

Abstract
Rapid species radiation due to adaptive changes or occupation of new ecospaces challenges our understanding of ancestral speciation and the relationships of modern species. At the molecular level, rapid radiation with successive speciations over short time periods—too short to fix polymorphic alleles—is described as incomplete lineage sorting. Incomplete lineage sorting leads to random fixation of genetic markers and hence, random signals of relationships in phylogenetic reconstructions. The situation is further complicated when you consider that the genome is a mosaic of ancestral and modern incompletely sorted sequence blocks that leads to reconstructed affiliations to one or the other relative, depending on the fixation of their shared ancestral polymorphic alleles. The laurasiatherian relationships among Chiroptera, Perissodactyla, Cetartiodactyla, and Carnivora present a prime example for such enigmatic affiliations. We performed whole-genome screenings for phylogenetically diagnostic retrotransposon insertions involving the representatives bat (Chiroptera), horse (Perissodactyla), cow (Cetartiodactyla), and dog (Carnivora), and extracted among 162,000 preselected cases 102 virtually homoplasy-free, phylogenetically informative retroelements to draw a complete picture of the highly complex evolutionary relations within Laurasiatheria. All possible evolutionary scenarios received considerable retrotransposon support, leaving us with a network of affiliations. However, the Cetartiodactyla–Carnivora relationship as well as the basal position of Chiroptera and an ancestral laurasiatherian hybridization process did exhibit some very clear, distinct signals. The significant accordance of retrotransposon presence/absence patterns and flanking nucleotide changes suggest an important influence of mosaic genome structures in the reconstruction of species histories.
The most challenging evolutionary diversification process for phylogenetic reconstructions is rapid radiation with fast speciation. Despite many attempts at resolution and growing amounts of whole-genome data, various rapidly radiating groups remain “anomaly zones” for deciphering phylogenetic relationships. In the worst case, these attempts lead to different, mutually exclusive, bifurcating trees depending on the chosen methods, sampling, or genomic areas analyzed. Such zones exist for deep coalescing animal groups (e.g., the early evolution of placentals ∼120 million years ago [mya]) (Churakov et al. 2009; Nishihara et al. 2009) as well as for more recent speciations (e.g., ursine bears 2–3 mya) (Kutschera et al. 2014).
With our analyses of increasing quantities of genomic information over the last decade, it became clear that the idea of unique bifurcating trees oversimplifies often complex ancestral demographic processes. Accumulating reports of discordance between gene and species trees indicate tangled evolutionary histories characterized by character conflicts due to phylogenetic hemiplasy (Avise and Robinson 2008) rather than to methodical errors or differences in taxonomic sampling (Bapteste et al. 2013).
Relationships among laurasiatherian orders present the most famous and still enigmatic example of controversial phylogenetic reconstructions among mammals. Laurasiatherians separated ∼81 mya from other boreotherians (Hallström and Janke 2010) and subsequently spread over the supercontinent of Laurasia. The superorder includes the six orders Eulipotyphla, Chiroptera, Perissodactyla, Cetartiodactyla, Carnivora, and Pholidota. Currently, only the basal position of Eulipotyphla (Murphy et al. 2001; Nishihara et al. 2006; Meredith et al. 2011) and the sister-group relationships of Carnivora and Pholidota (Murphy et al. 2001; Doronina et al. 2015) are well established. Close to the Cretaceous-Paleogene mass extinction (73–70 mya), Chiroptera, Perissodactyla, Cetartiodactyla, and Carnivora diversified from a common ancestor over a period of only 3 million years (Hallström and Janke 2010). The short speciation periods challenge the ability of polymorphic markers to be fixed in the population, and the existing variability beyond successive speciation events leads to incomplete lineage sorting (ILS). Many hopeful attempts have been made to reconstruct strict bifurcating phylogenetic trees for Laurasiatheria. Accordingly, all possible variants of tree topologies can be recovered in the literature. However, the sister group relationships of Perissodactyla and Carnivora (Zooamata clade) have received the most frequent support from both mitochondrial (Arnason et al. 2002) and large sets of nuclear data (Nery et al. 2012; Song et al. 2012). A competing hypothesis claiming the monophyly of Perissodactyla plus Cetartiodactyla (Euungulata clade) is supported by genome-wide nuclear studies (Zhou et al. 2012). Based on mitochondrial DNA (Arnason et al. 2002), combined mitochondrial and nuclear DNA (Murphy et al. 2001), and a large nuclear data set (Tsagkogeorga et al. 2013), Chiroptera is often placed as a second basal laurasiatherian group after Eulipotyphla, as a sister group to the Fereuungulata clade (Perissodactyla+Cetartiodactyla+Carnivora). However, some molecular data show various other results as well (e.g., the Chiroptera/Cetartiodactyla clade [Nery et al. 2012] or the weakly supported Chiroptera/Carnivora clade [Matthee et al. 2007]).
Nishihara et al. (2006) presented a pioneering work in laurasiatherian phylogenetic tree reconstruction based on retroelements. Shared retrotransposon insertions at orthologous genomic positions in different orders indicate their inheritance via a common ancestry. Unfortunately, only a limited number of phylogenetically informative retrotransposon presence/absence markers (long interspersed elements, LINEs, L1 elements) were accessible at that time. They found four such markers merging Chiroptera, Perissodactyla, and Carnivora in one monophyletic group (Pegasoferae), but they also extracted one confounding marker merging Cetartiodactyla, Perissodactyla, and Carnivora.
