamauchi S, Matsuyama N, Tamura Y, Satone A, Ide W, Hashimoto I, et al.. Histone deacetylase inhibitor, FK228, induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo. Acta Neuropathol 2004; 107:523-31; PMID:15024582; https://doi.org/ 10.1007/s00401-004-0841-3 [DOI] [PubMed] [Google Scholar]
- 123.Barker CA, Bishop AJ, Chang M, Beal K, Chan TA. Valproic acid use during radiation therapy for glioblastoma associated with improved survival. Int J Radiat Oncol Biol Phys 2013; 86:504-9; PMID:23523186; https://doi.org/ 10.1016/j.ijrobp.2013.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Weller M, Gorlia T, Cairncross JG, van den Bent MJ, Mason W, Belanger K, Brandes AA, Bogdahn U, Macdonald DR, Forsyth P, et al.. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 2011; 77:1156-64; PMID:21880994; https://doi.org/ 10.1212/WNL.0b013e31822f02e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med 2011; 17:330-9; PMID:21386836; https://doi.org/ 10.1038/nm.2305 [DOI] [PubMed] [Google Scholar]
- 126.Wadhwa E, Nicolaides T. Bromodomain inhibitor review: bromodomain and extra-terminal family protein inhibitors as a potential new therapy in central nervous system tumors. Cureus 2016; 8:e620; PMID:27382528; https://doi.org/ 10.7759/cureus.620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al.. Selective inhibition of BET bromodomains. Nature 2010; 468:1067-73; PMID:20871596; https://doi.org/ 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov 2014; 13:337-56; PMID:24751816; https://doi.org/ 10.1038/nrd4286 [DOI] [PubMed] [Google Scholar]
- 129.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al.. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011; 146:904-17; PMID:21889194; https://doi.org/ 10.1016/j.cell.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al.. Suppression of inflammation by a synthetic histone mimic. Nature 2010; 468:1119-23; PMID:21068722; https://doi.org/ 10.1038/nature09589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al.. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011; 478:529-33; PMID:21964340; https://doi.org/ 10.1038/nature10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan M, Brusq JM, Mordaunt JE, Grimes RM, Crowe M, Pineau O, et al.. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem 2013; 56:7501-15; PMID:24015967; https://doi.org/ 10.1021/jm401088k [DOI] [PubMed] [Google Scholar]
- 133.Berenguer-Daize C, Astorgues-Xerri L, Odore E, Cayol M, Cvitkovic E, Noel K, Bekradda M, MacKenzie S, Rezai K, Lokiec F, et al.. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer 2016; 139:2047-55; PMID:27388964; https://doi.org/ 10.1002/ijc.30256 [DOI] [PubMed] [Google Scholar]
- 134.Henssen A, Thor T, Odersky A, Heukamp L, El-Hindy N, Beckers A, Speleman F, Althoff K, Schafers S, Schramm A, et al.. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013; 4:2080-95; PMID:24231268; https://doi.org/ 10.18632/oncotarget.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, Graham RM, Allen B, Sarkaria JN, Komotar RJ, et al.. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 2014; 9:611-20; PMID:24496381; https://doi.org/ 10.4161/epi.27906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, Thompson RC, Muller S, Knapp S, Wang J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res 2013; 19:1748-59; PMID:23403638; https://doi.org/ 10.1158/1078-0432.CCR-12-3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pastori C, Kapranov P, Penas C, Peschansky V, Volmar CH, Sarkaria JN, Bregy A, Komotar R, St Laurent G, Ayad NG, et al.. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc Natl Acad Sci U S A 2015; 112:8326-31; PMID:26111795; https://doi.org/ 10.1073/pnas.1424220112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nervi C, De Marinis E, Codacci-Pisanelli G. Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenetics 2015; 7:127; PMID:26692909; https://doi.org/ 10.1186/s13148-015-0157-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR, et al.. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol 2009; 27:2052-8; PMID:19307505; https://doi.org/ 10.1200/JCO.2008.19.0694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et al.. Kruidenier et al. reply. Nature 2014; 514:E2; PMID:25279927; https://doi.org/ 10.1038/nature13689 [DOI] [PubMed] [Google Scholar]
- 141.Heinemann B, Nielsen JM, Hudlebusch HR, Lees MJ, Larsen DV, Boesen T, Labelle M, Gerlach LO, Birk P, Helin K. Inhibition of demethylases by GSK-J1/J4. Nature 2014; 514:E1-2; PMID:25279926; https://doi.org/ 10.1038/nature13688 [DOI] [PubMed] [Google Scholar]
- 142.Albert M, Schmitz SU, Kooistra SM, Malatesta M, Morales Torres C, Rekling JC, Johansen JV, Abarrategui I, Helin K. The histone demethylase Jarid1b ensures faithful mouse development by protecting developmental genes from aberrant H3K4me3. PLoS Genet 2013; 9:e1003461; PMID:23637629; https://doi.org/ 10.1371/journal.pgen.1003461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013; 502:489-98; PMID:24153302; https://doi.org/ 10.1038/nature12752 [DOI] [PubMed] [Google Scholar]
- 144.Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 2012; 338:349-54; PMID:22936566; https://doi.org/ 10.1126/science.1226339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Etchegaray JP, Lee C, Wade PA, Reppert SM. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 2003; 421:177-82; PMID:12483227; https://doi.org/ 10.1038/nature01314 [DOI] [PubMed] [Google Scholar]