Hallström et al. (2011) found 11 retrotransposon markers that provide evidence for a common ancestor of the various laurasiatherian orders. However, the group Pegasoferae was only moderately supported (three markers), and insertions supporting alternative phylogenetic relationships were also found (five markers). Using both sequence-based and retrotransposon-based analyses, they proposed that the phylogenetic relationships of laurasiatherian orders do not represent a simple bifurcating pattern, but rather a network. However, the question remains whether the early speciation process of Laurasiatheria left behind some traces of affiliations inside the network.
Recently it was proposed that genomes are composed of a mosaic of alleles or haplotype blocks of ∼5,000–200,000 nt from different times of origin and ancestors (Pääbo 2003). Therefore, different histories can be embedded in the genome, and depending on the region analyzed, can provide one or the other topology in tree reconstruction (e.g., 23% of the human genome does not show that chimpanzee is our closest relative) (Ebersberger et al. 2007). Thus, in clades with rapidly diversifying species and exposed mosaic genome structures, it is essential to conduct an exhaustive, genome-wide analysis and to use virtually homoplasy-free phylogenetic markers to overcome the incongruence between gene trees and species trees or to reveal a species network rather than a single bifurcating species tree.
Retrotransposons as phylogenetic markers proved to be a powerful source to examine controversial phylogenetic relationships (Shimamura et al. 1997; Shedlock and Okada 2000), to quantify ILS, and to filter out signals buried in the noise (Takahashi et al. 2001; Shedlock et al. 2004; Kuritzin et al. 2016). The retrophylogenomic approach was successfully applied to many rapidly diversifying groups of organisms (Churakov et al. 2009; Nishihara et al. 2009; Doronina et al. 2015; Suh et al. 2015). Nevertheless, all previous attempts to analyze the phylogenetic relationships within Laurasiatheria using retroelements provided confounding results (see also Fig. 4 in Gatesy et al. 2016). Due to their lack of extensive genomic data (Nishihara et al. 2006) or restriction of screening for insertions to only short introns (Nishihara et al. 2006; Hallström et al. 2011), neither of these studies was based on substantial amounts of data. In the present study, we performed exhaustive multigenome and multidirectional screening of phylogenetically diagnostic retrotransposon insertions to investigate the phylogenetic network of laurasiatherian orders and to explore relationships between the Chiroptera, Perissodactyla, Cetartiodactyla, and Carnivora orders.
Results
We derived two-way genome alignments and performed a genome-wide, multidirectional screening targeting the four laurasiatherian orders—Chiroptera, Perissodactyla, Cetartiodactyla, and Carnivora—and the subsequently manually added sequence information for the fifth order Pholidota (pangolin) and eulipotyphlan outgroup (shrew, hedgehog, or mole) (Methods). We identified a total of 162,000 retrotransposon insertions, from which we computationally extracted, under stringent conditions (Methods), 243 loci containing the following preliminary informative markers (shared by at least two of the investigated orders): 176 L1 elements, 47 long terminal repeats (LTRs), and 20 retropseudogenes. However, the short interspersed elements (SINEs) frequently used as phylogenetic markers within mammalian orders did not cross order boundaries in laurasiatherians and were therefore not suitable for the superorder Laurasiatheria phylogeny. All loci were carefully inspected manually (Methods), and those with exact orthologous insertions were taken as informative markers. This last criterion yielded 102 informative markers (76 LINEs, 23 LTRs, and three retropseudogenes).
We found retrotransposon insertions supporting all possible sister group relationships of the investigated orders (Fig. 1; Supplemental Table S1a). Four markers merged the Chiroptera and Cetartiodactyla orders, nine supported the Chiroptera–Carnivora sister group relationship, 14 were shared by Cetartiodactyla and Carnivora, 11 indicated the Perissodactyla–Carnivora sister group relationship, 11 merged Chiroptera and Perissodactyla, and 10 supported the Perissodactyla–Cetartiodactyla sister group relationship. We also found retrotransposon support for all combinations of triplets of orders. Our screen revealed 14 markers supporting Chiroptera as the most basal order, eight for Perissodactyla, nine for Cetartiodactyla, and 12 supporting Carnivora as the earliest diverged order (Fig. 1; Supplemental Table S1a).
Figure 1.
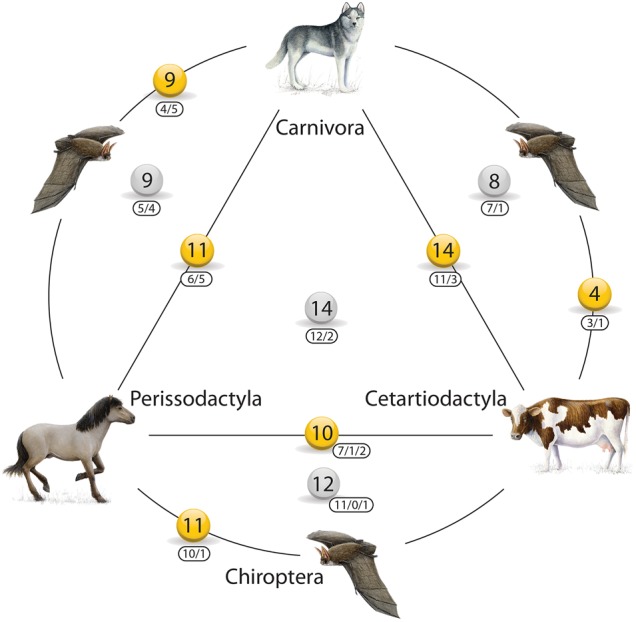
Retrotransposon-based network of the four investigated laurasiatherian orders. Numbers in balls represent the total number of retrotransposon insertions found for the respective order combination. Yellow balls represent markers merging two orders connected by the closest line. Gray balls represent markers merging the three orders arranged around the ball. The first framed number represents LINEs, the second LTRs, and the third (if present) the number of retropseudogenes.
Doronina et al. (2015) previously reported 10 shared retrotransposon insertions that significantly supported a phylogenetic association of Pholidota with Carnivora. Therefore, we did not initially include the pangolin genome in our underlying screening schema. Nevertheless, we were able to retrieve and align pangolin sequences to 144 of the 162 investigated loci. For 141 of these loci (including 92 loci from the final retrotransposon data set for SplitsTree and Dollop) the pattern of retrotransposon distribution in Pholidota was shared with Carnivora, supporting their close sister group relationship. However, three loci contained a confounding element pattern and were not included in the final retrotransposon data set for neighbor-net and parsimony analyses. These LINE insertions were present in Carnivora and some other laurasiatherian orders but clearly absent in Pholidota (Supplemental Table S1e), perhaps representing rare cases of the precise deletions of a retroelement in pangolin or reflecting the result of ILS.
However, because our genome-wide screening for diagnostic retrotransposon markers was performed under the highest possible stringency (Methods) to receive only the most reliable signals exempt of any homoplasy, and all previous screenings of diagnostic laurasiatherian markers were somehow much more relaxed or represented sequence information from different species (Nishihara et al. 2006; Hallström et al. 2011), these markers were not included among our 102 selected cases. It is worth mentioning, however, that under relaxed conditions (allowing [1] a 25-nt overlap of diagnostic elements and their flanking regions, instead of 10-nt overlaps, and [2] rejecting our strict criterion of complete 3′ regions for LINEs and searching for any parts of LINEs), we did retrieve seven of the previously published markers (Nishihara et al. 2006; Hallström et al. 2011; Supplemental Table S2).
In an additional screening for the common ancestry of the four investigated lineages (Chiroptera, Perissodactyla, Cetartiodactyla, Carnivora), we revealed 15 diagnostic monophyly markers and 88 markers for the higher-level monophyly of Laurasiatheria (Supplemental Table S1b).
Both neighbor-net analysis (SplitsTree) and the most parsimonious tree reconstruction (Dollop in Phylip) provided similar “tree” topologies (Fig. 2) for the four lineages in focus. We found a recurring tendency merging Carnivora and Cetartiodactyla (bootstrap support in SplitsTree 72%, in Dollop 57%) and placing Chiroptera at the basal position in our laurasiatherian tree (bootstrap support in SplitsTree 88%, in Dollop 63%). However, the neighbor-net analysis of retrotransposon presence/absence data also yielded some strong conflicting, mutually exclusive support for the Chiroptera–Perissodactyla sister group relationships (bootstrap support 77%).
Figure 2.
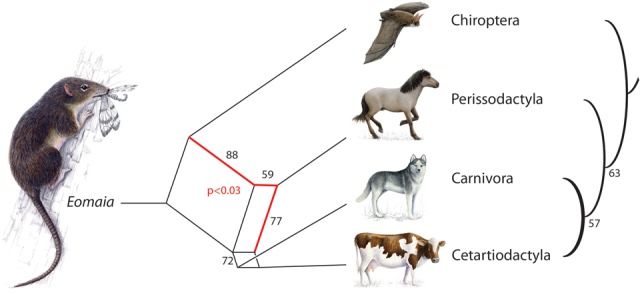
Phylogenetic network and phylogenetic tree reconstruction based on retrotransposon markers for the four investigated laurasiatherian orders. Neighbor-net (SplitsTree) analysis (left) and the most parsimonious tree reconstruction (Dollop in Phylip) (right) were conducted based on the presence/absence patterns of 102 retrotransposon insertions. Black numbers represent bootstrap values. The red number indicates the χ2 significance value from the four-lineage insertion likelihood test (Supplemental Material S1) favoring the hybridization scenario merging horse with the bat and dog or dog/cow ancestor (red lines in the SplitsTree). An imaginary reconstruction of the Eomaia, an assumed ancestor of placentals, based on a description of Ji et al. (2002) is presented at the root of the “tree.”
With an advanced and expanded mathematical diffusion model adjusted to four-lineage analyses and including hybridization scenarios (Supplemental Material S1) we found that the most likelihood binary tree topology agrees with the tree topology supported by our parsimony tree reconstruction (Fig. 2) indicating the dog/cow sister group relationship and bat as the first divergence (log L = 3.63; Hybridization model 3, binary tree); however, a χ2 test did not provide significant support for this tree topology (P > 0.1). The remaining hybridization models revealed tree topologies with significant support for two hybridization scenarios: (1) connecting the horse to the bat and dog ancestors (highest log L = 7.06, P < 0.029; Hybridization model 2); and (2) connecting the horse to the bat and a dog/cow ancestor at the point of dog/cow divergence (log L = 6.99; P < 0.031; Hybridization models 1 and 2) (for details, see Table 2 in Supplemental Material S1; Fig. 2, red lines).
To address the laurasiatherian sister group relationships using a different marker system, we performed two-way genome screens of phylogenetically informative deletions (Methods). We initially found 314,000 loci containing deletions, from which we extracted 457 potentially informative loci, and then identified 91 loci as phylogenetically informative. Similar to the data for retrotransposon markers, we found support for all possible combinations of order pairs but with different frequencies: (1) 36 markers for Cetartiodactyla+Carnivora; (2) 20 for Chiroptera+Carnivora; (3) 13 for Perissodactyla+Cetartiodactyla; (4) 12 for Chiroptera+Perissodactyla; (5) 8 for Chiroptera+Cetartiodactyla; and (6) 2 for Perissodactyla+Carnivora (Supplemental Table S3). Thus, the Cetartiodactyla–Carnivora sister group relationships again received strong support (99.9% bootstrap, neighbor-net; 96% bootstrap, Dollop) in agreement with the tree reconstructions based on retrotransposon markers.
For all included retrotransposon markers, we extracted ∼400 nt from their flanking regions and sorted and concatenated these sequences corresponding to the 10 different order affiliations derived from the presence/absence markers. Interestingly, for nine of the 10 combinations of affiliations, the neighbor-net analysis revealed the same phylogeny as previously shown for the retrotransposon presence/absence patterns (bootstrap >73%); in eight of these nine combinations, the support was very strong (bootstrap > 90%) (Supplemental Table S4). The neighbor-net analysis of the remaining concatenated data set, corresponding to the Chiroptera–Cetartiodactyla affiliation, shows their connection with a bootstrap support of just 57.5% (Supplemental Table S4).
An analysis of the chromosomal distribution of retrotransposon markers did not reveal any “phylogenetic” clusters of retroelement locations in the derived ideogram, showing that markers are distributed randomly across the entire genome (as exemplified for the dog ideogram) (Fig. 3).
Figure 3.

Ideogram representing the genomic locations of the 102 retrotransposon markers in the dog genome. The numbers to the left of the chromosomes represent the order pairs/triplets that are supported and serial number of the marker in that group. Six duplicated regions (but each with consensus presence/absence patterns) are indicated with lowercase letters (a–f). In addition, the colors of the numbers indicate the order pair/triplet affiliation to which the marker belongs, according to the legend at the bottom (Supplemental Table S1a): (Chi) Chiroptera, (Cet) Cetartiodactyla, (Car) Carnivora, (Per) Perissodactyla. The dog chromosome ideogram is based on the DAPI banded karyotype presented in Breen et al. (1999).
Discussion
With the continuously increasing amounts of genomic data accumulating today, new waves of discovering the evolutionary histories of many groups of organisms arise, but also reveal more and more incidences of ILS, challenging our phylogenetic perceptions. The extent of ILS can vary over a wide range. In certain lineages, ILS simply produces some background noise, while the phylogenetic signal remains dominant (e.g., Kutschera et al. 2014; Doronina et al. 2015). However, in other lineages, ILS leads to phylogenetic networks (e.g., Churakov et al. 2009; Nishihara et al. 2009; Suh et al. 2015), in which neither increased sampling nor application of different analyses help to reconstruct a uniform bifurcating species tree.
Laurasiatheria is a special group, in which confounding phylogenetic reconstructions have been the norm, leading to the consensus that it underwent a convoluted course of evolutionary speciations. Our retrotransposon presence/absence data contribute considerably to the current view of such a network connecting Chiroptera–Perissodactyla–Cetartiodactyla–Carnivora (Hallström et al. 2011). All 10 possible variants of relationships of pairs and triplets of orders found clear support from retrotransposon markers. Excitingly, and in agreement with data collected in birds (Matzke et al. 2012; Suh et al. 2015), ILS apparently occurred over a broad range of hierarchies of relationships within Laurasiatheria also. It occurred not only in terminal orders such as carnivores (Doronina et al. 2015) and was not only responsible for the confounding reconstructions of affiliations among the orders investigated here (Chiroptera, Perissodactyla, Cetartiodactyla, and Carnivora), but also arose in well-defined early divergences such as Eulipotyphla (in addition to 15 diagnostic markers for Chiroptera+Perissodactyla+Cetartiodactyla+Carnivora monophyly and 102 markers separating Eulipotyphla from other laurasiatherians, 13 cases supported relationships with various positions of Eulipotyphla inside Laurasiatheria).
Ebersberger et al. (2007) hypothesized that the structure of a genome is a composition of different haplotype blocks, following different genealogies (not necessarily according to the lineage evolutionary history). Incompletely sorted markers accumulating at different historical speciation points may lead to a mosaic of relationships in present reconstructions. Our retrotransposon data provide a virtually homoplasy-free illustration of such mosaics in Laurasiatheria. In our case, 102 diagnostic retrotransposons were randomly distributed across the genome (Fig. 3), rather than being spatially linked by their different phylogenetic topologies. We also investigated the sequence regions flanking our retrotransposon markers to determine whether the retrotransposons were embedded in phylogenetic mosaic units with consistent phylogenetic signals according to the flanking sequences. For nine of the 10 possible order combinations, a neighbor-net analysis (SplitsTree) of retrotransposon flanking concatenated sequences represented the same order relationships as given by the presence/absence information of the retrotransposons. The flanks of the tenth group (Chiroptera+Cetartiodactyla) represented only a slight signal for a mosaic consensus relationship. Because this was the group with the lowest number of supportive retrotransposon markers (four markers, with a length of concatenated flanking regions of 1600 nt), the weak support is possibly a result of random sequence variation. Hormozdiari et al. (2013) also found mostly consistent phylogenies for retrotransposon presence/absence patterns and flanking sequence signals in great apes. Of the tested retrotransposon loci, 84% revealed the same phylogenetic signals as the flanking sequences. They considered that this result underscores the virtually homoplasy-free origin of the studied retrotransposon insertions (free of random parallel insertions or deletions). Both studies provide significant evidence that ILS, rather than homoplasy, is the predominant reason for confounding presence/absence patterns of retrotransposon insertions and emphasize the mosaic structure of their historical signals in genomes.
Nevertheless, a closer phylogenetic examination of the laurasiatherian retrotransposon markers did reveal support for Chiroptera as the first divergence among the four investigated lineages (Dollop, four-lineage insertion likelihood test), followed by Perissodactyla (Dollop) and a Cetartiodactyla–Carnivora sister group relationship (Dollop and neighbor-net analyses, four-lineage insertion likelihood test) (Fig. 2; Supplemental Material S1). The basal position of Chiroptera among the four investigated orders, and correspondingly the monophyly of the Fereuungulata clade comprising Perissodactyla, Cetartiodactyla, and Carnivora, was also shown in some early studies (Pumo et al. 1998; Murphy et al. 2001; Waddell et al. 2001; Arnason et al. 2002) and found support in later studies of large data sets (Hallström et al. 2011; Song et al. 2012; Zhou et al. 2012). However, in one analysis, we found a possible Chiroptera–Perissodactyla order affiliation (neighbor-net analysis) (Fig. 2) that previously also found some support in nuclear+mitochondrial data set analysis (Murphy et al. 2007). The sister group relationships of Cetartiodactyla and Carnivora is a surprising result in disagreement with both the Zooamata (Perissodactyla–Carnivora) (Murphy et al. 2001; Nery et al. 2012) and Euungulata (Perissodactyla–Cetartiodactyla) (Waddell et al. 2001; Zhou et al. 2012) hypotheses. Only Prasad et al. (2008) presented tree reconstructions with a slight support of a Cetartiodactyla–Carnivora sister group relationship. Our test of this surprising Cetartiodactyla plus Carnivora signal with diagnostic random deletions as an alternative marker system confirmed this relationship. We propose the name “Cetartioferae” for this clade with the caveat that this group was not significantly supported by the hybridization models. We were lucky perhaps to detect this signal in rare genomic changes owing to their virtually homoplasy-free nature, nearly completely devoid of phylogenetic noise (Ray et al. 2006). However, it should be mentioned that despite their rarity, homoplasious retroposon markers cannot be completely excluded (e.g., Doronina et al. 2015). In addition, according to our analyses, the early diversification of laurasiatherians was accompanied by ancestral hybridization. The expanded four-lineage insertion likelihood test indicated that the horse is a result of fusion between the ancestral population of the bat and a dog or dog/cow ancestor. It should be considered that the ancestral laurasiatherian lineages diverged in a rapid succession, and that hybridization is a viable scenario under such conditions (Fig. 2, red lines).
Among the possible three-order combinations, we found nine markers supporting Pegasoferae, a clade merging Chiroptera, Perissodactyla, and Carnivora, but excluding Cetartiodactyla (Fig. 2; Nishihara et al. 2006, left species triplet), and 8, 12, and 14 markers supporting the other three three-order combinations. Interestingly, the analysis of our data that considered the possibility of ancestral hybridization indicated some affiliation between dog, horse, and bat ancestors that is somewhat similar to Pegasoferae. However, for our data, the two significantly supported hybridization scenarios and all binary tree reconstructions suggest that Chiroptera diverged earlier than Carnivora and Cetartiodactyla.
This is a good example of how important it is to apply whole genome–level analyses in complex, confounding zones of ancestral speciations. The heterogeneous genomic signals representing a legacy from a time of rapid speciation, incomplete lineage sorting of once polymorphic markers, and ancestral hybridization inevitably led to the confounding patterns of phylogenetic signals in modern Laurasiatheria. Thus, our extensive, whole-genome screens of the virtually homoplasy-free retrotransposon and deletion phylogenetic markers enabled us not only to reconstruct a homoplasy-free speciation network of laurasiatherian phylogenetic relationships but also to find some hidden phylogenetic signals that possibly resolve the early evolution of laurasiatherian orders. The detected mosaic nature of the order affiliations reflects the basic mosaic blocks of different sequence regions at the genomic level.
Methods
In this in silico high-throughput bioinformatics screening of derived two-way genome alignments from representatives of Chiroptera (bat), Perissodactyla (horse), Cetartiodactyla (cow), and Carnivora (dog), we mainly focused on two retrotransposon classes known to be active during the early laurasiatherian radiation, LINEs (L1s) and LTRs, and also compared phylogenetic sequence signals from their flanking regions. In addition, we conducted a genome-wide screening of phylogenetically informative deletions.
To test all 10 possible order affiliations, we screened for the following presence (+)/absence (−) patterns to determine possible sister group relationships: (1) +bat+cow−horse−dog, (2) +bat+dog−horse−cow, (3) +cow+dog−bat−horse, (4) +horse+dog−bat−cow, (5) +bat+horse−cow−dog, (6) +horse+cow−bat−dog. We also screened for retrotransposon loci merging triplets of orders: (1) +bat+cow+dog−horse, (2) +bat+horse+dog−cow, (3) +horse+cow+dog−bat, (4) +bat+horse+cow−dog.
To further verify previous data of Eulipotyphla being the first diverged laurasiatherian group (e.g., Murphy et al. 2001), we screened the aforementioned two-way genome alignments for the pattern +bat+horse+dog+cow, and subsequently manually aligned and checked representative eulipotyphlan sequences (shrew, hedgehog, or mole). To collect markers for laurasiatherian monophyly, we again queried two-way genome alignments, including one with the shrew genome, to search for the pattern +shrew+bat+horse+dog+cow, and then again manually checked the outgroup representatives.
Searching for informative retrotransposons
We performed an exhaustive, genome-wide screen for the genomic coordinates of phylogenetically informative retroelements and flanking regions based on custom-designed two-way alignments derived as described in Kent et al. (2003) and Hartig et al. (2013). We used the two-way genome alignments from the UCSC Genome Bioinformatics Center in Santa Cruz (Supplemental Material S2a). The bat (Myotis lucifugus), horse (Equus caballus), cow (Bos taurus), dog (Canis lupus familiaris), and shrew (Sorex araneus) RepeatMasker reports were downloaded from the sources described in Supplemental Material S2b.
We extracted coordinates of presence/absence patterns representing all combinations of the four tested species from two-way alignments corresponding to Doronina et al. (2015) and correlated these patterns with retrotransposon coordinates from the downloaded RepeatMasker reports. In total, we detected around 162,000 retrotransposon insertions. For these insertions, we applied stringent inclusion criteria, allowing only full-length LTR sequences (not more than 10-nt truncations from both sides) and only complete 3′ regions for LINEs (not more than 25-nt truncations from the 3′-end of L1 consensus sequences). For preanalysis, we considered loci to be potentially informative if at least 70% of the retroelement aligned to a gap in other species and if the elements did not overlap by more than 10 nt at both flanking sequences.
Based on the detected genomic coordinates, we extracted sequences with additional ∼500-nt flanking nucleotides of the 243 potentially informative loci from the genome sources described in Supplemental Material S2c.
The sequences of potentially informative loci were manually aligned and carefully inspected. The insertion orthology for each locus was defined by checking the element type and orientation, the exact position of insertion and target site duplications, and for L1 elements, the exact 5′ truncation point of the insertions. The last criterion was used as an additional diagnostic indication of orthologous insertions. Two species with the identical 5′ truncation of an inserted LINE most probably acquired this element from a common ancestor. Applying these criteria led to the exclusion of 77 of the 243 loci.
Using the National Center for Biotechnology Information (NCBI) BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and UCSC Genome Browser Database BLAT (http://genome.ucsc.edu/cgi-bin/hgBlat), we supplemented the alignments with at least one additional representative of Chiroptera, Perissodactyla, Cetartiodactyla, and Carnivora, with one representative of Eulipotyphla (Sorex araneus, Erinaceus europaeus, or Condylura cristata), and as far as available, complemented by pangolin genome information (Manis pentadactyla or Manis javanica). For each locus, two representatives from the following outgroups were added: Euarchontoglires, Afrotheria, and Xenarthra (Supplemental Tables S1a, S5). We selected only cases with support from two independent species per order (Chiroptera, Perissodactyla, Cetartiodactyla, and Carnivora) representing two different families to reduce the probability of falsely including homoplasious insertions into our analysis. Based on this criterion, four markers were excluded, because the Perissodactyla were represented only by horse, and the corresponding genome information for the white rhinoceros was not available, leaving us with 162 markers (Supplemental Table S1; Supplemental Materials S1, S3). In addition, the conflicting presence/absence pattern in pangolin (three loci) and the cases with missing sequences in the basal Eulipotyphla (44 loci) or incompatible presence patterns in Eulipotyphla (13 loci) were also excluded from our final data set for the neighbor-net analysis (SplitsTree) and the most parsimonious tree reconstruction (Dollop in Phylip), leaving us with a final total of 102 markers (Supplemental Table S1a; Supplemental Material S3a).
Additional markers for the “+bat+horse+dog+cow” pattern were manually supplemented by one of the eulipotyphlan species (S. araneus, E. europaeus, or C. cristata) and one outgroup representative from Euarchontoglires, Afrotheria, or Xenarthra. Only insertions showing the “-eulipotyphlan species +bat+horse+dog+cow” presence/absence pattern were taken as informative markers for the Eulipotyphla phylogenetic position within laurasiatherian orders (Supplemental Table S1b). Markers for laurasiatherian monophyly (+shrew+bat+horse+dog+cow pattern) were manually supplemented by human (Homo sapiens) sequences, and insertions with the “-human+shrew+bat+horse+dog+cow” pattern were taken as phylogenetically informative (Supplemental Table S1b).
Phylogenetic analysis
We built a presence/absence (1/0) data matrix for retrotransposon markers (Supplemental Table S1c) and reconstructed the most parsimonious tree using Dollop (Polymorphism Parsimony Program) in PHYLIP (Felsenstein 1989) version 3.695, applying standard parameters with a randomized input order of species (seven times to jumble, random seed “13131”). For each locus, data of two outgroups were pooled to create a single synthetic outgroup.
We also performed neighbor-net analysis of a presence/absence (1/0) data matrix in SplitsTree (version 4.13.1) (Huson and Bryant 2006) using the standard settings for net reconstruction and bootstrap analysis.
Because the KKSC insertion significance test of presence/absence data was originally designed for three-lineage relationships only (Kuritzin et al. 2016), it was not directly applicable to the four-lineage laurasiatherian comparison. Thus, we extended the previously derived mathematical diffusion model (Kuritzin et al. 2016) to find the tree topology with the most log-likelihood value and included different hybridization scenarios. We then used a χ2 approximation of log-likelihood ratios as proposed by Waddell et al. (2001), taking into account the degrees of freedom for the four-lineage tree topology to indicate the significance of the most probable tree. This expanded statistical model is available as Supplemental Material S1.
Sequence analysis of flanking regions
To compare the phylogenetic signals of the retrotransposon presence/absence data with phylogenetic signals of nucleotide changes in their flanking regions, we constructed concatenated alignments for each of the 10 possible groups of retrotransposon-derived affiliations (the 10 different tree topologies pooled in Fig. 1), representing the different competing relationships in Laurasiatheria. For each retrotransposon locus, sequences of ∼400 nt flanking the retroelement marker (for species with the diagnostic insertion) or empty insertion sites (species without the insertion) were extracted for all investigated species. The alignments of extracted sequences were concatenated for each of the 10 possible groups of affiliations (tree topologies) separately using the perl script “catfasta2phyml.pl” (https://github.com/nylander/catfasta2phyml). All additional repetitive elements in flanking regions not present in all analyzed species were removed from the concatenated data sets.
In SplitsTree (version 4.13.1) (Huson and Bryant 2006), neighbor-net analyses of the 10 concatenated alignments were performed using the standard settings for net reconstruction and bootstrap analyses (Supplemental Table S4; Supplemental Material S4).
Screening and analysis of informative deletions
To compare the retrotransposon presence/absence results of laurasiatherian sister group relationships to an independent data set, we screened two-way genome alignments for informative deletions in all six possible groups of order pairs. We screened for presence/absence patterns of deletions in two species with lengths in the range of 75–750 nt. We extracted the detected loci and checked for interfering repetitive sequences with a local RepeatMasker (http://www.repeatmasker.org/RepeatMasker-open-4-0-5.tar.gz) run. In the area of the deletion (in species with presence state), low complexity regions and repetitive elements occupying >20% of the deleted region were not included in potentially informative cases. The detected coordinates were used to extract genomic sequences using the same procedure as for retroelements. Potentially informative cases were carefully manually aligned and inspected. Species composition and outgroup selection are described above (Supplemental Table S3; Supplemental Material S5).
Data access
All alignments from Supplemental Material S3–S5 are available from the Dryad Digital Repository (http://datadryad.org/) under doi 10.5061/dryad.71s06.
Supplementary Material
Acknowledgments
We thank Marsha Bundman for editorial support and Jón Baldur Hlíðberg for the paintings of the bat, horse, dog, cow, and the restoration of the extinct Eomaia (in Figs. 1, 2). This publication was financially supported by the Deutsche Forschungsgemeinschaft (SCHM1469/3-2, SCHM1469/10-1).
Author contributions: L.D., G.C., and J. Schmitz designed the study. R.B. and H.C. built the two-way whole-genome alignments. G.C. performed the computational screenings of two-way alignments and extracted diagnostic markers. A.K. and G.C. developed the four-lineage insertion likelihood test. L.D. performed all manual alignments and carried out the data analyses. J. Shi assisted in manually aligning informative loci. L.D. and J. Schmitz wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.210948.116.
References
- Arnason U, Adegoke JA, Bodin K, Born EW, Esa YB, Gullberg A, Nilsson M, Short RV, Xu X, Janke A. 2002. Mammalian mitogenomic relationships and the root of the eutherian tree. Proc Natl Acad Sci 99: 8151–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avise JC, Robinson TJ. 2008. Hemiplasy: a new term in the lexicon of phylogenetics. Syst Biol 57: 503–507. [DOI] [PubMed] [Google Scholar]
- Bapteste E, van Iersel L, Janke A, Kelchner S, Kelk S, McInerney JO, Morrison DA, Nakhleh L, Steel M, Stougie L, et al. 2013. Networks: expanding evolutionary thinking. Trends Genet 29: 439–441. [DOI] [PubMed] [Google Scholar]
- Breen M, Bullerdiek J, Langford CF. 1999. The DAPI banded karyotype of the domestic dog (Canis familiaris) generated using chromosome-specific paint probes. Chromosome Res 7: 401–406. [DOI] [PubMed] [Google Scholar]
- Churakov G, Kriegs JO, Baertsch R, Zemann A, Brosius J, Schmitz J. 2009. Mosaic retroposon insertion patterns in placental mammals. Genome Res 19: 868–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronina L, Churakov G, Shi J, Brosius J, Baertsch R, Clawson H, Schmitz J. 2015. Exploring massive incomplete lineage sorting in arctoids (Laurasiatheria, Carnivora). Mol Biol Evol 32: 3194–3204. [DOI] [PubMed] [Google Scholar]
- Ebersberger I, Galgoczy P, Taudien S, Taenzer S, Platzer M, von Haeseler A. 2007. Mapping human genetic ancestry. Mol Biol Evol 24: 2266–2276. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1989. PHYLIP—Phylogeny inference package (version 3.2). Cladistics 5: 164–166. [Google Scholar]
- Gatesy J, Meredith RW, Janecka JE, Simmons MP, Murphy WJ, Springer MS. 2016. Resolution of a concatenation/coalescence kerfuffle: partitioned coalescence support and a robust family-level tree for Mammalia. Cladistics. 10.1111/cla.12170. [DOI] [PubMed] [Google Scholar]
- Hallström BM, Janke A. 2010. Mammalian evolution may not be strictly bifurcating. Mol Biol Evol 27: 2804–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallström BM, Schneider A, Zoller S, Janke A. 2011. A genomic approach to examine the complex evolution of laurasiatherian mammals. PLoS One 6: e28199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartig G, Churakov G, Warren WC, Brosius J, Makalowski W, Schmitz J. 2013. Retrophylogenomics place tarsiers on the evolutionary branch of anthropoids. Sci Rep 3: 1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Konkel MK, Prado-Martinez J, Chiatante G, Herraez IH, Walker JA, Nelson B, Alkan C, Sudmant PH, Huddleston J, et al. 2013. Rates and patterns of great ape retrotransposition. Proc Natl Acad Sci 110: 13457–13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Ji Q, Luo ZX, Yuan CX, Wible JR, Zhang JP, Georgi JA. 2002. The earliest known eutherian mammal. Nature 416: 816–822. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. 2003. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci 100: 11484–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuritzin A, Kischka T, Schmitz J, Churakov G. 2016. Incomplete lineage sorting and hybridization statistics for large-scale retroposon insertion data. PLoS Comput Biol 12: e1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutschera VE, Bidon T, Hailer F, Rodi JL, Fain SR, Janke A. 2014. Bears in a forest of gene trees: phylogenetic inference is complicated by incomplete lineage sorting and gene flow. Mol Biol Evol 31: 2004–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthee CA, Eick G, Willows-Munro S, Montgelard C, Pardini AT, Robinson TJ. 2007. Indel evolution of mammalian introns and the utility of non-coding nuclear markers in eutherian phylogenetics. Mol Phylogenet Evol 42: 827–837. [DOI] [PubMed] [Google Scholar]
- Matzke A, Churakov G, Berkes P, Arms EM, Kelsey D, Brosius J, Kriegs JO, Schmitz J. 2012. Retroposon insertion patterns of neoavian birds: strong evidence for an extensive incomplete lineage sorting era. Mol Biol Evol 29: 1497–1501. [DOI] [PubMed] [Google Scholar]
- Meredith RW, Janečka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, Goodbla A, Eizirik E, Simão TL, Stadler T, et al. 2011. Impacts of the cretaceous terrestrial revolution and KPg extinction on mammal diversification. Science 334: 521–524. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Eizirik E, O'Brien SJ, Madsen O, Scally M, Douady CJ, Teeling E, Ryder OA, Stanhope MJ, de Jong WW, et al. 2001. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 294: 2348–2351. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Pringle TH, Crider TA, Springer MS, Miller W. 2007. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res 17: 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery MF, Gonzalez DJ, Hoffmann FG, Opazo JC. 2012. Resolution of the laurasiatherian phylogeny: evidence from genomic data. Mol Phylogenet Evol 64: 685–689. [DOI] [PubMed] [Google Scholar]
- Nishihara H, Hasegawa M, Okada N. 2006. Pegasoferae, an unexpected mammalian clade revealed by tracking ancient retroposon insertions. Proc Natl Acad Sci 103: 9929–9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara H, Maruyama S, Okada N. 2009. Retroposon analysis and recent geological data suggest near-simultaneous divergence of the three superorders of mammals. Proc Natl Acad Sci 106: 5235–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pääbo S. 2003. The mosaic that is our genome. Nature 421: 409–412. [DOI] [PubMed] [Google Scholar]
- Prasad AB, Allard MW, Green ED. 2008. Confirming the phylogeny of mammals by use of large comparative sequence data sets. Mol Biol Evol 25: 1795–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumo DE, Finamore PS, Franek WR, Phillips CJ, Tarzami S, Balzarano D. 1998. Complete mitochondrial genome of a neotropical fruit bat, Artibeus jamaicensis, and a new hypothesis of the relationships of bats to other eutherian mammals. J Mol Evol 47: 709–717. [DOI] [PubMed] [Google Scholar]
- Ray DA, Xing J, Salem AH, Batzer MA. 2006. SINEs of a nearly perfect character. Syst Biol 55: 928–935. [DOI] [PubMed] [Google Scholar]
- Shedlock AM, Okada N. 2000. SINE insertions: powerful tools for molecular systematics. Bioessays 22: 148–160. [DOI] [PubMed] [Google Scholar]
- Shedlock AM, Takahashi K, Okada N. 2004. SINEs of speciation: tracking lineages with retroposons. Trends Ecol Evol 19: 545–553. [DOI] [PubMed] [Google Scholar]
- Shimamura M, Yasue H, Ohshima K, Abe H, Kato H, Kishiro T, Goto M, Munechika I, Okada N. 1997. Molecular evidence from retroposons that whales form a clade within even-toed ungulates. Nature 388: 666–670. [DOI] [PubMed] [Google Scholar]
- Song S, Liu L, Edwards SV, Wu S. 2012. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc Natl Acad Sci 109: 14942–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh A, Smeds L, Ellegren H. 2015. The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biol 13: e1002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Terai Y, Nishida M, Okada N. 2001. Phylogenetic relationships and ancient incomplete lineage sorting among cichlid fishes in Lake Tanganyika as revealed by analysis of the insertion of retroposons. Mol Biol Evol 18: 2057–2066. [DOI] [PubMed] [Google Scholar]
- Tsagkogeorga G, Parker J, Stupka E, Cotton JA, Rossiter SJ. 2013. Phylogenomic analyses elucidate the evolutionary relationships of bats. Curr Biol 23: 2262–2267. [DOI] [PubMed] [Google Scholar]
- Waddell PJ, Kishino H, Ota R. 2001. A phylogenetic foundation for comparative mammalian genomics. Genome Inform 12: 141–154. [PubMed] [Google Scholar]
- Zhou X, Xu S, Xu J, Chen B, Zhou K, Yang G. 2012. Phylogenomic analysis resolves the interordinal relationships and rapid diversification of the laurasiatherian mammals. Syst Biol 61: 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.